

Abstract
Transepithelial transducing cells, particularly the gastrin (G) cell, co-ordinate gastric acid secretion with the arrival of food in the stomach. Recent work suggests that multiple active products are generated from the gastrin precursor, and that there are multiple control points in gastrin biosynthesis. Biosynthetic precursors and intermediates (progastrin and Gly-gastrins) are putative growth factors; their products, the amidated gastrins, regulate epithelial cell proliferation, the differentiation of acid-producing parietal cells and histamine-secreting enterochromaffin-like (ECL) cells, and the expression of genes associated with histamine synthesis and storage in ECL cells, as well as acutely stimulating acid secretion. Gastrin also stimulates the production of members of the epidermal growth factor (EGF) family, which in turn inhibit parietal cell function but stimulate the growth of surface epithelial cells. Plasma gastrin concentrations are elevated in subjects with Helicobacter pylori, who are known to have increased risk of duodenal ulcer disease and gastric cancer. Studies of the physiology of gastrin may therefore contribute to an understanding of the mechanisms relevant to major upper gastrointestinal tract disease.
The interior face of the stomach is lined by one of the most highly organized populations of epithelial cells in the body. These cells are responsible for the secretion of acid, pepsinogen and mucus into the lumen. Epithelial function is regulated by systems that match secretion to the presence of food in the lumen, that control the expression of genes associated with secretory function, and that determine the renewal of cells and the proportions of different cell types. In recent years the application of molecular cell physiological approaches, including transgenesis, has shed new light on the relevant endocrine and paracrine control systems. This review focuses on recent progress in understanding how endocrine cell signals, and in particular the gastrins, influence the organization and function of the acid-secreting epithelium.
Architecture of the corpus mucosa
The gastric epithelium is folded into glands, which are the distinctive feature of the gastric mucosa. In the body or corpus of the stomach, the major differentiated glandular cell types are parietal (or oxyntic) cells secreting HCl, zymogen (chief or peptic) cells secreting pepsinogen, and surface epithelial/mucus neck cells secreting mucus glycoprotein. Intrinsic factor is secreted by parietal cells in man, and by chief cells in mouse. The corpus epithelium also contains endocrine cells that regulate acid secretion through paracrine mechanisms, the most important being histamine-releasing enterochromaffin-like (ECL) cells and somatostatin-releasing D-cells. In the glands of the pyloric antral part of the stomach, parietal and ECL cells are absent, but there is an additional endocrine cell type - the gastrin (G) cell (Walsh, 1994). In the proximal stomach of some species, e.g. the rat, there is squamous (or non-glandular) epithelium, which is not considered here.
A single multipotent class of stem cell gives rise to the various differentiated cell types of the epithelium. Proliferative cells are located in the isthmus region of the gland. Some cells migrate upwards, becoming mucus-secreting surface epithelial cells and surviving 2-3 days; other cells within the proliferative zone differentiate into parietal, chief or ECL cells and subpopulations migrate towards the base of the gland, surviving 50-190 days. The proliferation and differentiation of epithelial cells are determined partly by interactions between different cell types (Karam, 1995). Mature parietal cells, in particular, seem to determine the fate of other epithelial cells. For example, parietal cells have been selectively lesioned in transgenic mice by using the promoter of the H+,K+-ATPase β-subunit gene to direct expression to parietal cells of toxic agents such as diphtheria toxin, and herpes virus thymidine kinase - which generates a toxic product on treatment with the drug ganciclovir (Li et al. 1996; Canfield et al. 1996). The loss of parietal cells in these animals is also associated with loss of chief cells, even though the transgene is not expressed in zymogen cells, indicating interactions between these cells in determining their differentiation. In addition to these interactions, however, it is also clear that the pyloric antral hormone gastrin regulates not just acid secretion, but also the proliferation of gastric epithelial cells (Johnson, 1988; Ohning et al. 1996) and the relative numbers of different cell populations in the epithelium, thereby influencing secretory capacities (Koh et al. 1997; Friis-Hansen et al. 1998).
Antral-corpus interactions control acid secretion
The events leading to increased gastric acid secretion during digestion are initiated by cephalic stimuli that activate vagal efferent pathways. Thereafter, food in the stomach maintains acid secretion by activating both neural (local and vago-vagal) and endocrine reflexes. The endocrine systems of the corpus and antrum contribute in separate ways. Gastrin is the major output of the antral endocrine cell system and is a major stimulant of acid secretion. The G- and D-cells of the pyloric antral mucosa act in concert as transepithelial transducers monitoring luminal nutrients (protein and amino acids) and pH, respectively. Somatostatin is secreted from antral D-cells when luminal pH falls below 3·5; it acts by a paracrine mechanism to suppress G-cell function, thereby completing a negative feedback loop controlling acid secretion. A reduction in acid secretion, for example following the administration of proton pump inhibitors such as omeprazole, inhibits D-cells and so upregulates G-cells (Brand et al. 1988; Wu et al. 1990; Dimaline et al. 1991).
The two main endocrine cell types of the corpus differ from antral endocrine cells in that they seem not to respond directly to luminal chemicals. Instead, they act as integrators of neurohumoral signals. Thus gastrin releases histamine from ECL cells, which in turn increases acid secretion by acting at parietal cell histamine-H2 receptors. Both parietal and ECL cells are inhibited by somatostatin released from corpus D-cells in response to a variety of neurohumoral agents, e.g. noradrenaline, vasoactive intestinal polypeptide (VIP), calcitonin gene-related peptide (CGRP) and cholecystokinin (CCK). The somatostatin receptor type 2 (SSTR2) is important in mediating these effects, since mice in which the SSTR2 gene has been deleted exhibit increased basal and stimulated acid secretion (Martinez et al. 1998).
Multiple gastrins and receptors
It used to be thought that the main active forms of gastrin were the COOH-terminally amidated gastrins of 34 and 17 amino acid residues (G34 and G17) first characterized over 25 years ago (Gregory & Tracy, 1964, 1972). Recent studies indicate that this is an oversimplification: there is an unexpected variety both in the identity of the active products generated from the gastrin precursor during biosynthesis, and in the responses to these peptides (Fig. 1A and B).
Figure 1. Progastrin processing.
