

Abstract
The ionic mechanisms contributing to the rapid depolarization (RD) induced by in vitro ischaemia have been studied in dorsal vagal motoneurones (DVMs) of brainstem slices. Compared with CA1 hippocampal neurones, RD of DVMs was slower, generally occurred from a more depolarized membrane potential and was accompanied by smaller increases in [K+]o.
RD was not induced by elevation of [K+]o to values measured around DVMs during in vitro ischaemia or by a combination of raised [K+]o and 2–5 μM ouabain.
Neither TTX (5–10 μM) nor TTX combined with bepridil (10–30 μM), a Na+-Ca2+ exchange inhibitor, slowed RD. Block of voltage-dependent Ca2+ channels with Cd2+ (0.2 mM) and Ni2+ (0.3 mM) led to an earlier onset of RD, possibly because [K+]o was higher than that measured during in vitro ischaemia in the absence of divalent ions.
When [Na+]o was reduced to 11.25–25 mM, RD did not occur, although a slow depolarization was observed. RD was slowed (i) by 10 mM Mg2+ and 0.5 mM Ca2+, (ii) by a combination of TTX (1.5–5 μM), 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μM) and D-2-amino-5-phosphonovalerate (AP5, 50 μM) and (iii) by TTX (1.5–5 μM) and AP5 (50 μM).
Ni2+ at concentrations of 0.6 or 1.33 mM blocked RD whereas 0.6 mM Cd2+ did not. A combination of Cd2+ (0.2 mM), Ni2+ (0.3 mM), AP5 (50 μM) and bepridil (10 μM) was largely able to mimic the effects of high concentrations of Ni2+.
It is concluded that RD is due to Na+ entry, predominantly through N-methyl-D-aspartate receptor ionophores, and to Ca2+ entry through voltage-dependent Ca2+ channels. These results are consistent with known changes in the concentrations of extracellular ions when ischaemia-induced rapid depolarization occurs.
When neurones are deprived of oxygen, or oxygen and glucose, changes in membrane potential occur within 30–60 s. Initial changes are relatively small and may include an early short-lasting depolarization due to action potential-dependent neurotransmitter release (Fujiwara et al. 1987; Cowan & Martin, 1992). This is followed by either hyperpolarization or depolarization due to modulation of voltage-dependent or ATP-sensitive ion channels, predominantly K+ channels (reviewed by Martin et al. 1994). As a result there is an early rise in the extracellular K+ concentration, to which inhibition of the Na+-K+-ATPase may contribute, depending upon the severity of energy substrate deprivation (Martin et al. 1994). Ultimately, a rapid depolarization takes place, which is accompanied by an abrupt decrease in extracellular Na+, Cl− and Ca2+ concentrations and an increase in extracellular K+ concentration (Hansen, 1985).
The mechanisms underlying rapid depolarization (RD) induced by energy deprivation are poorly understood. It occurs in vivo and in in vitro slice preparations and superficially resembles the transient, spreading wave of cortical depression which is known as Leão's spreading depression (Leão, 1944). However, RD and Leão's spreading depression can be distinguished from each other by their different sensitivities to N-methyl-D-aspartate (NMDA) receptor antagonists. Whereas spreading depression is blocked by NMDA receptor antagonists, RD is not (Hernándéz-Cáceres et al. 1987; Marrannes et al. 1988; Haddad & Jiang, 1993).
Various drugs do alter the latency to onset of RD. Application of TTX sufficient to block action potentials delays its onset (Aitken et al. 1991; Xie et al. 1994). In contrast, blockers of voltage-dependent K+ channels bring its onset forward (Aitken et al. 1991), as does reduced extracellular Ca2+ (Young et al. 1991; but see Tanaka et al. 1997). Antagonists of the NMDA receptor have been reported to accelerate the onset of RD in some studies (Marrannes et al. 1988) but not in others (Hernándéz-Cáceres et al. 1987; Aitken et al. 1988; Tanaka et al. 1997). When AMPA/kainate receptors are blocked by 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzene(f)quinoxaline (NBQX) or 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), RD is delayed (Xie et al. 1995; Tanaka et al. 1997).
