

Abstract
ATP-sensitive potassium (KATP) channels are composed of pore-forming Kir6.2 and regulatory SUR subunits. ATP inhibits the channel by interacting with Kir6.2, while sulphonylureas block channel activity by interaction with a high-affinity site on SUR1 and a low-affinity site on Kir6.2. MgADP and diazoxide interact with SUR1 to promote channel activity.
We examined the effect of N-terminal deletions of Kir6.2 on the channel open probability, ATP sensitivity and sulphonylurea sensitivity by recording macroscopic currents in membrane patches excised from Xenopus oocytes expressing wild-type or mutant Kir6.2/SUR1.
A 14 amino acid N-terminal deletion (ΔN14) did not affect the gating, ATP sensitivity or tolbutamide block of a truncated isoform of Kir6.2, Kir6.2ΔC26, expressed in the absence of SUR1. Thus, the N-terminal deletion does not alter the intrinsic properties of Kir6.2.
When Kir6.2ΔN14 was coexpressed with SUR1, the resulting KATP channels had a higher open probability (Po = 0·7) and a lower ATP sensitivity (Ki = 196 μM) than wild-type (Kir6.2/SUR1) channels (Po = 0·32, Ki = 28 μM). High-affinity tolbutamide block was also abolished.
Truncation of five or nine amino acids from the N-terminus of Kir6.2 also enhanced the open probability, and reduced both the ATP sensitivity and the fraction of high-affinity tolbutamide block, although to a lesser extent than for the ΔN14 deletion. Site-directed mutagenesis suggests that hydrophobic residues in Kir6.2 may be involved in this effect.
The reduced ATP sensitivity of Kir6.2ΔN14 may be explained by the increased Po. However, when the Po was decreased (by ATP), tolbutamide was unable to block Kir6.2ΔN14/SUR1-K719A,K1385M currents, despite the fact that the drug inhibited Kir6.2-C166S/SUR1-K719A,K1385M currents (which in the absence of ATP have a Po of > 0·8 and are not blocked by tolbutamide). Thus the N-terminus of Kir6.2 may be involved in coupling sulphonylurea binding to SUR1 to closure of the Kir6.2 pore.
The ATP-sensitive potassium channel (KATP) serves to couple the metabolic status of the cell to its electrical activity (Ashcroft & Rorsman, 1989). One of the defining characteristics of this channel is that its open probability is decreased by intracellular ATP and enhanced by Mg2+ nucleotides, like MgADP. KATP channels are also blocked by sulphonylurea drugs, such as glibenclamide and tolbutamide, and activated by K+ channel openers, such as diazoxide (Zünckler et al. 1988b).
The β-cell KATP channel is a tightly associated hetero-octameric complex of an inwardly rectifying potassium channel (Kir6.2) and a regulatory subunit, the sulphonylurea receptor (SUR1) (Inagaki et al. 1995; Sakura et al. 1995; Aguilar-Bryan et al. 1995; Clement et al. 1997; Inagaki et al. 1997; Shyng & Nichols, 1997). Truncation of 26 amino acids from the C-terminus of Kir6.2 permits functional expression of this subunit in the absence of SUR1. Studies of this truncated subunit (Kir6.2ΔC26) have clearly demonstrated that Kir6.2 forms the pore of the channel and that it is inhibited by ATP, whereas SUR1 confers the stimulatory effects of MgADP and the K+ channel opener diazoxide, as well as the characteristic high-affinity block by sulphonylureas (Tucker et al. 1997; Babenko et al. 1998). Kir6.2ΔC26, in the absence of SUR1, can also be inhibited by high concentrations of sulphonylureas but this subunit is at least three orders of magnitude less sensitive than the high-affinity sulphonylurea binding site on SUR1 (Gribble et al. 1997b). SUR1 is able to ‘enhance’ or ‘sensitize’ the direct block of Kir6.2 by ATP, although the precise mechanism by which this occurs is as yet unresolved (Tucker et al. 1997; Trapp et al. 1998; Koster et al. 1999). Thus SUR1 plays a key role in determining the ATP sensitivity of Kir6.2, as well as transducing the effects of high-affinity sulphonylurea binding into the closure of the pore formed by Kir6.2. However, as yet there is little information about the precise regions involved in the functional coupling of SUR1 to Kir6.2.
A role for the intracellular N-domain of Kir6.2 in controlling the ATP sensitivity of the KATP channel has recently been reported. Mutations within this domain have been shown to have profound effects on the ATP sensitivity of the channel (Tucker et al. 1998; Proks et al. 1999), whilst truncation of the N-terminus reduces the ATP sensitivity indirectly, by altering the open probability of the channel in the presence of SUR1 (Koster et al. 1999; Babenko et al. 1999). In a previous study (Tucker et al. 1997), we showed that deletion of 14 amino acids from the N-terminus of Kir6.2 (Kir6.2ΔN14) was not sufficient to permit functional expression of this subunit in the absence of SUR1. In the present work, we demonstrate that functional expression of this N14 deletion can be achieved by additionally truncating the C-terminus (Kir6.2ΔC26-ΔN14). By expression of the Kir6.2ΔC26-ΔN14 deletion in the absence of SUR1, we demonstrate that deletion of the N-terminus has no effect on the intrinsic ATP sensitivity or gating kinetics of the Kir6.2ΔC26 subunit. In contrast, coexpression of this subunit with SUR1 produces KATP channels with a reduced ATP sensitivity, an effect that probably results from the increased open probability of this channel.
We also find that whilst Kir6.2ΔN14/SUR1 channels retain the ability to be stimulated by MgADP and by the K+ channel opener diazoxide they exhibit no high-affinity sulphonylurea block. Indeed, the Kir6.2ΔN14 deletion not only removes the ability of SUR1 to produce high-affinity sulphonylurea block of channel activity but also that of the related sulphonylurea receptor SUR2A. Smaller deletions of the N-terminus produce a graded decrease in high-affinity block. This effect is not simply the consequence of the altered channel kinetics. Therefore, in addition to a role for the N-terminus of Kir6.2 in the control of ATP sensitivity by SUR1 we propose a role for this domain in the functional coupling of sulphonylurea sensitivity between SUR1 and Kir6.2.
METHODS
Molecular biology
Mouse Kir6.2 (GenBank D50581; Inagaki et al. 1995; Sakura et al. 1995), rat SUR1 (GenBank L40624; Aguilar-Bryan et al. 1995; provided by Dr G. Bell, University of Chicago) and rat SUR2A (GenBank D83598; Inagaki et al. 1996; provided by Dr S. Seino, Chiba University School of Medicine) were used in this study.
A 26 or 36 amino acid C-terminal deletion of mouse Kir6.2 (Kir6.2ΔC) was made by introduction of a stop codon at the appropriate residue (Tucker et al. 1997). No differences were apparent in currents encoded by Kir6.2ΔC26 and Kir6.2ΔC36 except that Kir6.2ΔC36 expressed larger currents (Tucker et al. 1997). N-terminal deletions were made by using the polymerase chain reaction to replace the indicated residue with an initiator methionine residue. In this paper, we use the term ΔN5 to refer to the deletion of residues 2-5, ΔN9 to refer to the deletion of residues 2-9, and ΔN14 to refer to the deletion of residues 2-14. Site-directed mutagenesis of Kir6.2ΔC26, Kir6.2 and SUR1 was carried out by subcloning the appropriate fragments into the pALTER vector (Promega, Madison, WI, USA). We use the abbreviation SUR1-KAKM to refer to mutation of the Walker A lysines in both NBD1 (K719A) and NBD2 (K1385M) of SUR1. Synthesis of capped mRNA was carried out using the mMessage mMachine in vitro transcription kit (Ambion, Austin, TX, USA). Amino acids are indicated by the single-letter code.
