

Abstract
Using perforated-patch recordings, we have examined the part played by endogenous G-protein subunits in the α2-adrenoceptor-mediated inhibition of N-type Ca2+ currents in sympathetic neurones.
Two components of ICa inhibition by noradrenaline were recorded: a prominent, high affinity and voltage-dependent pertussis toxin (PTX)-sensitive pathway and a minor, low affinity and mostly voltage-insensitive PTX-resistant pathway.
PTX-sensitive inhibition was reduced by microinjection of antibodies against either GαoA,B or Gαi1,2. The voltage-dependent fraction of inhibition was reduced by anti-Gαo but not by anti-Gαi antibody.
Antisense depletion of GαoA led to a marked reduction of noradrenaline-induced inhibition and voltage dependence. By contrast, Gαi depletion attenuated noradrenergic modulation without affecting the voltage dependence.
Expression of the βγ-binding agents β-adrenergic receptor kinase 1 (C-terminus, βARK1C-ter) or Gαi1 with a Cys3 to Ser mutation partially prevented noradrenergic inhibition while α-transducin abolished it. Residual inhibition was mostly voltage independent in cells expressing βARK1C-ter but was strongly reversed by depolarization in Gαi1 Cys3Ser-expressing cells.
Expression of the PTX-resistant Gαi1 Cys351Ile mutant in cells treated with PTX restored α2-adrenoceptor inhibition. This restored inhibition was weakly reversed by depolarization. Both the degree and voltage dependence of inhibition were correlated with the level of expression of the Gαi1 Cys351Ile subunit.
Our findings identify βγ dimers associated with GαoA and Gαi as mediators of the PTX-sensitive α2-adrenoceptor-mediated inhibition of N-type Ca2+ channels. Different βγ combinations may account for the differential voltage-dependent effects of Go and Gi on ICa.
G-protein-coupled neurotransmitter receptors inhibit N-type (α1B) Ca2+ channels in sympathetic neurones via two biochemically and biophysically distinct routes. The most commonly documented is that mediated by receptors that rapidly inhibit ICa through activation of pertussis toxin (PTX)-sensitive G-proteins (Hille, 1994). This form of inhibition occurs in a voltage-dependent manner (Bean, 1989) and is mediated by G-protein βγ dimers released from their association with Gα subunits when the latter are activated by the receptor (Ikeda, 1996; Herlitze et al. 1996; Delmas et al. 1998a,b). The other main form of inhibition, which is one order of magnitude slower, occurs in a voltage-independent manner; it results from the coupling of neurotransmitter receptors to PTX-resistant G-proteins and the activation of subsequent diffusible messenger cascades (Bernheim et al. 1991; Beech et al. 1992). In this case, it is the G-protein α subunit (e.g. Gαq in the case of the M1 muscarinic acetylcholine receptor inhibition), rather than the βγ complex, that appears to mediate inhibition (Delmas et al. 1998a).
α2-Adrenoceptors represent a family of G-protein-coupled receptors that regulate effector function via activation of multiple members of Gi and Go G-protein families (Limbird, 1988). In rat superior cervical ganglion (SCG) neurones, the PTX-sensitive Go heterotrimer has been shown to participate in coupling α2-adrenoceptor(s) to Ca2+ channel inhibition (Caulfield et al. 1994). This inhibition occurs through the fast and voltage-dependent, presumed Gβγ-mediated pathway (Herlitze et al. 1996; Ikeda, 1996; Delmas et al. 1998b). However, the generality of this latter statement is not yet entirely clear since Gi-type G-proteins are involved (along with Go) in the PTX-sensitive adrenergic inhibition of N-type Ca2+ currents in chick sensory ganglion cells (Diversé-Pierluissi et al. 1995, 1997). Moreover, in this latter preparation, the roles of the α and βγ subunits seem to be reversed: the βγ subunits appear to mediate a voltage-insensitive inhibition (via stimulation of phospholipase C/protein kinase C), not a voltage-dependent inhibition like that found in SCG neurones, while GTP-γ-S-activated recombinant Gαo produces a voltage-dependent inhibition.
Accordingly, in the present study, we have investigated further the nature of the G-proteins and G-protein subunits that mediate noradrenergic inhibition of N-type Ca2+ channels in SCG neurones. Our data indicate that both Go- and Gi-type G-proteins couple α2-adrenoceptor(s) to ICa with associated βγ subunits acting as the most probable final transducers. However, we find that inhibitions apparently mediated by Goβγ and Giβγ display differential sensitivities to voltage and to different Gβγ-sequestering agents, suggesting that the endogenous βγ subunits associated with these two proteins are not equivalent in identity and/or action.
METHODS
DNA plasmids
The cloning and specificity of plasmids generating antisense RNA to various G-protein α subunits were described previously (Abogadie et al. 1997; Delmas et al. 1998a; Haley et al. 1998). Antisense sequences of rat GαoA (clone 207-8) and Gαq (clone C23-16, EMBL accession number Y17164) were subcloned into pCR3 expression vector (Invitrogen, NV Leek, The Netherlands). The antisense sequence of rat Gαicommon (clone 50-2) was subcloned into pCR3.1. This clone corresponds to nucleotides 1045-1215 of Gαi2 and shares approximately 80 % identity with Gαi1 and Gαi3. cDNA encoding the C-terminus of β-adrenergic receptor kinase 1 (βARK1 495-689) was subcloned in pCIN1 as described previously (Delmas et al. 1998b). The generation of PTX-resistant Gαi1 subunits (Gαi1 Cys351Ile and Gαi1 Cys351Arg, mutated Gαi1 subunits in which the cysteine 351 residue was replaced with isoleucine or arginine) was as detailed in Wise et al. (1997b) and Bahia et al. (1998). cDNAs encoding these mutants were subcloned into pCDNA3 (Invitrogen). Retinal Gα-transducin and the palmitoylation-negative Gαi1 Cys3Ser mutant (Wise et al. 1997a) were subcloned into pCDNA3. Plasmids were propagated in either XL1-Blue or DH5αEscherichia coli and purified using Qiagen maxiprep columns (Hilden, Germany).
Intranuclear injection of plasmids
Plasmids were diluted into calcium-free Krebs solution (290 mosmol l−1, pH 7.3) containing fluorescein isothiocyanate-conjugated dextran (FITC-dextran, 70 kDa, 0.5 %; Molecular Probes) to a final concentration of 10-600 μg ml−1 and then centrifugated and filtered (0.2 μm) to remove particles. Injection electrodes were pulled with a one-stage pull using a Flaming-Brown horizontal puller (P-87, Sutter Instruments) and had a series resistance of 50-80 MΩ when loaded (2-3 μl) with the plasmid-containing solution. Microinjection was performed under fluorescence microscopy (Nikon Diaphot 300) with the assistance of an Axoclamp-2B amplifier (Axon Instruments). Contact of the electrode with the cell and impalement were detected by passing hyperpolarizing current into the electrode. Injection was achieved by applying a positive pressure to the micropipette solution through the side arm of the pipette holder. Pressure was gentle in order to minimize nuclear swelling. Cells were returned to the incubator after microinjection.
Loading of antibodies
Antibodies were diluted into modified Krebs solution (KCl based and Ca2+ free) containing 0.5 % FITC-dextran and pressure-injected into the cytosol of SCG neurones (Caulfield et al. 1994; Delmas et al. 1998a). The resistance of the injecting electrodes was 40-60 MΩ. Injection was best achieved by positioning the electrode tangentially to the nucleus. Injection of antibody-free buffer (containing FITC-dextran) served as a control. Following injection, cells were maintained in culture for 3-4 h and identified for recording by fluorescence microscopy. Antibodies used were immunoglobulin G fractions raised against carboxyl terminal domains of GαoA/B and Gαi1/2 subunits (Goldsmith et al. 1987). The success of cytoplasmic antibody injections was routinely verified by immunostaining (see Fig. 3).
