

Abstract
The effects of tumour necrosis factor-α (TNF) on guinea-pig bronchial smooth muscle contractility were investigated.
The Ca2+-activated contractile response of permeabilized bronchial smooth muscle strips was significantly increased after incubation with 1 μg ml−1 TNF for 45 min. This TNF-induced effect was not due to a further increase in intracellular Ca2+.
The TNF-induced Ca2+ sensitization was, at least partly, the result of an increase in myosin light chain20 phosphorylation.
The intracellular signalling pathway involved in this effect of TNF was further investigated. Sphingomyelinase, a potential mediator of TNF, had no effect on Ca2+ sensitivity of permeabilized bronchial smooth muscle. Also, p42/p44 mitogen-activated protein kinase (p42/p44mapk), activated by TNF in some cell types, did not show an increased activation in bronchial smooth muscle after TNF treatment.
In conclusion, TNF may activate a novel signalling pathway in guinea-pig bronchial smooth muscle leading to an increase in myosin light chain20 phosphorylation and a subsequent increase in Ca2+ sensitivity of the myofilaments. This pathway does not appear to involve sphingomyelinase-liberated ceramides or activation of p42/p44mapk. Given the importance of TNF in asthma, this TNF-induced Ca2+ sensitization of the myofilaments may represent a mechanism responsible for airway hyper-responsiveness.
Bronchial asthma is a disease which is characterized by airway hyper-responsiveness (reviewed in Boushey et al. 1980) leading to an increased airway resistance. The pathophysiology of this disease is regulated by the release of cytokines from inflammatory cells such as neutrophils (Dubravec et al. 1990) and macrophages (Gosset et al. 1991). There is increasing evidence that one of these cytokines, tumour necrosis factor-α (TNF), is directly linked with airway inflammation and the hyper-responsiveness observed in asthma (Shah et al. 1995). TNF is elevated in the sputa of patients with bronchial asthma (Taki et al. 1991). In addition in vivo pretreatment of rat and human airways with TNF in aerosol form produced increases in bronchial airway resistance, similar to that observed in asthma, when challenged with endogenous agonists (Kips et al. 1992; Thomas et al. 1995). Pharmacological evidence has also pointed towards an important role for TNF in airway hyper-responsiveness. A TNF receptor fusion protein, which is a potent antagonist of TNF, was effective at reducing both the enhanced airway reactivity and neutrophil infiltration into the airways in two different asthmatic models (Renzetti et al. 1996). These observations suggest that TNF may be one of the primary components responsible for the bronchial smooth muscle hyperresponsiveness observed in asthma.
The mechanism of TNF-induced hyper-responsiveness remains unclear. The TNF effects may be indirect, via release of other inflammatory or bronchconstricting agents such as leukotrienes (Moore et al. 1991), or be a direct effect on the bronchial smooth muscle cells. TNF binds to specific TNF receptors of which there are two isoforms, p55TNF receptor and p75TNF receptor. These receptors are not 7-transmembrane G-protein coupled but are single transmembrane glycoproteins which form dimers upon activation (Heller & Kronke, 1994). Although bronchial smooth muscle cells do express TNF receptors (Amrani et al. 1996) very few studies have attempted to determine the direct effects of TNF on airway contractility. Amrani et al. (1995) have shown that incubation with TNF in cultured airway smooth muscle cells produced an enhanced increase in intracellular calcium following agonist stimulation. This increase may have been the result of an increased phosphoinositide turnover (Amrani et al. 1997) leading to an enhanced inositol 1,4,5-trisphosphate receptor-induced intracellular Ca2+ release. This is unlikely to be the only mechanism underlying TNF-induced airway hyperresponsiveness as the incubation with TNF was long (18 h) compared with incubations in in vivo studies (90 min; Kips et al. 1992). TNF receptor activation in airway smooth muscle cells may therefore have other, more rapid, mechanisms which can potentiate bronchoconstriction.
In the present study we have investigated the effects of TNF on permeabilized guinea-pig bronchial smooth muscle at a constant intracellular calcium concentration. We show for the first time that TNF can produce an increased sensitivity of the bronchial smooth muscle myofilaments to calcium. The time course of this effect (45 min) is consistent with previous in vivo studies (Kips et al. 1992) and is the result of an increase in myosin light chain20 (MLC20) phosphorylation. The intracellular pathways involved in this TNF-mediated effect does not appear to activate two known TNF effector pathways: the p42/p44 (extracellular signal-related kinases) mitogen-activated protein kinase isoforms or the sphingomyelinase/ceramide signalling pathways. Given the importance of TNF in asthma, this TNF-induced increase in calcium sensitivity may be an important mechanism involved in bronchial hyperresponsiveness.