Electrophysiology
Oocyte collection
Female Xenopus laevis were anaesthetized with MS222 (2 g l−1 added to the water; Sigma). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V-VI Xenopus oocytes were manually defolliculated and injected with 2 ng of mRNA encoding wild-type or mutant forms of Kir6.2ΔC26, or co-injected with ∼0·1 ng Kir6.2 (wild-type or mutant) and ∼2 ng of SUR. The final injection volume was ∼50 nl per oocyte. Control oocytes were injected with water. Isolated oocytes were maintained in Barth's solution and studied 1-4 days after injection (Gribble et al. 1997a).
Electrophysiology
Patch pipettes were pulled from thick-walled glass and had resistances of 250-500 kΩ when filled with pipette solution. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20-24°C (Gribble et al. 1997a). Currents were evoked by repetitive 3 s voltage ramps from -110 to +100 mV and recorded using an EPC7 patch-clamp amplifier (List Electronic). They were filtered at 5 kHz and stored on digital audiotape. Currents were subsequently filtered at 0·5 kHz, digitized at 1 kHz using a Digidata 1200 interface and analysed using pCLAMP software (Axon Instruments). Single-channel currents were recorded from small inside-out patches, filtered at 1 kHz and sampled at 3 kHz.
The pipette (external) solution contained (mM): 140 KCl, 1·2 MgCl2, 2·6 CaCl2, 10 Hepes (pH 7·4 with KOH). The intracellular (bath) solution contained (mM): 107 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7·2 with KOH; final K+ ∼140 mM) and nucleotides as indicated. Tolbutamide was made up as a 0·1 M stock solution in 0·15 M KOH and diazoxide as a 0·068 M stock solution in 0·1 M KOH. Solutions containing nucleotides were made up fresh each day. The pH of all solutions was checked and readjusted, if required, after drug and nucleotide addition. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Data analysis
The slope conductance was measured by fitting a straight line to the current-voltage relationship between -20 and -100 mV: the average of five consecutive ramps was calculated in each solution. ATP dose-response relationships were measured by alternating the control solution with a test ATP concentration. The conductance (G) was then expressed as a fraction of the mean of the value obtained in the control solution before and after exposure to ATP (Gc). Dose-response curves were fitted to the Hill equation:
| (1) |
where [ATP] is the ATP concentration, Ki is the concentration at which inhibition is half-maximal and h is the slope factor (Hill coefficient).
Tolbutamide dose-response curves were fitted to the following equation (Gribble et al. 1997a):
| (2) |
where x is a term describing the high-affinity site and y a term describing the low-affinity site.
| (3) |
| (4) |
where [Tolb] is the tolbutamide concentration; Ki1 and Ki2 are the tolbutamide concentrations at which inhibition is half-maximal at the high- and low-affinity sites, respectively; h1, h2 are the Hill coefficients (slope factors) for the high- and low-affinity sites, respectively; and L is the fractional conductance remaining when all of the high-affinity inhibitory sites are occupied.
Single-channel currents were analysed using a combination of pCLAMP and in-house software written by Dr P. A. Smith (Oxford University). Single-channel current amplitudes were calculated from an all-points amplitude histogram. Channel activity (NPo) was measured as the mean patch current divided by the single-channel current amplitude, for segments of the current records of ∼1 min duration. Open probability (Po) was calculated from NPo/N, where N is the number of channels in the patch and was estimated from the maximum number of superimposed events.
Data are given as means ± 1 s.e.m., and the symbols in the figures indicate the mean and the vertical bars indicate 1 s.e.m. (where this is larger than the symbol).
RESULTS
ATP sensitivity
Figure 1A shows that deletion of the first 14 amino acids from the N-terminus did not affect the ATP sensitivity of Kir6.2ΔC26. Half-maximal inhibition (Ki) of Kir6.2ΔC26-ΔN14 was produced by 109 ± 8 μM ATP (n = 7), while a Ki of 106 ± 4 μM (n = 7) was found for Kir6.2ΔC26. Coexpression of SUR1 with Kir6.2 enhances the channel ATP sensitivity, by a mechanism that is not yet clear (Tucker et al. 1998). Figure 1B shows that this effect was impaired when the N-terminus of Kir6.2 is truncated: the Ki for ATP block of Kir6.2ΔN14/SUR1 currents was 196 ± 17 μM (n = 10) compared with 28 ± 5 μM (n = 5) for Kir6.2/SUR1 currents. This suggests that the N-terminus of Kir6.2 may be involved in functionally coupling this subunit to SUR1.
Figure 1. Effects of ATP.
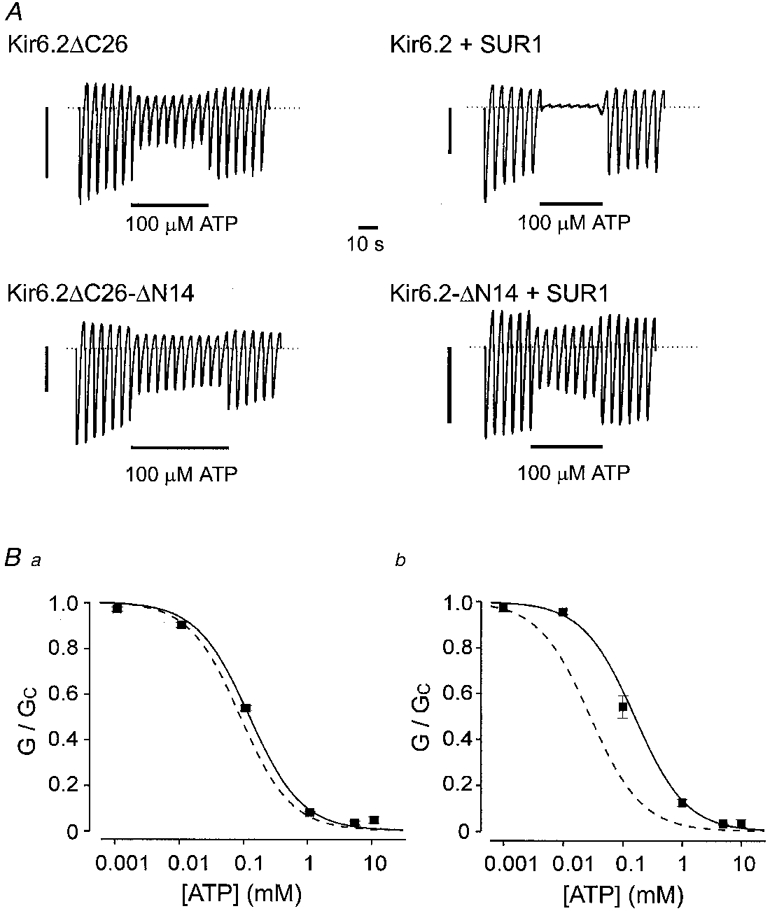
A, macroscopic currents recorded from inside-out patches excised from oocytes injected with mRNA encoding Kir6.2ΔC26, Kir6.2ΔC26-ΔN14, Kir6.2/SUR1, Kir6.2ΔN14/SUR1. Currents were elicited in response to a series of voltage ramps from -110 to +100 mV. ATP (100 μM) was added as indicated by the bars. The current scale bars are 0·5 nA for Kir6.2ΔC26 and 2 nA for the other currents. B, mean ATP dose-response relationships for Kir6.2ΔN14-ΔC26 currents (▪, n= 7) and Kir6.2ΔC26 (dashed line) (a); and for Kir6.2ΔN14 (▪, n= 10) or Kir6.2 (dashed line) coexpressed with SUR1 (b). The slope conductance (G) is expressed as a fraction of the mean of that obtained in control solution (Gc) before and after exposure to ATP. The continuous lines are the best fit of the data to the Hill equation using the mean values for Ki and h given in Table 1. The dashed lines are taken from Tucker et al. (1997) (a) and Gribble et al. (1997a) (b).
