

Abstract
Whole cell patch clamping of transfected HEK293 cells was used to examine the effects of a pentapeptide (KIFMK) containing the proposed inactivation particle of the Na+ channel on two mutations causing myotonia. One mutation (R1448P) is located in the voltage sensor IV/S4, and the other one (G1306E) near the postulated inactivation gate within the III-IV linker.
In the absence of peptide, currents of wild-type (WT) and mutant human muscle Na+ channels decayed monoexponentially with inactivation time constants that were 5-fold (R1448P) and 3-fold (G1306E) larger for the mutants. Upon intracellular application of KIFMK (0·3-1 mM) the current decay became biexponential with an additional fast decaying component that increased in amplitude with depolarization.
Furthermore, the peptide induced large tail currents upon repolarization, indicating that KIFMK prevents inactivation by blocking open Na+ channels. The peak of this tail current decreased only slowly with depolarizations of increasing duration. The voltage dependence of this decline indicated that the dissociation rate of the charged peptide decreased with depolarization. Increased external [Na+] ([Na+]e) antagonized block by KIFMK, consistent with a pore-blocking mechanism.
The results are discussed with regard to a three-state model for one open, an absorbing inactivated and one blocked state with voltage-dependent on- and off-rates for peptide binding. The peptide had qualitatively similar effects on WT and both mutants, indicating that the freely diffusible peptide accelerates the current decay in all three clones. However, for the R1448P mutation the affinity for KFIMK was decreased and the voltage dependence of peptide block was changed in a similar way to the voltage dependence of inactivation. These data suggest that the mutation R1448P affects the voltage-dependent formation of a receptor site for both the inactivation particle and KIFMK.
Voltage-gated Na+ channels are the basis for the generation and conduction of action potentials in nerve and muscle cells. Na+ channels open briefly upon depolarization and then close to a fast inactivated state from which they reopen only rarely. Thus, fast inactivation limits the duration of an action potential and initiates its repolarizing phase. The α-subunit constitutes both the gating and permeation machinery of the Na+ channel, and consists of four homologous domains (I-IV), each with six transmembrane segments (S1-S6). The S4 segments contain positively charged residues conferring voltage dependence to the channel protein, the S5-S6 loops contribute to the ion channel pore, and the intracellular loop linking domains III and IV (LIII-IV) contains structures required for channel inactivation (for a review, see Catterall, 1995). A current model for the molecular mechanism of fast Na+ channel inactivation proposes that an intracellular particle within LIII-IV consisting of three hydrophobic amino acids (isoleucine, phenylalanine and methionine; IFM) occludes the channel pore in a hinged-lid fashion (West et al. 1992). Further evidence for IFM forming a pore-closing particle comes from a recent study of Kellenberger et al. (1996). These authors showed that a cysteine substituted for the phenylalanine of IFM is only accessible for the thiol reagent MTSET in the hyperpolarized state, when channels are not inactivated. Vedantham & Cannon (1998) further showed that the voltage dependence of the reaction rate for MTSET with this cysteine mutation fits perfectly the steady-state inactivation curve of the channel.
A pentapeptide containing the IFM motif (lysine-isoleucine-phenylalanine-methionine-lysine = KIFMK) blocks a non-inactivating mutant lacking the natural IFM; other peptides not containing IFM, such as KIQMK or KAFAK, do not have this effect (Eaholtz et al. 1994). These data suggest a common binding site for the natural IFM and the peptide. Tang and coworkers tested the effects of KIFMK on two different, slowly inactivating Na+ channel mutants and concluded that the peptide is an open channel blocker acting in a different way from normal inactivation (Tang et al. 1996). Whereas Eaholtz and colleagues found a voltage-dependent association and a voltage-independent dissociation rate for KIFMK binding (Eaholtz et al. 1998), Tang et al. (1996) calculated voltage-independent rate constants for both binding and unbinding of the peptide.
In order to investigate the mechanism of action of KIFMK further, we chose two myotonia-causing mutations in different regions of the channel protein; both slow inactivation to a similar extent, but probably by different mechanisms. One mutation, glycine-1306-glutamate (G1306E), causes potassium-aggravated myotonia and is located only four amino acids away from IFM within LIII-IV. This mutation might slow inactivation by hindering the movement of the putative inactivation particle (Lerche et al. 1993; Mitrovic et al. 1995; Hayward et al. 1996). The other mutation, arginine-1448-proline (R1448P), causes paramyotonia congenita and is located at the extracellular surface of the voltage sensor IV/S4 (Wang et al. 1995; Lerche et al. 1996; Featherstone et al. 1998; Mitrovic et al. 1999; for a review of the Na+ channelopathies, see Lehmann-Horn & Rüdel, 1996). This voltage sensor plays an important role in the coupling of inactivation to activation (Chahine et al. 1994) and its outward movement (Yang et al. 1996) might therefore initiate the formation of a receptor site for the inactivation particle. Thus, R1448P should slow inactivation by affecting the conformation of the receptor for the inactivation gate, whereas G1306E should hinder the gate itself.
Hence, if the natural inactivation gate and the pentapeptide KIFMK had the same binding site, R1448P but not G1306E should affect channel block by KIFMK. Indeed, we found a difference in the affinity and voltage dependence of KIFMK block of the R1448P mutation resembling the altered voltage dependence of inactivation of this mutation, whereas KIFMK block for G1306E and wild-type (WT) channels was similar. In addition, our results complement the aforementioned studies (Eaholtz et al. 1994, 1998; Tang et al. 1996) and provide new insight into the mechanism of peptide block.