Interestingly, RD only occurred in 52 % of slices in the presence of Ni2+ and Co2+ (2 mM each) and in 3/8 slices when Ni2+ (2 mM), the NMDA receptor antagonist 3-((±)-2-carboxypiperazine-4-yl)-propyl-1-phosphonic acid (CPP, 10 μM) and the AMPA/kainate antagonist 6,7-dinitroquinoxaline-2,3-dione (DNQX) (10 μM) were present (Jing et al. 1993). A more recent study from the same laboratory reported that a cocktail of Ni2+, CPP and DNQX (concentrations as above) and TTX (1 μM) prevented RD in all CA1 pyramidal cells studied (Müller & Somjen, 1998). Although this result clearly refutes the idea that RD reflects development of a non-specific membrane leakiness, it does not provide full insights into the mechanisms of RD because 2 mM Ni2+ would be expected to have actions on voltage-dependent Ca2+ channels (Zamponi et al. 1996), the NMDA receptor (Mayer & Westbrook, 1985), the Na+-Ca2+ exchanger (Kimura et al. 1987) and possibly the Ca2+-ATPase of the plasma membrane (Enyedi et al. 1982).
In this study, the mechanisms underlying RD have been systematically explored in dorsal vagal motoneurones (DVMs) of the brainstem. In vivo ischaemia was mimicked in vitro by exposing the preparation to a perfusate depleted of oxygen and glucose. Initially it was shown that the characteristics of RD in DVMs are distinctly different from those in CA1 pyramidal neurones. Lowering the extracellular Na+ concentration or applying a high concentration of Ni2+ blocked RD of DVMs, although a slow depolarization still occurred. The reasons for the effectiveness of these treatments were explored using various combinations of drugs. The results demonstrate that ischaemia-induced RD primarily involves entry of Ca2+ through voltage-dependent Ca2+ channels and Ca2+ and Na+ through the NMDA receptor ionophore. Parts of the results have been published in abstract form (Martin, 1997, 1998).
METHODS
Preparation and solutions
Brainstem or hippocampal slices, 400 μm thick, were prepared from male Wistar rats aged 35–50 days using standard approaches (Cowan & Martin, 1992). Experimental procedures were approved by the Animal Experimentation Ethics Committee at the Australian National University. Rats were initially anaesthetized with halothane (4 % in O2) and when corneal and toe pinch reflexes were absent the skull was rapidly opened and a mid-collicular decerebration performed. For preparation of hippocampal slices the forebrain was removed, a midline transection was made and then both hemispheres glued, midline down, onto a chuck. For preparation of brainstem slices, the bone covering the brainstem was removed, a transection made at the spinomedullary junction, the brainstem turned out of the cranium and glued to a chuck. Throughout the dissection procedures ice-cold artificial cerebrospinal fluid (ACSF) was dripped onto the brain tissue of interest. The chuck and tissue was placed in ice-cold ACSF, whose composition had been modified from standard (see below) to ensure that its pH at 2°C was about 7.3 when bubbled with 95 % O2-5 % CO2 (NaCl reduced to 112.5 mM and NaHCO3 raised to 32 mM). After cutting (Vibroslice, Campden Instruments Ltd), one slice was immediately placed in a recording chamber at 34°C. The remainder were placed on a nylon net covering a Petri dish filled with ACSF. This dish was placed in an humidified, carbogenated holding chamber at 30°C and slices removed as required.
The standard ACSF contained (mM): 124.5 NaCl, 20 NaHCO3, 3 KCl, 1.25 NaH2PO4, 1.25 MgCl2, 2 CaCl2, together with 9.8 g glucose; when bubbled with 95 % O2-5 % CO2 the pH was about 7.35. ACSF was pumped to the recording chamber via a bubble trap containing a small reservoir of solution (< 0.3 ml), and a solution heater which was placed immediately beside the recording chamber. When in vitro ischaemia was induced by switching to standard ACSF bubbled with 95 % N2-5 % CO2 and lacking glucose, the moment at which the first drop of solution entered the bubble trap was marked on chart recordings (see below). The solution took about 20 s to travel from the bubble trap to the recording chamber. Previous measurements in this laboratory have shown that the PO2 of the perfusate in the recording chamber is 15–25 mmHg under these experimental conditions (Cowan & Martin, 1995).