In addition to enhancing the ability of ATP to inhibit Kir6.2, SUR1 confers sensitivity to the stimulatory effects of MgADP and diazoxide, and to the inhibitory effects of sulphonylureas (Ashcroft & Gribble, 1998). Therefore, we examined whether the N-terminus of Kir6.2 is also involved in transducing the interaction of these agents with SUR1 into changes in the gating of Kir6.2.
Effects of K+ channel activators
As shown in Fig. 2, both diazoxide and MgADP were able to increase Kir6.2ΔN14/SUR1 currents blocked by 100 μM ATP, as is found for the wild-type KATP channel (Kir6.2/SUR1). Diazoxide increased both currents to approximately the same amplitude but, possibly because ATP blocks Kir6.2ΔN14/SUR1 currents less potently, the relative increase in current was smaller than for Kir6.2/SUR1. Likewise, MgADP did not enhance Kir6.2ΔN14/SUR1 currents to the same extent as wild-type currents.
Figure 2. Effects of activators.
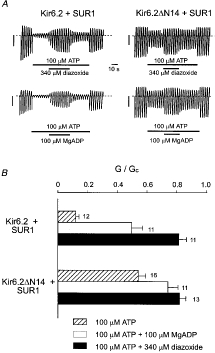
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes co-injected with SUR1 plus either Kir6.2 (left) or Kir6.2ΔN14 (right). ATP (0·1 mM), diazoxide (340 μM) or MgADP (100 μM) were added to the intracellular solution as indicated by the bars. The current scale bars are 2 nA in each case. B, mean slope conductance (G) recorded in the presence of the additions indicated, expressed as a percentage of that (Gc) in control solution (no additions), for Kir6.2/SUR1 and Kir6.2ΔN14/SUR1. The number of patches is given alongside each bar.
Sulphonylurea block
Tolbutamide interacts with a low-affinity site on Kir6.2 and a high-affinity site on SUR1 (Gribble et al. 1997b). Truncation of the N-terminus did not alter the low-affinity block of Kir6.2 (Fig. 3). When expressed in the absence of SUR1, the Ki for tolbutamide inhibition was 1·7 mM for Kir6.2ΔC36 (Gribble et al. 1997b) and 2·0 ± 0·1 mM (n = 5) for Kir6.2ΔC26-ΔN14. This indicates that the first 14 amino acids of the N-terminus do not form part of the low-affinity site for tolbutamide on Kir6.2.
Figure 3. Tolbutamide inhibition of Kir6.2 currents.
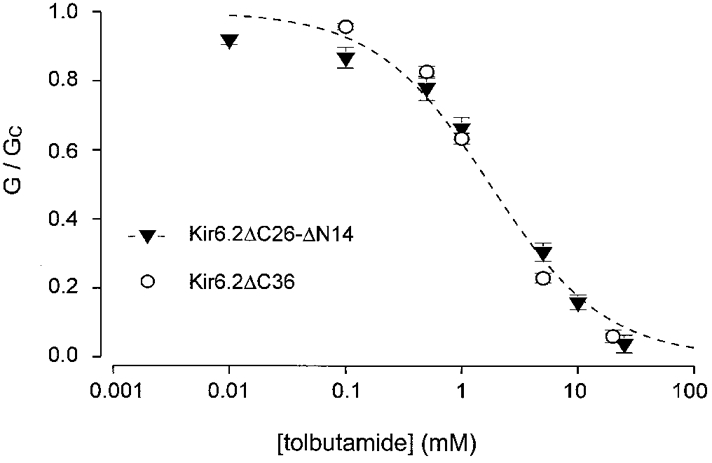
Relationship between tolbutamide concentration and the macroscopic KATP conductance, expressed as a fraction of its amplitude in the absence of the drug. ○, Kir6.2ΔC36 currents (n= 6); ▾, Kir6.2ΔC26-ΔN14 currents (n= 5). The line is the best fit of the data obtained for Kir6.2ΔC26-ΔN14 to eqn (4): Ki= 2 mM, h = 0·9. The data for Kir6.2ΔC36 are the same as those in Gribble et al. (1997b).
Truncation of the N-terminus profoundly affects the ability of SUR1 to cause high-affinity inhibition of Kir6.2. As previously reported (Gribble et al. 1997b), when SUR1 is coexpressed with Kir6.2 the dose-response curve for tolbutamide block is best fitted with two binding sites (Fig. 4). In contrast to the wild-type KATP channel, only a single low-affinity site was observed for Kir6.2ΔN14/SUR1 currents. The Ki of this low-affinity site (2·6 ± 0·2 mM, n = 5) was similar to that of wild-type KATP channels (2·4 ± 0·1 mM, n = 6) and of Kir6.2ΔC36 (Gribble et al. 1997b), indicating that it resides on Kir6.2. The Ki of the high-affinity site measured for Kir6.2/SUR1 was 1·4 ± 0·4 μM (n = 6), a value similar to that previously reported (Gribble et al. 1997b).
Figure 4. Tolbutamide block of Kir6.2/SUR1 and Kir6.2ΔN14/SUR1 currents.
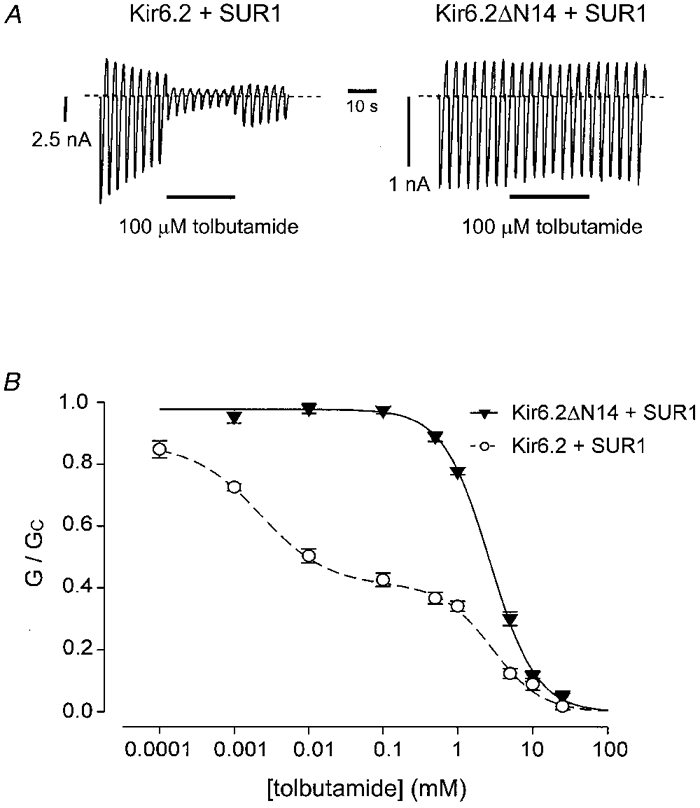
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes co-injected with SUR1 plus either Kir6.2 (left) or with Kir6.2ΔN14 (right). Tolbutamide (0·1 mM) was added to the intracellular solution as indicated by the bar. B, relationship between tolbutamide concentration and the macroscopic KATP conductance, expressed as a fraction of its amplitude in the absence of the drug. Kir6.2/SUR1 currents (○, n= 6) were fitted with eqn (1) and Kir6.2ΔN14/SUR1 currents (▾, n= 5) were fitted with eqn (4) using the parameters given in Table 1.