METHODS
Site-directed mutagenesis of both mutants using the Altered Sites system (Promega Corporation, Madison, WI, USA) has been reported previously (Mitrovic et al. 1995, 1999). Full-length WT and mutant constructs of the human muscle Na+ channel α-subunit cDNA were assembled in the mammalian expression vector pRC/CMV and transfected permanently into HEK293 cells as described (Mitrovic et al. 1994).
Standard whole-cell recording (Hamill et al. 1981) using an EPC-7 amplifier (List, Darmstadt, Germany) was performed on stable HEK293 cell lines expressing either the WT or one of the mutant alleles. Na+ currents ranged from 1 to 5 nA. The voltage error due to series resistance was always smaller than 5 mV (60-85 % compensation). Leakage and capacitative currents were automatically subtracted by means of a prepulse protocol (-P/4). Currents were low-pass filtered at 5 or 10 kHz (8-pole Bessel, -3 dB) and digitized at 25 or 50 kHz using pCLAMP (Axon Instruments). All tail currents were filtered at 10 kHz and sampled at 50 kHz. Data were analysed by a combination of pCLAMP, Excel (Microsoft), SigmaPlot (Jandel Scientific, San Rafael, CA, USA) and our own software. Data are shown as means ±s.e.m.
The pipette solution contained (mM): 110 CsCl, 30 NaCl, 2 MgCl2, 5 EGTA and 10 Hepes (pH 7.4). The bathing solution contained: 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 4 dextrose and 5 Hepes (pH 7.4). For a reversed Na+ gradient the pipette solution contained 100 NaCl and 40 CsCl and the bathing solution 30 NaCl and 110 CsCl. To slow the kinetics all recordings were performed at 15-16°C. Temperature was controlled via a water bath.
The pentapeptide KIFMK (Eaholtz et al. 1994) was synthesized by solid phase synthesis, acetylated at the N-terminus, amidated at the C-terminus and purified by high pressure liquid chromatography (HPLC) in the Department of Virology of the University of Ulm. It was added to the pipette solution in concentrations ranging from 0.1 to 1 mM. Kinetically stable currents were recorded within a few minutes of establishment of the whole cell configuration, recordings were made after at least 10 min. For control experiments, the same conditions were applied in the absence of peptide.
RESULTS
The altered gating of the two Na+ channel mutants R1448P and G1306E in comparison with that of the WT has been described previously (Lerche et al. 1993, 1996; Mitrovic et al. 1995, 1999; Hayward et al. 1996; Featherstone et al. 1998). The most important gating changes are as follows: R1448P slows inactivation 5-fold and strongly alters its voltage dependence, induces a slightly increased persistent current, shifts the steady-state inactivation curve by -7 mV and decreases its voltage dependence. G1306E slows inactivation 3-fold, increases the persistent current and shifts the steady-state inactivation curve by +15 mV without changing its voltage dependence.
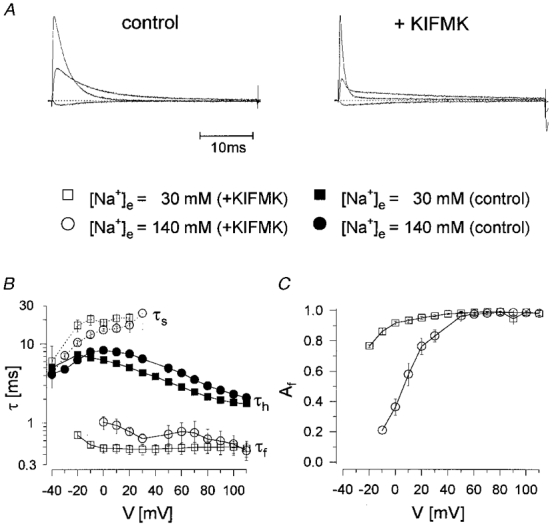
To elicit Na+ currents, cells were held at -85 mV, prepulsed to -120 mV for 300 ms and then depolarized to various test potentials. Figure 1 shows Na+ currents recorded in the absence and presence of KIFMK for WT and both mutants. At -10 mV and more hyperpolarized potentials, the peptide had little or no effect on the time course of inactivation. By contrast, for potentials more positive than -10 mV, KIFMK induced a fast decaying component to the slowly inactivating mutants that increased in amplitude with further depolarization. I-V curves were not significantly changed by the peptide, as shown in Fig. 2A and B. Currents were fitted to a Hodgkin-Huxley model, m4hx (where m and h are activation and inactivation particles, respectively; Hodgkin & Huxley, 1952). In the absence of peptide, the current was well described using a first-order exponential for inactivation, m4h, yielding a single inactivation time constant, τh. In the presence of peptide, WT currents were still well fitted to the same model throughout the whole voltage range; however, for the mutants the sum of two exponentials was required to describe the current decay at potentials more positive than -10 mV, m4h2:
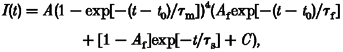 |
where τf and τs are the resulting fast and slow time constants describing the current decay, τm is the activation time constant, A is an amplitude factor, Af the relative amplitude of the fast component representing the percentage of initially blocked channels, t the time after onset of the depolarization, t0 the delay to activation of the channel and C a constant term. τm was similar for all clones and not altered by KIFMK (results not shown). All time constants describing the current decay, τh/f/s, are shown in Fig. 2C and D as a function of test potential for a peptide concentration of 0.5 mM. Both in the absence and presence of peptide, WT currents decayed most rapidly, and those of R1448P most slowly. In the presence of peptide, the fast time constant, τf, was faster and the slow one, τs, slower than τh in the absence of KIFMK. The relative amplitude of the fast component Af, in the presence of KIFMK, increased strongly with depolarization. Its voltage dependence was slightly shifted in the hyperpolarizing direction for G1306E compared with R1448P channels (Fig. 2E). Above +30 mV, the relative amplitude of the slow component was generally too small to evaluate τs accurately.