In some experiments extracellular Na+ ([Na+]o) was lowered to 25 mM or less by replacing the NaCl with N-methyl-D-glucamine (NMDG) and NaHCO3 with Hepes. When the pH was adjusted to 7.35 with 1 M HCl (volume dependent upon [Na+]o but around 100 ml) the osmolarity was similar to that of normal ACSF. These solutions were bubbled with either 100 % O2 or 100 % N2 (ischaemia). In a few cases extracellular Cl− was reduced to 25 mM by reducing NaCl to 15.5 mM and using 104 mM sodium gluconate to maintain osmolarity. When divalent ions were used to block ion channels, NaH2PO4 was omitted from the ACSF.
Drugs used were tetrodotoxin (TTX), CNQX, 2-amino-5-phosphonovalerate (D-AP5) (all from Tocris Cookson), bepridil (Research Biochemicals) and ouabain (Sigma).
Electrophysiological recording and experimental procedures
Intracellular recordings were made from DVMs or CA1 pyramidal cells using 3 M KCl-filled glass electrodes (aluminosilicate glass, SM100F-15, or borosilicate glass, GC100F-15, Clarke Electromedical Instruments). Membrane potential (Em) was measured relative to an agar-KCl electrode placed in the reticular formation of the brainstem or stratum radiatum. DVMs were identified on the basis of their locations, firing patterns and long after-hyperpolarization (see previous work from this laboratory, e.g. Cowan & Martin, 1992, 1995); locations and firing pattern were also used to identify CA1 pyramidal neurones (Costa et al. 1991; Andreasen & Lambert, 1995). Membrane potential was recorded in either current-clamp or discontinuous current-clamp mode using an Axoclamp 2A amplifier (Axon Instruments). Input resistance was monitored using small, hyperpolarizing current pulses. Data were digitized (TL-1 DMA Interface, Axon Instruments) at between 4 kHz and 30 kHz, depending upon the speed of the events being monitored, and processed using pCLAMP 5 software (Axon Instruments). Membrane potential was also displayed on a chart recorder (Yew) and on a MacIntosh computer using the MacLab system (digitization rate 4 Hz) with Chart software (ADInstruments, Australia).
When drugs were used for ion substitution experiments, a neurone was exposed to the treatment for a minimum of 10 min before switching to the oxygen- and glucose-depleted perfusate. In some experiments the efficacy of the treatment was tested electrophysiologically, e.g. tests were made for block of action potential by TTX, reduction in action potential height when [Na+]o was lowered, block of synaptic transmission by CNQX and AP5 or by high Mg2+-low Ca2+. To test for block of synaptic transmission, a bipolar stimulating electrode was placed ventral to the dorsal vagal motonucleus (DVMN) and the current was increased until a fast EPSP could be elicited. After application of the test solution, efficacy of treatment was tested at the same stimulus current and at double the stimulus current.
Ion-sensitive microelectrodes
On some occasions a K+-sensitive microelectrode was placed 200 μm into the contralateral DVMN or the CA1 pyramidal cell layer of the hippocampus. These electrodes were pulled from borosilicate glass which had been washed in nitric acid and thoroughly dried. A drop of 2,4 dichloromethylsilane was placed in the tip of each electrode and they were baked overnight at 200°C. Potassium ionophore I (Cocktail B, Fluka) was introduced into the tip of each silanized electrode using a fine plastic filler and then the electrodes were backfilled with 0.01 M KCl. Electrodes were tested for K+ sensitivity before and after each experiment using ACSF solutions containing from 1, 3, 10, 25 and 50 mM K+, maintained at 34°C. The relationship between electrode potential and the logarithm of K+ activity (aK) was highly linear (r2 > 0.98 in all cases), slopes ranged from 48 to 57 mV per log unit and the 10–90 % response time ranged from 25 to 30 s. Electrodes were tested for interference from Na+ and this was found to be negligible in well-silanized electrodes with tips of about 1 μm. Data concerning aK were only analysed in those experiments where electrodes had non-significant differences in K+ sensitivity before and after each experiment (comparison of slopes using F test).
