

Abstract
Whole-cell Na+ currents (holding potential, −80 mV; test potential, −30 mV) in rat myocytes were inhibited by 8,9-epoxyeicosatrienoic acid (8,9-EET) in a dose-dependent manner with 22 ± 4 % inhibition at 0.5 μM, 48 ± 5 % at 1 μM, and 73 ± 5 % at 5 μM (mean ± s.e.m., n = 10, P < 0.05 for each dose vs. control). Similar results were obtained with 5,6-, 11,12-, and 14,15-EETs, while 8,9-dihydroxyeicosatrienoic acid (DHET) was 3-fold less potent and arachidonic acid was 10- to 20-fold less potent.
8,9-EET produced a dose-dependent, hyperpolarized shift in the steady-state membrane potential at half-maximum inactivation (V½), without changing the slope factor. 8,9-EET had no effect on the steady-state activation of Na+ currents.
Inhibition of Na+ currents by 8,9-EET was use dependent, and channel recovery was slowed. The effects of 8,9-EET were greater at depolarized potentials.
Single channel recordings showed 8,9-EET did not change the conductance or the number of active Na+ channels, but markedly decreased the probability of Na+ channel opening. These results were associated with a decrease in the channel open time and an increase in the channel closed times.
Incubation of cultured cardiac myocytes with 1 μM [3H]8,9-EET showed that 25 % of the radioactivity was taken up by the cells over a 2 h period, and most of the uptake was incorporated into phospholipids, principally phosphatidylcholine. Analysis of the medium after a 2 h incubation indicated that 86 % of the radioactivity remained as [3H]8,9-EET while 13 % was converted into [3H]8,9-DHET. After a 30 min incubation, 1–2 % of the [3H]8,9-EET uptake by cells remained as unesterified EET.
These results demonstrate that cardiac cells have a high capacity to take up and metabolize 8,9-EET. 8,9-EET is a potent use- and voltage-dependent inhibitor of the cardiac Na+ channels through modulation of the channel gating behaviour.
Arachidonic acid is a precursor of many bioactive lipids that are involved in signal transduction and cellular regulatory mechanisms. In addition to the well-known and well-established cyclo-oxygenase and lipoxygenase pathways which generate important bio-mediators such as prostaglandins, thromboxanes and leukotrienes, the cytochrome P450 monoxygenase pathway has also emerged as an important source of bioactive arachidonic acid derivatives (McGiff, 1991). Cytochrome P450 mono-oxygenases convert arachidonic acid to four epoxyeicosatrienoic acid (EET) regioisomers: 5,6-, 8,9-, 11,12- and 14,15-EET, as well as to 19- and 20-hydroxyeicosatetraenoic acids (Oliw, 1994). EETs are potent endothelium-derived vasodilators that modulate vascular tone via enhancement of Ca2+-activated K+ channels in vascular smooth muscle, suggesting that these compounds are endothelium-derived hyperpolarizing factors (Gebremedhin et al. 1992; Hu & Kim, 1993; Campbell et al. 1996; Zou et al. 1996; Baron et al. 1997; Li et al. 1997; Li & Campbell, 1997). Studies in cultured vascular endothelial and smooth muscle cells indicate that EETs are avidly taken up and incorporated into cellular phospholipids, as well as being converted to their dihydroxyeicosatrienoic acid (DHET) derivatives (Fang et al. 1996; Weintraub et al. 1997). Thus, vascular cells are important biochemical sources and physiological targets of EETs.
Cytochrome P450 epoxygenase activity is present in rat, guinea-pig, rabbit and pig hearts (Comte & Gautheron, 1978; Guengerich & Mason, 1979; Abraham et al. 1987; McCallum et al. 1993). A human cytochrome P450 arachidonic acid epoxygenase (CYP2J2) was recently cloned and found to be constitutively expressed in the human heart (Wu et al. 1996). The same group subsequently reported the cloning and cDNA-directed expression of a rat P450 (CYP2J3) that is highly expressed in the heart, predominantly localized to atrial and ventricular cardiac myocytes and active in the mono-oxygenation of arachidonic acid (Wu et al. 1997). There are substantial amounts of EETs in the rat heart. 8,9-EET, the major regioisomer, accounts for 39 % of the total (Wu et al. 1997). Furthermore, 11,12-EET has been shown to enhance the recovery of cardiac function following global ischaemia (Wu et al. 1997). The cytochrome P450 pathway is therefore a potentially important component of the cardiac arachidonic acid cascade and may play a role in regulating the response of the heart to ischaemia. The direct effects of EETs on cardiac myocytes, however, remain to be established.
In the present study, we investigated the effects of EETs on cardiac ion channels. Na+ channels play a crucial role in cardiac electrogenesis and are a major determinant of impulse conduction. Na+ channel blocking antiarrhythmic drugs are the most commonly used pharmacological agents for the treatment of arrhythmias. Since the effects of EETs may be particularly important during ischaemia, and since the voltage-dependent Na+ channels are significantly modulated during cardiac ischaemia, we examined the effects of 8,9-EET on the Na+ channels in isolated rat cardiac myocytes. We found that 8,9-EET significantly inhibits the cardiac Na+ currents (INa) and that this inhibition is dose, voltage and frequency dependent by modulating channel gating behaviour. The other EET regioisomers are also potent INa inhibitors but 8,9-DHET is less effective. In addition, we found that cardiac myocytes have a high capacity for taking up 8,9-EET and incorporating it into phospholipids. These results demonstrate that the heart can metabolize EETs, which in turn may play an important role in modulating the electrophysiological properties of the heart.
METHODS
Solutions
Nominally Ca2+-free Tyrode solution contained (mM): NaCl, 138; KCl, 4.5; MgCl2, 0.5; Na2HPO4, 0.33; glucose, 5.5; Hepes, 10; pH 7.38. The solution was vigorously oxygenated for at least 30 min prior to use.
KB solution contained (mM): KOH, 70; KCl, 40; L-glutamic acid, 50; taurine, 20; MgCl2, 0.5; K2HPO4, 1.0; EGTA, 0.5; creatine, 5; pyruvic acid, 5; Na2ATP, 5; Hepes, 10; pH 7.38.
The bath solution for whole-cell Na+ channel recordings contained (mM): NaCl, 20; choline chloride, 130; CaCl2, 1; CoCl2, 2; MgCl2, 2; KCl, 4.5; glucose, 5.5; Hepes, 10; pH 7.38.
The pipette solution for whole-cell Na+ channel recordings contained (mM): CsCl, 130; CaCl2, 0.5; MgCl2, 2; Na2ATP, 5; GTP, 0.5; EGTA, 0.5; Hepes, 10; pH 7.25.
The bath solution for single Na+ channel recordings contained (mM): KCl, 140; MgCl2, 2; EGTA, 10; Hepes, 10; pH 7.38.
The pipette solution for single Na+ channel recordings contained (mM): NaCl, 250; CaCl2, 1; MgCl2, 2; Hepes, 10; pH 7.38.
The culture medium for neonatal rat cardiac myocytes consisted of Eagle's minimal essential medium (MEM) with 5.8 mM Hepes, 16 mM NaHCO3 and 2 mM L-glutamine. Unless stated otherwise, the medium was supplemented with 15 % horse serum plus 0.02 mg ml−1 gentamicin.
The potassium glutamate (KG) solution for neonatal rat cardiac myocyte culture contained (mM): potassium glutamate, 140; NaHCO3, 16; NaH2PO4, 0.5; Hepes, 25; glucose, 10.5; Phenol Red, 0.014.
EETs, 8,9-DHET and arachidonic acid were prepared in absolute ethanol as 5 mM stock solutions and were stored under nitrogen at −20°C. EETs and arachidonic acid were diluted to the various concentrations with buffer immediately prior to being used. The final concentration of ethanol was 0.1 % or less and had no effect on the cardiac INa.
Isolation of rat ventricular myocytes
Single ventricular myocytes from rat hearts were isolated by established techniques (Isenberg & Klockner, 1982). Briefly, Sprague-Dawley rats (200–250 g body weight) were anaesthetized with methoxyfluorane. The rat heart was rapidly excised and placed in ice-cold nominally Ca2+-free Tyrode solution. After the aorta was cannulated, the heart was perfused using a modified Langendorff apparatus with nominally Ca2+-free Tyrode solution containing 0.1 % (w/v) bovine serum albumin (BSA) for 5 min at 37°C. The perfusate was then changed to nominally Ca2+-free Tyrode solution containing 0.6 mg ml−1 collagenase (Worthington, CLS-2, 347 units mg−1) and 0.1 % (w/v) BSA for 7 min at 37°C. The ventricles were dissected and placed in 25 ml of fresh collagenase solution (0.6 mg ml−1) for 5 min. The myocardium was then cut into small pieces (approximately 1 mm cubes) and filtered through a medium mesh. After washing twice with nominally Ca2+-free Tyrode solution, single ventricular myocytes were maintained in KB solution until the time of the experiment.
Whole-cell Na+ current recordings
Voltage-dependent INa in isolated rat ventricular myocytes were recorded using patch-clamp techniques as previously described (Hamill et al. 1981; Lee et al. 1993). Bath solutions were superfused at 1–2 ml min−1 using a direct current-powered pump (Instech Laboratories, Inc., Plymouth Meeting, PA, USA) and solution exchanges were complete within 30–60 s. Whole-cell INa was recorded with an Axopatch 200 integrating amplifier (Axon Instruments), filtered with an eight-pole low-pass Bessel filter with a bandwidth (−3 dB) of 5 kHz and sampled at 50 kHz (12-bit resolution). Borosilicate glass capillaries (Corning 7052, Warner Instruments, Inc.) were used to make pipettes. Electrode resistance in the whole-cell pipette solution ranged from 0.5 to 1 MΩ, and seal resistance was 1–5 GΩ. Whole-cell series resistance was compensated more than 80 % of the uncompensated value. pCLAMP software (Axon Instruments) was used for generating voltage-clamp protocols and for the acquisition and analysis of INa. All cellular electrophysiology experiments were performed at room temperature (21–23°C). Spontaneous time-dependent changes of INa typically occurred in the first 10–20 min after rupture of the sealed membrane patch. Whole-cell INa recordings were started only after the current was stable.
Stimulus protocols
To measure the voltage-dependent activation of the Na+ channels, currents were elicited from a holding potential of −80 mV to a series of 20 ms test pulses from −80 to +35 mV in 5 mV increments and at 5 s intervals. The peak INa (INa,max) at different conditioning pulse voltages (Vm) was measured and the peak INa,max-Vm relationship was fitted with the following equation:
where Erev is the reversal potential. The voltage-dependent Na+ channel conductance (GNa) was fitted with a Boltzmann distribution equation:
where GNa,max is the maximum conductance, V½ is the membrane potential at half-maximal conductance and k is the slope factor.