Figure 1. KIFMK accelerates the current decay.
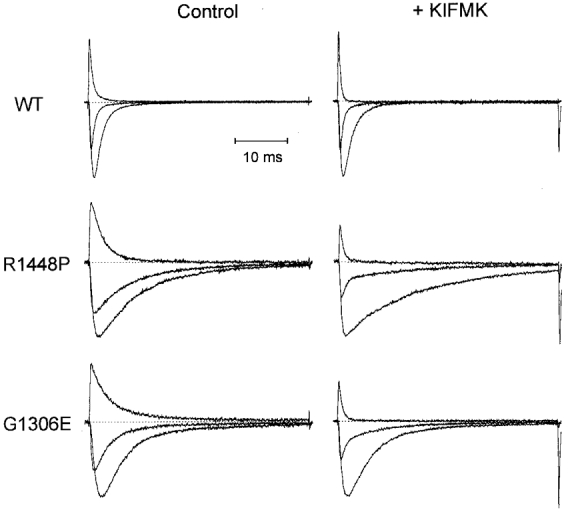
HEK cells transfected permanently with either wild-type (WT), R1448P or G1306E mutant Na+ channels were held at -85 mV, prepulsed to -120 mV for 300 ms and subsequently depolarized for 40 ms to various test potentials as indicated in Fig. 2A and B. Shown are normalized raw Na+ current traces at -10, +20 and +100 mV in the absence (Control, left panel) and presence (+ KIFMK, right panel) of peptide for WT (upper traces, 0.5 mM KIFMK), R1448P (middle traces, 1 mM KIFMK) and G1306E channels (lower traces, 0.5 mM KIFMK), respectively. Absolute current sizes at -10 mV are as follows: WT, 2.0 and 1.6 nA; R1448P, 1.2 and 1.3 nA; G1306E, 1.4 and 1.7 nA. Dotted lines indicate zero current.
Figure 2. Effects of KIFMK on inactivation kinetics.
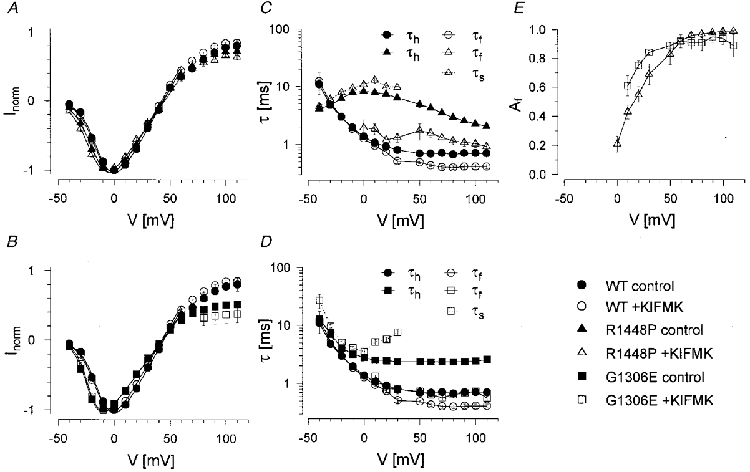
At the end of the 40 ms current traces in Fig. 1, large tail currents occur upon repolarization in the presence of peptide which are absent without KIFMK. This is shown in more detail in Fig. 3. Whereas without KIFMK, the decline of the tail current peak after prolonged depolarizations had a similar time constant as τh (Fig. 3A, upper trace), tail currents were much larger and the decay of their peaks dramatically slowed in the presence of KIFMK (Fig. 3A, lower traces). The peptide apparently prevents inactivation by blocking open Na+ channels, a ‘foot-in-the-door’ mechanism. The decay of peak tails was well fitted to a single exponential relaxation and was strongly voltage dependent. The time constants τptd (ptd = peak tail decay), are shown in Fig. 3B. In the absence of peptide, τptd reflected τh and decreased with further depolarization of the depolarizing test pulse (compare with Fig. 2C and D), whereas in the presence of peptide, τptd dramatically increased with depolarization, suggesting a voltage-dependent dissociation of KIFMK. Peak tails in the presence of peptide decayed with almost the same time constant as τs, as shown in the inset of Fig. 3. Whereas for WT and G1306E channels τptd in the presence of KIFMK increased exponentially with depolarization, the voltage dependence was markedly different for R1448P channels: τptd in the absence and presence of peptide was not significantly different up to +25 mV, and τptd increased only with further depolarization in the presence of peptide in a way similar to the two other clones.
Figure 3. Effects of KIFMK on tail currents.