METHODS
Male Duncan Hartley guinea-pigs, weight 350-550 g, were killed by cervical dislocation, the trachea and bronchi removed and placed in Hepes-buffered normal Krebs solution. Smooth muscle strips (200 μm wide, 3 mm long) were cut from first order bronchi and the epithelial layer removed. Strips were mounted on a ‘bubble’ plate and attached to a force transducer (AE801, Sensonor a.s., Alnwick, UK). After determining contractile responses to 143 mM K+ and 0·1 mM carbachol, the strips were incubated in normal relaxing solution (G1) (Ca2+ free, containing (mM): 1 EGTA, 30 Pipes, 10 creatine phosphate, 7·3 Na2ATP, 85·8 potassium methane sulphonate) and permeabilized for 1 h at 24°C with 80 μg ml−1Staphylococcus aureusα-toxin (List Biological Laboratories, Campbell, CA, USA). A23187 (10 μM) was added to release stored calcium. Details of solutions used have been described previously (Kobayashi et al. 1989).
Initially, the effects of recombinant human tumour necrosis factor-α (R & D Systems, Abingdon, UK) were examined in intact bronchial strips. Strips were stimulated with either 10−7, 10−6 or 10−4 m carbachol at intervals of 1 h until a consistent contractile response of the same magnitude was observed. This carbachol-induced contraction was repeated once more to establish a basal contractile response. The strip was incubated with 1 μg ml−1 (approximately 57 nM) TNF for 1 h at 24°C in Hepes-buffered Krebs solution and challenged with carbachol. The peak of the resulting contraction was expressed as a percentage of the original carbachol-induced response before incubation with TNF. Paired control experiments were treated identically but had no TNF added. Statistical analysis was carried out using 2-way ANOVA.
Strips of bronchi permeabilized with α-toxin were challenged with a maximally contracting Ca2+ concentration, pCa 4·5, to determine permeabilization and relaxed in G1 normal relaxing solution. Strips were further incubated with a submaximal concentration of Ca2+ which would typically induce 20-25 % of maximal contraction, usually pCa 6·5-6·3. After relaxing again in G1, strips were incubated for 45 mins with 1 μg ml−1 recombinant human TNF at 24°C in G1 (or in some experiments, 0·33 μg ml−1 sphingomyelinase). Following this incubation, strips were challenged with the same pCa solution as before. This second pCa response was expressed as a percentage of the initial pCa contractile response before TNF incubation. The tension measurements were taken as the height of contraction of the steady-state tonic component (plateau) from the calcium-activated contraction.
In some experiments with permeabilized strips, the submaximal pCa contractile response was immediately followed by the addition of 50 μM GTP and 0·1 mM carbachol to produce a Ca2+ sensitization. The strips were relaxed in G1 for 30 min to allow reversal of the carbachol-induced Ca2+ sensitization and this was confirmed by re-challenging with a submaximal pCa solution. The strip was incubated as before with TNF for 45 mins and challenged with the submaximal pCa solution followed by 50 μM GTP and 0·1 mM carbachol. Statistical analysis was carried out using an unpaired Student's t test.
In order to determine changes in MLC20 phosphorylation in permeabilized guinea-pig bronchial smooth muscle, strips were rapidly frozen in liquid Freon 22 either at the peak of contraction during TNF-induced Ca2+ sensitization or after tension reached a plateau in submaximal Ca2+ buffer. Strips were transferred to frozen acetone containing 10 % trichloroacetic acid. The acetone- trichloroacetic acid was exchanged for 100 % acetone and strips left to air dry at room temperature. Dried strips were homogenized in glycerol sample buffer (containing 1 % SDS, 10 % glycerol and 1 mM dithiothreitol) and subjected to 2-dimensional gel electrophoresis (Evans et al. 1999) consisting of isoelectric focussing (ampholyte range pH 4·5-5·5, Pharmacia Biotech, St Albans, UK) in the first dimension followed by SDS-polyacrylamide gel electrophoresis in the second dimension on 12 % polyacrylamide gels. Resolved proteins were transferred onto nitrocellulose (Biorad Labs Ltd., Hemel Hempstead, UK) and the membrane stained with colloidal gold solution to reveal protein spots of unphosphorylated, monophosphorylated and diphosphorylated MLC20. Phosphorylation of the myosin was quantitatively compared using Biorad GS690 densitometer.
To measure activation of p42/p44 mitogen-activated protein kinase (p42/p44mapk), a specific antibody (Santa Cruz Biotechnology Inc., Heidelberg, Germany) which recognizes only the phosphorylated (and therefore activated) form of this enzyme was utilized. Permeabilized or intact strips were incubated either in the presence or absence (control) of TNF as described above. Control and TNF-treated strips were placed in acetone containing 10 % trichloroacetic acid followed by acetone and dried in air. Dried strips were homogenized in sample buffer (1 % SDS, 10 % glycerol, 20 mM dithiothreitol, Bromophenol Blue) using a small glass hand-held homogenizer. Samples were split into two equal volumes. One half of the sample was used to measure p42/p44mapk activation with a phospho-p42/p44mapk antibody. The other half was used to determine the total amount of p42/p44mapk (activated and unactivated) using anti-p42/p44mapk antibody. As a positive control, primary cultured airways smooth muscle cells (passage 2, prepared as previously described, Pyne et al. 1997) were treated with either 1 μg ml−1 TNF or 0·1 mM 4-phorbol 12,13-dibutyrate (PDBu) and the reaction stopped by the addition of lysis buffer (2 % SDS, 70 mM Tris at pH 6·8). Control samples had no TNF or PDBu added. Samples were boiled for 5 min and subjected to SDS- polyacrylamide gel electrophoresis (12 %). Slab gels were transferred onto nitrocellulose membranes and incubated with primary antibody. Following incubation with the primary antibody, membranes were washed and incubated with a secondary antibody conjugated to horseradish peroxidase (Scottish Antibody Production Unit, Carluke, UK). Stained proteins were visualized by enhanced chemiluminescence (Amersham PLC, Little Chalfont, UK).