We next examined whether the N-terminus of Kir6.2 is also concerned in coupling this subunit to SUR2A, a sulphonylurea receptor that shares 68 % sequence identity with SUR1 (Inagaki et al. 1996). Although Kir6.2/SUR2A currents do not show high-affinity inhibition by tolbutamide, they are blocked by glibenclamide with a potency similar to that of SUR1 (Gribble et al. 1998b). Figure 5 shows that neither Kir6.2ΔN14/SUR1 nor Kir6.2ΔN14/SUR2A currents were blocked by glibenclamide at a concentration (100 nM) sufficient to cause maximal high-affinity inhibition of wild-type KATP channels. Thus an intact N-terminus of Kir6.2 is required for the inhibitory effect of both tolbutamide and glibenclamide, and SUR1 and SUR2A transduce the effects of sulphonylureas by a mechanism that involves common elements.
Figure 5. Effects of glibenclamide.
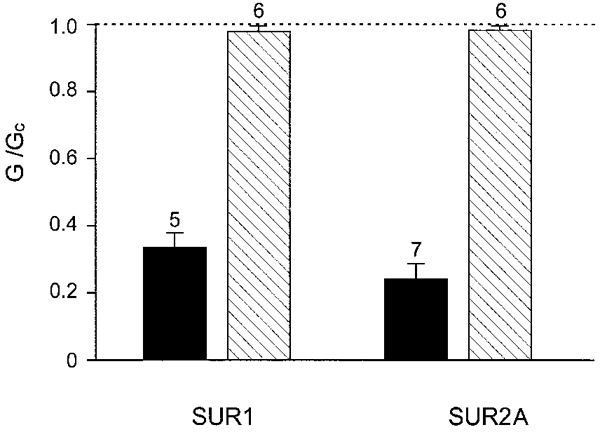
Mean slope conductance recorded in the presence of 100 nM glibenclamide, expressed as a percentage of that in the absence of the drug. The dotted line indicates the level of activity in the absence of glibenclamide. Currents were recorded from oocytes expressing Kir6.2 (▪) or Kir6.2ΔN14 ( ), with SUR1 or SUR2A, as indicated. The number of patches is given above each bar.
), with SUR1 or SUR2A, as indicated. The number of patches is given above each bar.
Mutations that affect coupling
To localize further residues within the N-terminus of Kir6.2 involved in coupling to SUR1, we made a number of additional truncations and mutations within this region. Deletion of residues 2-5 (ΔN5) or 2-9 (ΔN9) caused a progressive reduction in the amount of high-affinity tolbutamide inhibition (Fig. 6). The maximal inhibition produced at the high-affinity site was 54 % for wild-type KATP channels: this decreased to 38 %, 25 % and was finally completely abolished when the N-terminus of Kir6.2 was truncated by 5, 9 and 14 residues, respectively (Table 1). There was also a small shift in the Ki for the high-affinity site, but the Hill coefficient remained unchanged (Table 1).
Figure 6. Effect of tolbutamide on N-terminal deletions of different length.
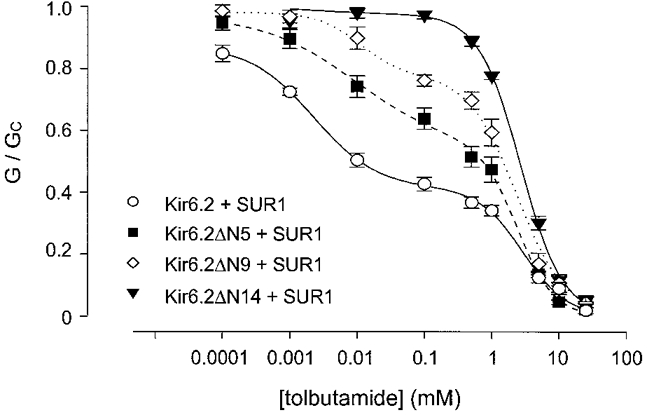
Relationship between tolbutamide concentration and the macroscopic KATP conductance, expressed as a fraction of its amplitude in the absence of the drug. Kir6.2/SUR1 (○, n= 6), Kir6.2ΔN5/SUR1 (▪, n= 6) and Kir6.2ΔN9/SUR1 (⋄, n= 4) currents were fitted with eqn (1). Kir6.2ΔN14/SUR1 currents (▾, n= 5) were fitted with eqn (4). The parameters used to fit the data are given in Table 1.
Table 1.
Tolbutamide block of KATP channels
| Clone* | Ki1(μM) | h1 | Ki2(mM) | h2 | L | n |
|---|---|---|---|---|---|---|
| Kir6.2 | 1.42 ± 0.35 | 0.92 ± 0.06 | 2.40 ± 0.1 | 1.20 ± 0.1 | 0.46 ± 0.03 | 6 |
| Kir6.2ΔN5 | 6.99 ± 2.02 | 1.01 ± 0.03 | 2.19 ± 0.33 | 1.45 ± 0.14 | 0.62 ± 0.03 | 6 |
| Kir6.2ΔN9 | 22.52 ± 5.56 | 0.99 ± 0.01 | 2.24 ± 0.35 | 1.37 ± 0.09 | 0.75 ± 0.02 | 4 |
| Kir6.2ΔN1 | — | — | 2.56 ± 0.17 | 1.34 ± 0.05 | — | 5 |
All coexpressed with SUR1. Data were fitted with a two-site model for tolbutamide inhibition (eqn (2)). Ki, tolbutamide concentration producing half-maximal block; h, Hill coefficient; L, maximum level of block produced by the high-affinity site
Figure 7 shows the extent of block produced by 100 μM tolbutamide for wild-type KATP currents, for the N-terminal truncations and for a number of mutations in the N-terminus of Kir6.2. No single mutation was sufficient to decrease tolbutamide block to that observed for Kir6.2ΔN14/SUR1: rather, the results suggest the involvement of a number of residues. Neutralization of the charged residues K5, E10 and E11 did not significantly alter the extent of tolbutamide block, suggesting that these amino acids do not interact with SUR1. Neutralization of the arginine at position 4, however, reduced the extent of block to that found for the ΔN5 truncation: thus R4 may be involved in functional interaction with SUR1. Hydrophobic interactions may also be important, since L2A, and the combination of V13N and L14N, also reduced tolbutamide inhibition. Furthermore, tolbutamide block was abolished when residues 7, 8, 13 and 14 were simultaneously mutated to asparagine. However, since multiple mutations may destroy the local structure of the protein, the precise involvement of these hydrophobic residues is hard to assess.
Figure 7. Effect of tolbutamide on wild-type and mutant KATP currents.