Data analysis
For data analysis, membrane potential was measured at minute intervals beginning 2 min before the onset of in vitro ischaemia. In DVMs the rate of rapid depolarization was estimated as the slope of line fitted, using the least squares linear regression technique, to the membrane potential trajectory between −40 and −20 mV, i.e. the most rapid phase of depolarization. This range was extended if the rapid depolarization clearly began at more negative membrane potentials. In some experiments the membrane potential did not cover the range −40 to −20 mV within 30 min of initiating an ischaemic insult. In these cases the final 40 % of the change in membrane potential (the last value being that at 30 min) was fitted with a linear regression to obtain the slope. This portion was used because in most experiments membrane potential changed from about −65 mV to −15 mV and therefore when the slope was fitted over 20 mV this represented 40 % of the data range. In hippocampal CA1 neurones, the rate of rapid depolarization was derived from the very obvious region of rapid membrane potential depolarization.
Because the moment at which RD occurred could not be definitively determined, the onset of RD was calculated as one-half of the time taken, t½, for the membrane potential to reach −20 mV. In some cases the trajectory of the membrane potential was extrapolated from its final point to −20 mV but this was only done if the membrane potential was within 5 mV of −20 mV. This analysis was employed with drug treatments which significantly slowed RD.
The effect of different drug treatments on the rate of ischaemia-induced rapid depolarization was analysed using a one-way analysis of variance (ANOVA) and Dunnett's procedure for multiple comparisons. Differences between DVMs and CA1 neurones, or between in vitro ischaemia and ouabain were tested using Student's unpaired t tests. Differences were considered significant when P < 0.05.
RESULTS
Differences in RD between CA1 neurones and DVMs
The rapid depolarization induced by in vitro ischaemia (RD) was significantly slower in DVMs than in CA1 neurones (P < 0.0001). In control DVMs, the mean rate of depolarization from digitized records was 12.3 ± 2.1 mV min−1 (n = 15, Fig. 1A). Such a rate of depolarization was not usually confused with loss of intracellular penetration. Nevertheless partial or complete recovery of Em when the perfusate was replaced with standard ACSF was used as evidence that the depolarization was ischaemia-induced (Fig. 2).
Figure 1. Changes in membrane potential and extracellular K+ activity induced by in vitro ischaemia.
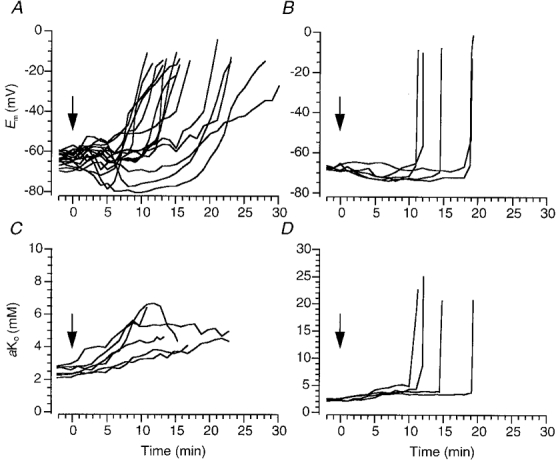
A and B, changes in membrane potential of dorsal vagal motoneurones (A) and CA1 pyramidal cells (B) induced by switching from a standard superperfusate to one bubbled with 95 % O2-5 % CO2 and lacking in glucose. Time zero (arrow) indicates when the new perfusate arrived at the bubble trap in the solution line (see Methods). C and D, potassium ion activity recorded extracellularly at the same time as some of the recordings in A or B, respectively. In studies of dorsal vagal motoneurones these measurements were made in the contralateral dorsal vagal motonucleus, and in studies of CA1 pyramidal cells they were made in the pyramidal cell layer.
Figure 2. Original records of rapid depolarization and recovery in dorsal vagal neurones.