All results are expressed as means ±s.e.m. where appropriate. P < 0·05 denotes statistical significance.
RESULTS
In intact strips of bronchial smooth muscle, a 1 h incubation with TNF produced a significant potentiation of the carbachol-induced steady state contraction compared with controls at all concentrations of carbachol examined (Fig. 1). The potentiation was greatest at lower concentrations of carbachol (approximately 270 % compared with controls). TNF alone did not produce any contractile response (data not shown). To investigate the nature of this potentiation further, the effects of TNF on permeabilized bronchial smooth muscle strips were examined. Under conditions with no added Ca2+ and 1 mM EGTA (G1 buffer), a 45 min incubation with TNF potentiated the plateau phase of the contractile response to a submaximal concentration of Ca2+, compared with control strips, by approximately 250 % (Fig. 2). A 20 min time course for the TNF incubation resulted in approximately half of the potentiation observed at 45 min (data not shown). The increase was not due to an effect on intracellular Ca2+ release because A23187 was added to deplete intracellular Ca2+ stores, suggesting that this is a Ca2+ sensitization of the myofilaments. This TNF potentiation of the Ca2+-activated contraction corresponded with an increase in MLC20 phosphorylation of approximately 20 % as measured by densitometry of colloidal gold stained membranes (Fig. 3).
Figure 1. Contractile effects of TNF on guinea-pig intact bronchial muscle stimulated with carbachol.
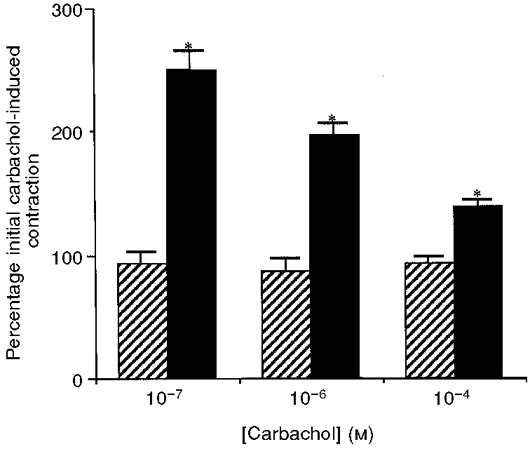
The force development is expressed as a percentage of the stabilized carbachol-induced contraction obtained before incubation of the bronchial muscle in TNF. In control samples ( ), the force is expressed as a percentage of the carbachol-induced contraction after a 1 h incubation in buffer with no TNF added. Following TNF treatment (▪), bronchial smooth muscle strips showed a significantly increased contractile response, compared with controls, at all concentrations of carbachol examined. * Statistical significance, P < 0·05. n= 4 for TNF-treated and control muscle strips.
), the force is expressed as a percentage of the carbachol-induced contraction after a 1 h incubation in buffer with no TNF added. Following TNF treatment (▪), bronchial smooth muscle strips showed a significantly increased contractile response, compared with controls, at all concentrations of carbachol examined. * Statistical significance, P < 0·05. n= 4 for TNF-treated and control muscle strips.
Figure 2. Contractile effects of TNF on Ca2+ sensitivity in permeabilized guinea-pig bronchial smooth muscle.

A, a typical tension recording of TNF-induced Ca2+ sensitization. B, mean increase in Ca2+ sensitization following incubation with TNF. ▪, TNF-treated, n= 27;  , control, n= 23. * Statistical significance, P < 0·05. The sensitized Ca2+-induced contractions are expressed as a percentage of the initial submaximal Ca2+ contractile response before TNF (or control) incubation. Permeabilized strips incubated with TNF for 45 min revealed a sensitized response to submaximally contracting calcium buffers.
, control, n= 23. * Statistical significance, P < 0·05. The sensitized Ca2+-induced contractions are expressed as a percentage of the initial submaximal Ca2+ contractile response before TNF (or control) incubation. Permeabilized strips incubated with TNF for 45 min revealed a sensitized response to submaximally contracting calcium buffers.
Figure 3. Effect of TNF on myosin light chain20 phosphorylation in permeabilized guinea-pig bronchial smooth muscle strips.

A typical colloidal gold stained 2-dimensional gel (n= 3) is shown. Permeabilized strips were incubated in a submaximal pCa buffer after incubation in G1 (control, A) or 1 μg ml−1 TNF in G1 (B) and frozen at the peak of steady-state contraction. Homogenized strips were subjected to 2-dimensional electrophoresis as described in Methods. Control strips were phosphorylated 17 ± 1 % of total MLC20 compared with TNF-treated strips which were phosphorylated 37 ± 2 % of total MLC20. Duplicate strips were used in each sample. Contractile responses of control strips to submaximal pCa buffer were 12 ± 2 % of maximal contraction and in TNF-treated strips were 34 ± 5 % of maximal contraction. NP, non-phosphorylated; P1, monophosphorylated; P2, diphosphorylated MLC20.