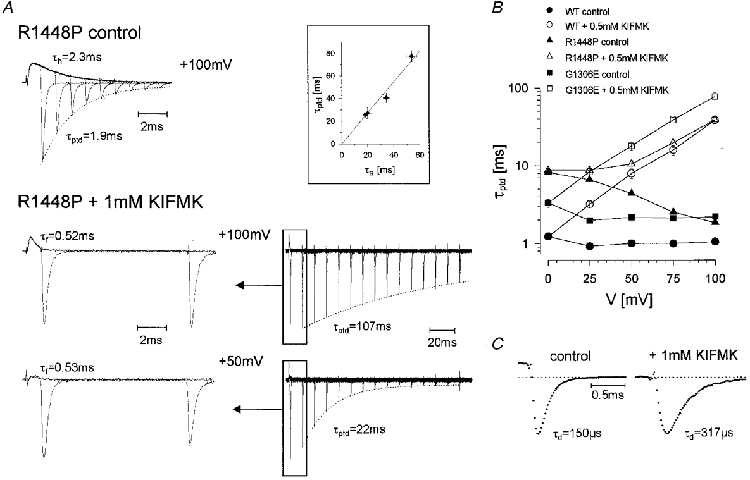
To record the decline of the tail current peak after a variable period of depolarization, cells were held at -85 mV, prepulsed to -120 mV, depolarized to various test potentials as indicated on the abcissa in B for increasing time intervals and finally repolarized to -85 mV to measure the tail current. The decay of the tail current peak with prolonged depolarizations was well fitted to a first-order exponential function yielding the time constant τptd. A, in the absence of peptide (upper trace, maximum tail current 4.5 nA), τptd was similar to τh, whereas in the presence of KIFMK (1 mM), τptd was dramatically prolonged (middle and lower traces show two different time scales for a better comparison with the upper trace; maximum tail currents 2.9 and 2.4 nA, respectively). τptd was almost identical to τs as could be shown for three cells at a reversed Na+ gradient, where currents were larger at the critical potentials (inset: peptide concentration 1 mM, voltage range 0 to +75 mV). B, voltage dependence of τptd for WT, G1306E and R1448P channels (0.5 mM KIFMK). C, tail current traces from B at a higher time resolution recorded upon repolarization from +100 to -85 mV for R1448P channels. The dotted line indicates zero current. The control current was recorded 1 ms after onset of the depolarization, while the current with KIFMK was recorded 11 ms after onset, since after 1 ms peptide block was not complete.
Tail current traces at a higher time resolution are shown in Fig. 3C for R1448P channels. Both the upstroke and the decay of the tail current were slowed by the peptide, suggesting that KIFMK interferes with the activation/deactivation gate, and that it must leave the pore before deactivation can occur. Whereas the rising phase of the tail current was not well enough resolved to be accurately fitted, its decay was well fitted to a first-order exponential function. The resulting deactivation time constants, τd, were increased about 2-fold by 1 mM KIFMK for R1448P (repolarizations from +100 to -85 mV, 0 vs. 1 mM KIFMK: τd = 146 ± 4 vs. 286 ± 11 μs, n = 5-6). With 0.5 mM KIFMK, the difference was less pronounced but was found for all three clones (same voltages, 0 vs. 0.5 mM KIFMK: WT, τd = 144 ± 8 vs. 205 ± 12 μs; G1306E, τd = 137 ± 8 vs. 194 ± 6 μs; R1448P, τd = 146 ± 4 vs. 222 ± 8 μs; n = 4-7).
Dose-response curves for peptide block are shown in Fig. 4 for R1448P channels. A concentration of 100 μM peptide had almost no effect on either the current decay or the peak tail decay. As expected, τf decreased and τptd increased with increasing peptide concentration, and the voltage dependence of the relative amplitude of the fast decaying component, Af, was shifted in the hyperpolarizing direction (Fig. 4).
Figure 4. Concentration dependence of peptide block.
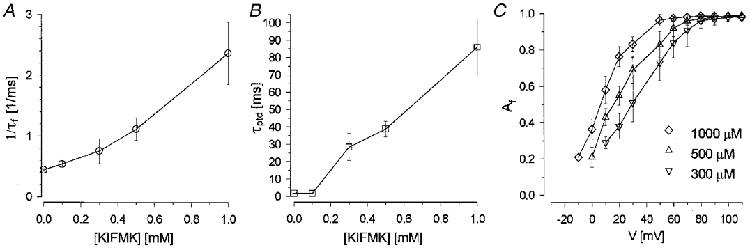
A and B, effects of 0.1-1 mM KIFMK on τf and τptd at a test potential of 100 mV. At high peptide concentrations, where τf << τs (=τptd), 1/τf varied approximately linearly with [KIFMK]. C, the relative amplitude of τf was shifted towards more hyperpolarized potentials with increasing peptide concentration.
If KIFMK is a pore blocker, its action should be antagonized by external Na+ ions. We therefore repeated some of the measurements with 30 mM Na+ in the external solution. Figure 5A shows such outward currents for R1448P channels in the absence and presence of KIFMK. The results are qualitatively the same as those measured with 140 mM external Na+. In the presence of KIFMK, we observed a fast decaying component increasing in amplitude with depolarization, and large tail currents upon repolarization. However, compared with a normal Na+ gradient, the voltage dependence of τf, and in particular that of Af, were shifted markedly in the hyperpolarizing direction (Fig. 5B and C) and τs (Fig. 5B) and τptd (not shown) were significantly larger. These results indicate that external Na+ antagonizes peptide block.