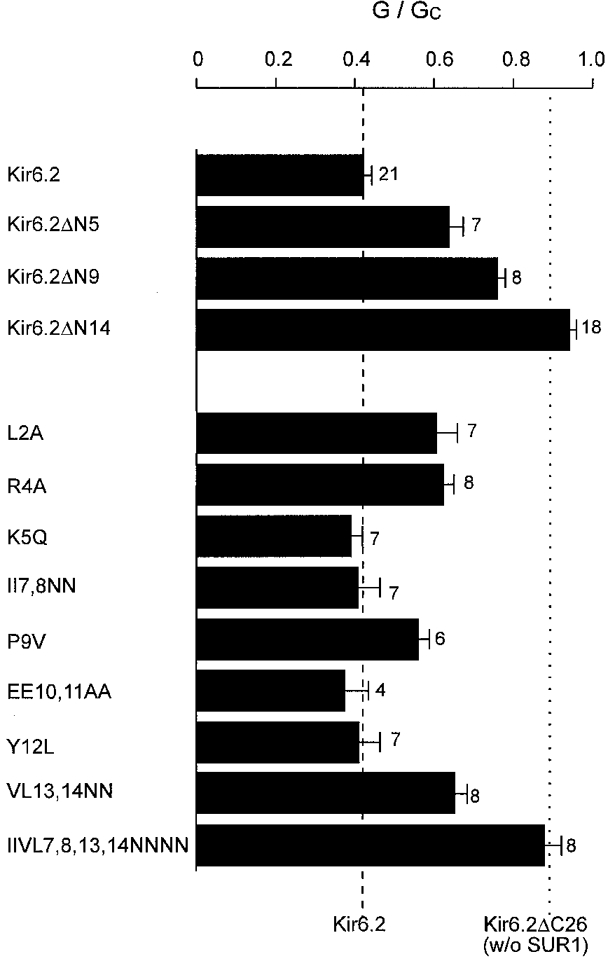
Mean current amplitude recorded in the presence of 100 μM tolbutamide (G), expressed as a fraction of that in the absence of the drug (Gc). Currents were recorded from oocytes expressing SUR1 and either wild-type or mutant Kir6.2ΔC26, as indicated. The number of patches is given alongside each bar. The mean values obtained for Kir6.2ΔC26 and Kir6.2/SUR1 channels are indicated by the dotted and dashed lines, respectively.
Single-channel currents
One mechanism by which truncation of the N-terminus may alter the channel ATP and tolbutamide sensitivity in the presence of SUR1 is by impairing the ability of the channel to close (Shyng et al. 1997a;Tucker et al. 1998). No difference was observed between the single-channel kinetics of Kir6.2ΔC26 and Kir6.2ΔC26-ΔN14 (Fig. 8, Table 2), indicating that the intrinsic gating of the channel is unaffected by removal of residues 2-14. The ability of SUR1 to enhance the open probability of Kir6.2 (Proks & Ashcroft, 1997), however, was much greater when the N-terminus of Kir6.2 was truncated. As Fig. 8 and Table 2 also show, when Kir6.2 containing the ΔN5 or ΔN14 truncation was coexpressed with SUR1, the open probability was greatly increased: the mean Po was 0·32 for the wild-type KATP channel, but rose to 0·44 for Kir6.2ΔN5/SUR1, 0·61 for Kir6.2ΔN9/SUR1 and 0·70 for Kir6.2ΔN14/SUR1. There was also a clear correlation between the increase in Po and that in the Ki for ATP inhibition (Table 2). Thus it seems likely that the reduced ATP sensitivity is a consequence of the altered gating kinetics.
Figure 8. Single-channel currents recorded from wild-type and mutant KATP channels.
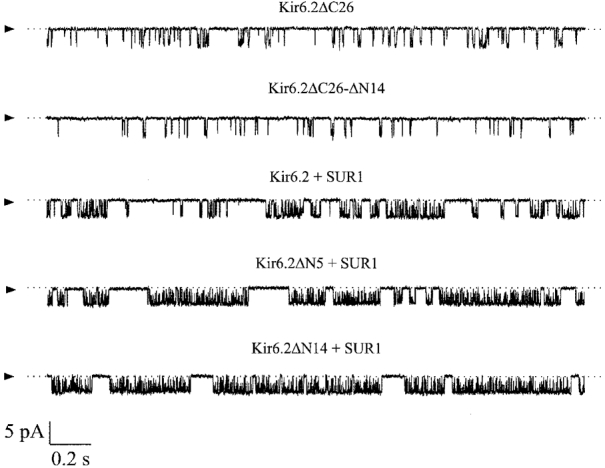
Single-channel currents recorded at -60 mV from inside-out patches excised from oocytes injected with mRNA(s) indicated. The arrowheads indicate the zero current level.
Table 2.
ATP block of KATP channels
| Clone | Ki(μM) | h1 | n | Po | n |
|---|---|---|---|---|---|
| Without SUR1 | |||||
| Kir6.2ΔC36 | 106 ± 4 | 1.2 ± 0.1 | 7 | 0.11 ± 0.03 | 4 |
| Kir6.2ΔC26-ΔN14 | 109 ± 8 | 0.9 ± 0.1 | 7 | 0.10 ± 0.02 | 4 |
| With SUR1 | |||||
| Kir6.2 | 28 ± 5 | 1.0 ± 0.01 | 5 | 0.32 ± 0.10 | 4 |
| Kir6.2ΔN5 | 50 ± 10 | 1.0 ± 0.1 | 5 | 0.44 ± 0.12 | 4 |
| Kir6.2ΔN9 | 96 ± 26 | 1.0 ± 0.1 | 6 | 0.61 ± 0.08 | 4 |
| Kir6.2ΔN14 | 196 ± 17 | 1.2 ± 0.1 | 10 | 0.70 ± 0.09 | 4 |
Ki, ATP concentration producing half-maximal block; h, Hill coefficient; Po, open probability.
How do N-terminal deletions alter tolbutamide sensitivity?
We next explored whether the reduced sulphonylurea block of Kir6.2ΔN14/SUR1 is also merely a consequence of the enhanced channel open probability. One way to test this idea would be to decrease Po by some independent means and then examine whether tolbutamide inhibition is affected. If the reduced sulphonylurea sensitivity of Kir6.2ΔN14/SUR1 results only from its altered kinetics, we would expect tolbutamide to block the channel when the Po was reduced prior to drug application: conversely, we predict that tolbutamide block would be independent of Po if deletion of the N-terminus of Kir6.2 impairs the mechanism by which sulphonylurea binding to SUR1 is translated into pore closure.
The simplest way to reduce the Po of the KATP channel is to apply ATP. Figure 9Aa shows that tolbutamide blocked Kir6.2ΔN14/SUR1 currents when applied in the presence of 100 μM ATP. This result requires careful interpretation, however, because of the interaction between Mg-nucleotides and sulphonylureas (Zünkler et al. 1988b; Gribble et al. 1997b). Like MgADP, MgATP interacts with the KATP channel at two sites, one that induces channel inhibition (on Kir6.2) and one that causes channel activation (on SUR1) (Gribble et al. 1998a). The overall action of MgATP is therefore a balance between these two opposing effects. It has been previously shown that tolbutamide abolishes the stimulatory action of MgADP, whilst leaving the inhibitory effect of the nucleotide on Kir6.2 intact (Gribble et al. 1997b). As a consequence, the extent of sulphonylurea block is greater when MgADP is present, because it results from the combined actions of MgADP and tolbutamide. Mutation of the Walker A lysines of SUR1 prevents MgATP binding (Ueda et al. 1997) and Mg-nucleotide stimulation of channel activity (Gribble et al. 1997b; Shyng et al. 1997b;Trapp et al. 1998). Figure 9Ab shows that the ability of tolbutamide to block the channel in the presence of MgATP was also abolished when both Walker A lysines were mutated (SUR1-K719A,K1385M, abbreviated SUR1-KAKM). This suggests that tolbutamide simply interferes with the stimulatory action of MgATP on Kir6.2ΔN14/SUR1 currents but has no direct blocking effect itself.