Experiments to determine if the TNF-induced Ca2+ sensitization was additive were carried out by examining carbachol-induced calcium sensitization in permeabilized bronchial smooth muscle strips. Strips were incubated with a submaximal concentration of Ca2+ until a steady-force development was obtained. Addition of GTP and 0·1 mM carbachol displayed a relatively fast contractile response (Ca2+ sensitization) (Fig. 4). The Ca2+ sensitization (GTP and carbachol) accounted for 77 ± 4 % (n= 3) of the total response (submaximal Ca2+ and GTP-carbachol contraction combined). Strips relaxed in G1 solution for 30 min and re-challenged with the same submaximal Ca2+-containing buffer showed that the carbachol-induced Ca2+ sensitization had reversed during this period (not shown). Following a further incubation in G1 with TNF, the strips displayed a sensitized contractile response to the submaximal Ca2+ buffer as previously seen. After a steady level of force was achieved, GTP-carbachol added to stimulate Ca2+ sensitization had a significantly reduced effect (Fig. 4) (31 ± 6 % sensitization of the total response, n= 3, P < 0·05), but the total tension recorded with the pCa solution and GTP-carbachol combined was similar to that observed before TNF incubation. In control strips, the proportion of the total contractile response produced by GTP-carbachol-induced Ca2+ sensitization was unchanged throughout the course of the experiment (78 ± 2 % of total response).
Figure 4. A typical tension recording of TNF-treated permeabilized guinea-pig bronchial smooth muscle showing non-additive effects of TNF-induced Ca2+ sensitization and carbachol-induced Ca2+ sensitization.

A typical trace (n= 3) is shown. CCh, carbachol. After incubation in submaximal Ca2+ buffer achieved a steady-state tension, GTP and CCh produced a Ca2+ sensitization in permeabilized strips. After 30 mins in G1 the sensitization had completely reversed (results not shown). Following TNF incubation, the submaximal Ca2+ response was increased. Addition of GTP and CCH resulted in a significantly reduced Ca2+ sensitization response.
In order to investigate a possible mechanism for the TNF-induced Ca2+ sensitization, sphingomyelinase was added to liberate ceramide products from the membrane. Experiments repeated with sphingomyelinase, under the same conditions which revealed TNF-induced Ca2+ sensitization, did not produce a potentiation of the submaximal Ca2+ activated contractile response (Fig. 5). However, the concentration of sphingomyelinase used (0·33 μg ml−1) was capable of producing a phasic contraction in intact bronchial smooth muscle, presumably via intracellular Ca2+ release (Kim et al. 1995), and therefore was biologically active and able to liberate lipid products from the membrane. Furthermore, this concentration of sphingomyelinase produces maximal stimulatory effects in other cell types (D. J. MacEwan, unpublished observations).
Figure 5. Lack of effect of sphingomyelinase on Ca2+ sensitization in permeabilized guinea-pig bronchial smooth muscle.

A typical trace (n= 4) is shown. After the sphingomyelinase incubation the submaximal pCa-induced contractile response was unchanged. A further incubation with TNF was still capable of increasing the Ca2+ sensitivity of the permeabilized bronchial smooth muscle strip.
The activation of signalling enzymes, which may be regulated by TNF, was examined. The activation of the p42/p44mapk isoforms was investigated using phospho-specific antibodies which recognizes the activated form of these enzymes. Homogenates of permeabilized and intact bronchial smooth muscle strips incubated with TNF in either G1 (permeabilized) for 45 min or Hepes-buffered Krebs solution (intact) for 1 h did not show an increased phosphorylation, compared with control strips, for p42/44mapk isoforms (Fig. 6A) when normalized for equal p42/p44mapk protein expression (Fig. 6B). Primary cultured unpermeabilized guinea-pig airway smooth muscle cells (passage 2) were incubated with PDBu to determine if p42/p44mapk could be activated in this cell type by known stimulators of MAPK isoforms. Cells treated with PDBu did produce an activation of p42/p44mapk of approximately 6-fold (n= 2; Fig. 7). These cells did not show any activation of MAPK isoforms after treatment with TNF.
Figure 6. Immunoblots of whole cell homogenates from permeabilized and intact bronchial smooth muscle strips showing activated p42/p44mapk.
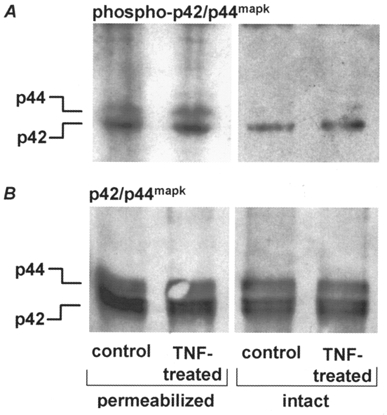
A typical immunoblot (n= 3) is shown. Strips were incubated with TNF; control strips had no TNF present. A, homogenates immunoblotted with anti-phospho p42/p44mapk antibody which recognizes the activated form of MAPK. B, homogenates immunoblotted with anti-p42/p44mapk antibody which recognizes both active and inactive forms for comparison of protein concentrations. There was no increased activation of p42/p44mapk by TNF in permeabilized or intact strips compared with controls.