Figure 5. Peptide block at a reversed Na+ gradient.
A, normalized raw current traces for R1448P channels at -40, +10 and +70 mV in the absence (left) and presence of KIFMK (1 mM) at a reversed Na+ gradient. The same voltage protocol as in Figs 1 and 2 was used. The absolute current size at +70 mV is 2.2 nA on the right and 2.4 nA on the left panel. Dotted lines indicate zero current. B and C, comparison of the time constants of the R1448P current decay and the relative amplitude of the fast component, Af, at a normal and a reversed Na+ gradient.
We also tried to induce a cumulative open channel block by KIFMK using high-frequency, repeated 10 ms depolarizations as described by Eaholtz et al. (1994) for rat brain II a Na+ channels. With depolarizations to +100 mV up to a frequency of 10 Hz we did not see any current reduction for either WT or R1448P (data not shown). This is compatible with a fast dissociation of KIFMK at the holding potential of -85 mV, as indicated by the rapidly rising tail current (Fig. 3C).
DISCUSSION
The pentapeptide KIFMK, containing the putative inactivation particle IFM of the voltage-gated Na+ channel, accelerated the current decay and induced large tail currents in a voltage-dependent manner in human skeletal muscle WT and two slowly inactivating mutant Na+ channels. We will discuss our data with regard to the mechanism of peptide block, providing the on and off binding rate constants for a simple kinetic scheme, followed by a comparison of peptide block with normal inactivation.
Mechanism of block by KIFMK
Our data suggest that KIFMK can only block open Na+ channels at depolarized membrane potentials. We assume that blocked channels are ‘frozen’ in the open state and can neither inactivate nor deactivate for the following reasons: (i) The current amplitude did not decrease during diffusion of the peptide from the pipette into the cell, but instead increased because of recovery from slow inactivation in the same way as in the absence of KIFMK (not shown). This is not the case for tetraalkylammonium ions, which also block closed channels (O'Leary & Horn, 1994). (ii) The peptide had no effect at hyperpolarized potentials, where most sodium channels are closed. (iii) Upon repolarization after depolarizing test pulses long enough to inactivate all channels in the absence of KIFMK, the peptide induced large tail currents, indicating prevention of inactivation. The shape of the tail currents was also different, with slowed rising and falling phases. Obviously, neither the inactivation gate nor the activation/deactivation gate can close until the peptide leaves. The effects on tail currents are similar to those described for quaternary strychnine (Cahalan & Almers, 1979). (iv) External Na+ antagonized peptide block compatible with a pore blocking mechanism, as was also found by Tang et al. (1996).
A simple kinetic scheme to describe an open channel peptide block is given by the three-state model shown in Fig. 6A, with one open, one inactivated and one blocked state. Comparable models were used by Tang et al. (1996) and Eaholtz et al. (1998). The rate constants for peptide binding and unbinding, kon and koff, were calculated using the appropriate differential equations and the experimentally determined time constants τh, τf, τs and τptd. It was assumed that τptd is equal to τs. The rate constants were as follows:
with Iss being the relative non-inactivating steady-state current.
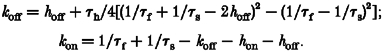 |
The rate from the inactivated back to the open state, hoff, was negligibly small compared with the other rate constants (Iss was between 0.01 and 0.03), and calculating with hon= 1/τh (i.e. without hoff) revealed almost the same results for kon and koff (not shown), indicating that the inactivated state can be considered as absorbing.
Figure 6. Rate constants for peptide block.
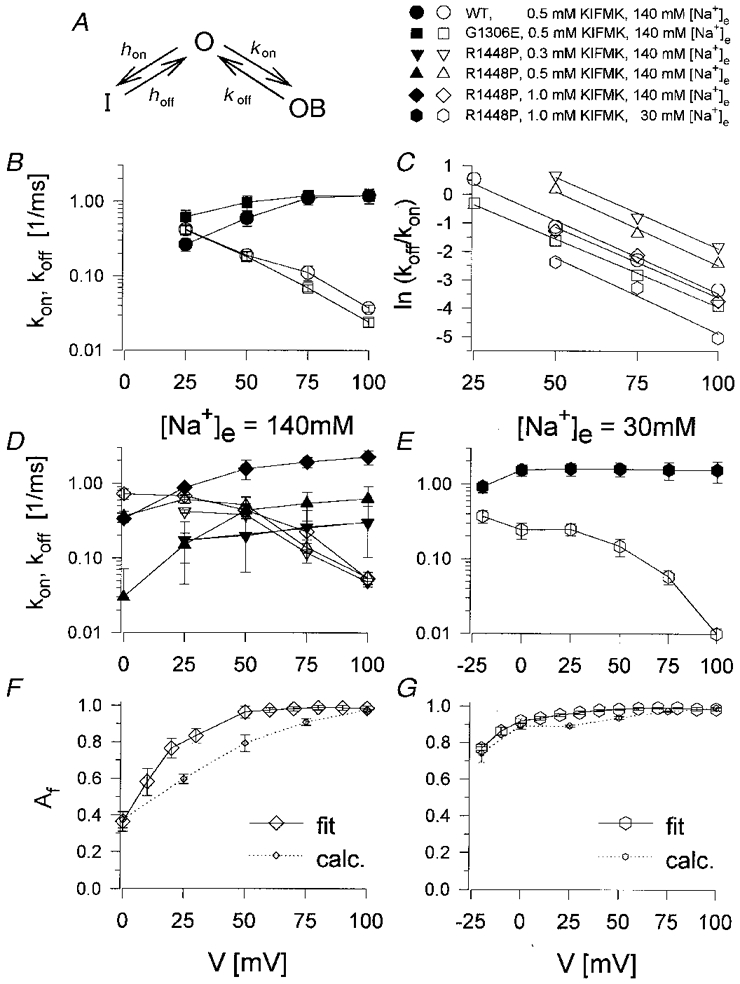
A, kinetic scheme for an open channel peptide block for one open (O), one inactivated (I) and one blocked state (OB). B, D and E, on- and off-rate constants for KIFMK binding, kon (filled symbols) and koff (open symbols). B, for WT and G1306E (0.5 mM KIFMK); D, for R1448P at 140 mM [Na+]e (0.3, 0.5 and 1 mM KIFMK); E, for R1448P at 30 mM [Na+]e (1 mM KIFMK). C, voltage dependence of ln(koff/kon). Lines are linear fits to the data points, which result from the mean values shown in B, D and E. The electrical distance, δ, that is traversed by KIFMK from the cytoplasmic face to reach its binding site, was calculated from the slopes to be 0.64 (WT), 0.60 (G1306E) and 0.62, 0.65, 0.62, 0.67 (R1448P), respectively. F and G, relative amplitude of the fast component, Af, as obtained from a Hodgkin-Huxley fit with a second-order exponential for the current decay (m4h2), as also shown in Fig. 5C (fit), and as calculated/predicted by the model (calc.), for 1 mM KIFMK using a normal (F) or a reversed Na+ gradient (G).