Voltage-dependent steady-state inactivation was determined using a two-pulse protocol consisting of a 500 ms conditioning pulse from −160 to −10 mV in 10 mV increments, followed by a test pulse of −20 mV. The steady-state inactivation curves obtained by normalizing currents to the maximal INa at −160 mV were fitted using a Boltzmann distribution equation:
where Vm is the conditioning pulse voltage, V½ is the voltage at half-inactivation and k is the slope factor. To examine the use-dependent block of INa by EET, trains of 20 depolarizing pulses (holding potential, −80 mV; test potential, −30 mV; 20 ms duration) at cycle lengths of 200 ms were used. Peak current amplitudes were plotted against time.
Recovery of INa from inactivation was determined using a two-pulse protocol. A 500 ms conditioning pulse from −80 to 0 mV was followed by a recovery period ranging from 1 ms to 2 s at −80 mV. A test pulse of 20 ms duration to −30 mV was then elicited. The amplitude of the peak INa during the test pulse was normalized to the value of INa after complete recovery from inactivation, INa,max, and this ratio was plotted against the recovery interval. Recovery curves were analysed using a two-exponential fit as previously reported (Lee et al. 1993):
where Amp1 and Amp2 represent the relative contributions of the fast (τ1) and slow (τ2) time constants of recovery and Amp1 + Amp2 = 1. Curve fitting was performed using a Marquardt-Levenberg least squares fitting procedure from Origin (MicroCal Software, Inc., Northampton, MA, USA) or Igor (WaveMetrics Inc., Lake Oswego, OR, USA) software.
Single Na+ channel recordings
Single channel recordings were performed with an improved patch clamp technique (Benndorf, 1995) in the cell-attached configuration. The patch pipettes were pulled from thick-walled borosilicate glass (Glass type 7740, Garner Glass Company). The pipette was coated with Sylgard 184 before being fire polished. The pipette resistance was 5–10 MΩ when filled with the pipette solution and the typical seal resistance was > 10 GΩ. Single channel Na+ currents were recorded with an Axopatch 200 amplifier and an eight-pole low-pass Bessel filter (902LPF, Frequency Devices) with bandwidth of 10 kHz and sampling rate of 100 kHz (12-bit resolution). The peak current amplitude of the single Na+ channel is greater than 2 pA at membrane potentials of −40 mV with 250 mM extracellular Na+. Events were easily distinguished from noise. Single channel Na+ current was identified by the amplitude and fast time course of the mean current. The capacitive transients and leak current were removed by subtraction of a sliding averaged blank formed from 10 empty traces out of the neighbourhood of the actual record. The subtracted traces were idealized at 10 kHz.
Opening and closing transitions were detected according to the conventional 50 % threshold of event amplitude. Computer-detected openings, confirmed by visual observation, were used to generate idealized records from which histograms of amplitude and closed- and open-time distributions were constructed. The closed states of the channel were determined by the last channel opening in order to separate them from inactivation states. The closed dwell time of events was fitted with a two-exponential equation. The open dwell time of events was fitted with an exponential equation. The channel open probability (Po) was calculated with the equation:
where I is the macroscopic Na+ current averaged from single channel recording, N is the number of functional channels in the patch, i is single channel current. Here i is identified by generation of amplitude histograms, which were fitted with Gaussian curves using a χ2non-linear regression routine.
Isolation and culture of neonatal rat cardiac myocytes
Neonatal rat cardiac myocytes were prepared and cultured as previously described (Atkins et al. 1992). Healthy rat pups aged 1–3 days old were decapitated. The hearts were isolated and placed in modified Eagle's MEM. After excess blood was rinsed off, the hearts were placed in KG solution, minced into 1 mm3 sized pieces and incubated with collagenase (3 mg ml−1) at 37°C for 20 min. Cells were isolated by multiple-cycle trypsinization. The cardiac fragments were digested with trypsin (1 mg ml−1) at 37°C with gentle mechanical stirring for 15 min cycle−1 for three to six cycles. The dispersed cells were pooled and placed in modified Eagle's MEM. Fibroblasts and other non-myocardial cells were allowed to settle and attach to the bottom of a tissue culture flask by incubation for 60 min at 37°C. The unattached cells were recovered, an aliquot was counted and the remaining cells were seeded into 6-well plates in culture medium at 37°C. Histological staining confirmed that greater than 96 % of the cultured cells were cardiac myocytes.
Synthesis of [3H]8,9-EET
[3H]8,9-EET was synthesized from [3H]arachidonic acid as previously described (VanRollins et al. 1995). In brief, [3H]arachidonic acid was esterified with ethereal diazomethane, and the [3H]methyl arachidonate was purified by reverse-phase high-performance liquid chromatography (HPLC) and extracted into CH2Cl2. Methyl arachidonate was then epoxygenated using meta-chloroperoxybenzoic acid. The individual [3H]EET regioisomers were isolated as methyl esters by preparative, normal-phase HPLC. Shortly before use, the methyl ester of [3H]8,9-EET was saponified, extracted into water-saturated ethyl acetate, and [3H]8,9-EET was isolated using semi-preparative normal-phase HPLC. The specific activity of [3H]8,9-EET was 929 d.p.m. ng−1.
Incubation of neonatal rat cardiac myocytes with radiolabelled 8,9-EET
Twenty-four hours after isolation of neonatal rat cardiac myocytes, the medium was removed and replaced with 1 ml of fresh modified Eagle's MEM containing 1 μM [3H]8,9-EET and supplemented with 1 % horse serum. After 30 min to 4 h, the medium was removed and saved for analysis (see below). The cells were washed with 2 ml of Dulbecco's phosphate-buffered saline (4°C), detached by scraping gently with a rubber policeman and transferred to a test tube. Eight millilitres of CHCl3-CH3OH (2:1, v/v) and 1.75 ml of 4 mM HCl-154 mM NaCl were added, and the cell suspension was vortex-mixed. The phases were allowed to separate overnight at 4°C. After transferring the lower phase to a new test tube, 1 ml of CHCl3-CH3OH-154 mM NaCl (86:14:1, v/v/v) was added to the top phase and the contents were mixed. After phase separation, the lower phase was transferred to the original CHCl3 extract. The CHCl3 was evaporated from the combined extractant using N2 at 37°C, and the lipids were resuspended in 200 μl of CHCl3-CH3OH (2:1, v/v). A 10 μl aliquot was removed for determination of radioactivity by liquid scintillation counting, and the remainder was stored for analysis (see below).
An aliquot of the incubation medium was removed for determination of radioactivity by liquid scintillation counting, and the remainder of the medium was acidified to ∼pH 5 with H3PO4-H2O (1:50, v/v). Lipids were extracted twice using 4 ml of ice-cold water-saturated ethyl acetate, followed by centrifugation and phase separation. After evaporating the ethyl acetate under a stream of N2, the extract was resuspended in 250 μl of CH3CN.
To analyse the medium-associated lipids, reverse-phase high-performance liquid chromatography (HPLC) was performed using a Gilson System equipped with an automatic sample injector and a 4.6 mm × 150 mm column containing 3 μm spherical particles of EQC C18 (Alltech) as previously described (Fang et al. 1996; Weintraub et al. 1997). A mobile phase system was employed, consisting of water adjusted to pH 3.4 with phosphoric acid and an acetonitrile gradient increasing from 30–100 % over 60 min at a flow rate of 0.7 ml min−1. Radioactivity was measured by combining the column effluent with Budget Solve scintillation solution (0.7:2.5, v/v) and passing the mixture through a Radiomatic Flo-One Beta isotope detector.
The cell lipid extracts were separated by thin layer chromatography (TLC) as previously described (Fang et al. 1996; Weintraub et al. 1997). Phospholipids were separated with chloroform-methanol-40 % methylamine (60:36:5) or with chloroform-methanol-water (60:35:8), a solvent system that separates unesterified 8,9-EET from phospholipids and glycerides. The distribution of radioactivity on the TLC plate was determined with a gas flow proportional scanner (Radiomatic model R), using radioactive lipid standards applied to each plate as described previously (Fang et al. 1996).
Materials
EETs and 8,9-DHET were obtained from Cayman Chemical Co. (Ann Arbor, MI, USA), Eagle's MEM was from Gibco, horse serum was from HyClone Laboratories (Logan, UT, USA), TLC plates were from Alltech Associates Inc. (Deerfield, IL, USA) and the rest of the chemicals were from Sigma Chemical Co.
Statistical analysis
All data were expressed as means ± s.e.m. Student's paired t test was used to compare data obtained before and after intervention. A one-way ANOVA followed by contrast testing was used to compare data from multiple groups. Statistically significant differences are defined as P < 0.05.
RESULTS
Effect of 8,9-EET on whole-cell INa in isolated rat ventricular myocytes
Dose-dependent effects
Whole-cell INa in isolated rat ventricular myocytes was measured with reduced external Na+ to ensure adequate voltage control as previously described (Matsuda et al. 1992; Lee et al. 1993). Cardiac INa was rapidly inhibited by 8,9-EET in a dose-dependent manner. Figure 1 shows the results of a typical experiment. Exposure to 0.5, 1.0 and 5.0 μM 8,9-EET produced rapid inhibition of INa (holding potential = −80 mV; test potential = −30 mV), which reached a new steady state in 2–3 min at each dose. The effect of 8,9-EET was at least partially reversible upon washout. Reversibility of the effects of 8,9-EET was not enhanced by washout with buffer containing 0.1 % BSA. Figure 1A shows the representative current tracings before and after application of 0.5, 1.0 and 5 μM 8,9-EET. Figure 1B shows the time course of the peak INa amplitudes. Composite data (Fig. 2A) showed that whole-cell INa in rat cardiac myocytes was inhibited by 22 ± 4, 48 ± 5 and 73 ± 5 % with 0.5, 1.0 and 5.0 μM 8,9-EET, respectively (mean ± s.e.m., n = 10, P < 0.05vs. control for all interventions). At 0.01 μM, 8,9-EET had no effect on the INa. The other EET regioisomers produced similar effects in isolated rat ventricular myocytes. INa was inhibited by 18 ± 10, 35 ± 5 and 49 ± 5 % with 0.5, 1.0 and 5.0 μM 5,6-EET, respectively (n = 4); by 19 ± 5, 40 ± 5 and 65 ± 6 % with 0.5, 1.0 and 5.0 μM 11,12-EET, respectively (n = 4); and by 17 ± 5, 35 ± 4 and 49 ± 5 % with 0.5, 1.0 and 5.0 μM 14,15-EET, respectively (n = 5) (P < 0.05vs. control for all interventions). We found that 8,9-DHET (1 μM) was also an inhibitor of the cardiac INa (15 ± 3 %, n = 3, P < 0.05vs. control), but was not as effective as its epoxide precursor. Further assessment of the effects of EET was performed using mainly the 8,9-regioisomer.