Figure 7. Activation of p42/p44mapk in whole cell homogenates from primary cultured airway smooth muscle cells treated after control, TNF or PDBu incubations.

Each lane contains 40 μg total protein. A, samples immunoblotted with anti-phospho p42/p44mapk antibody to show activated enzyme (n= 3). B, anti-p42/p44mapk antibody to show protein. TNF-treated cells showed no increased activation of p42/p44mapk compared with control samples, but PDBu-treated cells revealed an approximately 6-fold increase in activation.
DISCUSSION
There is considerable in vivo evidence of an important role for TNF in the airway hyper-responsiveness observed in asthma (Kips et al. 1992; Thomas et al. 1995; Renzetti et al. 1996). The present study describes a novel cellular mechanism which may be, at least partly, responsible for producing the airway hyper-responsiveness observed as a result of agonist stimulation following TNF treatment. Permeabilized bronchial smooth muscle strips incubated with TNF for 45 min showed a significantly increased Ca2+ sensitivity of the myofilaments. This increase in the Ca2+ sensitivity corresponds with an increase in MLC20 phosphorylation. The mock intracellular buffer used during the TNF incubation (G1) maintains the intracellular Ca2+ concentration at less than pCa 8·0. Under these conditions in permeabilized cells, it is unlikely that the bronchial smooth muscle will be able to produce or release other factors which may affect contractility, such as prostaglandins or leukotrienes, although other mediators of cellular signalling, such as arachidonic acid, may be produced. The TNF effects on contractility of the permeabilized bronchial smooth muscle are, therefore, likely to be direct effects produced by TNF-receptor activation of intracellular signalling pathways and not the result of TNF releasing other mediators which in turn act on the bronchial smooth muscle cells. Furthermore, as this effect is observed by increasing the intracellular calcium exogenously after TNF incubation in permeabilized cells, it is presumed in vivo that any agonist (demonstrated here with carbachol) which increases intracellular Ca2+ through either Ca2+ release or influx will produce this effect. This is therefore not an effect restricted to muscarinic agonists but is a general effect of all vasocontractile agonists.
The TNF-induced Ca2+ sensitization of the myofilaments revealed by these in vitro techniques provides a potential mechanism for TNF-induced airway hyper-responsiveness observed in vivo. The TNF incubation times used in this study are comparable with those used for in vivo studies (Kips et al. 1992). The concentrations of TNF (1 μg ml−1) are higher than those normally used to invoke other known TNF effects in cell lines, e.g. cell death (MacEwan, 1996), which is in the order of 1-100 ng ml−1. The concentration used here is similar to the in vivo studies which have observed TNF effects on airway resistance (Kips et al. 1992). Although these relatively higher concentrations were required to observe the TNF-induced Ca2+ sensitization, they may be relevant in pathophysiological terms in vivo where local increases in TNF release from cells adjacent to smooth muscle cells during inflammatory responses will certainly reach higher concentrations than those measured in serum (Yoshida et al. 1996).
The TNF-induced Ca2+ sensitization observed in this study has distinct differences from the agonist-induced Ca2+ sensitization in smooth muscle previously described in other studies (Bradley & Morgan, 1987; Kitazawa et al. 1991a; Somlyo & Somlyo, 1994). Most of the agonists to date which have been shown to induce Ca2+ sensitization in smooth muscle, including airways, are vasocontractile agonists and are members of the superfamily of 7-transmembrane hetero-trimeric G-protein coupled receptors (Kitazawa et al. 1991a; Parsons et al. 1996; Iizuka et al. 1997). TNF is not a vasocontractile agonist itself and is not an agonist at 7-transmembrane heterotrimeric G-protein coupled receptors. TNF receptors consist of single transmembrane glycoprotein subunits which dimerize on binding TNF thereby activating the receptor (Heller & Kronke, 1994). There are two predominant subtypes of the TNF receptor; the p55TNF and p75TNF receptors. Most of the known TNF responses occur by activation of the p55 receptor (Weigmann et al. 1992), including effects of TNF on airways smooth muscle (Amrani et al. 1996). To date there is little evidence that TNF receptors are directly coupled to heterotrimeric G-proteins in smooth muscle cells. This suggests that the mechanism of TNF-induced Ca2+ sensitization in bronchial smooth muscle is at least partly different from the 7-transmembrane receptor-activated Ca2+ sensitization. Another difference in the TNF-induced Ca2+ sensitization is the time course of effects. In this study 45 min was found to give the maximum sensitization and this increased Ca2+ response was not reversed for at least 1 h after removal of TNF (results not shown). Ca2+ sensitization induced by vasocontractile agonists activating 7-transmembrane receptors occurs usually within a few seconds of receptor activation and reverses within 5-15 min. The TNF effects observed here may reflect the very high affinity and stability of the TNF-receptor complex (slow off-rate; Grell et al. 1998) and involve several different pathways which require longer activation times.