Figure 9. Tolbutamide block of mutant KATP channels.
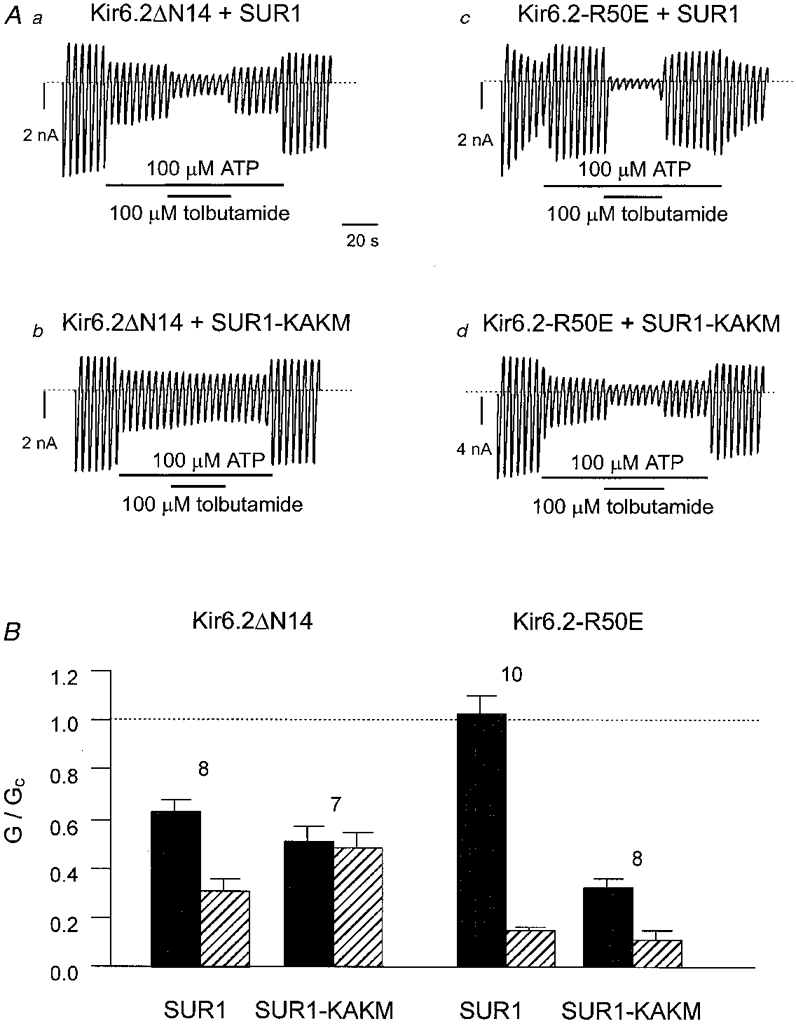
Aa-d, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes with the indicated mRNAs. Tolbutamide (0·1 mM) or ATP (0·1 mM) were added to the intracellular solution as indicated by the bars. B, mean slope conductance recorded in the presence of 100 μM ATP (▪) or in 100 μM ATP + 100 μM tolbutamide ( ), expressed as a fraction of that in control solution (lacking drugs and nucleotide). Currents were recorded from oocytes expressing Kir6.2ΔN14, or Kir6.2-R50E, together with either SUR1 or SUR1-KAKM, as indicated. The number of patches is given above each bar. The current level in control solution is indicated by the dotted line.
), expressed as a fraction of that in control solution (lacking drugs and nucleotide). Currents were recorded from oocytes expressing Kir6.2ΔN14, or Kir6.2-R50E, together with either SUR1 or SUR1-KAKM, as indicated. The number of patches is given above each bar. The current level in control solution is indicated by the dotted line.
When SUR1-KAKM is coexpressed with full-length Kir6.2, rather than Kir6.2ΔN14, tolbutamide causes channel inhibition in the absence of ATP (Gribble et al. 1997b). To confirm that this is also the case in the presence of ATP, we used a mutant form of Kir6.2 (Kir6.2-R50E) which is blocked by ATP to a similar extent as Kir6.2ΔC26-ΔN14 (Proks et al. 1999). This is because both the wild-type channel and Kir6.2/SUR1-KAKM are highly sensitive to ATP and the stimulatory effect of MgATP is not observed at low ATP concentrations (Gribble et al. 1998a).
As Fig. 9Ac shows, when Kir6.2-R50E and SUR1 were coexpressed, the resulting channel was not inhibited by 100 μM ATP: indeed in some patches, as shown here, the channel was actually activated. This may be attributed to the stimulatory effect of MgATP mediated by the nucleotide binding domains of SUR1, because ATP was an effective inhibitor of the channel when the Walker A lysines in both NBDs were mutated (Fig. 9Ad). Addition of tolbutamide in the presence of ATP produced a much more profound block of Kir6.2-R50E/SUR1 than was observed for Kir6.2ΔN14/SUR1. Moreover, the drug remained able to block the channel when the stimulatory effect of MgATP was abolished by coexpression of Kir6.2-R50E with SUR1-KAKM (Fig. 9Ad), in marked contrast to the lack of tolbutamide effect on Kir6.2ΔN14/SUR1-KAKM (Fig. 9Ab).
Taken together these results suggest that the ability of tolbutamide to uncouple the potentiatory effect of MgATP is not impaired by deletion of the N-terminus of Kir6.2. They further argue that the loss of tolbutamide block (most evident in the absence of nucleotide) involves a functional uncoupling of Kir6.2ΔN14 from SUR1, rather than being simply a secondary consequence of the enhanced Po.
To confirm this hypothesis, we used a mutant form of Kir6.2 that shows a markedly enhanced open probability. Mutation of cysteine 166 to serine (C166S) increases the Po to 0·8, and concomitantly reduces the ATP sensitivity and abolishes inhibition by 0·1 mM tolbutamide (Trapp et al. 1998). Figure 10A demonstrates that when the Po of Kir6.2-C166S/SUR1-KAKM was decreased to 50 % by addition of ATP, 100 μM tolbutamide caused significant inhibition of the current. In contrast, Kir6.2ΔN14/SUR1-KAKM currents were unaffected when the Po was reduced. Mean data for the effect of tolbutamide at different ATP concentrations are given in Fig. 10B, and show that as the ATP concentration was increased, tolbutamide block of Kir6.2-C166S/SUR1-KAKM was enhanced, while the drug continued to remain ineffective on Kir6.2ΔN14/SUR1-KAKM. Indeed, there was a reciprocal correlation between the extent of ATP block and that of tolbutamide inhibition in the presence of ATP for Kir6.2-C166S/SUR1-KAKM, but not for Kir6.2ΔN14/SUR1-KAKM (Fig. 10C). These data provide additional support for the idea that the lack of tolbutamide sensitivity of Kir6.2ΔN14/SUR1 is not the result of the enhanced Po.