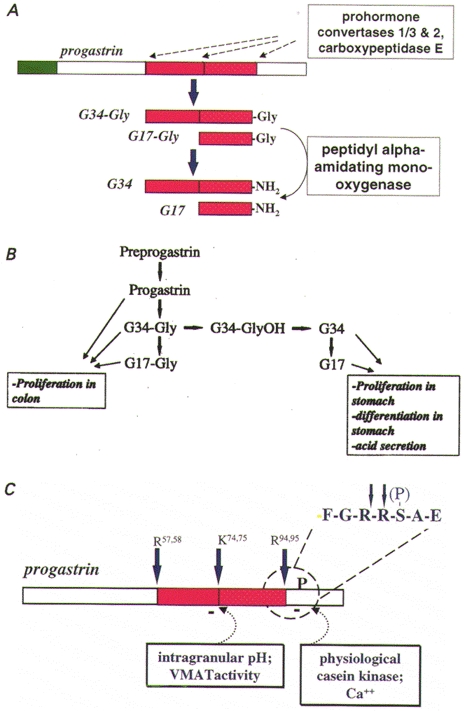
A, schematic of the main products of preprogastrin processing. The signal sequence (green) of preprogastrin is cleaved in the endoplasmic reticulum and progastrin then passes through the Golgi complex to secretory vesicles where it is cleaved (dashed arrows) and the COOH-terminus amidated; processing enzymes are indicated in the boxes. B, the pathways of post-translational processing of rat preprogastrin as revealed by pulse-chase kinetic studies are shown together with a summary of the main biological properties of the various progastrin-derived peptides. C, putative mechanisms regulating progastrin processing are shown: selected cleavages are inhibited by raising intravesicular pH and by phosphorylation by physiological casein kinase. The inset shows the primary sequence of the cleavage/phosphorylation site in the single-letter notation. Cleavage sites (Arg57,58, Lys74,75, Arg94,95) are also identified in the single-letter notation.
The gastrin family of peptides
The rate of clearance of G17 from the circulation is about five times that of G34 (Walsh et al. 1974). The release of G34 therefore leads to changes in plasma concentrations that are higher and more persistent compared with the secretion of equimolar amounts of G17. The COOH-terminal amide moiety of these peptides has long been thought to be essential for the stimulation of acid secretion and cell proliferation (Johnson, 1988; Walsh, 1994). This view has now been revised and extended with the observation that gastrins with COOH-terminal glycine (G-Gly), and intact progastrin, also stimulate proliferation (Seva et al. 1994; Wang et al. 1996; Koh et al. 1999). In addition, G-Gly increases expression of the parietal cell proton pump gene (Todisco et al. 1995), but neither G-Gly nor progastrin is an acid secretagogue on its own. Because G-Gly and progastrin are biosynthetic precursors of the amidated peptides, the control of progastrin processing allows the production of multiple, alternative, active products from a single precursor molecule (Dockray et al. 1996) (Fig. 1B).
Gastrin biosynthesis
Experiments using pulse-chase labelling protocols show that like other peptide hormones, the gastrin precursor, preprogastrin is translated at the endoplasmic reticulum (ER) where the signal peptide is rapidly removed (Varro & Dockray, 1993). Progastrin then progresses through the Golgi complex. In the trans-Golgi network (TGN), tyrosine-86 may be sulphated and serine-96 may be phosphorylated (Varro et al. 1994). The cleavage of progastrin at two pairs of Arg residues (positions 57 and 58, and 94 and 95) occurs relatively rapidly after exit from the TGN (t½∼10 min) to secretory vesicles (Varro et al. 1995); further cleavage at a pair of Lys residues (positions 74 and 75) occurs more slowly (t½∼60 min). These cleavages are thought to be mediated by the prohormone convertases PC1/3 and PC2 (Macro et al. 1997) (Fig. 1A). The cleavage products terminate in a pair of COOH-terminal basic residues that are removed by carboxypeptidase E (CPE). A mis-sense mutation in the CPE gene in Cpefat/Cpefat mice leads to substantially reduced tissue concentrations of the amidated gastrins and an accumulation of processing intermediates terminating in -Arg-Arg (Udupi et al. 1997).
The products of proteolysis in G-cells are peptides of 18 and 35 residues with a COOH-terminal Gly residue (i.e. G17-Gly and G34-Gly). The COOH-terminal Gly-gastrins are converted to amidated gastrins by the action of the bifunctional enzyme peptidyl α-amidating mono-oxygenase (PAM). The mono-oxygenase domain of PAM first converts the Gly residue to Gly-OH in a reaction requiring copper, ascorbate and molecular oxygen (Fig. 2). This intermediate is then converted by the lyase domain of PAM to the corresponding COOH-terminal amide (Eipper et al. 1993). In rat G-cells, amidation continues up to about 80 min after the arrival of progastrin in secretory vesicles and then stops, even though about 50 % of the substrate remains unconverted at this time (Varro et al. 1995). Kinetic studies suggest that in the rat the major intermediate, G34-Gly, is either converted via G34-GlyOH to G34, or alternatively cleaved to G17-Gly (Fig. 1B). The amidated peptide G34 may in turn be cleaved to G17, but the evidence suggests that G17-Gly is not normally converted to G17 (Varro et al. 1995).
Figure 2. Schematic representation of a secretory vesicle containing progastrin and processing enzymes indicating the major steps and requirements for processing.
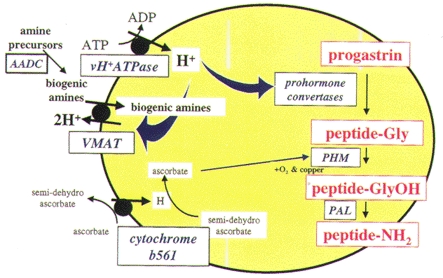
Internal pH is approximately 5·5 due to the activity of the vacuolar proton pump (vH+-ATPase), but proton extrusion via vesicular monoamine transporters (VMAT) may raise intragranular pH and inhibit cleavage. Biogenic amines transported by VMATs are generated from their precursors by aromatic amino acid decarboxylase (AADC). The bifunctional enzyme peptidyl α-amidating mono-oxygenase (PAM) converts peptides with COOH-terminal -Gly into the corresponding amide in a 2-step reaction; the peptidyl α-hydroxylating mono-oxygenase (PHM) domain of PAM generates a hydroxy-Gly intermediate that is the substrate for the peptidyl α-hydroxyglycine α-amidating lyase (PAL) domain of the enzyme.
Modulation of progastrin processing
Because the cleavage and amidation of progastrin-derived peptides occur in vesicles of the regulatory pathway of exocytosis, there is impaired post-translational processing in cells in which these vesicles are absent. In such cells, progastrin passes directly from the TGN to the cell surface via the constitutive route of secretion (Dockray et al. 1996). This is a feature of colon cancer cells, which often express the gastrin gene (Nemeth et al. 1993; Nakata et al. 1998). Progastrin and G-Gly produced in these cells may act as autocrine growth factors, since antisense inhibition of gastrin gene expression, or gastrin immunoneutralization, inhibits cell growth (Watson et al. 1996; Singh et al. 1996).