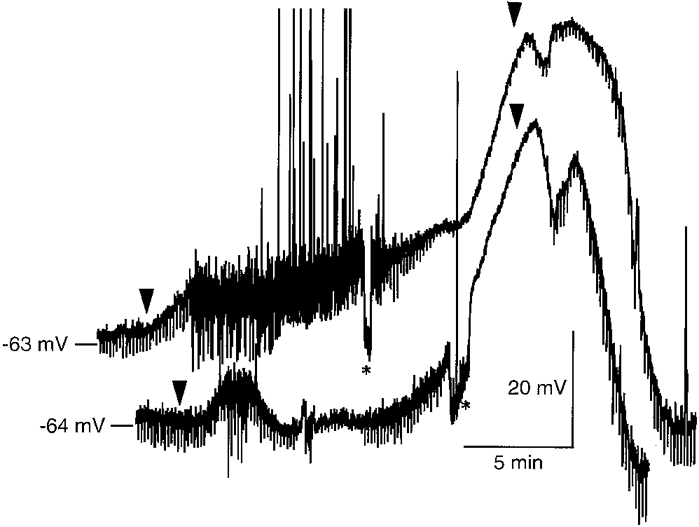
Raw data, as recorded with the MacLab system, showing response of two DVMs to in vitro ischaemia, and their subsequent recovery on return to standard perfusate. Arrowheads indicate when the new perfusate arrived at the bubble trap in the solution line (see Methods). Asterisks mark periods of each trace when hyperpolarizing current was injected into the neurones to return membrane potential to initial values for monitoring of input resistance. Positive-going shifts in membrane potential are action potentials, which were often truncated by the slow digitizing rate (4 samples s−1).
In contrast, RD in CA1 neurones was so fast that it was indistinguishable from the sudden loss of membrane potential that occurs when an electrode ‘jumps’ out of a neurone (Fig. 1B). Thus, in order to distinguish RD from loss of an intracellular recording it was necessary to show some recovery of Em when the perfusate was replaced with standard ACSF (recovery is not shown in Fig. 1B). The mean rate of depolarization was 6.3 ± 1.1 mV s−1 (n = 5), very similar to values reported by Tanaka et al. (1997).
In DVMs, changes in Em induced by in vitro ischaemia were accompanied by small increases in aKo from about 3 mM to 5–6 mM. The increase in aKo began shortly after the onset of ischaemia and usually rose steadily throughout the period of oxygen and glucose deprivation (n = 6, Fig. 1C). There was no rapid increase in aKo at the time of RD. These data contrast with measurements made in the CA1 cell layer where a dramatic rise in aKo to about 25 mM occurred (n = 4, Fig. 1D).
Role of [K+]o in RD
Experiments were undertaken to ascertain if elevation of [K+]o to values recorded in the DVMN during ischaemia was sufficient to initiate a spiral of rapid depolarization in DVMs. However, even when [K+]o was increased to 10 mM (using KCl or K2SO4), either as an hyperosmotic solution or with isosmotic replacement of NaCl, DVMs only depolarized by about 10–12 mV and Em remained steady thereafter (n = 5, Fig. 3A). Since the depolarization was substantially less than that predicted by the shift in EK this result suggests that the membrane of DVMs has a substantial resting permeability to Cl−.
Figure 3. Effects of raised [K+]o and ouabain on membrane potential and extracellular K+ activity.
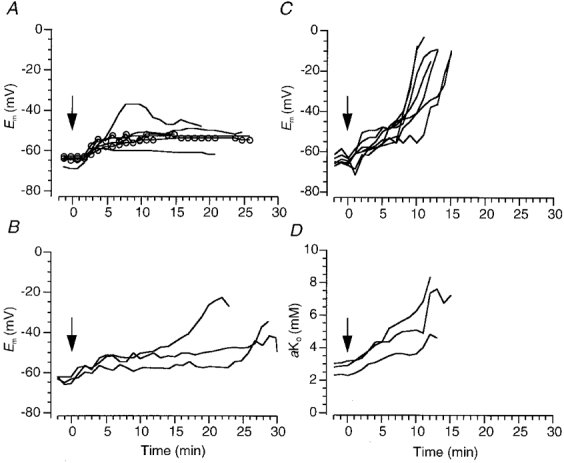
A, changes in membrane potential of DVMs induced by switching the superperfusate from one containing 3 mM K+ to one containing 10 mM KCl (−) or 10 mM K2SO4 ( ) at time zero (arrow; see Methods). B, effects on membrane potential of 2–5 μM ouabain when [K+]o was raised to 10 mM. C and D, changes in membrane potential (C) and contralaterally measured extracellular K+ activity (D) induced by 25 μM ouabain.