Figure 10. Tolbutamide block of mutant KATP channels.
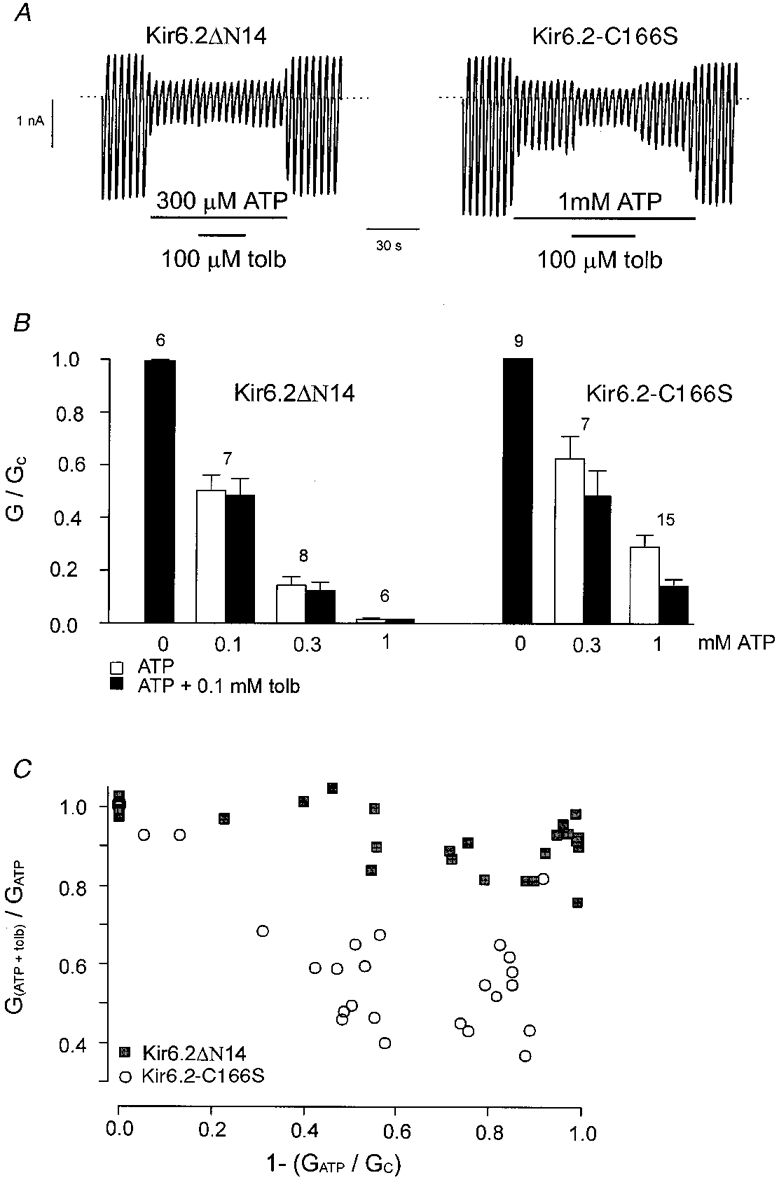
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes expressing SUR1-KAKM together with either Kir6.2ΔN14 (ΔN14) or Kir6.2-C166S. Tolbutamide (tolb; 0·1 mM) or ATP were added to the intracellular solution as indicated by the bars. B, mean current amplitude recorded in the presence of various concentrations of ATP (□), or various concentrations of ATP + 0·1 mM tolbutamide (▪), expressed as a fraction of that in control solution (lacking drugs and nucleotide). Currents were recorded from oocytes expressing Kir6.2ΔN14, or Kir6.2-C166S, together with SUR1-KAKM. The number of patches is given above each bar. C, relationship between the fractional block by ATP and the fractional block by tolbutamide in the presence of ATP, measured in the same patch. ▪, Kir6.2ΔN14/SUR1-KAKM; ○, Kir6.2-C166S/SUR1-KAKM. Data were obtained at different ATP concentrations. The conductance in the presence of ATP is expressed as a fraction of its value in ATP-free solution (1 - (GATP/Gc)). The conductance in the presence of ATP and 100 μM tolbutamide is expressed as a fraction of its value in ATP-containing solution (G(ATP + tolb)/GATP).
DISCUSSION
Our results show that partial deletion of the N-terminus of Kir6.2 has no effect on the intrinsic properties of Kir6.2, but influences KATP channel behaviour when Kir6.2 is coexpressed with SUR1. In particular, the open probability is enhanced, the sensitivity to the inhibitory effects of ATP and sulphonylureas is decreased and there is a reduction in the stimulatory action of diazoxide and MgADP. These data argue that the first 14 amino acids of Kir6.2 are involved in functional coupling to SUR1.
Gating
The open probability of Kir6.2ΔC26 is not affected by N-terminal deletions of up to 14 amino acids, indicating that these residues are not involved in the intrinsic gating of the Kir6.2 subunit. When wild-type Kir6.2 is coexpressed with SUR1, however, the open probability (Po) of the KATP channel is enhanced - from a Po of ∼0·1 to ∼0·3 (Proks & Ashcroft, 1997; Table 2). The effect of SUR1 is magnified if between 5 and 14 residues are deleted from the N-terminus of Kir6.2, the Po increasing in parallel with the length of the deletion. These data argue that the N-terminus of Kir6.2 may exert a negative effect on the ability of SUR1 to promote KATP channel activity. They also demonstrate that the ability of SUR1 to enhance the Po (Proks & Ashcroft, 1997) cannot involve the first 14 residues of Kir6.2. Finally, our results raise the possibility that SUR1 may physically interact with the N-terminus of Kir6.2 to influence channel gating.
ATP sensitivity
There is accumulating evidence that ATP interacts with Kir6.2 to mediate KATP channel inhibition. This derives from the observations that a C-terminally truncated form of Kir6.2 (Kir6.2ΔC), expressed in the absence of SUR1, is blocked by ATP (Tucker et al. 1997), that mutations within Kir6.2 affect the channel ATP sensitivity (Shyng et al. 1997a;Tucker et al. 1997, 1998; Drain et al. 1998), and that Kir6.2 can be photo-affinity labelled with 8-azido-[32P]ATP (Tanabe et al. 1999). The results presented here demonstrate that the first 14 residues of Kir6.2 are not involved in ATP binding since deletion of these residues was without effect on ATP inhibition.
A marked decrease in ATP sensitivity was observed when Kir6.2ΔN14 was coexpressed with SUR1. A similar finding has been reported for N-terminal truncations of 10-30 residues by others (Koster et al. 1999). Although we cannot exclude a direct involvement of the N-terminus of Kir6.2 in the mechanism by which SUR1 enhances the ATP sensitivity of the KATP channel (Tucker et al. 1997), we favour the idea that the decrease in ATP sensitivity of Kir6.2ΔN14/SUR1 is a secondary consequence of the reduced channel open probability. It has been shown that mutations which increase Po also increase the Ki for ATP inhibition (Shyng et al. 1997a;Trapp et al. 1998). Our results demonstrate that there is a reciprocal increase in Po and a decrease in ATP sensitivity for N-terminal truncations of different length. Thus the reduced efficacy of ATP may be an indirect result of the altered gating kinetics. A similar conclusion was reached by Koster et al. (1999).