The rate constants for peptide binding and unbinding, kon and koff, are shown in Fig. 6B,D and E. For all three clones, kon (filled symbols) showed little and koff (open symbols) showed a strong voltage dependence. For a simple first-order binding process of a charged particle to a site within the membrane electric field, the rate constants should be exponential functions of voltage. Whereas this was not exactly the case for kon at highly depolarized potentials, the semilogarithmic plot of koffversus voltage showed the expected straight line for WT and G1306E channels (Fig. 6B). The rate constants were very similar for these two clones (Fig. 6B). In contrast, for R1448P channels (Fig. 6D and E) kon was decreased and koff increased relative to the two other clones, indicating a decreased affinity to KIFMK for this mutation. In addition, koff deviated from the expected exponential voltage relationship in the voltage range more negative than +50 mV, as already suggested by the different voltage dependence of τptd for R1448P channels (Fig. 3B). The data indicate that both inactivation and peptide block were affected by the R1448P mutation.
Using the voltage dependence of the rate constants, we were able to calculate how far the charged peptide KIFMK moves into the membrane electric field. The electrical distance δ (0….1) that is traversed by KIFMK from the cytoplasmic side is determined by the equation:
with koff(0) and kon(0) being the rate constants at 0 mV, z the number of elementary charges, e0, of KIFMK (2e0 for the two lysines, which are almost completely protonated at pH 7.4), k the Boltzmann constant, T the absolute temperature in kelvins and V the membrane voltage (Woodhull, 1973). Thus,
with m being the slope of a linear fit to ln[koff/kon](V). Such fits are shown in Fig. 6C using the mean values from Fig. 6B,D and E. For WT and G1306E channels this relationship was almost identical and clearly linear over the whole voltage range where the rate constants could be calculated: δ was determined to be 0.64 and 0.60 for WT and G1306E, respectively. We obtained similar results for R1448P channels (0.62-0.67), when only the voltage range of 50-100 mV was considered, for which ln[koff/kon](V)was fairly linear (Fig. 6C). According to this result, KIFMK should traverse about two-thirds of the membrane electric field, hence entering deeply into the channel pore. However, these calculations have to be interpreted with caution, because they assume the simplification of KIFMK being a point charge within the membrane electric field instead of a large peptide with two positively charged lysines, one at each end.
The kinetic scheme shown in Fig. 6A is consistent with two more results: (i) kon depended strongly on peptide concentration while koff was concentration independent (Fig. 6D); (ii) the relative amplitude of the fast decaying component, Af, representing the fraction of initially blocked channels was fairly well predicted by the model (Fig. 6F and G).
Blocked channels cannot inactivate
The decrease of the tail current amplitude with prolonged depolarization is explained by an absorbing inactivated state. Peptide block is faster than inactivation, which is why almost all channels first reach the blocked state at highly depolarized potentials. However, the natural inactivated state is much more stable than the blocked state. Therefore, with prolonged depolarization, more and more channels reach the inactivated state - by briefly passing through the open state. Once inactivated, the channels hardly ever reopen. We do not think that blocked channels can inactivate. First, it is difficult to imagine that a large, bulky peptide can occlude the pore and inactivation occur on top of it. Second, if this were the case, further depolarization should favour inactivation of blocked channels and therefore accelerate the decrease of the tail current peak with prolonged depolarizations, yielding the opposite voltage dependence for τptd to that observed.