Figure 1. Effect of 8,9-EET on the Na+ currents in isolated rat cardiac myocytes.
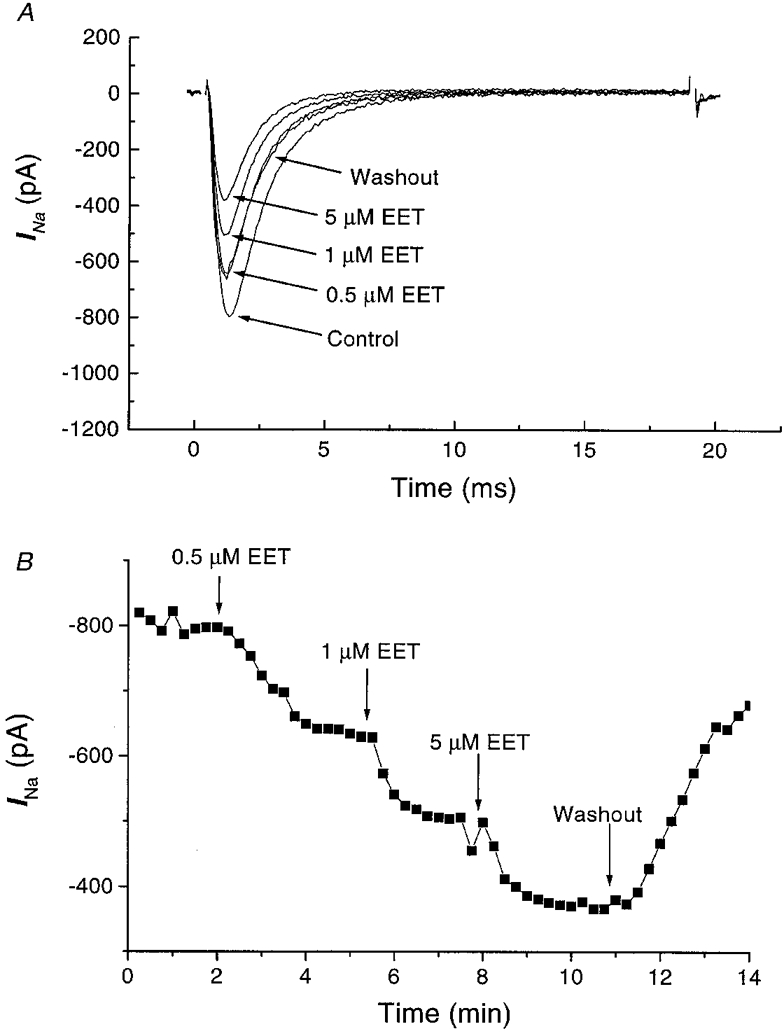
Whole-cell INa was elicited from a holding potential of −80 mV to a test potential of −30 mV in the presence of 20 mM [Na+]o at room temperature. A represents raw tracings of Na+ currents before (control) and after application of 0.5, 1.0 and 5.0 μM 8,9-EET, followed by the partial reversal of the EET effect upon washout. The capacitive transients are eliminated for cosmetic reasons. B represents the peak whole-cell Na+ current in the same cell plotted versus time with pulses elicited at 15 s intervals. The arrows indicate the onsets of the interventions.
Figure 2. Dose-dependent inhibition of the Na+ currents by 8,9-EET (A) and arachidonic acid (B).
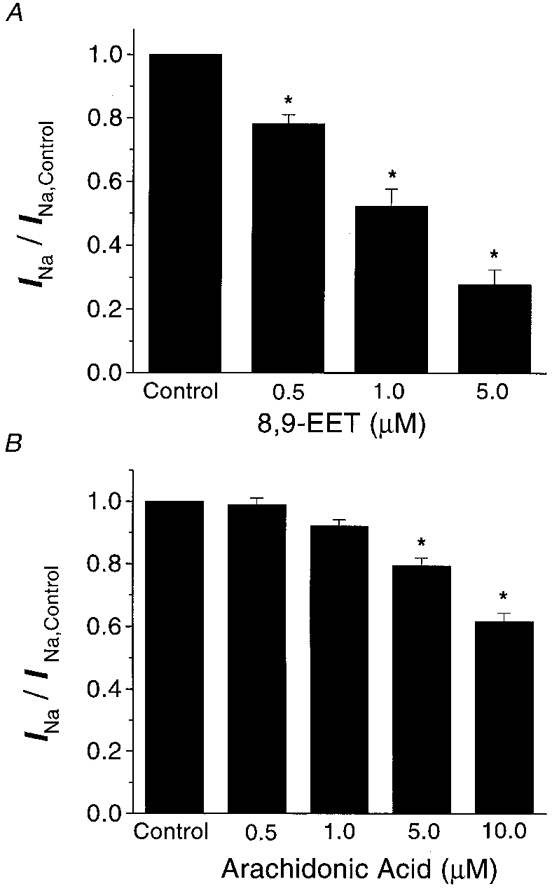
A shows the magnitude of Na+ current inhibition by 0.5, 1.0 and 5.0 μM 8,9-EET as a ratio of Na+ currents under control conditions. Na+ currents were evoked from a resting potential of −80 mV to a test potential of −30 mV (n = 10, *P < 0.05vs. control). B shows the magnitude of Na+ current inhibition by 0.5, 1.0, 5.0 and 10.0 μM arachidonic acid as a ratio of control Na+ currents under conditions similar to those shown in A (n = 11, * P < 0.05).
The effects of 8,9-EET on the rat cardiac INa were much more potent than those of its parent compound, arachidonic acid (Fig. 2). Arachidonic acid inhibited the rat cardiac INa by 1 ± 2, 8 ± 2, 21 ± 3 and 38 ± 3 % with 0.5, 1.0, 5.0 and 10.0 μM, respectivley (n = 11, P = n.s. for 0.5 and 1 μM vs. control; P < 0.05 for 5.0 and 10.0 μM vs. control). These results suggest that the epoxy groups in EETs enhance their properties as Na+ channel inhibitors, since the IC50 for 8,9-EET was around 1 μM, whereas the IC50 for arachidonic acid was greater than 10 μM.
Voltage-dependent effects
Figure 3A shows the effects of 0.5, 1.0 and 5.0 μM 8,9-EET on the current-voltage (I-V) relationship of whole-cell INa normalized to the peak amplitude in the I-V relationship under control conditions. These results show that inhibition of INa by 8,9-EET occurred throughout the INa activation range and in a dose-dependent manner. There was significant reduction of INa by 1.0 and 5.0 μM 8,9-EET at membrane potentials between −50 and +20 mV (n = 5, P < 0.05vs. control).
Figure 3. Effect of 8,9-EET on whole-cell (A) Na+ current-voltage relations and activation curves (B).
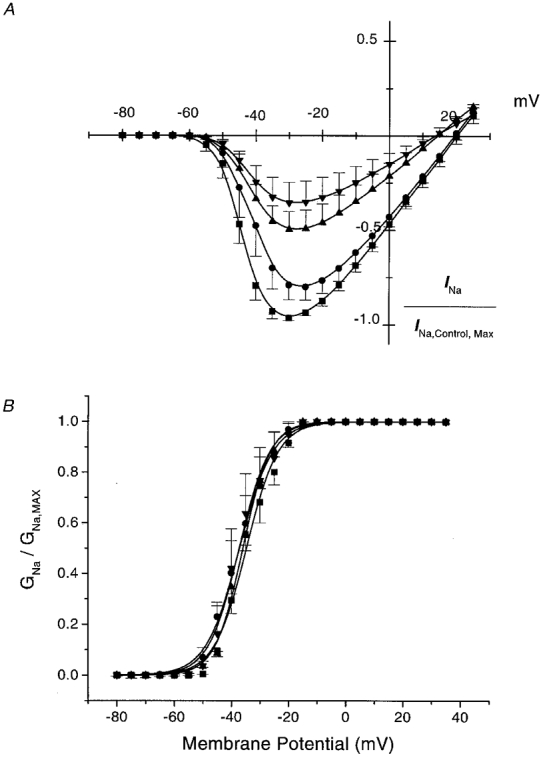
A represents the current-voltage relations in five cells, in the presence of 0 (control, ▪), 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET. Holding potentials are at −80 mV with pulse durations of 20 ms. Pulses are repeated at 5 mV increments at 5 s intervals. Values are normalized to the peak INa amplitude under control conditions and are expressed as means ± s.e.m.B represents the whole-cell Na+ current activation curve showing the conductance-voltage relations at baseline (control, ▪) and in the presence of 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET. Values are normalized to maximal conductance (GNa,max) and are expressed as means ± s.e.m. The data are curve-fitted using a Boltzmann equation. V½ is the half-activation value and k is the slope factor. Differences between control values and those with the various doses of 8,9-EET are not statistically significant.
To determine whether these effects of 8,9-EET were due to changes in the INa activation, we analysed the voltage activation of INa by determining channel conductance from the current elicited and the Na+ electrochemical driving force using Ohm's law as described in Methods. The conductance-voltage (G-V) relationships under control conditions and in the presence of 0.5, 1.0 and 5.0 μM of 8,9-EET are shown in Fig. 3B. The half-activation values (V½) were −34.9 ± 1.5, −36.5 ± 3.0, −35.6 ± 0.6 and −36.6 ± 3.6 mV for control, 0.5, 1.0 and 5.0 μM 8,9-EET, respectively (n = 5, n.s.) The slope factors (k) were 5.0 ± 0.4, 4.8 ± 0.7, 4.3 ± 0.2 and 4.2 ± 0.6 for control 0.5, 1.0 and 5.0 μM 8,9-EET, respectively (n = 5, n.s.). These results suggest that 8,9-EET had no significant effect on the activation of INa in rat cardiac myocytes.