Nucleotide activation
Our results argue that deletion of the N-terminus of Kir6.2 does not alter the ability of MgADP or MgATP/diazoxide to enhance channel activity. Although the percentage increase in channel activity is less for Kir6.2ΔN14/SUR1 than for wild-type channels, the final current amplitude in the presence of the nucleotide is similar (or even enhanced). Since the channel has an upper limit of activity (Po ≈ 0·9), the smaller increase in current observed for Kir6.2ΔN14/SUR1 is probably an inevitable consequence of the enhanced basal Po.
Tolbutamide modulation of MgADP sensitivity
The ability of tolbutamide to abolish the stimulatory effects of MgATP was unaffected by deletion of the first 14 residues of Kir6.2. This result indicates that tolbutamide binding to SUR1 is unaffected by the deletion of the N-terminus of Kir6.2. It also indicates that the distal N-terminus is not required for this action of tolbutamide.
Sulphonylureas might interfere with the potentiatory effects of Mg-nucleotides either by reducing nucleotide binding (to the NBDs of SUR1) or by preventing the translation of binding into channel activation. Because glibenclamide is able to block the co-operative binding of MgATP and MgADP to SUR1 (Ueda et al. 1999), the former explanation seems more likely. The fact that removal of the N-terminus of Kir6.2 does not affect the ability of tolbutamide to inhibit MgADP activation is consistent with this idea.
Tolbutamide sensitivity
Removal of the first 14 residues of Kir6.2 did not alter the low-affinity block by tolbutamide, indicating that these amino acids do not contribute to the low-affinity binding site on Kir6.2. When Kir6.2ΔN14 was coexpressed with either SUR1 or SUR2A, however, high-affinity inhibition by sulphonylureas was completely abolished. Indeed, truncation of the N-terminus of Kir6.2 by as few as five amino acids greatly decreased the amount of block. There is good evidence that the high-affinity sulphonylurea binding site lies on SUR (Aguilar-Bryan et al. 1995; Tucker et al. 1997). It therefore seems unlikely that N-terminal truncations of Kir6.2 influence the affinity of the sulphonylurea binding site on SUR. This idea is supported by the fact that tolbutamide is still able to abolish the stimulatory effects of Mg-nucleotides.
We have considered two possible mechanisms by which N-terminal deletion of Kir6.2 might affect the efficacy of sulphonylurea inhibition. First, it may be a secondary consequence of the greater open probability of Kir6.2ΔN14/SUR1 channels; second, it may result because the N-terminus is involved in transducing sulphonylurea binding to SUR1 into closure of the pore of Kir6.2. It is undeniable that a reduction of the channel open probability will concomitantly decrease sulphonylurea sensitivity: as previously described, Kir6.2-C166S/SUR1 channels have an open probability ∼4-fold greater than wild-type and are unaffected by 100 μM tolbutamide, a concentration specific for the high-affinity site on SUR1 (Trapp et al. 1998). Thus the enhanced Po of Kir6.2ΔN14/SUR1 may be expected to reduce sulphonylurea inhibition. Our results show, however, that this effect masks a more interesting phenomenon. When the Po is lowered by other means, tolbutamide is still unable to block Kir6.2ΔN14/SUR1 currents, despite causing marked inhibition of Kir6.2-C166S/SUR1 channels. We therefore conclude that the N-terminus of Kir6.2 is involved in coupling sulphonylurea binding to SUR1 into closure of the Kir6.2 pore. Whether this results from a direct physical interaction of the N-terminus of Kir6.2 with SUR1, or whether it involves an allosteric interaction between the two subunits, will require biochemical investigation.
Acknowledgments
We thank Yamanouchi Research Institute International, The Wellcome Trust and the British Diabetic Association for support. F. R. is a Yamanouchi Research Institute Fellow. S. J. T. is a Wellcome Trust Research Fellow.
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, Boyd AE, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends in Neurosciences. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Progress in Biophysics and Molecular Biology. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Aguilar-Bryan L, Bryan J. A view of sur/KIR6.X, KATP channels. Annual Review of Physiology. 1998;60:667–687. doi: 10.1146/annurev.physiol.60.1.667. [DOI] [PubMed] [Google Scholar]
- Babenko AP, Gonzalez G, Bryan J. The N-terminus of KIR6.2 limits spontaneous bursting and modulates the ATP-inhibition of KATP channels. Biochemical and Biophysical Research Communications. 1999;255:231–238. doi: 10.1006/bbrc.1999.0172. [DOI] [PubMed] [Google Scholar]
- Clement JP, IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Drain P, Li L, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Auml;mmälä C, Ashcroft FM. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. The Journal of Physiology. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. The Journal of Physiology. 1997b;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the β-cell KATP channel by interaction with its SUR1 subunit. Proceedings of the National Academy of Sciences of the USA. 1998a;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Seino S, Ashcroft FM. Tissue specificity of sulphonylureas: studies on cloned cardiac and β-cell KATP channels. Diabetes. 1998b;47:1412–1418. doi: 10.2337/diabetes.47.9.1412. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulphonylurea receptors determines the sensitivity of the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic β-cell ATP-sensitive K+ channel. FEBS Letters. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Shyng SL, Nichols CG. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. The Journal of Physiology. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P, Ashcroft FM. Phentolamine block of KATP channels is mediated by Kir6.2. Proceedings of the National Academy of Sciences of the USA. 1997;94:11716–11720. doi: 10.1073/pnas.94.21.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proks P, Gribble FM, Adhikari R, Tucker SJ, Ashcroft FM. Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. The Journal of Physiology. 1999;514:19–25. doi: 10.1111/j.1469-7793.1999.019af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Auml;mmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Letters. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Control of rectification and gating of KATP channels by the Kir6.2 subunit. Journal of General Physiology. 1997a;110:141–153. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Journal of General Physiology. 1997b;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Octameric stochiometry of the KATP channel complex. Journal of General Physiology. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, Amach IT, Ueda K. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-azido-ATP. Journal of Biological Chemistry. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. Journal of General Physiology. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proceedings of the National Academy of Sciences of the USA. 1997;94:8872–8877. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO Journal. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+-channels in the absence of the sulphonylurea receptor. Nature. 1997;378:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Ueda K, Inagaki N, Seino S. MgADP antagonism to Mg-independent ATP binding of the sulfonylurea receptor SUR1. Journal of Biological Chemistry. 1997;272:22983–22986. doi: 10.1074/jbc.272.37.22983. [DOI] [PubMed] [Google Scholar]
- Ueda K, Komine J, Matsuo M, Seino S, Amachi T. Cooperative binding of ATP and MgADP in the sulphonylurea receptor is modulated by glibenclamide. Proceedings of the National Academy of Sciences of the USA. 1999;96:1268–1272. doi: 10.1073/pnas.96.4.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zünckler BJ, Lenzen S, Männer K, Panten U, Trube G. Concentration-dependent effects of tolbutamide, meglitinide, glipizide, glibenclamide and diazoxide on ATP-regulated K+ currents in pancreatic B-cells. Naunyn-Schmiedeberg's Archives of Pharmacology. 1988a;337:225–230. doi: 10.1007/BF00169252. [DOI] [PubMed] [Google Scholar]
- Zünckler BJ, Lins S, Ohno-Shosaku T, Trube G, Panten U. Cytosolic ADP enhances the sensitivity of tolbutamide of ATP-dependent K+ channels from pancreatic β-cells. FEBS Letters. 1988b;239:241–244. doi: 10.1016/0014-5793(88)80925-6. [DOI] [PubMed] [Google Scholar]