) at time zero (arrow; see Methods). B, effects on membrane potential of 2–5 μM ouabain when [K+]o was raised to 10 mM. C and D, changes in membrane potential (C) and contralaterally measured extracellular K+ activity (D) induced by 25 μM ouabain.
These results also indicate that the [K+]o found in the DVMN during ischaemia does not single-handedly initiate RD. However, elevation of [K+]o in combination with reduced activity of the Na+-K+-ATPase as a result of the reduced availability of ATP during ischaemia could initiate RD. When 2–5 μM ouabain was used to block the Na+-K+-ATPase in the presence of 10 mM extracellular K+, RD did not occur within 20 min, i.e. within the time frame usually observed for ischaemia-induced RD (n = 3, Fig. 3B). In 2/3 neurones a relatively slow depolarization started to occur but it was not possible to follow the membrane potential until depolarization occurred because the intracellular recordings always became unstable. At 2–5 μM, ouabain would be expected to block the α2 and α3 isoforms of the Na+-K+-ATPase but probably not the α1 isoform (for review see Ewart & Klip, 1995). All these isoforms of the Na+-K+-ATPase are present in the brain.
In contrast, 25 μM ouabain consistently induced a rapid depolarization within 9–13 min, even when [K+]o was normal (mean rate of depolarization 12.2 ± 1.9 mV min−1, n = 7, Fig. 3C). During perfusion with ouabain, aKo rose from 3 to 5–9 mM (n = 3, Fig. 3D). These results also suggest that the principle reason for ouabain-induced rapid depolarization is not elevation of aKo, although this may initiate contributing events.
Role of Ca2+ in RD
Switching to ACSF without added Ca2+ (nominally ‘Ca2+-free’ perfusate) at the onset of in vitro ischaemia did not change the rate of RD but tended to cause an earlier time of onset (n = 2). At the time of RD there was an unusually large elevation of aKo to 10 mM. In control neurones subjected to removal of extracellular Ca2+ only, the membrane depolarized but aKo increased by less than 1–2 mM. Thus, in the absence of extracellular Ca2+, in vitro ischaemia induced an unusual rise in aKo and for this reason additional experiments were not undertaken.
To investigate further the role of Ca2+ in RD a pharmacological approach was used. DVMs possess a variety of voltage-dependent Ca2+ channels (Sah, 1995) which can be blocked by a combination of 0.2 mM Cd2+ and 0.3 mM Ni2+ (A. I. Cowan & R. L. Martin, unpublished observations). While the onset of RD was significantly earlier than in control cells (4.4 ± 0.8 min (n = 4), compared with 8.4 ± 0.8 min (n = 15) in controls; ANOVA followed by Dunnett's test) the rate of depolarization was similar to that in controls (Fig 4A and Fig 6). Again aKo tended to rise abruptly and to higher levels (n = 3, Fig. 4B).
Figure 4. Effects of low concentrations of divalent ions on rapid depolarization and extracellular K+ activity.
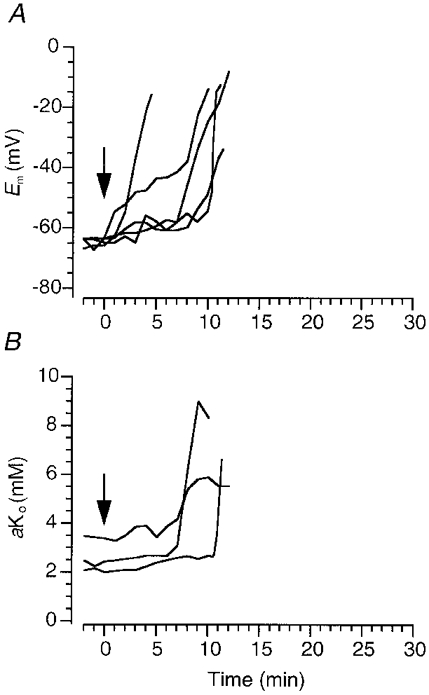
A, changes of membrane potential in DVMs induced by in vitro ischaemia in the presence of 0.2 mM Cd2+ and 0.3 mM Ni2+. Perfusate switched at time zero (arrow; see Methods). B, in some experiments extracellular K+ activity was simultaneously measured in the contralateral dorsal vagal motonucleus.