The downstream target of 7-transmembrane receptor G-protein-induced Ca2+ sensitization is, in most cases, the inhibition of the smooth muscle myosin phosphatase (Kitazawa et al. 1991b). This may be mediated in part by agonist-induced activation of a monomeric G-protein, p21rhoA (Gong et al. 1996; Otto et al. 1996) and subsequent activation of a rho-associated kinase, p160ROCK (Matsui et al. 1996). p160ROCK inhibits the myosin phosphatase activity (Kimura et al. 1996) leading to an increased MLC20 phosphorylation (Kurieshi et al. 1997). Arachidonic acid (produced via an activation of phospholipase A2) has also been shown to inhibit the myosin phosphatase leading to an increased MLC20 phosphorylation in smooth muscle (Gong et al. 1992). The TNF-induced Ca2+ sensitization in airways smooth muscle observed here is, at least in part, due to an increase in MLC20 phosphorylation. However, the increased Ca2+ sensitivity after TNF incubation observed in this study was not additive with Ca2+ sensitization following muscarinic receptor activation, suggesting a possible downstream convergence of the signalling pathway activated by TNF incubation, possibly at the level of the myosin light chain phosphorylation. The signalling pathways activated by TNF receptors are still being elucidated but are known to involve several interacting signal transduction mechanisms (Heller & Kronke, 1994). In a study by Guy et al. (1992), TNF was observed to mimic the actions of okadaic acid (a phosphatase inhibitor) on myosin light chain phosphorylation in fibroblasts, although direct evidence to date of a TNF inhibitory effect on the myosin phosphatase has not been demonstrated. A common downstream signalling pathway of both TNF and 7-transmembrane receptor agonists may therefore be an inhibition of the myosin phosphatase. It is unlikely that similar upstream pathways of 7-transmembrane receptor-activated Ca2+ sensitization will be stimulated due to the different nature of the signalling pathways for these different receptors. No reports have yet described activation of p21rhoA by TNF in a similar time course to the experiments in this study, although this remains a possibility. TNF may stimulate PLA2 activity (Jayadev et al. 1994) via activation of p42/p44 MAPK (Veitor et al. 1991; Lin et al. 1993). This would lead to production of arachidonic acid which could produce an inhibition of the myosin phosphatase. We have examined p42/p44mapk activation in guinea-pig bronchial smooth muscle. Although these MAPK isoforms are present in bronchial smooth muscle and can be activated by phorbol esters in airway smooth muscle cells, under the conditions used here to reveal TNF-induced Ca2+ sensitization in permeabilized strips, no activation of p42/p44mapk was observed after TNF incubation. This lack of activation was not a result of the permeabilization procedure as TNF did not activate p42/p44mapk in intact bronchial smooth muscle strips. This suggests that TNF-induced Ca2+ sensitization in bronchial smooth muscle strips does not involve activation of p42/p44mapk and possibly subsequent activation of phospholipase A2.
In several cell types, evidence has suggested that TNF may activate a sphingomyelinase, to release ceramide from membrane lipids (Hannun, 1994). Although the effects of ceramide lipids on smooth muscle contractility have yet to be assessed, they can activate kinases in other cells (Hannun, 1994). The TNF-induced Ca2+ sensitization observed here does not appear to be signalling via a ceramide pathway, as revealed by a lack of effect of sphingomyelinase. It is possible that other signalling enzymes may be activated in the conditions used in this study. TNF may activate phosphatidylcholine-specific phospholipase C (Schutze et al. 1991) leading to diacylglycerol production and subsequent activation of protein kinase C isoforms. Phorbol esters can induce a Ca2+ sensitization in airways smooth muscle (Iizuka et al. 1997) although this has a much faster time course than that observed with TNF. This is currently the subject of further investigation.
In conclusion, TNF can produce an increase in the Ca2+ sensitivity of the myofilaments by increasing MLC20 phosphorylation in permeabilized bronchial smooth muscle. This effect differs from Ca2+ sensitization induced by 7-transmembrane receptor vasocontractile agonists due to the nature of TNF receptors and in the time course of the sensitization. The intracellular signalling pathways remain to be elucidated but do not appear to involve activation of sphingolipid pathways or p42/p44mapk. The ability of TNF to activate a Ca2+ sensitization mechanism in bronchial smooth muscle may explain the TNF-induced hyperresponsiveness of the airways observed in vivo. This pathway may represent a primary event of the bronchoconstriction associated with asthma.
Acknowledgments
The authors would like to thank Helen Anderson for assistance with primary culture. This work was supported by The Royal Society and The Wellcome Trust. J. R. M. P. is funded by the Sir Dudley Spurling Scholarship, Bank of Butterfield, Bermuda.