To determine whether the effects of EET were due to changes in INa inactivation, we examined the effects of prepulse potential on the suppression of the cardiac INa by 8,9-EET (Fig. 4). Figure 4A shows that 8,9-EET reduced the steady-state Na+ channel availability in a dose-dependent manner. In Fig. 4B, which shows the normalized steady-state inactivation curves, there was a dose-dependent shift in the half-inactivation value (V½) from −76.1 ± 2.0 mV for control, to −77.8 ± 1.5 mV with 0.5 μM, to −82.1 ± 1.4 mV with 1.0 μM and to −89.2 ± 2.4 mV with 5.0 μM 8,9-EET (n = 9, P < 0.05 for 1.0 and 5.0 μM 8,9-EET vs. control). On the other hand, the slope factor was not affected by 8,9-EET (6.06 ± 0.39 for control, 6.07 ± 0.42 for 0.5 μM, 6.52 ± 0.46 for 1.0 μM and 6.50 ± 0.53 for 5.0 μM 8,9-EET; n = 9, n.s. vs. control). The inhibition of INa in rat cardiac myocytes by 8,9-EET was voltage dependent. At a prepulse potential of −120 mV, whole-cell INa values were 81.7 ± 1.3, 67.1 ± 3.1 and 62.0 ± 8.8 % of control values in the presence of 0.5, 1.0 and 5.0 μM 8,9-EET, respectively. At a prepulse potential of −90 mV, whole-cell INa values were 79.0 ± 2.2, 54.7 ± 5.8 and 37.9 ± 10.8 % of control values in the presence of 0.5, 1.0 and 5.0 μM 8,9-EET, respectively. At a prepulse potential of −70 mV, whole-cell INa values were 56.9 ± 11.9, 31.1 ± 10.4 and 12.7 ± 8.0 % of control values in the presence of 0.5, 1.0 and 5.0 μM 8,9-EET, respectively. Taken together, these results indicated that 8,9-EET is a potent inhibitor of the cardiac INa at both physiological and depolarized membrane potentials.
Figure 4. Effect of prepulse potential on the inhibition of Na+ current by 8,9-EET.
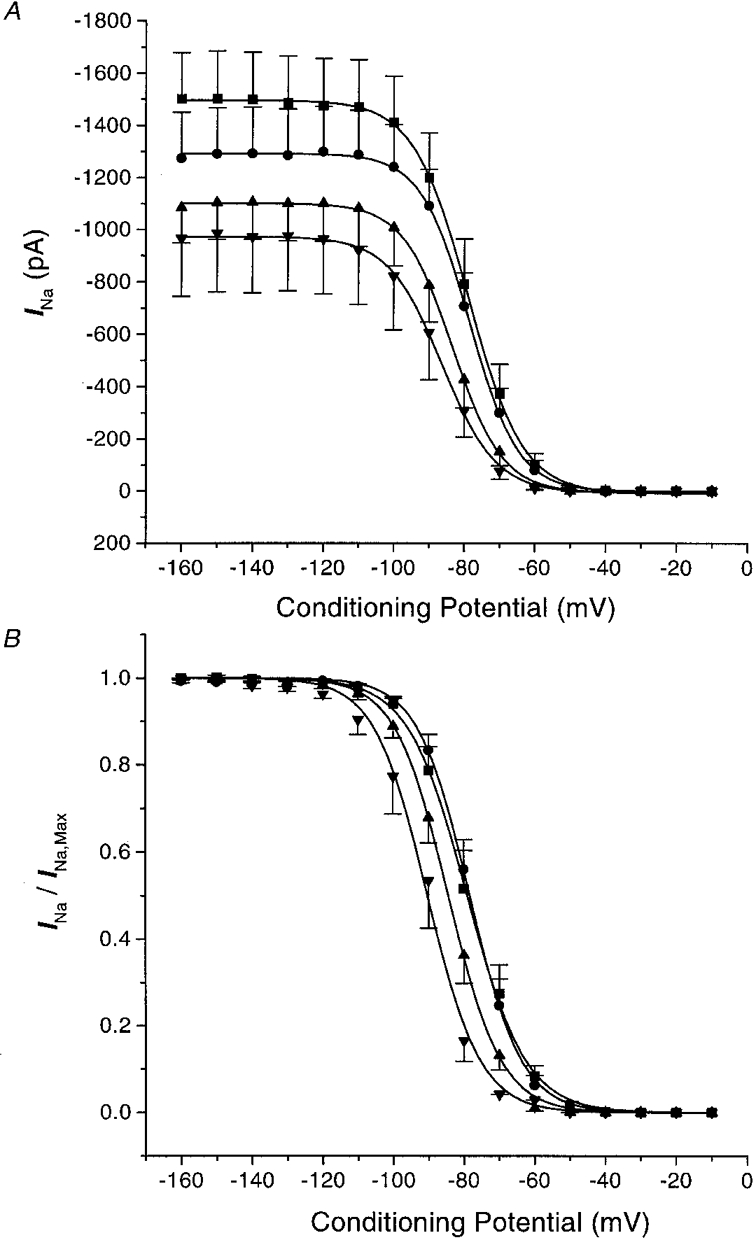
A shows the amplitudes of the Na+ currents (test potentials of −20 mV) plotted against the indicated prepulse potentials (500 ms) under control conditions (▪) and after application of 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET. Each point represents the means ± s.e.m. of nine experiments. B shows the effects of 8,9-EET on the whole-cell Na+ current steady-state inactivation curves. The data in A are normalized to maximal Na+ current and plotted versus prepulse potentials for control (▪) and after application of 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET. Data are expressed as means ± s.e.m. The data are fitted with a Boltzmann equation where V½ represents the half-inactivation potential and k is the slope factor. The V½ values are significantly different for 1.0 and 5.0 μM 8,9-EET versus control (P < 0.05). The differences in k values are not statistically significant.
Frequency-dependent effects
To characterize further the cardiac INa inhibition by 8,9-EET, we determined use-dependent INa block using trains of depolarizing pulses. Under control conditions, a train of 20 depolarizing pulses at 200 ms interpulse intervals with a holding potential of −80 mV and a test potential of −30 mV produced a modest amount of accumulation of channel inactivation. The current amplitude of the last pulse of the train was 86.8 ± 0.5 % of that of the first pulse of the train. In the presence of 8,9-EET, the same pulse protocol resulted in the development of significantly greater amounts of use-dependent INa block (Fig. 5A). The current amplitudes of the last pulse were 81.7 ± 2.2, 74.7 ± 2.9 and 72.1 ± 2.6 % of that of the first pulse with 0.5, 1.0 and 5.0 μM 8,9-EET respectively (n = 6, P < 0.05vs. control for 0.5, 1.0 and 5.0 μM 8,9-EET). The mechanism of frequency-dependent reduction of INa under control conditions and in the presence of 8,9-EET can be explained by the kinetics of Na+ channel repriming as previously reported (Lee et al. 1993). We examined the effects of 8,9-EET on the recovery of steady-state inactivation of INa by using the two-pulse protocol described in Fig. 5B. 8,9-EET produced dose-dependent slowing of the INa recovery from inactivation, resulting in the development of further frequency-dependent inhibition of INa. The time course of INa recovery showed two exponential components: a fast component with an amplitude (Amp1) of 0.87 ± 0.03 and a time constant (τ1) of 41.4 ± 7.4 ms, and a slow component with an amplitude (Amp2) of 0.13 ± 0.03 and a time constant (τ2) of 128.1 ± 20.2 ms (Table 1). Both τ1 and the τ2 of INa recovery were prolonged by 8,9-EET in a dose-dependent manner. At 5.0 μM 8,9-EET, τ1 was 2-fold (80.9 ± 0.06 ms) and τ2 was 3-fold (401.3 ± 64.5 ms) greater than the control values (n = 6, P < 0.05vs. control for both).
Figure 5. Effect of 8,9-EET on the frequency-dependent inhibition of the cardiac Na+ currents (A) and the recovery of the Na+ currents from inactivation (B).
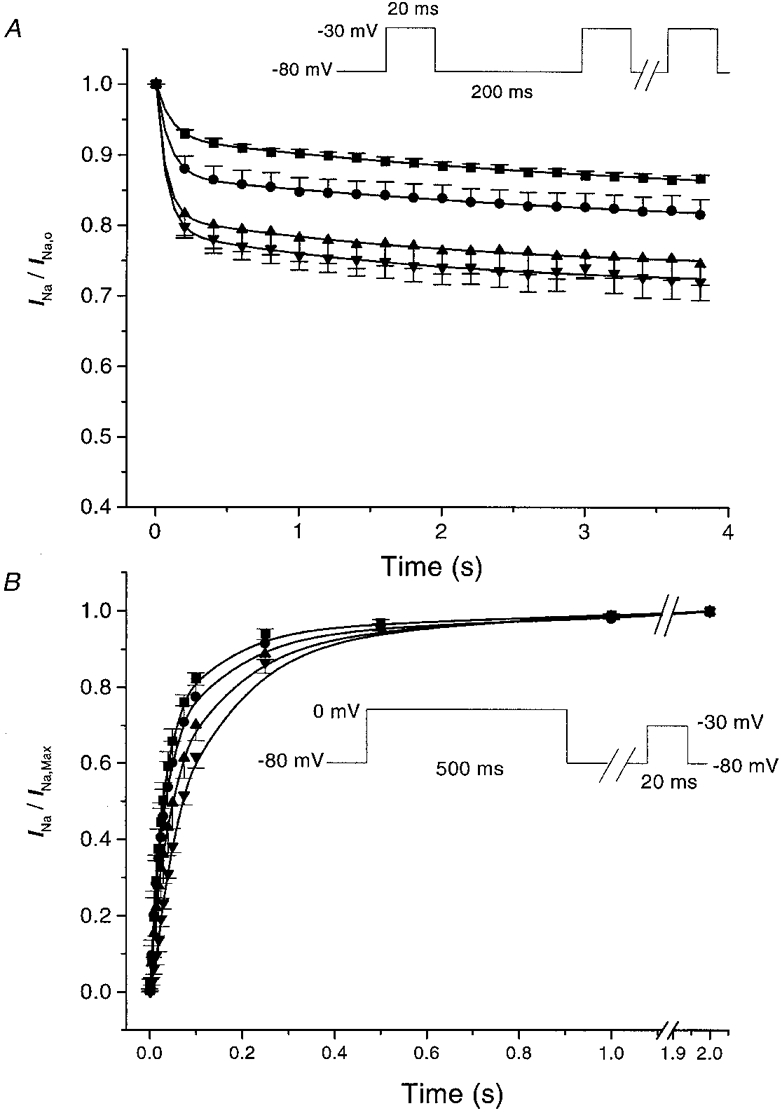
A shows the peak whole-cell Na+ current amplitudes during trains of 20 depolarizing current pulses at a cycle length of 200 ms with holding potentials of −80 mV and test potentials of −30 mV (inset). Results are normalized against the amplitude of the first pulse and are expressed as means ± s.e.m. Responses in the presence of 0 (control, ▪), 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET are plotted against time (n = 6). B shows the effect of 8,9-EET on the recovery of the Na+ current from inactivation using a two-pulse protocol. Conditioning pulses at 0 mV of 500 ms duration are followed by test pulses at −30 mV of 20 ms duration with an intertrain interval of 5 s (inset). Holding potentials are −80 mV with interpulse recovery intervals between 1 and 2000 ms. The results are expressed as means ± s.e.m. for 0 (control, ▪), 0.5 μM (•), 1.0 μM (▴) and 5.0 μM (▾) 8,9-EET (n = 6).