Several different mechanisms have now been identified which determine the post-translational processing of progastrin-derived peptides in endocrine cells (Fig. 1C). (a) A proportion of progastrin is normally phosphorylated at Ser-96 (Dockray et al. 1987). Phosphorylation appears to be mediated by a TGN-resident kinase which resembles physiological casein kinase; depletion of calcium stores inhibits kinase activity, suggesting a possible mechanism to modulate prohormone phosphorylation (Fig. 1C). Site-directed mutagenesis of either Ser-96 (to Ala), or of the kinase recognition site (Glu-98 to Ala), to abolish phosphorylation, increased cleavage at Arg-94/95, so that phosphorylation of Ser-96 has inhibitory effects on subsequent cleavage (Bishop et al. 1998). (b) In antral G-cells, there is regulated expression of the genes encoding the prohormone convertases PC1/3 and PC2, which mediate progastrin cleavage. Inhibition of acid secretion with proton pump inhibitors leads to upregulation of the mRNAs encoding PC1/3 and PC2, as well as gastrin. This is associated with rapid cleavage at Lys74/75, but not at Arg57/58 or Arg94/95 (Macro et al. 1996, 1997). (c) Endocrine cell secretory vesicles are thought to have an internal pH of about 5·5, and the prohormone convertases that act in these vesicles have acidic pH optima (Fig. 1C). Raising intravesicular pH inhibits cleavage of progastrin-derived peptides at Lys-74/75, but not Arg-57/58 or Arg-94/95 (Voronina et al. 1997). G-cell secretory granules express the vesicular monoamine transporter type 1 (VMAT1), which transports biogenic amines like dopamine and serotonin into secretory vesicles in exchange for two protons, and amine loading leads to inhibition of Lys-74/75 cleavage (Hussain et al. 1999). Dietary amines such as tyramine, which are membrane permeable in their uncharged form, also inhibit G34 cleavage, although this effect is not reversed by VMAT inhibitors. Together these observations suggest that dietary and biogenic amines modulate the cleavage of progastrin-derived peptides secondary to raising intragranular pH (Figs 1C and 2).
How many receptors for gastrin-related peptides?
The amidated gastrins act at the gastrin/CCK-B receptor which is a member of the G-protein-coupled, seven transmembrane domain, receptor family. This receptor is expressed by gastric epithelial cells and by neurons in the central nervous system (Kopin et al. 1992; Wank et al. 1992a). It has similar affinities for gastrin and the structurally related brain-gut peptide, CCK; the latter is the main endogenous ligand in the CNS, and gastrin is considered to be the main peripheral ligand. In contrast, gastrins have low affinity, and CCK has high affinity, at the CCK-A receptor found on gall bladder and pancreas (Wank et al. 1992b). Activation of gastrin/CCK-B receptors increases in both ECL and parietal cells (Prinz et al. 1993, 1994; Hersey & Sachs, 1995). This is thought to be a major signalling pathway in parietal cells, but in ECL cells there is evidence that gastrin also stimulates the p42/44 MAP kinase pathway (Kinoshita et al. 1998). Unlike parietal cells, ECL cells (at least in rodents) retain the capacity to proliferate in response to gastrin, perhaps due to activation of the p42/44 MAP kinase pathway.
In addition to the gastrin/CCK-B receptor, there is evidence for at least two other putative receptors for progastrin-derived peptides. First, some colon cancer cell lines express a 78 kDa protein structurally related to fatty acid oxidation enzymes, which is reported to function as a receptor for both amidated and Gly-gastrins, but its physiological significance is uncertain (Baldwin, 1994). Second, pharmacological and binding studies in several cell lines indicate receptors that have high affinity for G-Gly (Seva et al. 1994; Singh et al. 1995; Hollande et al. 1997). The receptors in this class have not yet been cloned, and there remains some doubt as to their relative affinity for amidated gastrin. They are considered interesting, however, because they may mediate the trophic effects of progastrin derivatives in various gastrointestinal cancers.
Gastrin in genetically modified mice
The physiology of progastrin-derived peptides has recently been clarified by studies in which the gastrin gene is over-expressed in transgenic mice, or in which the gastrin (GAS-KO) or gastrin/CCK-B receptor (GAS/CCK-BR-KO) genes have been subjected to targeted deletion. In transgenic mice expressing the gastrin gene in pancreatic β-cells (INS-GAS mice), the product is processed in a similar way to antral G-cells but synthesis and release are independent of the normal control mechanisms. In mice expressing the progastrin gene in the liver (H-GAS mice), the main product is intact progastrin (Wang et al. 1996). INS-GAS mice exhibit increased cell proliferation and thickness in both stomach and colon, whereas H-GAS mice exhibit increased growth of the colonic mucosa, but not of the stomach (Wang et al. 1996).
There are complementary gastric phenotypes in GAS-KO and GAS/CCK-BR-KO mice. In both cases, there are reduced numbers of parietal and ECL cells and reduced acid secretion (Nagata et al. 1996; Langhans et al. 1997; Koh et al. 1997; Friis-Hansen et al. 1998). Acid secretion in GAS-KO mice is refractory to acute administration of gastrin, acetylcholine and histamine (Friis-Hansen et al. 1998), suggesting that gastrin is required for the maturation of parietal cells. In keeping with this idea, infusion of G17 for 7 days induced acid secretion in GAS-KO mice.
It has been clear for many years that exogenous gastrin stimulates cell proliferation (Johnson, 1988). Moreover, immunoneutralization of gastrin in rats inhibits the increased gastric mucosal proliferation after feeding (Ohning et al. 1996) and, as already mentioned, the moderate hypergastrinaemia in INS-GAS mice is associated with increased gastric epithelial proliferation (Wang et al. 1996). It was therefore somewhat surprising to find that rates of gastric epithelial cell proliferation were similar in GAS-KO and wild-type mice fed ad libitum (Koh et al. 1997). There are, however, differences in the proportions of the different cell types: thus, while parietal and ECL cells are decreased in GAS-KO and GAS/CCK-BR-KO mice, the mucus-secreting neck and surface epithelial cells are increased. It seems, then, that gastrin influences cell kinetics in two ways: it is one of a variety of factors that increase proliferation, but in addition it plays a role in determining the differentiation of parietal and ECL cells from a precursor pool (Fig. 3A). Because parietal cells are terminally differentiated and do not divide, the regulatory action of gastrin in determining the numbers of these cells presumably includes events that lead to inhibition of cell division and stimulation of the expression of genes characteristic of parietal cells. Further work is now needed to define how gastrin might stimulate the proliferation of some epithelial cells while inhibiting proliferation and promoting differentiation of others.
Figure 3. Cellular interactions in the gastric epithelium.
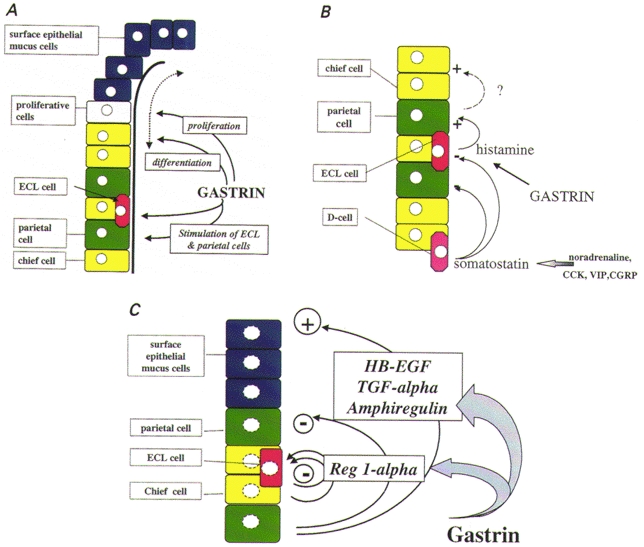
A, schematic representation of an acid-secreting gastric gland, showing the main classes of epithelial cell and sites of action of gastrin. Proliferation in the isthmus region followed by cell migration gives rise to mucus-secreting neck and surface cells (blue), and the main glandular cell types, i.e. parietal (green), chief (yellow) and enterochromaffin-like (ECL) cells (red). Gastrin stimulates proliferation and the differentiation of parietal and ECL cells; it also stimulates acid secretion from parietal cells, at least in part by increasing histamine synthesis and release in ECL cells. B, cellular interactions in the gastric epithelium. Gastrin stimulates histamine release from ECL cells which acts on parietal cells. Various neurohumoral agents (noradrenaline, cholecystokinin (CCK), vasoactive intestinal polypeptide (VIP) and calcitonin gene-related peptide (CGRP) stimulate somatostatin release from D-cells, which inhibits ECL and parietal cells. By an unknown mechanism, the latter also regulate chief cell numbers. C, remodelling of gastric glands with prolonged stimulation by gastrin. Hypergastrinaemia increases the expression of EGF family members such as heparin-binding EGF (HB-EGF), amphiregulin and transforming growth factor (TGF)-α in parietal cells, and Reg-1α in chief and ECL cells. EGF-related peptides inhibit acid secretion and depress parietal cell numbers, but increase surface epithelial cell numbers. Inactivating mutations of Reg-1α occur in ECL cell tumours, suggesting that this peptide also has autocrine growth inhibitory effects, although it is a stimulant of the growth of surface mucus cells.