Figure 3. Antibody loading.

Anti-rabbit immunoreactivity in a rat SCG neurone microinjected with rabbit anti-Gαo antibody. The neurone was fixed with acetone 3 h following cytoplasmic microinjection. Note the staining of distal neurites. Scale bar, 20 μm.
Cell culture
Sympathetic neurons were isolated from SCG of young rats (15-19 days old) as described previously (Delmas et al. 1998b). The rats were killed by exposure to a rising concentration of carbon dioxide, followed by decapitation, according to the Animals (Scientific Procedures) Act 1986. Cells were seeded at a density of 50 cells mm−2 onto laminin-coated glass coverslips for immunocytochemistry. Glass slides were imprinted with squares for later location of injected cells.
Electrophysiological recordings
Calcium currents were measured using the amphotericin B perforated-patch method largely as described by Delmas et al. (1998a, b). Electrodes were fabricated from borosilicate glass capillaries (Clark Electromedical Instruments) with the use of a Flaming-Brown (P-87) micropipette puller (Sutter Instruments) and fire polished. Pipettes had a resistance of 1-2 MΩ when filled with the following intracellular solution (mM): CsCl, 30; caesium acetate, 110; MgCl2, 1; and Hepes, 10 (pH 7.2-7.3 with CsOH; 295-300 mosmol l−1) and 0.1 mg ml−1 amphotericin B. The external solution consisted of (mM): NaCl, 130; KCl, 3; MgCl2, 1; Hepes, 10; tetrodotoxin (TTX), 0.0005; CaCl2, 2; and glucose, 11 (pH 7.3 with NaOH, ∼300 mosmol l−1). Access resistances after permeabilization (10-20 min at 32-33°C) ranged between 6 and 10 MΩ. Cell membrane capacitance and series resistance compensations (∼80 %) were applied. For whole-cell (‘ruptured-patch’) recording, pipettes (1.5-2.5 MΩ) were filled with an intracellular solution consisting of (mM): CsCl, 130; MgCl2, 1; BAPTA, 10-20; CaCl2, 0.1-0.5; Na2ATP, 2; Na3GTP, 0.12; and Hepes 10 (pH 7.2-7.3 with CsOH). Recordings were obtained with an Axopatch 200A patch clamp amplifier (Axon Instruments), filtered at 2-5 kHz and corrected for leak and capacitive currents using the leak subtraction procedure (P/6) of pCLAMP 6 software (Axon Instruments). pCLAMP 6 software was used to collect and analyse the data. Calcium currents were recorded at 32-33°C in relatively small (17-40 pF) SCG neurones. Space clamp quality was assessed by examining activation and tail current time constants evoked by positive voltage pulses. Data are from cells which exhibited graded voltage-dependent current activation and tail current time constants < 1 ms at 0 mV. Calcium current-voltage relationships determined in the perforated-patch configuration displayed a similar voltage dependence to those obtained in the classical whole-cell recording configuration. Inward Ca2+ currents activated near -30 mV, reached a maximum amplitude near +5 mV and approached a zero current asymptote at about +50 mV (with internal Cs+). Approximately 90 % of high voltage-activated (HVA) Ca2+ currents appeared sensitive to ω-conotoxin GVIA (500 nM, n = 5) (in accordance with previous data: see Plummer et al. 1989). No significant rundown of ICa was observed using the perforated patch over > 1 h recording whereas rundown was relatively rapid (25-35 % in < 20 min) in whole-cell recording. The amplitude of ICa was measured isochronally 4 ms after the onset of a test pulse after subtracting the current remaining in the presence of 500 μM Cd2+. The voltage dependence of inhibition was examined using a three-pulse voltage protocol consisting of two test pulses (P1 and P2) to 0 mV separated by a conditioning prepulse to +90 mV (see Fig. 4). Facilitation was then determined as the P2/P1 ratio of the current amplitudes. The ratio of inhibition (IR) was defined as the ratio of inhibitions determined before and after the conditioning pulse (with 100 - 100/IR = percentage of voltage-dependent component). Data are expressed as the mean ±s.e.m. Student's unpaired t test and analysis of variance were applied to determine statistical significance. Differences were considered significant if P < 0.05.
Figure 4. Both anti-Gαo and anti-Gαi antibodies reduce PTX-sensitive noradrenergic inhibition.

Left panels: calcium current inhibition induced by 1 μM noradrenaline in neurones cytoplasmically injected with either FITC-dextran (A) or antibodies raised against Gαo (B) or Gαi (C) subunits. Currents were elicited by the use of the three-pulse voltage protocol as illustrated. In B and C - and subsequent figures - the outward currents elicited by the +90 mV voltage pulses (at break) are omitted for clarity. Right panels: calcium current amplitude (•) and facilitation (○) plotted as a function of time for the corresponding cells shown in the left panels. All neurones were recorded using the perforated-patch method 3-4 h after cytoplasmic microinjection.
Immunocytochemistry
Immunocytochemistry was performed essentially as described previously (Abogadie et al. 1997). Briefly, following electrophysiological recordings, SCG neurones were fixed in acetone (10-20 min at room temperature). The cells were then incubated (1 h at room temperature) with polyclonal antibodies raised against Gαo (sc-387, reactive with GαoA and GαoB, 1 : 1000 dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA), Gαi3 (sc-262, reactive with Gαi1, Gαi2 and Gαi3, 1 : 1000 dilution; Santa Cruz Biotechnology) and Gαq (IQB, antiserum generated against a synthetic peptide corresponding to amino acids 119-134 of Gαq, 1 : 1000 dilution; Milligan et al. 1993). Bound antibodies were detected using biotinylated Fab2 swine anti-rabbit IgG antibody (Dako, Denmark) conjugated with alkaline phosphatase (1 : 500 dilution). The specificity of the staining was assessed by competing out with the respective antigenic peptides (typically 10-fold excess) (Delmas et al. 1998a; Haley et al. 1998).
Drugs and chemicals
Cells were superfused at 10 ml min−1 during recording. The solutions containing test agents were applied to neurones through a large-bore tube (∼1 mm i.d.) placed 2-3 mm away from the neurone under study. Noradrenaline (Sigma) was prepared daily from frozen stock solutions (10 mM). When used, Bordetellapertussis toxin (PTX, 1 μg ml−1; Sigma) was added to the culture medium for 24-28 h (37°C, 5 % CO2). All other chemical compounds were from Sigma, except oxotremorine-M (RBI).
RESULTS
PTX-sensitive and PTX-insensitive components of noradrenergic inhibition
A dose-response curve for the inhibition of high voltage-activated Ca2+ currents (HVA ICa) by noradrenaline recorded using the amphotericin B perforated-patch method is illustrated in Fig. 1 (○). Inhibition was detectable with 30 nM noradrenaline and reached maximum levels at concentrations > 1 μM. The data could be fitted by an equation of the form y =a/(1 + IC50/[NA]) (where y is percentage inhibition, a is maximal percentage inhibition and [NA] is the concentration of noradrenaline), giving an IC50 of 150 ± 50 nM (n = 7). This was significantly less than that obtained using whole-cell patch recording (IC50= 500 ± 40 nM, n = 8; Fig. 1, ▵). In addition to the increase in sensitivity to noradrenaline, maximum suppression of ICa was also enhanced in perforated-patch recording (79 ± 4 % inhibition with 10 μM noradrenaline versus 66 ± 5 % in whole-cell recording). By contrast, the rate of desensitization of noradrenaline-mediated responses was similar under the two recording configurations (10-20 % within 2 min of exposure to 10 μM noradrenaline). Pretreatment with PTX reduced the noradrenaline-mediated inhibition of ICa by 73 ± 4 % in 11 out of 17 neurones and abolished the response in the remaining six cells. Following PTX treatment, inhibition of ICa required a higher concentration of noradrenaline (IC50 3.3 ± 0.3 μM; Fig. 1). Hence, in agreement with the observations made by Beech et al. (1992), Shapiro et al. (1994) and Zhou et al. (1997) using whole-cell recording, the action of noradrenaline on rat SCG neurones is mediated by at least two routes: a ubiquitously distributed, high affinity PTX-sensitive pathway and a rarer, low affinity PTX-resistant pathway.