Table 1.
Parameters of sodium channel recovery from inactivation
| Amp1 | τ1 (ms) | Amp2 | τ2 (ms) | |
|---|---|---|---|---|
| Control | 0.87 ± 0.03 | 41.4 ± 7.4 | 0.13 ± 0.03 | 128.1 ± 20.2 |
| 0.5 μm 8,9-EET | 0.80 ± 0.03 | 46.3 ± 11.6 | 0.20 ± 0.03 | 203.5 ± 35.8 |
| 1.0 μm 8,9-EET | 0.76 ± 0.04 | 60.4 ± 13.2 | 0.25 ± 0.04 | 264.1 ± 45.6* |
| 5.0 μm 8,9-EET | 0.80 ± 0.06 | 80.9 ± 12.8* | 0.20 ± 0.05 | 401.3 ± 64.5* |
Data from Fig. 5B were analysed using a two-exponential fit with an equation of the following form: f = Amp1(1 – e−t/τ1) + Amp2(1 – e−t/τ2). Amp1 and Amp2 represent the relative contribution (as a ratio) and τ1 and τ2 represent the time constants (in ms) of the fast and slow components of INa recovery. n = 6
P < 0.05 compared with control values.
Effects of 8,9-EET on single channel INa in isolated rat cardiac myocytes
To examine the effects of 8,9-EET on the single channel Na+ currents in isolated rat cardiac myocytes, experiments were conducted in the cell-attached configuration. In these experiments, the pipette contained 250 mM of Na+ to enhance the size of unitary current and to improve signal-to-noise ratios. Figure 6 shows the raw current tracings of cell-attached patches that contained a single Na+ channel elicited from a holding potential of −100 mV to various test potentials. The unitary Na+ current amplitudes were plotted against membrane potentials and the results were fitted with a linear regression equation. The single Na+ channel conductance (γ) was calculated to be 27 pS, under the conditions of our recordings, and 5 μM 8,9-EET did not alter γ.
Figure 6. Effect of 8,9-EET on single Na+ channel conductance.
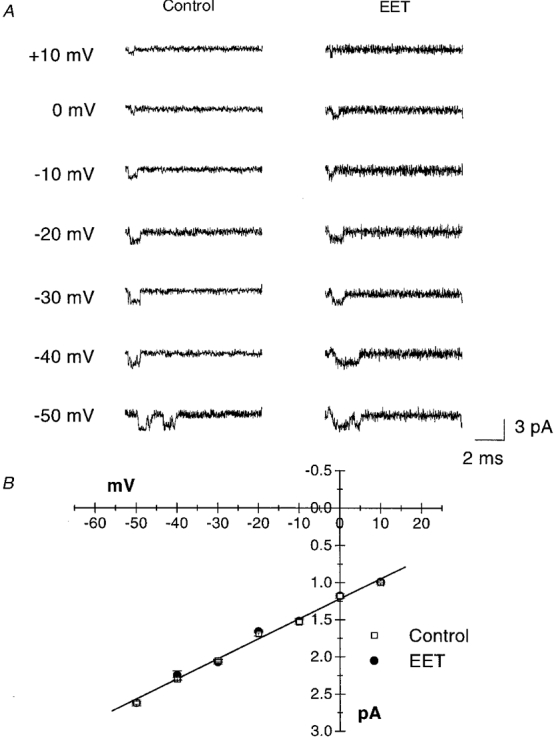
Currents from single Na+ channels in rat ventricular myocytes are recorded in cell-attached configuration from a holding potential of −100 mV to various test potentials as indicated. The raw current tracings under control conditions or after exposure to 5 μM 8,9-EET are shown in A. In B, the unitary Na+ current amplitudes are plotted against membrane potentials and the results are fitted with a linear regression equation. The single Na+ channel conductance (γ) is 27 pS and 5 μM 8,9-EET does not alter γ (n = 4).
Figure 7A shows characteristic raw tracings of single Na+ channel recordings, with a holding potential of −100 mV and a test potential of −40 mV, obtained under control conditions and after the application of 5 μM 8,9-EET. The number of active Na+ channels in each membrane was not altered by 8,9-EET. However, the probability of Na+ channel opening was dramatically reduced by 8,9-EET. Figure 7B shows the ensemble-averaged current from an experiment with 500 sweeps in the presence and absence of 8,9-EET. Figure 7C shows the collective data in bar graphs. Under control conditions, the probability of Na+ channel opening (Po) was 0.270 ± 0.027 (n = 7). However, exposure to 5 μM 8,9-EET markedly reduced Po to 0.048 ± 0.026 (P < 0.05vs. control). Thus, 8,9-EET did not reduce the number of Na+ channels, but instead reduced the probability that the Na+ channel would be open.
Figure 7. Effect of 8,9-EET on single Na+ channel opening probability.
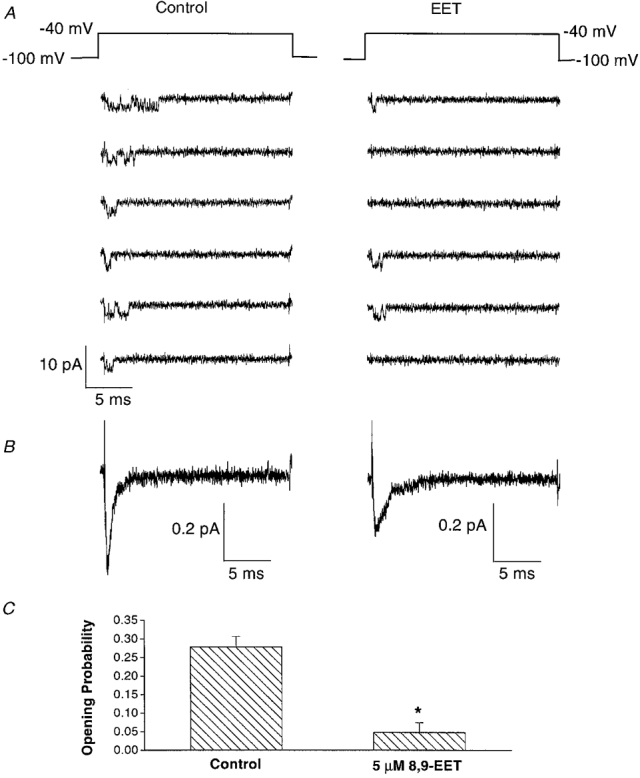
Representative single channel recordings in cell-attached membrane patches of rat ventricular myocytes with a holding potential of −100 mV and a test potential of −40 mV before (control) and after application of 5 μM 8,9-EET are shown in A. In the presence of EET, the number of null sweeps is significantly increased. The ensemble-averaged currents from 500 sweeps before (control) and after application of 5 μM 8,9-EET are shown in B. C represents the results from seven experiments measuring single Na+ channel opening probability (Po) under control conditions and in the presence of 5 μM 8,9-EET. Po is 0.270 ± 0.027 for the controls, and 8,9-EET markedly reduced Po to 0.048 ± 0.026.
Analysis of the single Na+ channel kinetics is presented in Fig. 8. The courses of both channel open dwell time and channel closed dwell time were fitted with exponential curves. Figure 8A shows the Na+ channel open-duration histogram and the distribution could be fitted with a single exponential equation. 8,9-EET shortened the channel open time constant from 163 μs under control conditions to 100 μs. The Na+ channel closed-duration histograms are shown in Fig. 8B, where the distribution was described by a two-exponential equation. 8,9-EET prolonged both time constants; τ1 was increased from 22 μs to 108 μs, and τ2 from 1.4 ms to 1.95 ms in the presence of 5 μM 8,9-EET. These results indicate that 8,9-EET inhibits the cardiac Na+ channels by shortening the duration of channel opening and prolonging the duration of channel closing. Thus, 8,9-EET alters the gating behaviour of the Na+ channel.
Figure 8. Effect of 8,9-EET on the kinetics of single Na+ channels.
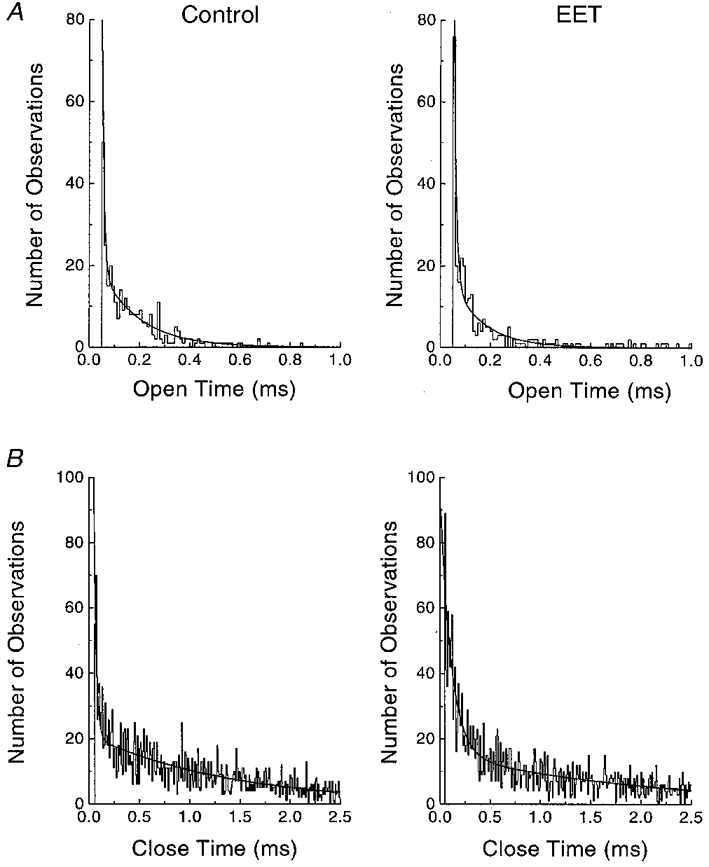
Single Na+ channel currents are recorded in cell-attached patches with holding potentials of −100 mV and test potentials of −40 mV. A shows the Na+ channel open duration histograms for 0 (control) and 5 μM 8,9-EET. The open dwell time distributions are fitted with a single exponential equation with time constants (τ) of 163 μs for control and 100 μs for 8,9-EET. B shows the Na+ channel closed-duration histograms for control and 5 μM 8,9-EET treatment. The mean closed time of the Na+ channel recordings is fitted with a two-exponential equation. Both τ1 and τ2 are prolonged by 8,9-EET. τ1 is 22 μs for control and 108 μs for EET and τ2 is 1.4 ms for control and 1.95 ms for EET.