Enterochromaffin-like cells
Gastrin rapidly releases histamine from isolated enterochromaffin-like (ECL) cells (Prinz et al. 1993, 1994). In the perfused rat stomach, histamine secretion is decreased after 2-3 h of gastrin stimulation, presumably due to depletion of stores (Ronning et al. 1996). The maintenance of continued secretion over longer periods, and the replenishment of stores, therefore requires histamine synthesis. This is achieved by increased histidine decarboxylase activity (HDC), which converts histidine to histamine (Kahlson & Rosengren, 1971), probably secondary to regulation at both translational and transcriptional levels. Increased HDC mRNA abundance is detectable 0·5-2 h after raising plasma gastrin by refeeding in fasted rats (Dimaline & Sandvik., 1991; Dimaline et al. 1993b). Over several days, raised plasma gastrin concentrations lead to parallel increases in the abundance of mRNA species encoding HDC and vesicular monoamine transporter type 2 (VMAT2), which transports histamine from cytosol into secretory vesicles (Dimaline & Struthers, 1996), and chromogranin A (CGA), which is a major vesicle protein and a putative precursor for auto- or paracrine inhibitor peptides including pancreastatin (Dimaline et al. 1993a).
Gastrin stimulates HDC and CGA gene expression by different mechanisms. In the human gastric cancer cell line AGS-B (transfected with the gastrin/CCK-B receptor), gastrin increases expression of human HDC promoter- reporter constructs via a protein kinase C-mediated pathway acting at a novel enhancer-like response element +2 to +24 bases downstream of the transcriptional start site (Zhang et al. 1996). In contrast, stimulation of chromogranin A gene expression by gastrin in the same system is mediated by a cAMP response element (CRE) and phosphorylation of the CRE binding protein (CREB) (Hocker et al. 1998).
Gastrin and ECL cell growth
Some ECL cells are produced in the absence of gastrin, e.g. in GAS-KO and GAS/CCK-BR-KO mice, but gastrin is nevertheless an important regulator of ECL cell number. In hypergastrinaemia there is ECL cell hyperplasia and in extreme cases, the development of ECL cell carcinoid tumours (Bordi et al. 1995). Hyperplasia of ECL cells is detectable in rats within 9 days of hypergastrinaemia due to treatment with omeprazole (Tielemans et al. 1989), and is reported in patients receiving omeprazole for up to 5 years (Lamberts et al. 1993). The development of ECL cell carcinoid tumours occurs after 12-18 months in rats with hypergastrinaemia due to continuous administration of certain H2-receptor antagonists or proton pump inhibitors (Bordi et al. 1995). These tumours also occur spontaneously in the African rodent Mastomys, in which three species-specific point mutations in the gastrin/CCK-B receptor lead to its constitutive activation (Schaffer et al. 1998).
Patients with pernicious anaemia typically have hypergastrinaemia secondary to achlorhydria due to the loss of parietal cells. Approximately 5 % of these patients develop ECL cell carcinoid tumours, which often resolve after surgical resection of the antrum to lower plasma gastrin levels (Bordi et al. 1995; Higham et al. 1998). ECL cell carcinoid tumours also occur in up to 30 % of patients with hypergastrinaemia due to a gastrin-producing tumour as part of the multiple endocrine neoplasia type 1 syndrome (MEN1) (Debelenko et al. 1997). Mutations of the menin gene account for MEN1 syndrome and the protein is thought to act as a tumour suppressor; however the nature of the physiological relationship between menin and gastrin remains unclear (Chandrasekharappa et al. 1997).
A second putative tumour suppressor for ECL cell carcinoids has recently been identified; mutations of the gene encoding Reg1-α have been found in several ECL cell carcinoid tumours (Higham et al. 1999). Reg1-α is normally expressed in pancreas and is upregulated during differentiation of islet β-cells. Recent studies indicate it is also a growth factor for gastric mucus cells (Fukui et al. 1998). In rat, it is expressed in ECL cells, in man in ECL and chief cells, and in both species its expression is increased with hypergastrinaemia (Asahara et al. 1996; Higham et al. 1999). In some ECL cell carcinoid tumours there is a mutation of the initiator methionine which disrupts the signal peptide and prevents sequestration into the secretory pathway. Together these observations are consistent with the idea that Reg1-α might be an autocrine inhibitor of ECL cell growth and a stimulant of mucus cell growth, and might therefore mediate the effects of gastrin in modelling the gastric epithelium (Fig. 3C).
Parietal cell function
The question of whether gastrin stimulates acid secretion by acting directly on parietal cells or indirectly via histamine release from ECL cells (Fig. 3B) has attracted intense debate over many years (Black & Shankley, 1987). Gastrin receptors are expressed by parietal cells, gastrin modestly increases aminopyrine accumulation in isolated parietal cells (equivalent to acid secretion), and this is enhanced by histamine (Soll et al. 1984). However, quantitative pharmacological studies suggest that in the intact mucosa in vivo the predominant action of gastrin on acid secretion is secondary to histamine release (Black & Shankley, 1987). Aside from the importance of the acute effect of gastrin on parietal cells for acid secretion, the observations in GAS-KO mice cited above indicate that gastrin may act over longer periods to sensitize parietal cells to other acid secretagogues (Friis-Hansen et al. 1998).
Acid secretion and parietal cell growth and differentiation
The stimulation of acid secretion depends on the translocation into an apical canalicular system of the H+,K+-ATPase (Forte & Yao, 1996). The proton pump is subsequently retrieved into tubulovesicles on cessation of stimulation. Retrieval of the H+,K+-ATPase β-subunit depends on a cytoplasmic tyrosine-based endocytosis signal. The expression in transgenic mice of a mutated version of this sequence, which inhibits retrieval, leads to chronic acid hypersecretion, the development of peptic ulcers and an increased thickness of the mucosa (Courtois-Coutry et al. 1997). Intracellular pH is controlled in part by Na+-H+ exchanger type 2 (NHE2) in the basolateral membrane. This isoform is stimulated at an extracellular alkaline pH and so is expected to be active during acid secretion when bicarbonate is released at the basolateral membrane via Cl−-HCO3− exchange. In mice in which the NHE2 gene has been disrupted in exon 2, there are reduced numbers of parietal cells and zymogen cells (Schultheis et al. 1998). Thus, the homeostatic mechanisms that maintain parietal cell function during and after stimulation of acid secretion also have consequences over longer periods for maintaining parietal cell numbers and therefore epithelial organization more generally.