Figure 1. Perforated-patch recording of α2-adrenoceptor-mediated inhibition of HVA Ca2+ currents.
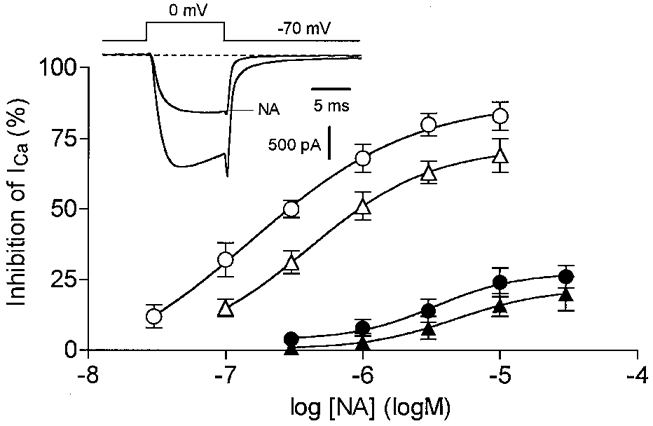
Concentration-inhibition curves for noradrenaline (NA) determined using the perforated-patch (circle) or whole-cell (triangle) methods, in SCG neurones pretreated (filled symbols) or not (open symbols) with PTX (1 μg ml−1 for 24 h). Each point represents the mean ±s.e.m. of 4-9 cells. The data were fitted to the equation y =a/(1 + IC50/[NA]) where y is percentage inhibition and a is maximal percentage inhibition. Calculated values for a and IC50 were: ○, 79 % and 150 nM; ▵, 67 % and 500 nM; •, 27 % and 3.3 μM; and ▴, 22 % and 4.7 μM, respectively. Inset, superimposed HVA ICa traces recorded using the perforated-patch method, in the presence or absence of 1 μM noradrenaline.
The onset rate for ICa inhibition exerted by noradrenaline in perforated-patch recordings was measured at saturating concentrations (30 μM) using the protocol shown in Fig. 2A. Four successive depolarizing steps to a test potential of -10 mV were applied to the cell from a holding potential of -70 mV. This stimulatory sequence was reiterated every 100 ms and caused very little cumulative inactivation of ICa; it was chosen to estimate the time constant of Cd2+ block, which was 40-80 ms. Flow of noradrenaline-containing solution was begun randomly and turned off after inhibition reached steady-state level. The onset rate of noradrenaline-induced inhibition was then obtained by subtracting the time constant of the action of Cd2+ from that of noradrenaline. Both the reduction of HVA ICa (Fig. 2B) and recovery (not shown) from noradrenaline inhibition developed exponentially. In cells not treated with PTX, time constants of noradrenaline inhibition were 380 ± 25 ms (n = 4) for the onset and 1.7 ± 0.5 s (n = 4) for the offset, whereas inhibition in PTX-treated cells displayed ‘slower’ time constants of 950 ± 40 ms (n = 4) and 4.5 ± 0.5 s (n = 3), respectively. For comparison, ICa inhibition through the slow M1 muscarinic acetylcholine receptor (M1 mAChR)-mediated pathway (in response to 10 μM oxotremorine-M in PTX-treated cells) developed with a 6 ± 0.5 s time constant (Fig. 2B, n = 3) and recovered (often incompletely) with a time constant of 22 ± 6 s (n = 3).
Figure 2. Time course of α2-adrenoceptor-mediated inhibition of Ca2+ currents.
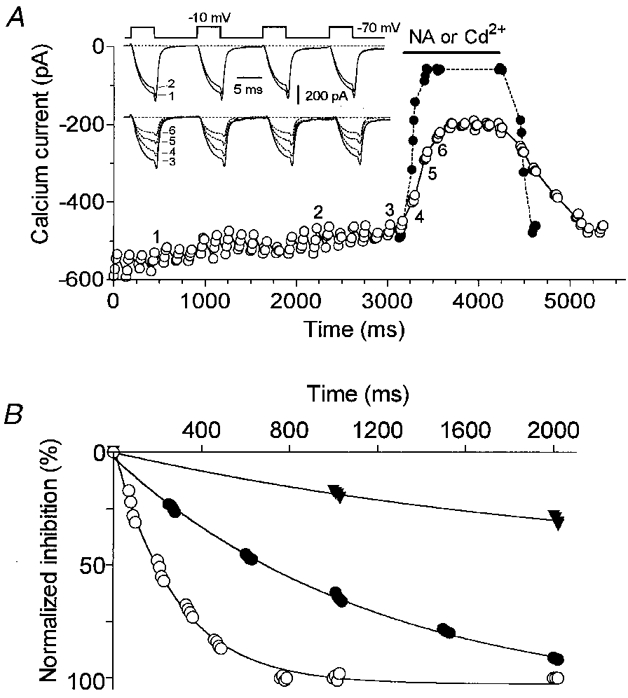
A, changes in peak Ca2+ currents induced by applying noradrenaline (30 μM) or cadmium (500 μM, applied at the end of the experiment; superimposed symbols, •). The horizontal bar indicates the time and duration of application of the drugs. Inset, superimposed current traces selected at the times indicated by the corresponding numbers in A. ICa was elicited by applying repetitive sequences consisting of 4 successive depolarizing steps to -10 mV. Cd2+-sensitive currents were subtracted off-line. B, time course of inhibition of HVA ICa by 10 μM noradrenaline: SCG neurones pretreated (•) or not (○) with PTX (1 μg ml−1 for 24 h). Compare with the time course of inhibition induced by 10 μM oxotremorine-M (M1 mAChR pathway, ▾) in a neurone incubated with PTX. Inhibition was normalized to the inhibition measured at steady state. Smooth curves are exponential fits to the data.
Neutralization of Go- and Gi-type G-proteins differentially alters the PTX-sensitive inhibition
One micromolar noradrenaline was used to study the noradrenergic PTX-sensitive pathway in isolation (see Fig. 1). In confirmation, treatment with PTX (1 μg ml−1 for 24 h) completely prevented the inhibitory effects of 1 μM noradrenaline (3 ± 1 %, n = 8). This inhibition exhibited the trademark features of voltage dependence as previously reported in whole-cell recordings (Bean, 1989; Beech et al. 1992), such that the inhibition was less at large that at moderate depolarizations. For example, noradrenaline showed a mean inhibition of 20 ± 1.5 % (n = 6) (cf. 15 and 30 % in Bean, 1989, and Beech et al. 1992, respectively) following a pulse to +120 mV compared with 61.1 ± 0.8 % (n = 20) at moderate depolarizations. Modulated HVA currents also showed slowing of activation kinetics and reversal of inhibition (‘facilitation’; Ikeda, 1991) following a conditioning membrane depolarization. Facilitation occurred only in the presence of noradrenaline, not in its absence, and was maximal (facilitation 1.9 ± 0.15, n = 16) at voltages near the point of peak Ca2+ current. In agreement with tail current recordings, reversal of inhibition using the three-pulse protocol was always incomplete, leaving part of the current still inhibited (24.1 ± 0.7 %, n = 16) following the intercalating depolarization. PTX-insensitive inhibition produced by higher concentrations of noradrenaline (10-30 μM; n = 6) was largely refractory to modulation by voltage (data not shown).