Incubation of cultured cardiac myocytes with [3H]8,9-EET
To examine the capacity of neonatal rat cardiac myocytes to take up and metabolize 8,9-EET, cells were incubated with 1 μM [3H]8,9-EET for 30 min to 4 h, after which the medium- and cell-associated lipids were individually extracted and analysed. Following a 2 h incubation with [3H]8,9-EET, about 75 % of the applied radioactivity was present in the medium, while 25 % was associated with the cells (Fig. 9A). During a 4 h incubation, the cells took up more radioactivity (234 ± 18 pmol well−1 (4 h incubation) vs. 178 ± 16 pmol well−1 (2 h incubation), n = 3, P < 0.05). Following incubations of 2–4 h duration, analysis of cell lipids by thin-layer chromatography (TLC) indicated that virtually all the radioactivity was contained in phospholipids, principally phosphatidylcholine (not shown). After a 30 min incubation, most of the uptake was also in phospholipids, but approximately 1–2 % of the radioactivity associated with the cells remained as unesterified 8,9-EET (Fig. 9B).
Figure 9. Distribution of radioactivity following incubation of rat neonatal myocytes with [3H]8,9-EET.
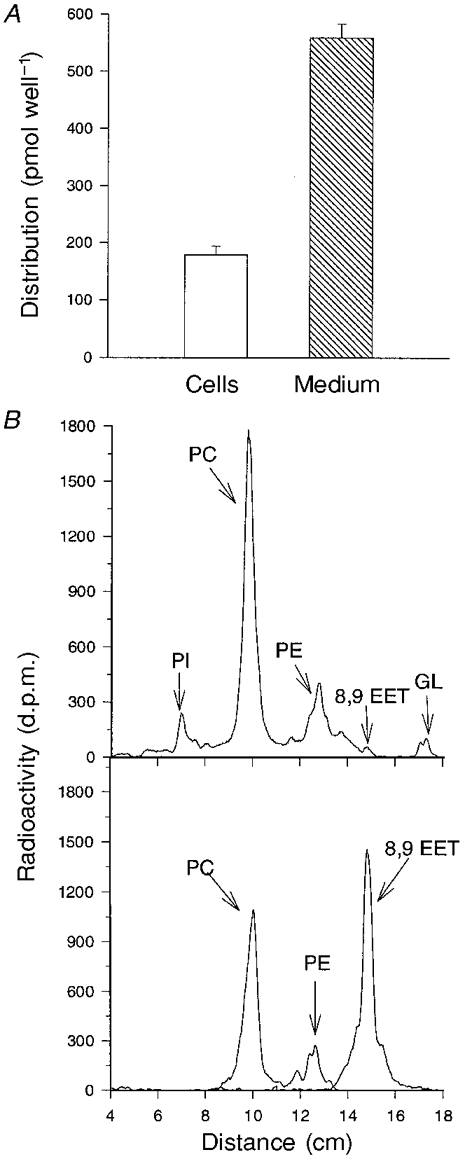
Cells were incubated in medium containing 1 μM [3H]8,9-EET for 30 min to 4 h, after which the cell- and medium-associated lipids were extracted and analysed. The distribution of radioactivity between the cells and medium (quantified by liquid scintillation counting) following a 2 h incubation with [3H]8,9-EET is shown in A. The values are expressed as means ± s.e.m., n = 3. In B, a representative TLC chromatogram (upper panel) shows the distribution of radioactivity in cell lipids following a 30 min incubation with [3H]8,9-EET. PI, phosphatidylinositol; PC, phosphatidylcholine; PE, phosphatidylethanolamine; GL, glycerides. Similar results were obtained from a duplicate culture. Migration of radiolabelled phospholipid standards (PC and PE) and 8,9-EET are shown in the lower panel.
Analysis of the lipids extracted from the medium following a 2 h incubation with [3H]8,9-EET indicated that 86 % of the radioactivity remained as [3H]8,9-EET, while 13 % was converted to a more polar metabolite that co-migrated with radiolabelled 8,9-DHET (Fig. 10A). After a 4 h incubation, 56 % of the medium-associated radioactivity was in the form of [3H]8,9-EET, while 32 % was present as [3H]8,9-DHET (Fig. 10B). In addition, another metabolite was detected which eluted from the column before [3H]8,9-DHET and accounted for about 13 % of the medium-associated radioactivity. The chemical identity of this metabolite remains to be determined.
Figure 10. Metabolites of [3H]8,9-EET found in the medium of cultured rat neonatal myocytes during 2 h (A) and 4 h (B) incubations.
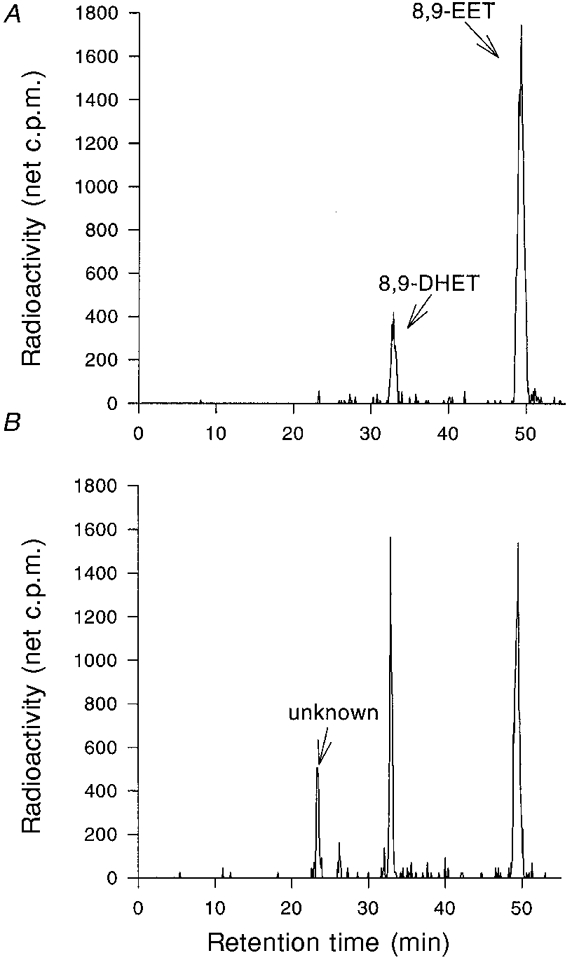
Following incubation, lipids were extracted from the medium and separated by reverse-phase HPLC, and the radioactivity was assayed with an on-line flow scintillation detector. In A, after a 2 h incubation, the radiolabelled components co-migrated with 8,9-EET and 8,9-DHET standards. In B, after a 4 h incubation, a major unidentified product formed from [3H]8,9-EET is apparent. Both panels contain a chromatogram from a single culture, but similar results were obtained from a duplicate culture in each case.
DISCUSSION
In this study, we have demonstrated that EETs are potent inhibitors of the voltage-dependent Na+ channels in rat cardiac myocytes. The inhibition of INa by 8,9-EET is dose, voltage and use dependent through modulation of the Na+ channel gating behaviour. In addition, we have demonstrated that cardiac myocytes can avidly take up 8,9-EET and incorporate it into cellular phospholipids. These results suggest that EETs may play an important role in the modulation of cardiac electrophysiology.
The cytochrome P450-derived EETs are potent vasodilators in multiple vascular beds, including the coronary (Gebremedhin et al. 1992; Campbell et al. 1996; Chataigneau et al. 1998; Miura & Gutterman, 1998; Oltman et al. 1998). Direct electrophysiology studies showed that EETs activate Ca2+-dependent K+ channels and are, by definition, endothelium-derived hyperpolarizing factors (Hu & Kim, 1993; Zou et al. 1996; Baron et al. 1997; Li et al. 1997; Li & Campbell, 1997). In addition, EETs are known to modulate ion channels in non-vascular smooth muscle. For example, EETs inhibit a Ca2+-insensitive Cl− channel (Salvail et al. 1998) and activate Ca2+-activated K+ channels in bovine tracheal smooth muscle (Dumoulin et al. 1998). Therefore, it is apparent that ion channels constitute major effector targets of EETs. However, the effects of EETs on cardiac ion channels have not been studied in detail.
Cytochrome P450 is ubiquitous, and cytochrome P450-dependent mono-oxygenase activity has been detected in mammalian hearts (Comte & Gautheron, 1978; Guengerich & Mason, 1979; Abraham et al. 1987; McCallum et al. 1993). The newly identified human and rat isoforms of cytochrome P450, CYP2J2 and CYP2J3, respectively (Wu et al. 1996, 1997), are highly expressed in hearts and are active in the mono-oxgenation of arachidonic acid. The recombinant human CYP2J2 and the rat CYP2J3 have the capacity to generate all four EET regioisomers (Wu et al. 1996, 1997). Moreover, direct quantification showed that the rat heart contains substantial amounts of endogenous EETs, with 8,9-EET being the major regioisomer, constituting 39 % of the total. These results provide strong evidence that EETs can be actively synthesized in situ in the heart. Our results further suggest that cardiac myocytes may also take up unesterified EETs and incorporate them into cellular phospholipids. Cardiac myocytes have a high capacity for EET uptake, incorporating 25 % into phospholipids over 2 h. The fact that cardiac myocytes continue to accumulate radioactive EET over a 4 h period suggests that the heart is a site where EETs can be stored in esterified form in phospholipids. The bound EETs could then be released upon phospholipase activation, as was recently shown for cardiac endothelial cells (Weintraub et al. 1997). However, even though most of the uptake is incorporated into phospholipids, the small amount of unesterified 8,9-EET (1–2 %) in the cell is capable of modulating the Na+ channel function by interacting with the channel protein. Our results, therefore, support the possibility that EETs are important products of arachidonic acid metabolism in the heart.