Corpus epithelial organization is likely to be influenced by reflex changes in G-cell function, e.g. hypergastrinaemia secondary to decreased parietal cell function, and interlinked changes in the production of soluble growth factors. Thus, gastrin stimulates the production of members of the epidermal growth factor (EGF) family, notably transforming growth factor (TGF)-α, amphiregulin and heparin-binding EGF (HB-EGF) (Murayama et al. 1995; Tsutsui et al. 1997; Miyazaki et al. 1999). Overexpression of TGF-α in transgenic mice produces a phenotype that resembles the human condition of Menetrier's disease, characterized by an increased growth of the gastric mucosa and an expansion of the mucus cell population of the gastric pit, but also by the inhibition of acid secretion, a decrease in mature parietal and chief cells and in the expression of the H+,K+-ATPase and pepsinogen (Takagi et al. 1992; Sharp et al. 1995). Direct evidence linking endogenous gastrin to the control of EGF-related peptides in the stomach is provided by the observation that the moderate hypergastrinaemia in INS-GAS mice initially (for the first 3 months) leads to increased acid secretion and parietal cell numbers, but in older INS-GAS mice (after 12 months) there is a progressive depression of acid secretion, loss of parietal cells, increased expression of EGF-family growth factors, and increased expansion of the surface epithelial cell population, which eventually progresses to an invasive gastric carcinoma (Wang et al. 1999). This sequence of events is accelerated by infection with Helicobacter. The loss of parietal cells with prolonged hypergastrinaemia resembles the human condition of atrophic gastritis. It is widely appreciated that reduced acid secretion due to the loss of parietal cells leads in turn to increased plasma gastrin secondary to depression of the acid-inhibitory control mechanisms working on G-cells. In INS-GAS mice, however, moderate hypergastrinaemia precedes the development of atrophic gastritis. At first sight, it would seem to be paradoxical to suggest a causal relationship between elevated plasma gastrin and atrophic gastritis, given the actions of gastrin in increasing parietal cell numbers. It is, however, compatible with the idea that while gastrin initially stimulates parietal cells, with prolonged stimulation there is an upregulation of members of the EGF family of growth factors. The latter provide a local mechanism restraining acid secretion and decreasing parietal cell numbers, while stimulating the growth of surface epithelial cells (Fig. 3C).
Overview and implications
The products of the G-cell act as transepithelial transducers, responsible both for the acute control of gastric secretion and for maintaining the organization of the acid-secreting epithelium. Thus the proliferation and relative proportions of different epithelial cell types are determined by gastrin either directly, or indirectly via locally produced auto- and paracrine growth regulators. There is now a growing recognition that these mechanisms are a component in the pathology of the upper gastrointestinal tract, particularly in conditions involving Helicobacter pylori. Most patients with H. pylori are asymptomatic, but some develop duodenal ulcers and others gastric cancer. Infection with H. pylori is associated with modestly elevated plasma gastrin concentrations (Levi et al. 1989). In part this is likely to be secondary to decreased somatostatin synthesis, since there is reduced somatostatin mRNA abundance in H. pylori infection, which is thought to be attributable to the release of inflammatory mediators such as tumour necrosis factor (TNF)-α that inhibit antral D-cell function (Calam et al. 1997). The elevated plasma gastrin stimulates acid secretion, which is a feature of the infection in patients with duodenal ulcers. In other patients, however, moderately elevated plasma gastrin is accompanied by decreased acid secretion, which leads to infection of the corpus and the development of atrophic gastritis, which predisposes to gastric cancer (El-Omar et al. 1997). The results now emerging from physiological studies of the control of gastric epithelial cell function should facilitate an understanding of the mechanisms responsible for major upper gastrointestinal tract diseases.
References
- Asahara M, Mushiake S, Shimada S, Fukui H, Kinoshita Y, Kawanami C, Watanabe T, Tanaka S, Ichikawa A, Uchiyama Y, Narushima Y, Takasawa S, Okamoto H, Tohyama M, Chiba T. Reg gene expression is increased in rat gastric enterochromaffin-like cells following water immersion stress. Gastroenterology. 1996;111:45–55. doi: 10.1053/gast.1996.v111.pm8698224. [DOI] [PubMed] [Google Scholar]
- Baldwin GS. Antiproliferative gastrin/cholecystokinin receptor antagonists target the 78-kDa gastrin-binding protein. Proceedings of the National Academy of Sciences of the USA. 1994;91:7593–7597. doi: 10.1073/pnas.91.16.7593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bishop L, Dimaline R, Blackmore C, Deavall D, Dockray GJ, Varro A. Modulation of the cleavage of the gastrin precursor by phosphorylation. Gastroenterology. 1998;115:1154–1162. doi: 10.1016/s0016-5085(98)70086-1. [DOI] [PubMed] [Google Scholar]
- Black JW, Shankley NP. How does gastrin act to stimulate oxyntic cell secretion. Trends in Pharmacological Sciences. 1987;8:486–490. [Google Scholar]
- Bordi C, D'adda T, Azzoni C, Pilato FP, Carbuana P. Hypergastrinemia and gastric enterochromaffin-like cells. American Journal of Surgical Pathology. 1995;19:S8–19. [PubMed] [Google Scholar]
- Brand SJ, Stone DL. Reciprocal regulation of antral gastrin and somatostatin gene expression by omeprazole-induced achlorhydria. Journal of Clinical Investigation. 1988;82:1059–1066. doi: 10.1172/JCI113662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calam J, Gibbons A, Healey ZV, Bliss P, Arebi N. How does Helicobacter pylori cause mucosal damage? Its effect on acid and gastrin physiology. Gastroenterology. 1997;113:S43–49. doi: 10.1016/s0016-5085(97)80010-8. [DOI] [PubMed] [Google Scholar]