Previous experiments using microinjection of antisera against Gα subunits showed that Go heterotrimers contributed to PTX-sensitive noradrenergic inhibition (McFadzean et al. 1989; Caulfield et al. 1994). However, it is not clear from these experiments which particular G-protein and G-protein subunit(s) is (are) responsible for voltage-dependent inhibition (see Introduction). To address this further, we injected antibodies (IgGs) against C-terminal decapeptide sequences of GαoA/B (anti-Gαo) or Gαi1/2 (anti-Gαi) into the cytoplasm and examined the modulation of calcium currents 3-4 h later. To ensure antibody loading in recorded cells, the efficiency of the injection was assessed immunocytochemically. An example of a SCG neurone preinjected with Gαo antibody is shown in Fig. 3 where the microinjected Gα antibody could be detected even in very distal processes. As reported previously (Delmas et al. 1998a), antibodies did not alter the kinetic properties of ICa; however, the injection per se slightly reduced the calcium current density. In agreement with the work by Caulfield et al. (1994), we found that the anti-Gαo antibody significantly reduced noradrenergic inhibition from 58 ± 4 % (cells microinjected with antibody-free buffer) to 34.1 ± 3.3 % (Fig. 4B and Fig. 5A). We observed, however, that the anti-Gαi antibody, though less effective, also attenuated noradrenergic inhibition (to 44.2 ± 2.1 %, n = 5) (Fig. 4C and Fig. 5A). The specificity of the effects of the antibodies was assessed on Go-dependent M4 mAChR inhibition of ICa (Delmas et al. 1998a) and Gq-dependent M1 mAChR inhibition of M-type potassium currents (Haley et al. 1998). Anti-Gαi antibody did not alter M4 muscarinic modulation of calcium currents (Fig. 5A), nor did Gαo antibody affect M1 muscarinic inhibition of M-type K+ currents (see Delmas et al. 1998a).
Figure 5. Anti-Gα antibodies alter the voltage dependence of inhibition.

A, scatter plot showing ICa inhibition by 1 μM noradrenaline (open symbols) or 10 μM oxotremorine-M (filled symbols) in individual neurones as indicated (Control, neurones microinjected with FITC-dextran). The horizontal bars indicate the mean values. ICa was evoked using the three-pulse voltage protocol as in Fig. 4 and inhibition was calculated from the baseline current in the presence of 500 μM Cd2+ as (1 - INA/Icontrol) × 100. * P = 0.015; ** P = 0.0012. Voltage-dependent M4 mAChR inhibition was recorded in the whole-cell (patch-ruptured) mode using high BAPTA internal solution (see Methods). B, summary of facilitation (means and s.e.m.) observed in uninjected neurones or neurones injected with anti-Gα antibodies (Gα ab) in the presence (+) or absence (-) of 1 μM noradrenaline. n.s. (not significant), P = 0.42; ** P = 0.0039. C, mean inhibition ratio (and s.e.m.) under the different conditions indicated. Inhibition ratio was calculated as the ratio of inhibition before conditioning depolarization to inhibition after conditioning depolarization.
Noradrenaline-induced inhibition was then examined using the three-pulse voltage protocol in order to assay effects of antibodies on the voltage dependence of inhibition. As shown in Fig. 4A, strong depolarization in control cells restored most of the modulated ICa, in that inhibition decreased from 61 ± 2.5 to 22 ± 2 % (n = 16) following depolarization. In cells loaded with anti-Gαo antibody, modulated ICa was largely resistant to facilitation, the inhibition after the conditioning step (23.1 ± 3 %) being little different from the overall inhibition (34 %, see above). Thus, the mean facilitation measured at 0 mV was 1.16 ± 0.17 (n = 6) compared with 1.88 ± 0.12 (n = 8) and 1.7 ± 0.2 (n = 3) in uninjected cells and cells injected with antibody-free buffer solution, respectively (Fig. 4B). By contrast, residual inhibition in cells loaded with anti-Gαi antibody appeared strongly voltage dependent with a facilitation of 1.70 ± 0.2 (n = 5) (Fig. 4C and Fig. 5B). However, because the facilitation depends upon both basal modulation and the degree of inhibition, it does not provide a good estimate of the voltage-dependent action of neurotransmitters. Hence, we chose to take the inhibition ratio (IR, see Methods) as a much more appropriate index for voltage dependence. This clearly revealed that most (68 %) of the residual inhibition in the presence of the anti-Gαo antibody was not reversed by depolarization whereas it was strongly reversed (by 82 %) in cells injected with the anti-Gαi antibody (Fig. 5C).
Depletion of GαoA subunits, but not Gαi, depresses the voltage-dependent fraction of the PTX-sensitive inhibition
GαoA and Gαi were selectively depleted by expressing specific antisense RNAs. SCG neurones were injected intranuclearly with expression plasmids (400 μg ml−1) designed to drive production of high levels of antisense RNA complementary to either the 3′ untranslated sequence of GαoA (the more abundant Gαo isoform in SCG neurones) or a coding sequence common to Gαi1-3 subunits. Expression of these antisense RNAs specifically decreased immunoreactivity for their targeted Gα proteins (Delmas et al. 1998a; Haley et al. 1998). Inhibition of calcium currents by 1 μM noradrenaline was then examined 2 days later when the effects of the antisense RNAs reach a plateau (Haley et al. 1998).
As reported previously (Delmas et al. 1998a), basal facilitation was high in neurones expressing GαoA antisense RNA (Fig. 6Ab). This effect was ascribed to the action of Gβγ dimers released from GoA heterotrimers (as GαoA is depleted) and was not observed in cells expressing Gαq or Gαicommon antisense RNAs (Fig. 6Aa and Ac). Depletion of GαoA subunits reduced the noradrenergic inhibition from 61.1 ± 0.8 to 17 ± 4 % (n = 7). The residual inhibition seen in these cells was mostly voltage independent, with an inhibition ratio of 1.12 ± 0.12 (Fig. 6B). In cells expressing the Gαicommon antisense RNA, inhibition was slightly reduced to 48.7 ± 2.8 % (n = 6) but, in contrast with the GαoA antisense RNA, was still strongly reversed by depolarization (IR = 4.8 ± 0.4) (Fig. 6B). The Gαq antisense RNA had no significant effects on noradrenaline-induced inhibition.
Figure 6. Noradrenergic inhibition in cells depleted of either Gαi or GαoA subunits.
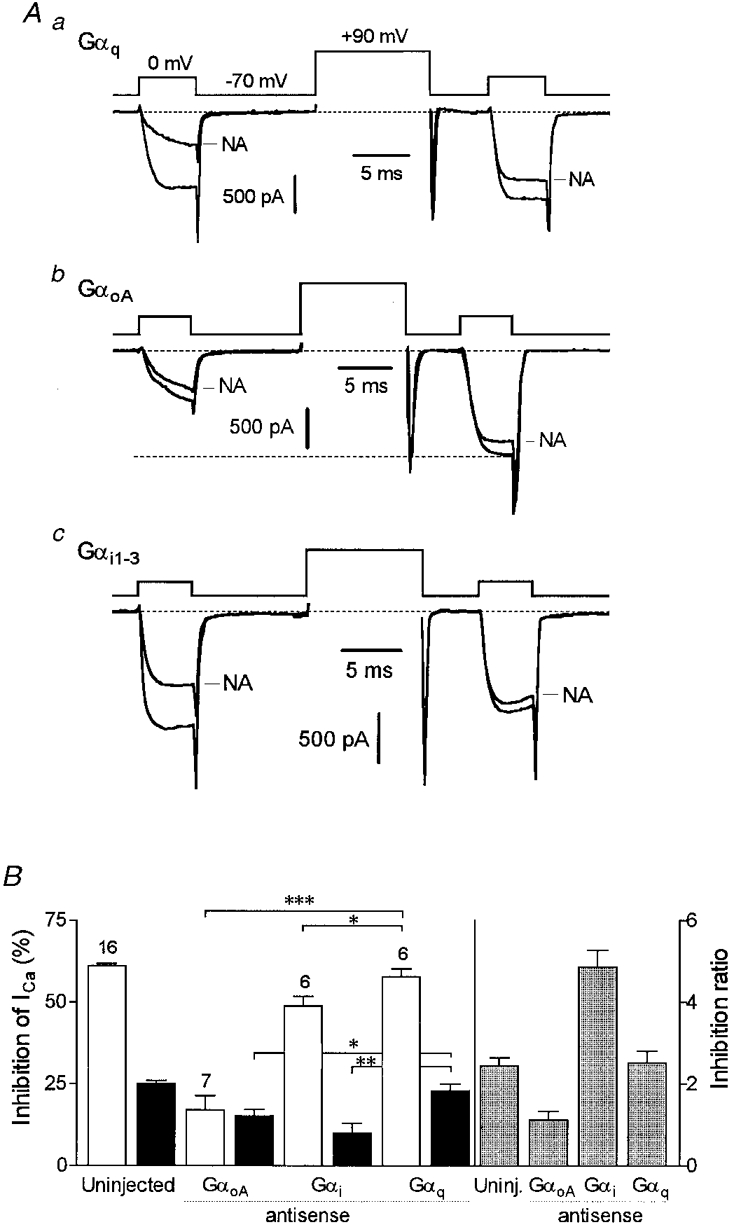
A, superimposed Ca2+ current traces recorded in the absence and presence of noradrenaline (1 μM) in neurones preinjected intranuclearly with cDNA plasmids generating antisense RNA specific to Gαq (a), GαoA (b) and Gαi1-3 (c) subunits. Note the tonic facilitation (delineated by the dashed line) of basal ICa in the cell expressing GαoA antisense RNA (b). B, summary of calcium current inhibition (inhibition before (□) and after (▪) the conditioning depolarization) and ratio of inhibition (grey bars) in uninjected cells and cells expressing different Gα antisense RNAs. * P < 0.05; ** P < 0.005; *** P < 0.0001.