Cardiac myocytes can also metabolize 8,9-EET to 8,9-DHET, similar to the pathway observed in arterial smooth muscle (Fang et al. 1996). Our finding that 8,9-DHET is not as effective as 8,9-EET as a Na+ channel inhibitor suggests the presence of a metabolic pathway for ‘degrading’ EET, hence providing further support that 8,9-EET is functionally active in cardiac myocytes. This could be a mechanism for removal or regulating the level of EET in the heart. After 4 h of incubation, accumulation of a polar metabolite of 8,9-EET was observed but its identity is at present unknown. This substance could be from a further step into the degradative pathway, and might be a chain-shortened product such as dihydroxyhexadecadienoic acid (DHHD) which has been previously identified in arterial smooth muscle cells (Fang et al. 1996). It is currently unknown whether this polar EET metabolite has any effects on INa or other cardiac ion channels.
Moffat et al. (1993) examined the effects of EETs in isolated guinea-pig hearts and in ventricular myocytes. They found that EETs produced no effects on the contractility of normal isolated hearts. In hearts subjected to 60 min of low-flow ischaemia, however, 5,6- and 11,12-EET significantly delayed the recovery of myocardial function during early reperfusion. Furthermore, both 5,6- and 11,12-EET increased cell shortening and intracellular Ca2+ concentrations in isolated cardiac myocytes, suggesting that during ischaemia EETs may lead to intracellular Ca2+ overload. Recently, a calcium current (ICa) in isolated rat ventricular myocytes was shown to be augmented by 11,12-EET and suppressed by P450 inhibitors and by anti-P450 antibodies (Xiao et al. 1998a). These results suggest that endogenously produced or exogenously administered EETs augment ICa via modulation of cellular cAMP levels, because 11,12-EET significantly increased, whereas P450 inhibitors reduced, the cAMP content of rat ventricular myocytes. Yet, EETs do not affect the cAMP levels in human platelets (Fitzpatrick et al. 1986), suggesting that the effects of EET on cellular cAMP contents are probably tissue specific.
In the present study, we found that 8,9-EET and other EET regioisomers are potent inhibitors of the cardiac Na+ channel. It is unlikely that the effects of 8,9-EET on INa were mediated through increased concentration of cAMP because we have previously reported that β-adrenergic stimulation and cAMP-mediated phosphorylation enhance INa (Matsuda et al. 1992; Lu et al. 1998). Moreover, long chain fatty acids have been reported to suppress INa in cardiac myocytes (Xiao et al. 1995) and in HEK293 cells transfected with the α-subunit of the human (Xiao et al. 1998b) or rat cardiac Na+ channel (Bendahhou et al. 1997). However, the present results indicate that 8,9-EET is at least ten times more potent than arachidonic acid in inhibiting INa, suggesting the epoxide group confers an enhanced capacity to blockade the Na+ channel. 8,9-EET produces significant inhibition of INa at 0.5 μM, with an IC50 around 1.0 μM, concentrations similar to those required to produce vasodilatation (Campbell et al. 1996; Fang et al. 1996; Weintraub et al. 1997) and to activate ion channels in smooth muscle cells (Li & Campbell, 1997; Li et al. 1997; Dumoulin et al. 1998; Salvail et al. 1998).
The mechanism of inhibition of the cardiac INa by 8,9-EET was determined from single channel studies. Our results suggest that the single Na+ channel conductance and the number of active channels are not altered by the effects of 8,9-EET. Instead, the probability of channel opening was dramatically reduced, with an increase in the number of null sweeps. These findings are similar to the inhibition of INa by lidocaine (lignocaine; Nilius et al. 1987; Grant et al. 1989). 8,9-EET alters the gating behaviour of Na+ channels by abbreviating the mean open time, as has been reported for lidocaine (Nilius et al. 1987). This could be interpreted as indicating that 8,9-EET directly interacts with the open conformation of the Na+ channel protein. In addition, 8,9-EET prolongs the Na+ channel mean closed time. This, together with the negative shift in the steady-state inactivation curve, suggests that 8,9-EET binding also results in accumulation of Na+ channels in an absorbing inactivated state.
Inhibition of the cardiac INa by 8,9-EET exhibits both tonic and use-dependent blocks. The basic requirements for use dependence are that there is development of channel blockade with each depolarization, and that the interval between depolarizations is not sufficient to allow complete recovery from this block. Our results indicate that there is a frequency-dependent increase in accumulation of channel inactivation in the presence of 8,9-EET, which slows channel recovery from inactivation. The behaviour of 8,9-EET as a Na+ channel inhibitor is similar to that of local anaesthetics and Class I antiarrhythmics, which are thought to interact with the Na+ channel through a ‘modulated receptor’ mechanism (Hondeghem & Katzung, 1977). The basic elements of the modulated receptor hypothesis are: (i) the Na+ channel has a specific receptor for interaction with the drug or inhibitor; (ii) the voltage-induced conformational changes in the channel result in different affinities of the receptor for the drug in each of the channel states; (iii) drug-bound channels can continue to gate; (iv) the rate of recovery from inactivation is modified due to a change or shift of inactivation kinetics for drug-bound channels; and (v) drug-bound channels may not conduct ionic current (Snyders et al. 1992). The effects of 8,9-EET on the cardiac INa embody these features. Therefore, it would be reasonable to infer that 8,9-EET exerts its effects through binding to a specific site on the Na+ channel with specific binding and unbinding rate constants, rather than through mechanisms such as modulation of membrane fluidity as a result of incorporation into phosphatidylcholine. In support of this possibility, a small amount of unesterified 8,9-EET was detected in the cardiac myocyte cell lipids after a 30 min incubation. Indeed, Kang & Leaf (1996) have reported evidence that some polyunsaturated fatty acids bind directly to the Na+ channel proteins. Whether EETs bind to the same site is unclear at present.
The effects of 8,9-EET on INa are voltage dependent, with greater inhibition of INa at more depolarized potentials. This finding has potential pathophysiological implications since the suppressive effects of EETs on cardiac impulse conduction and tissue excitability would be amplified during conditions of ischaemia or myocardial infarction (Kleber et al. 1995). Interestingly, ischaemia-induced phospholipolysis occurs in many tissues including the heart (Hazen & Gross, 1992), leading to release of arachidonic acid. The increase in arachidonic acid levels may promote the in situ formation of EETs through the cytochrome P450 mono-oxygenase pathway. But perhaps a more important source is the hydrolytic release of EETs esterified to phospholipids, the location of 99 % of the EETs present in cells and tissues (Karara et al. 1991, 1992; Nakamura et al. 1997). We have shown in this study that cardiac myocytes have a high capacity to take up unesterified 8,9-EET and incorporate it into cellular phospholipids. In addition, the formation of esterified EET in phospholipids has been shown to be dramatically increased (59-fold increase) in the presence of oxidative stress, a condition that induces free radical oxidation of membrane phospholipids (Nakamura et al. 1997). Hence, during conditions of ischaemia, the cellular concentrations of unesterified EETs may be enhanced through three different mechanisms: increased arachidonic acid release with subsequent cytochrome P450 epoxygenation, non-enzymatic oxidation and hydrolysis of membrane phospholipids through lipid peroxidation, and release of esterified EETs through activation of phospholipases. Indeed, the amount of EETs released from phospholipids through activation of phospholipase A2 has been estimated to reach the micromolar range in human platelets (Zhu et al. 1995). These findings, together with the voltage-dependent effects on INa, strongly suggest that EETs may play an important role in the modulation of cardiac electrophysiology during ischaemia.
Acknowledgments
We thank Beena Padanilam and Papri Chatterjee for technical assistance. This work was supported by a Merit Review Award from the Department of Veterans Affairs and by a Program Project Grant from the National Institute of Health HL-49264. Dr VanRollins is supported by the American Heart Association (Grant-in Aid, 96012380) and by the National Institute of Health (RO1 HL-56670). Dr Weintraub is a Clinician-Scientist awardee of the American Heart Association. Dr Shibata is an Established Investigator of the American Heart Association.