- Canfield V, West AB, Goldenring JR, Levenson R. Genetic ablation of parietal cells in transgenic mice: A new model for analyzing cell lineage relationships in the gastric mucosa. Proceedings of the National Academy of Sciences of the USA. 1996;93:2431–2435. doi: 10.1073/pnas.93.6.2431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandrasekharappa SC, Guru SC, Manickam P, Olufemi S-E, Collins FS, Emmert-Buck MR, Debelenko LV, Zhuang Z, Lubensky IA, Liotta LA, Crabtree JS, Wang Y, Roe BA, Weisemann J, Boguski MS, Agarwal SK, Kester MB, Kim YS, Heppner C, Dong Q, Spiegel AM, Burns AL, Marx SJ. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science. 1997;276:404–407. doi: 10.1126/science.276.5311.404. [DOI] [PubMed] [Google Scholar]
- Courtois-Coutry N, Roush D, Rajendran V, McCarthy JB, Geibel J, Kashgarian M, Caplan MJ. A tyrosine-based signal targets H/K-ATPase to a regulated compartment and is required for the cessation of gastric acid secretion. Cell. 1997;90:501–510. doi: 10.1016/s0092-8674(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Debelenko LV, Emmert-Buck MR, Zhuang Z, Epshteyn E, Moskaluk CA, Jensen RT, Liotta LA, Lubensky IA. The multiple endocrine neoplasia type I gene locus is involved in the pathogenesis of type II gastric carcinoids. Gastroenterology. 1997;113:773–781. doi: 10.1016/s0016-5085(97)70171-9. [DOI] [PubMed] [Google Scholar]
- Dimaline R, Evans D, Forster ER, Sandvik AK, Dockray GJ. Control of gastric corpus chromogranin A messenger RNA abundance in the rat. American Journal of Physiology. 1993a;264:G583–588. doi: 10.1152/ajpgi.1993.264.3.G583. [DOI] [PubMed] [Google Scholar]
- Dimaline R, Evans D, Varro A, Dockray GJ. Reversal by omeprazole of the depression of gastrin cell function by fasting in the rat. The Journal of Physiology. 1991;433:483–493. doi: 10.1113/jphysiol.1991.sp018439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaline R, Sandvik AK. Histidine decarboxylase gene expression in rat fundus is regulated by gastrin. FEBS Letters. 1991;281:20–22. doi: 10.1016/0014-5793(91)80348-7. [DOI] [PubMed] [Google Scholar]
- Dimaline R, Sandvik AK, Evans D, Forster ER, Dockray GJ. Food stimulation of histidine decarboxylase messenger RNA abundance in rat gastric fundus. The Journal of Physiology. 1993b;465:449–458. doi: 10.1113/jphysiol.1993.sp019686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaline R, Struthers J. Expression and regulation of a vesicular monoamine transporter (VMAT2) in rat stomach: a putative histamine transporter. The Journal of Physiology. 1996;490:249–256. doi: 10.1113/jphysiol.1996.sp021140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dockray GJ, Varro A, Desmond H, Young J, Gregory H, Gregory RA. Post-translational processing of the porcine gastrin precursor by phosphorylation of the COOH-terminal fragment. Journal of Biological Chemistry. 1987;262:8643–8647. [PubMed] [Google Scholar]
- Dockray GJ, Varro A, Dimaline R. Gastric endocrine cells: gene expression, processing and targeting of active products. Physiological Reviews. 1996;76:767–798. doi: 10.1152/physrev.1996.76.3.767. [DOI] [PubMed] [Google Scholar]
- Eipper BA, Milgram SL, Husten EJ, Yun HY, Mains RE. Peptidylglycine α-amidating monooxygenase: a multifunctional protein with catalytic processing, and routing domains. Protein Science. 1993;2:489–497. doi: 10.1002/pro.5560020401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Omar EM, Oien K, El-Nujumi A, Gillen D, Wirz A, Dahill S, Williams C, Ardill JES, McColl KEL. Helicobacter pylori infection and chronic gastric acid hyposecretion. Gastroenterology. 1997;113:15–24. doi: 10.1016/s0016-5085(97)70075-1. [DOI] [PubMed] [Google Scholar]
- Forte JG, Yao X. The membrane-recruitment-and-recycling hypothesis of gastric HCl secretion. Trends in Cell Biology. 1996;6:45–48. doi: 10.1016/0962-8924(96)81009-9. [DOI] [PubMed] [Google Scholar]
- Friis-Hansen L, Sundler F, Li Y, Gillespie PJ, Saunders TL, Greenson JK, Owyang C, Rehfeld JF, Samuelson LC. Impaired gastric acid secretion in gastrin-deficient mice. American Journal of Physiology. 1998;274:G561–568. doi: 10.1152/ajpgi.1998.274.3.G561. [DOI] [PubMed] [Google Scholar]
- Fukui H, Kinoshita Y, Maekawa T, Okada A, Waki S, Hassan MDS, Okamoto H, Chiba T. Regenerating gene protein may mediate gastric mucosal proliferation induced by hypergastrinemia in rats. Gastroenterology. 1998;115:1483–1493. doi: 10.1016/s0016-5085(98)70027-7. [DOI] [PubMed] [Google Scholar]
- Gregory RA, Tracy HJ. The constitution and properties of two gastrins extracted from hog antral mucosa. Gut. 1964;5:103–114. [PMC free article] [PubMed] [Google Scholar]
- Gregory RA, Tracy HJ. Isolation of two ‘big gastrins’ from Zollinger-Ellison tumour tissue. Lancet. 1972;2:797–799. doi: 10.1016/s0140-6736(72)92151-4. [DOI] [PubMed] [Google Scholar]
- Hersey SJ, Sachs G. Gastric acid secretion. Physiological Reviews. 1995;75:155–189. doi: 10.1152/physrev.1995.75.1.155. [DOI] [PubMed] [Google Scholar]
- Higham A, Bishop AE, Dimaline R, Blackmore C, Dobbins AC, Varro A, Thompson DG, Dockray GJ. Mutations of Reg1α in enterochromaffin-like cell tumors in patients with hypergastrinemia. Gastroenterology. 1999 doi: 10.1016/s0016-5085(99)70495-6. in the Press. [DOI] [PubMed] [Google Scholar]
- Higham A, Dimaline R, Varro A, Attwood S, Armstrong G, Dockray GJ, Thompson DG. Octreotide suppression test predicts beneficial outcome from antrectomy in a patient with gastric carcinoid tumor. Gastroenterology. 1998;114:817–822. doi: 10.1016/s0016-5085(98)70596-7. [DOI] [PubMed] [Google Scholar]
- Hocker M, Raychowdhury R, Plath T, Wu H, O'Connor DT, Wiedenmann B, Rosewicz S, Wang TC. Sp 1 and CREB mediate gastrin-dependent regulation of chromogranin A promoter activity in gastric carcinoma cells. Journal of Biological Chemistry. 1998;273:34000–34007. doi: 10.1074/jbc.273.51.34000. [DOI] [PubMed] [Google Scholar]
- Hollande F, Imdahl A, Mantamadiotis T, Ciccotosto GD, Shulkes A, Baldwin G. Glycine-extended gastrin acts as an autocrine growth factor in a nontransformed colon cell line. Gastroenterology. 1997;113:1576–1588. doi: 10.1053/gast.1997.v113.pm9352860. [DOI] [PubMed] [Google Scholar]
- Hussain I, Bate GW, Henry J, Djali PK, Dimaline R, Dockray GJ, Varro A. Identification of vesicular monoamine transporter type 1 (VMAT1) in G-cells and its role in modulating cleavage of G34. The Journal of Physiology. 1999;517:495–505. doi: 10.1111/j.1469-7793.1999.00495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LR. Regulation of gastrointestinal mucosal growth. Physiological Reviews. 1988;68:456–502. doi: 10.1152/physrev.1988.68.2.456. [DOI] [PubMed] [Google Scholar]