Comparison with other studies
As discussed above, our data suggest an open channel block by KIFMK with a weakly voltage-dependent on-rate and a strongly voltage-dependent off-rate, while inactivation and peptide block exclude each other. The results extend those of two other groups (Eaholtz et al. 1994, 1998; Tang et al. 1996). Whereas Eaholtz and colleagues (1998) found a voltage-dependent on-rate, in the study of Tang et al. the on-rate was voltage independent. This difference can be readily explained by the different external Na+ concentrations used. The first group used a normal Na+ gradient and a voltage range from -30 to +30 mV, the second group used a reversed Na+ gradient and a voltage range from +20 to +60 mV. Hence, both observations fit perfectly with our results regarding the voltage dependence of kon in Fig. 6B,D and E. In addition, Tang et al. also found a highly voltage-dependent current decay using a normal instead of a reversed Na+ gradient, but they did not calculate the rate constants for this experiment.
Both groups found the off-rate to be voltage independent. Eaholtz et al. (1998) used a very slowly inactivating mutant (F1489Q, replacing the crucial phenylalanine of the putative inactivation particle IFM by a glutamine (West et al. 1992)) to explore the action of KIFMK. This allowed them to simplify the calculations of the rate constants by fitting the currents with a single exponential function plus a constant term. In the study of Tang et al. (1996), using a reversed Na+ gradient, the current decay was well fitted to a single exponential function throughout the whole voltage range. However, according to the kinetic scheme shown in Fig. 6A, a biexponential decay is expected. Using the decay of the tail current peak, we were able to determine both exponential time constants. The results revealed a strongly voltage-dependent off-rate in contrast to the other studies.
Does KIFMK mimic inactivation?
Eaholtz et al. (1994) proposed that KIFMK can restore inactivation in the non-inactivating Na+ channel mutant F1489Q (the phenylalanine from IFM, amino acid position 1489 from the rat brain II a sodium channel corresponding to F1311 in the human skeletal muscle isoform). Evidence that the peptide really mimics inactivation by binding to the same site as the natural inactivation particle came from the fact that other peptides not containing IFM - KIQMK and KAFAK - did not show any effect on the non-inactivating mutant. Tang et al. (1996) argued against a common binding site for the natural IFM and KIFMK, because of several differences in their experiments between normal inactivation and peptide block. The most important points were: (i) that KIFMK had similar effects on two mutants (MM/QQ and MM/AA within the IV/S4-S5 loop) which differed markedly in their inactivation kinetics, (ii) that it was a poor blocker of WT channels, and (iii) that external [Na+] ([Na+]e) antagonized peptide block but not normal inactivation. In contrast, in our study KIFMK was an equivalent blocker of WT and mutant channels, as could be shown using a tail current analysis. The most important finding in our study concerning a similarity between inactivation and peptide block was a difference between G1306E and WT channels on the one side, and R1448P channels on the other side, as will be explored below.
We chose two myotonia-causing mutants for our experiments, one located near IFM within LIII-IV (G1306E), and the other one in the voltage sensor IV/S4 (R1448P). Both mutants slow inactivation, probably by different mechanisms: whereas G1306E might hinder the movement of the inactivation gate (West et al. 1992; Lerche et al. 1993; Mitrovic et al. 1995), R1448P should act by slowing the outward movement of IV/S4 (Chahine et al. 1994; Lerche et al. 1996; Yang et al. 1996; Mitrovic et al. 1999). The latter suggests that the formation of a receptor site for the putative inactivation particle IFM is coupled to the movement of the voltage sensor, which explains the coupling between activation and inactivation (Chahine et al. 1994; O'Leary et al. 1995; Yang et al. 1996; Lerche et al. 1997; McPhee et al. 1998; Tang et al. 1998; Filatov et al. 1998). Hence, R1448P may delay the formation of a receptor site for the inactivation particle.
According to this idea, and if the peptide KIFMK bound to the same site as the natural IFM, we should expect that R1448P but not G1306E would affect channel block by KIFMK. Indeed, for WT and G1306E we found almost identical parameters of peptide block, whereas for R1448P the affinity for KIFMK was decreased and the voltage dependence of block was changed in comparison to the two other clones. In addition, the R1448P mutation affects both the voltage dependence of inactivation and that of peptide block in a similar voltage range (compare Figs 2, 3, 5 and 6). Hence, our data suggest that the R1448P mutation affects the voltage-dependent formation of both a receptor site for the natural IFM and for the pentapeptide KIFMK. However, it cannot be definitely concluded from our data if those binding sites are identical. The peptide may still bind to a different site than IFM, while exposure of both sites is influenced by the outward movement of IV/S4 upon activation of the channel.
Acknowledgments
We thank Dr Th. Ruppert and Ms J. Neckermann for providing the peptide, Drs R. Horn, W. Melzer and S. Grissmer for insightful discussions and Ms U. Pika-Hartlaub for performing expert cell culture. This study was supported by the Deutsche Forschungsgemeinschaft (Le481/3-3 to F. L. H.), the Muscular Dystrophy Association and the Deutsche Gesellschaft für Muskelkranke.