References
- Abraham NG, Pinto A, Mullane K. Identification of heme oxygenase and cytochrome P-450 in the rabbit heart. Journal of Molecular and Cellular Cardiology. 1987;19:73–81. doi: 10.1016/s0022-2828(87)80546-1. [DOI] [PubMed] [Google Scholar]
- Atkins DL, Rosenthal JK, Krumm PA, Marvin WJ., Jr Differential growth of neonatal WYK and SHR ventricular myocytes within sympathetic co-cultures. Journal of Molecular and Cellular Cardiology. 1992;24:1479–1489. doi: 10.1016/0022-2828(92)91088-m. [DOI] [PubMed] [Google Scholar]
- Baron A, Frieden M, Beny JL. Epoxyeicosatrienoic acids activate a high-conductance Ca2+-dependent K+ channel on pig coronary artery endothelial cells. The Journal of Physiology. 1997;504:537–543. doi: 10.1111/j.1469-7793.1997.537bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendahhou S, Cummins TR, Agnew WS. Mechanism of modulation of the voltage-gated skeletal and cardiac muscle sodium channels by fatty acids. American Journal of Physiology. 1997;272:C592–600. doi: 10.1152/ajpcell.1997.272.2.C592. [DOI] [PubMed] [Google Scholar]
- Benndorf K. Low-noise recording. In: Sakman B, Neher E, editors. Single-Channel Recording. 2. New York: Plenum Press; 1995. pp. 129–145. [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Chataigneau T, Feletou M, Duhault J, Vanhoutte PM. Epoxyeicosatrienoic acids, potassium channel blockers and endothelium-dependent hyperpolarization in the guinea-pig carotid artery. British Journal of Pharmacology. 1998;123:574–580. doi: 10.1038/sj.bjp.0701629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comte J, Gautheron DC. The markers of pig heart mitochondrial sub-fractions. I. The dual location of NADPH-cytochrome c reductase in outer membrane and microsomes. Biochemie. 1978;60:1289–1298. [PubMed] [Google Scholar]
- Dumoulin M, Salvail D, Gaudreault SB, Cadieux A, Rouseau E. Epoxyeicosatrienoic acids relax airway smooth muscles and directly activate reconstituted KCa channels. American Journal of Physiology. 1998;275:L423–431. doi: 10.1152/ajplung.1998.275.3.L423. [DOI] [PubMed] [Google Scholar]
- Fang X, Kaduce TL, Weintraub NL, VanRollins M, Spector AA. Functional implications of a newly-characterized pathway of 11,12-epoxyeicosatrienoic acid metabolism in arterial smooth muscle. Circulation Research. 1996;79:784–793. doi: 10.1161/01.res.79.4.784. [DOI] [PubMed] [Google Scholar]
- Fitzpatrick FA, Ennis MD, Baze ME, Wynalda MA, McGee JE, Liggett WF. Inhibition of cyclooxygenase activity and platelet aggregation by epoxyeicosatrienoic acids. Influence of sterochemistry. Journal of Biological Chemistry. 1986;261:15334–15338. [PubMed] [Google Scholar]
- Gebremedhin D, Ma Y-H, Falck JR, Roman RJ, VanRollins M, Harder DR. Mechanism of action of cerebral epoxyeicosatrienoic acids on cerebral arterial smooth muscle. American Journal of Physiology. 1992;32:H519–525. doi: 10.1152/ajpheart.1992.263.2.H519. [DOI] [PubMed] [Google Scholar]
- Grant AO, Dietz MA, Gilliam R, III, Starmer CF. Blockade of cardiac sodium channels by lidocaine. Single-channel analysis. Circulation Research. 1989;65:1247–1262. doi: 10.1161/01.res.65.5.1247. [DOI] [PubMed] [Google Scholar]
- Guengerich FP, Mason PS. Immunological comparison of hepatic and extrahepatic cytochrome P-450. Molecular Pharmacology. 1979;15:154–164. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hazen SL, Gross RW. Principles of membrane biochemistry and their application to the pathophysiology of cardiovascular disease. In: Fozzard HA, Haber E, Jennings RB, Katz AM, Morgan HE, editors. The Heart and Cardiovascular System. 2. New York: Raven Press; 1992. pp. 839–860. [Google Scholar]
- Hondeghem LM, Katzung BG. Time- and voltage-dependent interactions of antiarrhythmic drugs with cardiac sodium channels. Biochimica et Biophysica Acta. 1977;472:373–398. doi: 10.1016/0304-4157(77)90003-x. [DOI] [PubMed] [Google Scholar]
- Hu S, Kim HS. Activation of K+ channel in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. European Journal of Pharmacology. 1993;230:215–221. doi: 10.1016/0014-2999(93)90805-r. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Klockner U. Calcium tolerant ventricular myocytes prepared by preincubation in a ‘KB Medium’. Pflügers Archiv. 1982;395:6–18. doi: 10.1007/BF00584963. [DOI] [PubMed] [Google Scholar]
- Kang JX, Leaf A. Evidence that free polyunsaturated fatty acids modify Na+ channels by directly binding to the channel proteins. Proceedings of the National Academy of Sciences of the USA. 1996;93:3542–3546. doi: 10.1073/pnas.93.8.3542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karara A, Dishman E, Falck JR, Capdevila JH. Endogenous epoxyeicosatrienoyl-phospholipids. A novel class of cellular glycerolipids containing epoxidized arachidonate moieties. Journal of Biological Chemistry. 1991;266:7561–7569. [PubMed] [Google Scholar]
- Karara A, Wei S, Spady D, Swift L, Capdevila JH, Falck JR. Arachidonic acid epoxygenase: structural characterization and quantification of epoxyeicosatrienoates in plasma. Biochemical and Biophysical Research Communications. 1992;182:1320–1325. doi: 10.1016/0006-291x(92)91877-s. [DOI] [PubMed] [Google Scholar]
- Kleber AG, Fleischhauer J, Cascio WE. Ischemia-induced propagation failure in the heart. In: Zipes DP, Jalife J, editors. Cardiac Electrophysiology. From Cell to Bedside. 2. Philadelphia: W. B. Saunders Co.; 1995. pp. 174–182. [Google Scholar]
- Lee H, Matsuda JJ, Reynertson SI, Martins JB, Shibata EF. Reversal of lidocaine effects on sodium currents by isoproterenol in rabbit hearts and heart cells. Journal of Clinical Investigation. 1993;91:693–701. doi: 10.1172/JCI116250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circulation Research. 1997;80:877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- Li P, Zou A, Campbell WB. Regulation of potassium channels in coronary arterial smooth muscle by endothelium-derived vasodilators. Hypertension. 1997;29:262–267. doi: 10.1161/01.hyp.29.1.262. [DOI] [PubMed] [Google Scholar]
- Lu T, Lee H, Shibata EF. Enhancement of rat cardiac sodium current by G-protein α-subunit. Biophysical Journal. 1998;74:A328. [Google Scholar]
- McCallum GP, Horton JE, Falkner KC, Bend JR. Microsomal cytochrome P450 1A1 dependent mono-oxygenase activity in guinea pig heart: induction, inhibition, and increased activity by addition of exogenous NADPH-cytochrome P450 reductase. Canadian The Journal of Physiology and Pharmacology. 1993;71:151–156. doi: 10.1139/y93-021. [DOI] [PubMed] [Google Scholar]
- McGiff JC. Cytochrome P-450 metabolism of arachidonic acid. Annual Review of Pharmacology and Toxicology. 1991;31:339–369. doi: 10.1146/annurev.pa.31.040191.002011. [DOI] [PubMed] [Google Scholar]
- Matsuda JJ, Lee H, Shibata EF. Enhancement of rabbit cardiac sodium channels by beta-adrenergic stimulation. Circulation Research. 1992;70:199–207. doi: 10.1161/01.res.70.1.199. [DOI] [PubMed] [Google Scholar]
- Miura H, Gutterman DD. Human coronary arteriolar dilation to arachidonic acid depends on cytochrome P-450 monooxygenase and Ca2+-activated K+ channels. Circulation Research. 1998;83:501–507. doi: 10.1161/01.res.83.5.501. [DOI] [PubMed] [Google Scholar]
- Moffat MP, Ward CA, Bend JR, Mock T, Farhangkhoee P, Karmazyn M. Effects of epoxyeicosatrienoic acids on isolated hearts and ventricular myocytes. American Journal of Physiology. 1993;264:H1154–1160. doi: 10.1152/ajpheart.1993.264.4.H1154. [DOI] [PubMed] [Google Scholar]
- Nakamura T, Bratton DL, Murphy RC. Analysis of epoxyeicosatrienoic and monohydroxyeicosatetraenoic acids esterified to phospholipids in human red blood cells by electrospray tandem mass spectometry. Journal of Mass Spectrometry. 1997;32:888–896. doi: 10.1002/(SICI)1096-9888(199708)32:8<888::AID-JMS548>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- Nilius B, Benndorf K, Markwardt F. Effects of lidocaine on single cardiac sodium channels. Journal of Molecular and Cellular Cardiology. 1987;19:865–874. doi: 10.1016/s0022-2828(87)80615-6. [DOI] [PubMed] [Google Scholar]
- Oliw EH. Oxygenation of polyunsaturated fatty acids by cytochrome P450 monooxygenases. Progress in Lipid Research. 1994;33:329–354. doi: 10.1016/0163-7827(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Oltman CL, Weintraub NL, VanRollins M, Dellsperger KC. Epoxyeicosatrienoic acids and dihydroxyeicosatrienoic acids are potent vasodilators in the canine coronary microcirculation. Circulation Research. 1998;83:932–939. doi: 10.1161/01.res.83.9.932. [DOI] [PubMed] [Google Scholar]
- Salvail D, Dumoulin M, Rousseau E. Direct modulation of tracheal Cl− channel activity by 5,6- and 11,12-EET. American Journal of Physiology. 1998;275:L432–441. doi: 10.1152/ajplung.1998.275.3.L432. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Bennett PB, Hondeghem LM. Mechanisms of drug-channel interaction. In: Fozzard HA, Haber E, Jennings RB, Katz AM, Morgan HE, editors. The Heart and Cardiovascular System. 2. New York: Raven Press; 1992. pp. 2165–2193. [Google Scholar]
- VanRollins M, Kochanek PM, Evans RW, Schilding JK, Nemoto EM. Optimization of epoxyeicosatrienoic acid syntheses to test their effects on cerebral blood flow in vivo. Biochimica et Biophysica Acta. 1995;1256:263–274. doi: 10.1016/0005-2760(95)00029-c. [DOI] [PubMed] [Google Scholar]
- Weintraub NL, Fang X, Kaduce TL, VanRollins M, Chatterjee P, Spector AA. Potentiation of endothelium-dependent relaxation by epoxyeicosatrienoic acids. Circulation Research. 1997;81:258–267. doi: 10.1161/01.res.81.2.258. [DOI] [PubMed] [Google Scholar]
- Wu S, Chen W, Murphy E, Gabel S, Tomer KB, Foley J, Steenbergen C, Falck JR, Moomaw CR, Zeldin DC. Molecular cloning, expression, and functional significance of a cytochrome P450 highly expressed in rat heart myocytes. Journal of Biological Chemistry. 1997;272:12551–12559. doi: 10.1074/jbc.272.19.12551. [DOI] [PubMed] [Google Scholar]
- Wu S, Moomaw CR, Tomer KB, Falck JR, Zeldin DC. Molecular cloning and expression of CYP2J2, a human cytochrome P450 arachidonic acid epoxygenase highly expressed in heart. Journal of Biological Chemistry. 1996;271:3460–3468. doi: 10.1074/jbc.271.7.3460. [DOI] [PubMed] [Google Scholar]
- Xiao Y-F, Huang L, Morgan JP. Cytochrome P450: a novel system modulating Ca2+ channels and contraction in mammalian heart cells. The Journal of Physiology. 1998a;508:777–792. doi: 10.1111/j.1469-7793.1998.777bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y-F, Kang JX, Morgan JP, Leaf A. Blocking effects of polyunsaturated fatty acids on Na+ channels of neonatal rat ventricular myocytes. Proceedings of the National Academy of Sciences of the USA. 1995;92:11000–11004. doi: 10.1073/pnas.92.24.11000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Y-F, Wright SN, Wang GK, Morgan JP, Leaf A. Fatty acids suppress voltage-gated Na+ currents in HEK293t cells transfected with the α-subunit of the human cardiac Na+ channel. Proceedings of the National Academy of Sciences of the USA. 1998b;95:2680–2685. doi: 10.1073/pnas.95.5.2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Schieber B, McCiff JC, Balazy M. Identification of arachidonate P-450 metabolites in human platelet phospholipids. Hypertension. 1995;25:854–859. doi: 10.1161/01.hyp.25.4.854. [DOI] [PubMed] [Google Scholar]
- Zou A, Fleming JT, Falck JR, Jacobs ER, Gebremedhin D, Harder DR, Roman RJ. Stereospecific effects of epoxyeicosatrienoic acids on renal vascular tone and K+-channel activity. American Journal of Physiology. 1996;270:F822–832. doi: 10.1152/ajprenal.1996.270.5.F822. [DOI] [PubMed] [Google Scholar]