- Kahlson G, Rosengren E. Biogenesis and Physiology of Histamine. London: Edward Arnold; 1971. [PubMed] [Google Scholar]
- Karam SM. New insights into the stem cells and the precursors of the gastric epithelium. Nutrition. 1995;11:607–613. [PubMed] [Google Scholar]
- Kinoshita Y, Nakata H, Kishi K, Kawanami C, Sawada M, Chiba T. Comparison of the signal transduction pathways activated by gastrin in enterochromaffin-like and parietal cells. Gastroenterology. 1998;115:93–100. doi: 10.1016/s0016-5085(98)70369-5. [DOI] [PubMed] [Google Scholar]
- Koh TJ, Dockray GJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Wang TC. Overexpression of glycine-extended gastrin in transgenic mice results in increased colonic proliferation. Journal of Clinical Investigation. 1999;103:1119–1126. doi: 10.1172/JCI4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh TJ, Goldenring JR, Ito S, Mashimo H, Kopin AS, Varro A, Dockray GJ, Wang TC. Gastrin deficiency results in altered gastric differentiation and decreased colonic proliferation in mice. Gastroenterology. 1997;113:1015–1025. doi: 10.1016/s0016-5085(97)70199-9. [DOI] [PubMed] [Google Scholar]
- Kopin AS, Lee MY, McBride EW, Miller LJ, Lu M, Lin YH, Kolakowski LF, Jr, Beinborn M. Expression cloning and characterization of the canine parietal cell gastrin receptor. Proceedings of the National Academy of Sciences of the USA. 1992;89:3605–3609. doi: 10.1073/pnas.89.8.3605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamberts R, Creutzfeldt W, Struber HG, Brunner G, Solcia E. Long term omeprazole therapy in peptic ulcer disease: gastrin, endocrine cell growth and gastritis. Gastroenterology. 1993;104:1356–1370. doi: 10.1016/0016-5085(93)90344-c. [DOI] [PubMed] [Google Scholar]
- Langhans N, Rindi G, Chiu M, Rehfeld JF, Ardman B, Beinborn M, Kopin AS. Abnormal gastric histology and decreased acid production in cholecystokinin-B/gastrin receptor-deficient mice. Gastroenterology. 1997;112:280–286. doi: 10.1016/s0016-5085(97)90000-7. [DOI] [PubMed] [Google Scholar]
- Levi S, Haddad G, Ghosh P, Beardshall K, Playford RJ, Calam J. Campylobacter pylori and duodenal ulcers: the gastrin link. Lancet. 1989;1:1167–1168. doi: 10.1016/s0140-6736(89)92752-9. [DOI] [PubMed] [Google Scholar]
- Li Q, Karam SM, Gordon JI. Diphtheria toxin-mediated ablation of parietal cells in the stomach of transgenic mice. Journal of Biological Chemistry. 1996;271:3671–3676. [PubMed] [Google Scholar]
- Macro JA, Bate GW, Varro A, Vaillant C, Seidah NG, Dimaline R, Dockray GJ. Regulation by gastric acid of the processing of progastrin-derived peptides in rat antral mucosa. The Journal of Physiology. 1997;502:409–419. doi: 10.1111/j.1469-7793.1997.409bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macro JA, Dimaline R, Dockray GJ. Identification and expression of prohormone-converting enzymes in the rat stomach. American Journal of Physiology. 1996;279:G87–93. doi: 10.1152/ajpgi.1996.270.1.G87. [DOI] [PubMed] [Google Scholar]
- Martinez V, Curi AP, Torkian B, Schaeffer JM, Wilkinson HA, Walsh JH, Tache Y. High basal gastric acid secretion in somatostatin receptor subtype 2 knockout mice. Gastroenterology. 1998;114:1125–1132. doi: 10.1016/s0016-5085(98)70417-2. [DOI] [PubMed] [Google Scholar]
- Miyazaki Y, Shinomura Y, Tsutsui S, Zuchi S, Higashimoto Y, Kanayama S, Taniguchi N, Matsuzawa Y. Gastrin induces heparin-binding epidermal growth factor-like growth factor in rat gastric epithelial cells transfected with gastrin receptor. Gastroenterology. 1999;116:78–89. doi: 10.1016/s0016-5085(99)70231-3. [DOI] [PubMed] [Google Scholar]
- Murayama Y, Miyagawa J, Higashiyama S, Kondo S, Yabu M, Isozaki K, Kayanoki Y, Kanayama S, Shinomura Y, Taniguchi N, Matsuzawa Y. Localization of heparin-binding epidermal growth factor-like growth factor in human gastric mucosa. Gastroenterology. 1995;109:1051–1059. doi: 10.1016/0016-5085(95)90562-6. [DOI] [PubMed] [Google Scholar]
- Nagata A, Ito M, Iwata N, Kuno J, Takano H, Minowa O, Chihara K, Matsui T, Noda T. G protein-coupled cholecystokinin-B/gastrin receptors are responsible for physiological cell growth of the stomach mucosa in vivo. Proceedings of the National Academy of Sciences of the USA. 1996;93:11825–11830. doi: 10.1073/pnas.93.21.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakata H, Wang S-L, Chung DC, Westwick JK, Tillotson LG. Oncogenic ras induces gastrin gene expression in colon cancer. Gastroenterology. 1998;115:1144–1153. doi: 10.1016/s0016-5085(98)70085-x. [DOI] [PubMed] [Google Scholar]
- Nemeth J, Taylor B, Pauwels S, Varro A, Dockray GJ. Identification of progastrin derived peptides in colorectal carcinoma extracts. Gut. 1993;34:90–95. doi: 10.1136/gut.34.1.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohning GV, Wong HC, LLoyd KCK, Walsh JH. Gastrin mediates the gastric mucosal proliferative response to feeding. American Journal of Physiology. 1996;271:G470–476. doi: 10.1152/ajpgi.1996.271.3.G470. [DOI] [PubMed] [Google Scholar]
- Prinz C, Kajimura M, Scott DR, Mercier F, Helander HF, Sachs G. Histamine secretion from rat enterochromaffinlike cells. Gastroenterology. 1993;105:449–461. doi: 10.1016/0016-5085(93)90719-s. [DOI] [PubMed] [Google Scholar]
- Prinz C, Scott DR, Hurwitz D, Helander HF, Sachs G. Gastrin effects on isolated rat enterochromaffin-like cells in primary culture. American Journal of Physiology. 1994;267:G663–675. doi: 10.1152/ajpgi.1994.267.4.G663. [DOI] [PubMed] [Google Scholar]
- Ronning K, Sandvik AK, Waldum HL. The fade of gastrin-stimulated gastric acid secretion in the rat is due to depletion of releasable mucosal histamine. Acta Physiologica Scandinavica. 1996;157:487–491. doi: 10.1046/j.1365-201X.1996.506272000.x. [DOI] [PubMed] [Google Scholar]
- Schaffer K, McBride EW, Beinborn M, Kopin AS. Interspecies polymorphisms confer constitutive activity to the Mastomys cholecystokinin-B/gastrin receptor. Journal of Biological Chemistry. 1998;273:28779–28784. doi: 10.1074/jbc.273.44.28779. [DOI] [PubMed] [Google Scholar]