Both Go and Gi signals are reduced by βγ-sequestering agents
The above experiments show that Go and Gi heterotrimers are responsible for two biophysically distinct forms of inhibition: that mediated by Go is voltage dependent whereas Gi-mediated inhibition is not. We next wanted to test whether βγ subunits are involved in these components. One approach to defining whether ICa modulation is transduced via Gβγ dimers is to express peptides containing Gβγ-binding domains (βγ-sequestering agents) thereby competing with the Ca2+ channel for free βγ dimers. There are inherent concerns with this strategy as Gβγ-sequestering agents may not bind all βγ dimers and may interfere with membrane receptor-G-protein interaction. To overcome this problem we over-expressed three distinct putative βγ scavengers (200 μg ml−1): the carboxyl terminal domain of βARK1 (βARK1C-ter), which can effectively neutralize released Gβγ (Koch et al. 1994; Delmas et al. 1998a,b; Stephens et al. 1998) but is thought to not bind all βγ subunits (Daaka et al. 1997); a mutationally modified (palmitoylation-negative) Cys3 to Ser Gαi1 subunit, which binds Gβγ dimers but does not interact with the α2A-adrenoceptor (Wise et al. 1997a); and retinal α-transducin. To assay specificity, the effects of these peptides were examined in parallel on Gq-mediated M1 mAChR inhibition, which does not involve βγ subunits (Delmas et al. 1998a).
Gαi1 Cys3Ser, βARK1C-ter and α-transducin had no noticeable effect on basal calcium current properties. However, they strongly attenuated calcium current inhibition by 1 μM noradrenaline to 39.5 ± 1.5 % (n = 5), 29 ± 2 % (n = 6) and 3 % (n = 5), respectively. Residual inhibition was mostly voltage insensitive in cells expressing βARK1C-ter (IR = 1.3 ± 0.2) whereas it was strongly reversed by depolarization in cells expressing Gαi1 Cys3Ser (IR = 2.9 ± 0.3) (Fig. 7). βARK1C-ter expression had no significant effects on M1 mAChR-mediated inhibition (PTX-treated cells; see Delmas et al. 1998a) but Gαi1 Cys3Ser and α-transducin slightly depressed M1 mAChR responses by 17 ± 4 % (n = 4) and 34 ± 7 % (n = 3), respectively (data not shown).
Figure 7. Gβγ-binding agents.

Calcium current inhibition by noradrenaline (1 μM) in neurones expressing the C-terminal domain of βARK1 (A), a mutationally modified form of Gαi1 (Gαi1 Cys3Ser) (B) or α-transducin (C). The filled symbols indicate the inhibition ratio upon noradrenaline application. Insets show superimposed current traces collected at the indicated times (*). D, inhibition of ICa plotted against inhibition ratio in uninjected neurones (○) and in neurones expressing either the C-terminal domain of βARK1 (•) or Gαi1 Cys3Ser (▴). Cells recorded 24 h after intranuclear injection. Plasmid concentration, 200 μg ml−1.
Reconstitution of the α2-adrenoceptor-Gi pathway by the use of PTX-resistant Gαi mutants
To assess further the role of the Gi heterotrimer in mediating a component of noradrenergic inhibition, neurones were injected with cDNAs encoding PTX-resistant forms of Gαi1 (Wise et al. 1997b; Bahia et al. 1998). Cells were subsequently treated with PTX to eliminate interactions of α2-adrenoceptor(s) with the endogenous Gi/o population. As anticipated, there was no inhibition of ICa by 1 μM noradrenaline in control cells after PTX treatment (Fig. 8A). By contrast, in cells expressing the PTX-resistant Gαi1 Cys351Ile mutant, noradrenaline depressed ICa by 39.2 ± 4 % (n = 10) (Fig. 8B). Expression of another PTX-resistant Gαi1 mutant (Gαi1 Cys351Arg) that could not be activated by the α2A-adrenoceptor (Bahia et al. 1998) did not reconstitute noradrenergic inhibition (Fig. 8C). Interestingly, in most (7 out of 10) of the cells expressing Gαi1 Cys351Ile, noradrenaline-mediated inhibition was only weakly reversed by depolarization, as shown by the relatively low inhibition ratio (1.24 ± 0.05; Fig. 8D). In the other three cells the inhibition ratios were 1.9, 2.1 and 2.3, so about half the inhibition in these particular cells was voltage sensitive (Fig. 8D) - i.e. similar to control cells. Interaction of the Gαi1 Cys351Ile mutant with the α2-adrenoceptor(s) appeared to be very specific since expression of this subunit had no significant effects on M1 muscarinic inhibition of ICa (49 ± 4 % (n = 4) in control neurones and 56 ± 3 % (n = 5) in neurones expressing Gαi1 Cys351Ile) and failed to reconstitute the fast M4 mAChR inhibition in PTX-treated cells (n = 5) (Fig. 8B).
Figure 8. PTX-resistant Gαi1 mutants rescue noradrenergic inhibition.

A-C, cells pretreated with 1 μg ml−1 PTX for 24 h. Superimposed calcium current traces in the presence or absence of noradrenaline (1 μM) in an uninjected neurone (A) and in neurones expressing either Gαi1 Cys351Ile (B) or Gαi1 Cys351Arg (C) (100 μg ml−1). Lower panels in A and B show the time course of noradrenaline and oxotremorine-M (1 μM) actions. Note the absence of fast muscarinic (M4 mAChR) inhibition in B, inhibition being slow in onset. D, plot of inhibition of ICa in response to 1 μM noradrenaline against inhibition ratio in uninjected neurones (○, not treated with PTX) and in neurones expressing Gαi1 Cys351Ile (•, treated with PTX).