References
- Amrani Y, Krymskaya V, Maki C, Panettieri RA. Mechanisms underlying TNF-α effects on agonist-mediated calcium homeostasis in human airway smooth muscle cells. American Journal of Physiology. 1997;17:L1020–1028. doi: 10.1152/ajplung.1997.273.5.L1020. [DOI] [PubMed] [Google Scholar]
- Amrani Y, Martinet N, Bronner C. Potentiation by tumour necrosis factor-α of calcium signals induced by bradykinin and carbachol in human tracheal smooth muscle cells. British Journal of Pharmacology. 1995;114:4–5. doi: 10.1111/j.1476-5381.1995.tb14896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amrani Y, Panettieri RA, Frossard N, Bronner C. Activation of the TNF-α-p55 receptor induces myocyte proliferation and modulates agonist-evoked calcium transients in cultured human tracheal smooth-muscle cells. American Journal of Respiratory Cell and Molecular Biology. 1996;15:55–63. doi: 10.1165/ajrcmb.15.1.8679222. [DOI] [PubMed] [Google Scholar]
- Boushey HA, Holtzman MJ, Sheller JR, Nadel JA. Bronchial hyperreactivity. American Review of Respiratory Disease. 1980;121:389–413. doi: 10.1164/arrd.1980.121.2.389. [DOI] [PubMed] [Google Scholar]
- Bradley AF, Morgan KG. Alterations in cytoplasmic calcium sensitivity during porcine coronary artery contractions as detected by aequorin. The Journal of Physiology. 1987;385:437–448. doi: 10.1113/jphysiol.1987.sp016500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubravec DB, Spriggs DR, Mannick JA, Rodrick ML. Circulating human peripheral blood granulocytes synthesize and secrete tumour necrosis factor α. Proceedings of the National Academy of Sciences of the USA. 1990;87:6758–6761. doi: 10.1073/pnas.87.17.6758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans AM, Cobban HJ, Nixon GF. ETA receptors are primary mediators of myofilament calcium sensitization induced by ET-1 in rat pulmonary artery smooth muscle: a tyrosine kinase independent pathway. British Journal of Pharmacology. 1999;127:153–160. doi: 10.1038/sj.bjp.0702548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong MC, Fuglsang A, Alessi D, Kobayashi S, Cohen P, Somlyo AV, Somlyo AP. Arachidonic acid inhibits myosin light chain phosphatase and sensitizes smooth muscle to calcium. Journal of Biological Chemistry. 1992;267:21492–21498. [PubMed] [Google Scholar]
- Gong MC, Iizuka I, Nixon GF, Browne JP, Hall A, Eccleston JF, Sugai M, Kobayashi S, Somlyo AV, Somlyo AP. Role of guanine nucleotide binding proteins in calcium sensitisation of smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1996;93:1340–1345. doi: 10.1073/pnas.93.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gosset P, Tsicopoulos A, Wallaert B, Vannimenus C, Joseph M, Tonnel AB, Capron A. Increased secretion of tumor-necrosis factor-α and interleukin-6 by alveolar macrophages consecutive to the development of the late asthmatic reaction. Journal of Allergy and Clinical Immunology. 1991;88:561–571. doi: 10.1016/0091-6749(91)90149-i. [DOI] [PubMed] [Google Scholar]
- Grell M, Wajant H, Zimmermann G, Scheurich P. The type 1 receptor (CD120a) is the high-affinity receptor for soluble tumor necrosis factor. Proceedings of the National Academy of Sciences of the USA. 1998;95:570–575. doi: 10.1073/pnas.95.2.570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guy GR, Cao SP, Chua SP, Tan YH. Okadaic acid mimics multiple changes in early protein phosphorylation and gene expression induced by tumour necrosis factor or interleukin-1. Journal of Biological Chemistry. 1992;267:1846–1852. [PubMed] [Google Scholar]
- Hannun YA. The sphingomyelin cycle and the second messenger function of ceramide. Journal of Biological Chemistry. 1994;269:3125–3128. [PubMed] [Google Scholar]
- Heller RA, Kronke M. Tumor necrosis factor receptor-mediated signalling pathways. Journal of Cell Biology. 1994;126:5–9. doi: 10.1083/jcb.126.1.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iizuka K, Dobashi K, Yoshii A, Horie T, Suzuki H, Nakazawa T, Mori M. Receptor-dependent G protein-mediated Ca2+ sensitization in canine airway smooth muscle. Cell Calcium. 1997;22:21–30. doi: 10.1016/s0143-4160(97)90086-5. [DOI] [PubMed] [Google Scholar]
- Jayadev S, Linardic CM, Hannun YA. Identification of arachidonic acid as a mediator of sphingomyelin hydrolysis in response to tumour necrosis factor. Journal of Biological Chemistry. 1994;269:5755–5763. [PubMed] [Google Scholar]
- Kim S, Lakhani V, Costa DI, Sharara AI, Fitz JG, Huang LW, Peters KG, Kindman LA. Sphingolipid-gated Ca2 + release from intracellular stores of endothelial-cells is mediated by a novel Ca2+-permeable channel. Journal of Biological Chemistry. 1995;270:5266–5269. doi: 10.1074/jbc.270.10.5266. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng JH, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by rho and rho-associated kinase. Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kips JC, Tavernier J, Pauwels RA. Tumour necrosis factor causes bronchial hyperresponsiveness in rats. American Review of Respiratory Diseases. 1992;145:332–336. doi: 10.1164/ajrccm/145.2_Pt_1.332. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denny GH, Somlyo AP. G-protein mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. Journal of Biological Chemistry. 1991a;266:1708–1715. [PubMed] [Google Scholar]