References
- Cahalan MD, Almers W. Block of sodium conductance and gating current in squid giant axons poisoned with quaternary strychnine. Biophysical Journal. 1979;27:57–74. doi: 10.1016/S0006-3495(79)85202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA. Structure and function of voltage-gated ion channels. Annual Review of Biochemistry. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Na+ channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Scheuer T, Catterall WA. Restoration of inactivation and block of open sodium channels by an inactivation gate peptide. Neuron. 1994;12:1041–1048. doi: 10.1016/0896-6273(94)90312-3. 10.1016/0896-6273(94)90312-3. [DOI] [PubMed] [Google Scholar]
- Eaholtz G, Zagotta WN, Catterall WA. Kinetic analysis of block of open sodium channels by a peptide containing the isoleucine, phenylalanine and methionine (IFM) motif from the inactivation gate. Journal of General Physiology. 1998;111:75–82. doi: 10.1085/jgp.111.1.75. 10.1085/jgp.111.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Featherstone DE, Fujimoto E, Ruben PC. A defect in skeletal muscle sodium channel deactivation exacerbates hyperexcitability in human paramyotonia congenita. The Journal of Physiology. 1998;506:627–638. doi: 10.1111/j.1469-7793.1998.627bv.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filatov GN, Nguyen TP, Kraner SD, Barchi RL. Inactivation and secondary structure in the D4/S4-5 region of the SkM1 sodium channel. Journal of General Physiology. 1998;111:703–715. doi: 10.1085/jgp.111.6.703. 10.1085/jgp.111.6.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon S C. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. Journal of General Physiology. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kellenberger S, Scheuer T, Catterall WA. Movement of the sodium channel inactivation gate during inactivation. Journal of Biological Chemistry. 1996;271:30971–30979. doi: 10.1074/jbc.271.48.30971. [DOI] [PubMed] [Google Scholar]
- Lehmann-Horn F, Rüdel R. Molecular pathophysiology of voltage-gated ion channels. Reviews in Physiology, Biochemistry and Pharmacology. 1996;128:195–268. doi: 10.1007/3-540-61343-9_9. [DOI] [PubMed] [Google Scholar]
- Lerche H, Heine R, Pika U, George AL, Jr, Mitrovic N, Browatzki M, Weiss T, Rivet-Bastide M, Franke C, Lomonaco M, Ricker R, Lehmann-Horn F. Human Na+ channel myotonia: slowed channel inactivation due to substitutions for a glycine within the III-IV linker. The Journal of Physiology. 1993;470:13–22. doi: 10.1113/jphysiol.1993.sp019843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerche H, Mitrovic N, Dubowitz V, Lehmann-Horn F. Paramyotonia congenita: the R1448P Na+ channel mutation in adult human skeletal muscle. Annals of Neurology. 1996;39:599–608. doi: 10.1002/ana.410390509. [DOI] [PubMed] [Google Scholar]
- Lerche H, Peter W, Fleischhauer R, Pika-Hartlaub U, Malina T, Mitrovic N, Lehmann-Horn F. Role in fast inactivation of the IV/S4-S5 loop of the human muscle Na+ channel probed by cysteine mutagenesis. The Journal of Physiology. 1997;505:345–352. doi: 10.1111/j.1469-7793.1997.345bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPhee JC, Ragsdale DS, Scheuer T, Catterall WA. A critical role for the S4-S5 intracellular loop in domain IV of the sodium channel α-subunit in fast inactivation. Journal of Biological Chemistry. 1998;273:1121–1129. doi: 10.1074/jbc.273.2.1121. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Heine R, Wagner S, Pika U, Hartlaub U, Zhou M, Lerche H, Fahlke C, Lehmann-Horn F. K+-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. The Journal of Physiology. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. The Journal of Physiology. 1995;487:107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Rüdel R, Lehmann-Horn F, Lerche H. Mutant channels contribute less than 50 % to Na+ current in paramyotonia congenita muscle. Brain. 1999 doi: 10.1093/brain/122.6.1085. in the Press. [DOI] [PubMed] [Google Scholar]
- O'Leary ME, Chen LQ, Kallen RG, Horn R. A molecular link between activation and inactivation of sodium channels. Journal of General Physiology. 1995;106:641–658. doi: 10.1085/jgp.106.4.641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary ME, Horn R. Internal block of human heart sodium channels by symmetrical tetraalkylammoniums. Journal of General Physiology. 1994;104:507–522. doi: 10.1085/jgp.104.3.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Chehab N, Wieland SJ, Kallen RG. Glutamine substitution at alanine1649 in the S4-S5 cytoplasmic loop of domain 4 removes the voltage sensitivity of fast inactivation in the human heart sodium channel. Journal of General Physiology. 1998;111:639–652. doi: 10.1085/jgp.111.5.639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang L, Kallen RG, Horn R. Role of an S4-S5 linker in sodium channel inactivation probed by mutagenesis and a peptide blocker. Journal of General Physiology. 1996;108:89–104. doi: 10.1085/jgp.108.2.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedantham V, Cannon SC. Slow inactivation does not affect movement of the fast inactivation gate in voltage-gated sodium channels. Journal of General Physiology. 1998;111:83–93. doi: 10.1085/jgp.111.1.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J, Dubowitz V, Lehmann-Horn F, Ricker K, Ptacek L, Hoffman EP. In vivo structure/function studies: consecutive Arg1448 changes to Cys, His, and Pro at the extracellular surface of IVS4. Society of General Physiology Series. 1995;50:77–88. [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+ channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992;91:12785–12789. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. Journal of General Physiology. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, George AL, Jr, Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]