Degree and voltage dependence of inhibition as a function of Gαi1 plasmid concentration
To test whether the variability in the magnitude as well as the voltage dependence of Gi1 protein-mediated inhibition may result from different levels of expression of Gαi1 subunits from cell to cell, we injected SCG neurones with various concentrations (10-600 μg ml−1) of cDNA encoding Gαi1 Cys351Ile. Figure 9A and B clearly shows that the degree of inhibition as well as the fraction of voltage-dependent inhibition increased as a function of the concentration of the Gαi1 Cys351Ile plasmid. Increasing the plasmid concentration up to 600 μg ml−1, however, tended to block the inhibition (13 ± 4 %, n = 4), probably due to the excess of Gαi1 mutant with respect to activated Gβγ. The onset rate of the inhibition was also dependent upon Gαi1 Cys351Ile plasmid concentration, with time constants of 2.47 ± 0.2 s (n = 3) and 0.67 ± 0.17 s (n = 4) at 10 and 300 μg ml−1 plasmid concentration, respectively (Fig. 9D). Interestingly, the voltage-dependent fraction of Gi-mediated inhibition (with 300 μg ml−1 Gαi1 Cys351Ile plasmid) was largely prevented by co-expressing βARK1C-ter (200 μg ml−1) (Fig. 9C) while the time course of inhibition in these cells was not significantly altered (0.75 ± 0.2 s, n = 4) (Fig. 9D). Gαi1 Cys351Ile-mediated inhibition was mostly prevented by co-expression of α-transducin (7 ± 2 %, n = 4).
Figure 9. Degree, voltage dependence and onset rate of inhibition as a function of Gαi level.
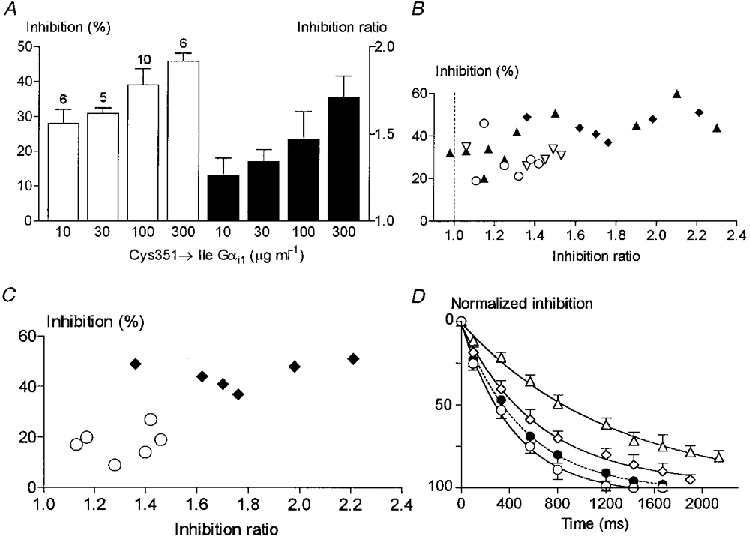
A, degree of inhibition in response to 1 μM noradrenaline (□) and inhibition ratio (▪) in neurones preinjected with various concentrations (10-300 μg ml−1 as indicated) of cDNA encoding Gαi1 Cys351Ile. B, relationship between inhibition and inhibition ratio in individual cells preinjected with 10 (○), 30 (▿), 100 (▴) and 300 μg ml−1 (♦) Gαi1 Cys351Ile-encoding plasmids. C, the same as in B in cells expressing Gαi1 Cys351Ile alone (300 μg ml−1, ♦) or together with the C-terminal domain of βARK1 (200 μg ml−1, ○). D, time course of inhibition in cells preinjected with Gαi1 Cys351Ile-encoding plasmid alone (10 μg ml−1, ▵; 30 μg ml−1, ⋄; 300 μg ml−1, ○) or together with βARK1C-ter (300 μg ml−1 Gαi1 Cys351Ile + 200 μg ml−1βARK1C-ter, •). Continuous lines are mono-exponential fits to the data.
DISCUSSION
In agreement with previous studies (Beech et al. 1992; Shapiro et al. 1994; Zhou et al. 1997), but now using perforated-patch recording, we have detected two pathways leading to ICa inhibition in rat SCG neurones following noradrenergic stimulation: a predominant one mediated via PTX-sensitive G-proteins that responds to low concentrations of noradrenaline (IC50∼150 nM); and one, more marginal, that needs higher concentrations of noradrenaline (IC50∼3 μM) and is resistant to PTX. These two inhibitory pathways could also be differentiated kinetically, in that the PTX-resistant pathway had slower onset kinetics (τ∼1 s) than the PTX-sensitive one (τ∼400 ms). Zhou et al. (1997) have suggested that these two adrenergic signals may correspond to the contribution of low and high affinity receptors, although there is no direct evidence so far that a G-protein-coupled receptor(s) is actually involved in the ‘low affinity’ PTX-insensitive response. A possible candidate for mediating PTX-insensitive noradrenergic inhibition would be Gz; however, reconstituted Gz coupling to Ca2+ channels in SCG neurones appeared to be voltage dependent and as much as 10-fold slower in onset/offset (Jeong & Ikeda, 1998) than the PTX-insensitive component determined here.
Further dissection of the PTX-sensitive noradrenergic inhibition suggests that more than one G-protein is involved in coupling the receptor(s) to the channel. Microinjection of antibodies directed against the C-terminal decapeptide sequence of Gα subunits revealed that both Go and Gi heterotrimers couple α2-adrenoceptors to Ca2+ channels. The effects of the anti-Gαi antibody cannot be ascribed to a non-specific action on Go-type G-proteins since this antibody had no significant effects on M4 mAChR inhibition of ICa which involves GoA (Delmas et al. 1998a). Since the anti-Gαo antibody was about twice as effective as the anti-Gαi antibody, the Go heterotrimer is likely to be the principle transducer (in agreement with the previous conclusion of Caulfield et al. 1994, from experiments using whole-cell recording). This inference is supported by the results obtained on expressing antisense RNAs to GαoA and Gαi subunits, which decreased α2-adrenoceptor inhibition with a similar potency ratio (Go > Gi) to the antibodies.
The participation of multiple PTX-sensitive G-proteins in α2-adrenoceptor modulation in rat SCG neurones is in agreement with a previous report showing that both Go and Gi heterotrimers are involved in adrenergic inhibition of Ca2+ currents in chick ganglion neurones (Diversé-Pierluissi et al. 1995), although the mechanistic basis may differ (see below). Earlier biochemical studies using exogenously expressed α2A-adrenoceptors have also shown that these receptors are tightly coupled to GoA and Gi1-3 with little discrimination (Grassie & Milligan, 1995; Wise et al. 1997b). Whether the two G-proteins actually couple to the same α2-adrenoceptor subtype in SCG neurones, or whether a different receptor subtype specifically interacts with each type of G-protein remains to be established.
As previously reported (Bean, 1989; Beech et al. 1992), reversal of α2-adrenergic inhibition by depolarization is incomplete, leaving part of the current still modulated (by 10-30 %) even at very positive voltages. According to the two-state ‘willing-reluctant’ model (see Jones & Elmslie, 1997), this ‘voltage-insensitive’ component of inhibition may reflect low affinity binding of activated G-protein (e.g. βγ) to the open state of the Ca2+ channel, thus being phenomenologically identical to the voltage-dependent mechanism. Alternatively, this may result from an intrinsically distinct intracellular pathway like that described by Diversé-Pierluissi et al. (1997). The experiments presented here suggest that incomplete reversal of inhibition largely reflects two mechanistically different actions: a voltage-dependent action of Go and a ‘voltage-independent’ (or less voltage-dependent) action of Gi. First, antibody neutralization of Gi heterotrimers and antisense depletion of Gαi subunits both induced a substantial increase in the proportional amount of voltage-dependent inhibition, indicative of a major role for the Go heterotrimer in this component. Conversely, neutralization of GoA heterotrimers strongly reduced the voltage-dependent fraction of inhibition, suggesting that Gi mainly acts in a ‘voltage-resistant’ manner. Second, when activation of endogenous G-proteins was prevented with PTX, the reconstituted inhibition obtained on expressing a PTX-resistant form of Gαi1 was clearly less voltage dependent than that normally seen when endogenous GαoA is allowed to participate in the inhibition (see Fig. 9).