- Schultheis PJ, Clarke LL, Meneton P, Harline M, Boivin GP, Stemmermann G, Duffy JJ, Doetschman T, Miller ML, Shull GE. Targeted disruption of the murine Na+/H+ exchanger isoform 2 gene causes reduced viability of gastric parietal cells and loss of net acid secretion. Journal of Clinical Investigation. 1998;101:1243–1253. doi: 10.1172/JCI1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seva C, Dickinson CJ, Yamada T. Growth-promoting effects of glycine-extended progastrin. Science. 1994;265:410–412. doi: 10.1126/science.8023165. [DOI] [PubMed] [Google Scholar]
- Sharp R, Babyatsky MW, Takagi H, Tagerud S, Wang TC, Bockman DE, Brand SJ, Merlino GT. Transforming growth factor I disrupts the normal program of cellular differentiation in the gastric mucosa of transgenic mice. Development. 1995;121:149–161. doi: 10.1242/dev.121.1.149. [DOI] [PubMed] [Google Scholar]
- Singh P, Owlia A, Espeijo R, Dai B. Novel gastrin receptors mediate mitogenic effects of gastrin and processing intermediates of gastrin on Swiss 3T3 fibroblasts. Journal of Biological Chemistry. 1995;270:8429–8438. doi: 10.1074/jbc.270.15.8429. [DOI] [PubMed] [Google Scholar]
- Singh P, Owlia A, Varro A, Dai B, Rajaraman S, Wood T. Gastrin gene expression is required for the proliferation and tumorigenicity of human colon cancer cells. Cancer Research. 1996;56:4111–4115. [PubMed] [Google Scholar]
- Soll AH, Amirian DA, Thomas LP, Reedy TJ, Elashoff JD. Gastrin receptors on isolated canine parietal cells. Journal of Clinical Investigation. 1984;73:1434–1447. doi: 10.1172/JCI111348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takagi H, Jhappan C, Sharp R, Merlino GT. Hypertrophic gastropathy resembling Menetrier's disease in transgenic mice overexpressing transforming growth factor I in the stomach. Journal of Clinical Investigation. 1992;90:1161–1167. doi: 10.1172/JCI115936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tielemans Y, Hakanson R, Sundler F, Willems G. Proliferation of enterochromaffinlike cells in omeprazole-treated hypergastrinemic rats. Gastroenterology. 1989;96:723–729. [PubMed] [Google Scholar]
- Todisco A, Takeuchi Y, Seva C, Dickinson CJ, Yamada T. Gastrin and glycine-extended progastrin processing intermediates induce different programs of early gene activation. Journal of Biological Chemistry. 1995;270:28337–28341. doi: 10.1074/jbc.270.47.28337. [DOI] [PubMed] [Google Scholar]
- Tsutsui S, Shinomura Y, Higashiyama S, Higashimoto Y, Miyazaki Y, Kanayama S, Hiraoka S, Minami T, Kitamura S, Murayama Y, Miyagawa J, Taniguchi N, Matsuzawa Y. Induction of heparin binding epidermal growth factor-like growth factor and amphirefulin mRNAs by gastrin in the rat stomach. Biochemical and Biophysical Research Communications. 1997;235:520–523. doi: 10.1006/bbrc.1997.6824. [DOI] [PubMed] [Google Scholar]
- Udupi V, Gomez P, Song L, Varlamov O, Reed JT, Leiter EH, Fricker LD, Greeley GH., Jr Effect of carboxypeptidase E deficiency on progastrin processing and gastrin messenger ribonucleic acid expression in mice with the fat mutation. Endocrinology. 1997;138:1959–1963. doi: 10.1210/endo.138.5.5113. [DOI] [PubMed] [Google Scholar]
- Varro A, Dockray GJ. Post-translational processing of progastrin: inhibition of cleavage, phosphorylation and sulphation by brefeldin A. Biochemical Journal. 1993;295:813–819. doi: 10.1042/bj2950813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varro A, Henry J, Vaillant C, Dockray GJ. Discrimination between temperature- and brefeldin A-sensitive steps in the sulfation, phosphorylation, and cleavage of progastrin and its derivatives. Journal of Biological Chemistry. 1994;269:20764–20770. [PubMed] [Google Scholar]
- Voronina S, Henry J, Vaillant C, Dockray GJ, Varro A. Amine precursor uptake and decarboxylation: significance for processing of the rat gastrin precursor. The Journal of Physiology. 1997;501:363–374. doi: 10.1111/j.1469-7793.1997.363bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JH. Gastrin. In: Walsh JH, Dockray GJ, editors. Gut Peptides. New York: Raven Press; 1994. pp. 75–121. [Google Scholar]
- Walsh JH, Debas HT, Grossman MI. Pure human big gastrin: immunochemical properties, disappearance half time and acid stimulating action in dogs. Journal of Clinical Investigation. 1974;54:477–485. doi: 10.1172/JCI107783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TC, Dangler CA, Chen D, Goldenring JR, Koh TJ, Raychowdhury R, Coffey RJ, Ito S, Varro A, Dockray GJ, Fox JG. Hypergastrinaemia leads to atrophy and invasive gastric cancer in transgenic mice. Gastroenterology. 1999;116:A527. doi: 10.1016/s0016-5085(00)70412-4. [DOI] [PubMed] [Google Scholar]
- Wang TC, Koh TJ, Varro A, Cahill RJ, Dangler CA, Fox JG, Dockray GJ. Processing and proliferative effects of human progastrin in transgenic mice. Journal of Clinical Investigation. 1996;98:1918–1929. doi: 10.1172/JCI118993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wank SA, Harkins R, Jensen RT, Shapira H, De Weerth A, Slattery T. Purification, molecular cloning, and functional expression of the cholecystokinin receptor from rat pancreas. Proceedings of the National Academy of Sciences of the USA. 1992b;89:3125–3129. doi: 10.1073/pnas.89.7.3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wank SA, Pisegna JR, De Weerth A. Brain and gastrointestinal cholecystokinin receptor family: structure and functional expression. Proceedings of the National Academy of Sciences of the USA. 1992a;89:8691–8695. doi: 10.1073/pnas.89.18.8691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson SA, Michaeli D, Grimes S, Morris TM, Robinson G, Varro A, Justin TA, Hardcastle JD. Gastrimmune raises antibodies that neutralize amidated and glycine-exended gastrin-17 and inhibit the growth of colon cancer. Cancer Research. 1996;56:880–885. [PubMed] [Google Scholar]
- Wu SV, Giraud A, Mogard M, Sunii K, Walsh JH. Effects of inhibition of gastric secretion on antral gastrin and somatostatin gene expression in rats. American Journal of Physiology. 1990;258:G788–793. doi: 10.1152/ajpgi.1990.258.5.G788. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Hocker M, Koh TJ, Wang TC. The human histidine decarboxylase promoter is regulated by gastrin and phorbol 12-myristate 13-acetate through a downstream cis-acting element. Journal of Biological Chemistry. 1996;271:14188–14197. [PubMed] [Google Scholar]