- Kitazawa T, Masuo M, Somlyo AP. G-protein mediated inhibition of myosin light chain phosphatase in vascular smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1991b;88:9307–9310. doi: 10.1073/pnas.88.20.9307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi S, Kitazawa T, Somlyo AV, Somlyo AP. Cytosolic heparin inhibits muscarinic and adrenergic Ca2+ release in smooth muscle. Journal of Biological Chemistry. 1989;264:17997–18004. [PubMed] [Google Scholar]
- Kureishi Y, Kobayashi S, Amano M, Kimura K, Kanaide H, Nakano T, Kaibuchi K, Ito K. Rho-associated kinase directly induces smooth muscle contraction through myosin light chain phosphorylation. Journal of Biological Chemistry. 1997;272:12257–12260. doi: 10.1074/jbc.272.19.12257. [DOI] [PubMed] [Google Scholar]
- Lin LL, Wartmann M, Lin AY, Knopf JL, Seth A, Davis RJ. cPLA2 is phosphorylated and activated by MAP kinase. Cell. 1993;72:269–278. doi: 10.1016/0092-8674(93)90666-e. [DOI] [PubMed] [Google Scholar]
- MacEwan DJ. Elevated cPLA2 levels as a mechanism by which the p70 TNF and p75 NGF receptors enhance apoptosis. FEBS Letters. 1996;379:77–81. doi: 10.1016/0014-5793(95)01495-0. [DOI] [PubMed] [Google Scholar]
- Matsui T, Amano M, Yamamoto T, Chihara K, Nakafuku M, Ito M, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Rho-associated kinase, a novel serine threonine kinase, as a putative target for small GTP binding protein rho. EMBO Journal. 1996;15:2208–2216. [PMC free article] [PubMed] [Google Scholar]
- Moore KP, Sheron N, Ward P, Taylor GW, Alexander GIM, Williams R. Leukotriene and prostaglandin production after infusion of tumor-necrosis-factor in man. Eicosanoids. 1991;4:115–118. [PubMed] [Google Scholar]
- Otto B, Stuesloff A, Just I, Aktories K, Pfitzer G. Role of rho proteins in carbachol induced contractions in intact and permeabilized guinea-pig intestinal smooth muscle. The Journal of Physiology. 1996;496:317–320. doi: 10.1113/jphysiol.1996.sp021687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsons SJW, Summer MJ, Garland CJ. Phospholipase A2 and protein kinase C contribute to myofilament sensitization to 5-HT in rabbit mesenteric artery. The Journal of Physiology. 1996;491:447–453. doi: 10.1113/jphysiol.1996.sp021228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyne NJ, Tolan D, Pyne S. Bradykinin stimulates cAMP synthesis via mitogen-activated protein kinase-dependent regulation of cytosolic phospholipase A2 and prostaglandin E2 release in airway smooth muscle. Biochemistry Journal. 1997;328:689–694. doi: 10.1042/bj3280689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renzetti LM, Paciorek PM, Tannu SA, Rinaldi NC, Tocker JE, Wasserman MA, Gater PR. Pharmacological evidence for tumour necrosis factor as a mediator of allergic inflammation in the airways. Journal of Pharmacology and Experimental Therapeutics. 1996;278:847–853. [PubMed] [Google Scholar]
- Schutze S, Berkovic D, Tomsing O, Unger C, Kronke M. Tumor necrosis factor induced rapid production of 1,2-diacylglycerol by a phosphatidylcholine-specific phospholipase C. Journal of Experimental Medicine. 1991;174:975–988. doi: 10.1084/jem.174.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah A, Church MK, Holgate ST. Tumour necrosis factor α: a potential mediator of asthma. Clinical and Experimental Allergy. 1995;25:1038–1044. doi: 10.1111/j.1365-2222.1995.tb03249.x. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Taki F, Torii K, Ikuta N. Increased levels of TNF concentrations in sputa of patients with bronchial asthma. American Review of Respiratory Diseases. 1991;143:A13. [Google Scholar]
- Thomas PS, Yates DH, Barnes PJ. Tumor-necrosis-factor-α increases airway responsiveness and sputum neutrophilia in normal human subjects. American Journal of Respiratory Critical Care Medicine. 1995;152:76–80. doi: 10.1164/ajrccm.152.1.7599866. [DOI] [PubMed] [Google Scholar]
- Vietor I, Schwenger P, Li W, Schlessinger J, Vilcek J. Tumour necrosis factor-induced activation and increased tyrosine phosphorylation of mitogen activated protein kinase in human fibroblasts. Journal of Biological Chemistry. 1991;268:18994–18999. [PubMed] [Google Scholar]
- Weigmann K, Schutze S, Kampen E, Himmler A, Machleidt T, Kronke M. Human p55-kDa receptor for tumor necrosis factor coupled to signal transduction cascades. Journal of Biological Chemistry. 1992;267:17997–18001. [PubMed] [Google Scholar]
- Yoshida S, Hashimoto S, Nakayama T, Kobayashi T, Koizumi A, Horie T. Elevation of serum soluble tumour necrosis factor (TNF) receptor and IL-1 receptor antagonist levels in bronchial asthma. 1996. [DOI] [PMC free article] [PubMed]