There is now compelling evidence suggesting that the voltage-dependent inhibition of calcium currents produced by neurotransmitters results from the action of G-protein βγ subunits rather than the Gα subunits (Ikeda, 1996; Herlitze et al. 1996; Zamponi et al. 1997; Delmas et al. 1998a,b; Stephens et al. 1998; Zamponi & Snutch, 1998; see however Diversé-Pierluissi et al. 1997). The present results provide confirmatory evidence that this is true for Go-mediated inhibition by noradrenaline in SCG neurones. However, they further suggest that the less voltage-sensitive Gi-mediated noradrenergic inhibition is also mediated by βγ subunits, and that the two components of inhibition may involve different βγ subunits. The obligatory role of Gβγ is demonstrated by the finding that expression of α-transducin virtually abolished inhibition, whether mediated through endogenous G-proteins or through exogenously expressed Gi, whereas it had only a minor effect on M1 mAChR/Gαq inhibition. We also found that the expression of βARK1C-ter and Gαi1 Cys3Ser differentially reduced the magnitude and the voltage dependence of inhibition. The greater attenuation of inhibition and the parallel reduction of the voltage-dependent fraction by βARK1C-ter is compatible with the idea that it preferentially suppresses the predominant voltage-dependent Goβγ pathway. Conversely, the Gαi1 Cys3Ser mutant had the smallest effects on overall inhibition but enhanced the voltage-dependent fraction of inhibition, consistent with an action mainly directed against the ‘voltage-resistant’ Giβγ pathway. The simplest interpretation of this is that different βγ pairs bind to Gαo and Gαi respectively, and that the two βγ pairs, when released, act in different ways on the Ca2+ channels. The latter inference is supported by our previous observations showing that only βγ dimers liberated from GαoA (following antisense depletion), not from Gαi, induce (tonic) voltage-dependent inhibition (Delmas et al. 1998a; see also Fig. 6A). The participation of different βγ pairs is also substantiated by the finding that Gαi1 Cys351Ile-mediated inhibition could be resolved into a voltage-dependent component sensitive to both βARK1C-ter and α-transducin and a ‘voltage-independent’ component only sensitive to α-transducin. The increasing amount of (βARK1C-ter-sensitive) voltage-dependent inhibition with increasing Gαi1 Cys351Ile expression may be explained by the forced interaction of excess Gαi1 with some ‘inappropriate’βγ dimers: under more physiological conditions Gαi seems to couple preferentially to βARK1C-ter-insensitive βγ dimers.
Such a specificity between α and βγ pairs has been documented in earlier studies using antisense oligonucleotides. For example, in the rat pituitary cell line GH3, the M4 mAChR couples to the G-protein trimer consisting of αoAβ3γ4, the somatostatin receptor couples to the trimer αoBβ1γ3 and the galanin receptor preferentially couples to the trimer αoAβ2γ2 (Schneider et al. 1997). However, because of the difficulty of distinguishing the different β and γ isoforms immunocytochemically we did not attempt to define further the subunit composition of Go and Gi heterotrimers using antisense strategies. Nevertheless, since βARK1 is known to bind Gβ1 and Gβ2 subunits (Daaka et al. 1997), the sensitivity of the voltage-dependent Go-mediated inhibition to βARK1C-ter suggests βγ dimers containing β1 and/or β2 as possible transducers of this component of inhibition. This is supported by the recent observation that voltage-dependent inhibition of Ca2+ currents is replicated by exogenous expression of Gβ1 and Gβ2 subunits but not Gβ3 and Gβ4 subunits (García et al. 1998).
Voltage-dependent inhibition of neuronal N-type (α1B) and P/Q-type (α1A) Ca2+ channels is thought to result from the direct binding of some Gβγ dimers to the pore-forming α1 subunit. At least three putative sites of interaction have been identified: two of them, including the QXXER sequence common to many Gβγ-binding proteins, are located in the intracellular loop that connects transmembrane domains I and II of the Ca2+ channel α1 subunit (De Waard et al. 1997; Zamponi et al. 1997), and a third is located in the C-terminal domain (Qin et al. 1997). Since voltage-dependent and ‘voltage-independent’βγ-mediated inhibitions were both rapid, with indistinguishable onset rates, it seems likely that both resulted from a direct action of the different βγ dimers on the Ca2+ channel. The question then arises whether these biophysically distinct inhibitions involve different Gβγ binding sites or whether the same Gβγ binding site(s) recognizes different βγ combinations varying in their binding affinity and voltage dependence of their dissociation rates - i.e. whether Ca2+ channels exhibit different binding affinities for defined βγ dimers.
In conclusion, our data indicate that the βγ dimers derived from GoA or Gi heterotrimers are not equally effective in promoting voltage-dependent inhibition. They therefore favour the view that the intrinsic composition of the G-protein βγ complex (see also García et al. 1998) plays an important role in defining the voltage-dependent characteristics of Ca2+ current inhibition by membrane receptors.
Acknowledgments
We thank Dr Carol Harris and Dr Carol Scorer (Receptor Systems, Glaxo Wellcome) for the gift of the βARK1 minigene and Ms M. Dayrell for expert technical assistance. This work was supported by The Wellcome Trust and the UK Medical Research Council.
References
- Abogadie FC, Vallis Y, Buckley NJ, Caulfield MP. Use of antisense-generating plasmids to probe the function of signal transduction proteins in primary neurons. In: Challis RAJ, editor. Receptor Signal Transduction Protocols. Totowa, NJ, USA: Humana Press; 1997. pp. 217–225. [DOI] [PubMed] [Google Scholar]
- Bahia DS, Wise A, Fanelli F, Lee M, Rees S, Milligan G. Hydrophobicity of residue351 of the G-protein Gi1α determines the extent of activation by the α2A-adrenoceptor. Biochemistry. 1998;37:11555–11562. doi: 10.1021/bi980284o. [DOI] [PubMed] [Google Scholar]
- Bean BP. Neurotransmitter inhibition of neuronal calcium channels by changes in channel voltage dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bernheim L, Hille B. Pertussis toxin and voltage dependence distinguish multiple pathways modulating calcium channels of sympathetic neurons. Neuron. 1992;8:97–106. doi: 10.1016/0896-6273(92)90111-p. [DOI] [PubMed] [Google Scholar]
- Bernheim L, Beech DJ, Hille B. A diffusible second messenger mediates one of the pathways coupling receptors to calcium channels in rat sympathetic neurons. Neuron. 1991;6:859–867. doi: 10.1016/0896-6273(91)90226-p. [DOI] [PubMed] [Google Scholar]
- Caulfield MP, Jones S, Vallis Y, Buckley NJ, Kim G-D, Milligan G, Brown DA. Muscarinic M-current inhibition via Gαq/11 and α-adrenoceptor inhibition of Ca2+ current via Gαo in rat sympathetic neurones. The Journal of Physiology. 1994;477:415–422. doi: 10.1113/jphysiol.1994.sp020203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Pitcher JA, Richardson M, Stoffel RH, Robishaw JD, Lefkowitz RJ. Receptor and Gβγ isoform-specific interactions with G protein-coupled receptor kinases. Proceedings of the National Academy of Sciences of the USA. 1997;94:2180–2185. doi: 10.1073/pnas.94.6.2180. 10.1073/pnas.94.6.2180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Abogadie FC, Dayrell M, Haley JE, Milligan G, Caulfield MP, Brown DA, Buckley NJ. G-proteins and G-protein subunits mediating cholinergic inhibition of N-type calcium currents in sympathetic neurons. European Journal of Neuroscience. 1998a;10:1654–1666. doi: 10.1046/j.1460-9568.1998.00170.x. 10.1046/j.1460-9568.1998.00170.x. [DOI] [PubMed] [Google Scholar]
- Delmas P, Brown DA, Dayrell M, Abogadie FC, Caulfield MP, Buckley NJ. On the role of endogenous G-protein βγ subunits in Ca2+ current inhibition by neurotransmitters in rat sympathetic neurones. The Journal of Physiology. 1998b;506:319–329. doi: 10.1111/j.1469-7793.1998.319bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, Liu H, Walker D, Scott VES, Gurnett CA, Campbell KP. Direct binding of G-protein βγ complex to voltage-dependent calcium channels. Nature. 1997;385:446–450. doi: 10.1038/385446a0. 10.1038/385446a0. [DOI] [PubMed] [Google Scholar]
- Diversé-Pierluissi M, Goldsmith PK, Dunlap K. Transmitter-mediated inhibition of N-type calcium channels in sensory neurons involves multiple GTP-binding proteins and subunits. Neuron. 1995;14:191–200. doi: 10.1016/0896-6273(95)90254-6. 10.1016/0896-6273(95)90254-6. [DOI] [PubMed] [Google Scholar]
- Diversé-Pierluissi M, Remmers AE, Neubig RR, Dunlap K. Novel form of crosstalk between G protein and tyrosine kinase pathways. Proceedings of the National Academy of Sciences of the USA. 1997;94:5417–5421. doi: 10.1073/pnas.94.10.5417. 10.1073/pnas.94.10.5417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García DE, Li B, García-Ferreiro RE, Hernández-Ochoa EO, Yan K, Gautam N, Catterall WA, Mackie K, Hille B. G-protein β-subunit specificity in the fast membrane-delimited inhibition of Ca2+ channels. Journal of Neuroscience. 1998;18:9163–9170. doi: 10.1523/JNEUROSCI.18-22-09163.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldsmith P, Gierschick P, Milligan G, Unson CG, Vinitsky R, Malech H, Spiegel AM. Antibodies directed against synthetic peptides distinguish between GTP-binding proteins in neutrophil and brain. Journal of Biological Chemistry. 1987;262:14683–14688. [PubMed] [Google Scholar]
- Grassie MA, Milligan G. Analysis of the relative interactions between the α2C10 adrenoceptor and the guanine-nucleotide-binding proteins Go1α and Gi2α following co-expression of these polypeptides in rat 1 fibroblasts. Biochemical Journal. 1995;306:525–530. doi: 10.1042/bj3060525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haley JE, Abogadie FC, Delmas P, Dayrell M, Vallis Y, Milligan G, Caulfield MP, Brown DA, Buckley NJ. The α subunit of Gq contributes to muscarinic inhibition of the M-type potassium current in sympathetic neurons. Journal of Neuroscience. 1998;18:4521–4531. doi: 10.1523/JNEUROSCI.18-12-04521.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, García DE, Mackie K, Hille B, Scheuer T, Catterall WA. Modulation of Ca2+ channels by G-protein βγ subunits. Nature. 1996;380:258–262. doi: 10.1038/380258a0. 10.1038/380258a0. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends in Neurosciences. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. The Journal of Physiology. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda SR. Voltage-dependent modulation of N-type calcium channels by G-protein βγ subunits. Nature. 1996;380:255–258. doi: 10.1038/380255a0. 10.1038/380255a0. [DOI] [PubMed] [Google Scholar]
- Jeong S-W, Ikeda SR. G protein α subunit Gαz couples neurotransmitter receptors to ion channels in sympathetic neurons. Neuron. 1998;21:1201–1212. doi: 10.1016/s0896-6273(00)80636-4. 10.1016/S0896-6273(00)80636-4. [DOI] [PubMed] [Google Scholar]
- Jones SW, Elmslie KS. Transmitter modulation of neuronal calcium channels. Journal of Membrane Biology. 1997;155:1–10. doi: 10.1007/s002329900153. 10.1007/s002329900153. [DOI] [PubMed] [Google Scholar]
- Koch WJ, Hawes BE, Inglese J, Luttrell LM, Lefkowitz RJ. Cellular expression of the carboxyl terminus of a G protein-coupled receptor kinase attenuates Gβγ-mediated signaling. Journal of Biological Chemistry. 1994;269:6193–6197. [PubMed] [Google Scholar]
- Limbird L. The Alpha 2 Adrenergic Receptors. Clifton, NJ, USA: Humana Press; 1988. [Google Scholar]
- McFadzean I, Mullaney I, Brown DA, Milligan G. Antibodies to GTP binding protein, Go, antagonize noradrenaline-induced calcium current inhibition in NG 108-15 hybrid cells. Neuron. 1989;3:177–182. doi: 10.1016/0896-6273(89)90030-5. 10.1016/0896-6273(89)90030-5. [DOI] [PubMed] [Google Scholar]
- Milligan G, Mullaney I, McCallum F. Distribution and relative levels of expression of the phosphoinositidase-C-linked G-proteins Gqα and G11α: absence of G11α in human platelets and haemopoietically derived cell lines. Biochemica et Biophysica Acta. 1993;1179:208–212. doi: 10.1016/0167-4889(93)90143-d. 10.1016/0167-4889(93)90143-D. [DOI] [PubMed] [Google Scholar]
- Plummer MR, Logothetis DE, Hess P. Elementary properties and pharmacological sensitivities of calcium channels in mammalian peripheral neurons. Neuron. 1989;2:1453–1463. doi: 10.1016/0896-6273(89)90191-8. [DOI] [PubMed] [Google Scholar]
- Qin N, Platano D, Olcese R, Stefani E, Birnbaumer L. Direct interaction of Gβγ with a C-terminal Gβγ-binding domain of the Ca2+ channel α1 subunit is responsible for channel inhibition by G protein-coupled receptors. Proceedings of the National Academy of Sciences of the USA. 1997;94:8866–8871. doi: 10.1073/pnas.94.16.8866. 10.1073/pnas.94.16.8866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider T, Igelmund P, Hescheler J. G protein interaction with K+ and Ca2+ channels. Trends in Pharmacological Sciences. 1997;18:8–11. doi: 10.1016/s0165-6147(96)01001-2. 10.1016/S0165-6147(96)01001-2. [DOI] [PubMed] [Google Scholar]
- Shapiro MS, Wollmuth LP, Hille B. Modulation of Ca2+ channels by PTX-sensitive G-proteins is blocked by N-ethylmaleimide in rat sympathetic neurons. Journal of Neuroscience. 1994;14:7109–7116. doi: 10.1523/JNEUROSCI.14-11-07109.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens GJ, Brice NL, Berrow NS, Dolphin AC. Facilitation of rabbit α1B calcium channels: involvement of endogenous Gβγ subunits. The Journal of Physiology. 1998;509:15–27. doi: 10.1111/j.1469-7793.1998.015bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise A, Grassie MA, Parenti M, Lee M, Rees S, Milligan G. A cysteine-3 to serine mutation of the G-protein Gi1α abrogates functional activation by the α2A-adrenoceptor but not interactions with the βγ complex. Biochemistry. 1997a;36:10620–10629. doi: 10.1021/bi9702997. [DOI] [PubMed] [Google Scholar]
- Wise A, Watson-Koken MA, Rees S, Lee M, Milligan G. Interactions of the α2A-adrenoceptor with multiple Gi-family G-proteins: studies with pertussis toxin-resistant G-protein mutants. Biochemical Journal. 1997b;321:721–728. doi: 10.1042/bj3210721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Nelson D, Nargeot J, Snutch TP. Crosstalk between G proteins and protein kinase C mediated by the calcium channel α1 subunit. Nature. 1997;385:442–446. doi: 10.1038/385442a0. 10.1038/385442a0. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Snutch TP. Decay of prepulse facilitation of N type calcium channels during G protein inhibition is consistent with binding of a single Gβγ subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:4035–4039. doi: 10.1073/pnas.95.7.4035. 10.1073/pnas.95.7.4035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J, Shapiro MS, Hille B. Speed of Ca2+ channel modulation by neurotransmitters in rat sympathetic neurons. Journal of Neurophysiology. 1997;77:2040–2048. doi: 10.1152/jn.1997.77.4.2040. [DOI] [PubMed] [Google Scholar]