Figure 6. Rates of ischaemia-induced rapid depolarization during various treatments.
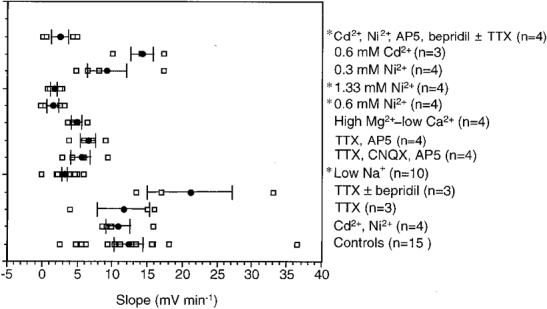
The size of the symbols prevents all data points from being visible. Means ± s.e.m. are indicated by filled circles with bars. A one-way ANOVA demonstrated a highly significant effect of drug treatment on the rate of rapid depolarization (P < 0.0001). The asterisks mark those treatments demonstrating significant differences from controls (Dunnett's procedure): low [Na+]o, 0.6 mM Ni2+, 1.33 mM Ni2+ and combined 0.2 mM Cd2+, 0.3 mM Ni2+, 50 μM AP5 and 10 μM bepridil ± 3–5 μM TTX.
These data indicate that when Ca2+ entry into neurones during in vitro ischaemia is limited by either lowering extracellular Ca2+ or by blocking voltage-dependent Ca2+ channels with Cd2+ the rise in aKo is potentiated. The cause of this was not investigated but the observation may explain why other studies have noted an earlier onset of RD in the presence of Cd2+ (Young et al. 1991).
Role of Na+ in RD
Block of TTX-sensitive voltage-dependent and persistent Na+ currents (if present in DVMs) with 5–10 μM TTX, did not obviously change RD (mean rate of depolarization 11.6 ± 3.9 mV min−1, n = 3, Fig 5A and Fig 6). This result confirms data reported in numerous previous studies. A combination of 10–30 μM bepridil (n = 3), to block forward Na+-Ca2+ exchange, and TTX (3–5 μM) did not slow RD either (mean rate of depolarization 21.1 ± 6.0 mV min−1, n = 3, Fig 5A and Fig 6).
Figure 5. Effects of TTX, low [Na+]o and glutamate receptor antagonists on rapid depolarization.
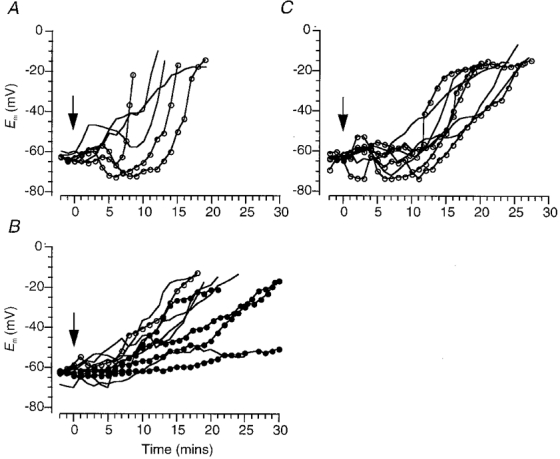
Changes in membrane potential of DVMs induced by in vitro ischaemia in the presence of: 5–10 μM TTX (−) or 3–5 μM TTX with 10–30 μM bepridil () (A); when the extracellular Na+ concentration was reduced to 11.25 (), 18 (−) or 25 mM ( ) (B); in the presence of 3–5 μM TTX and 50 μM AP5, with (−) and without 10 μM CNQX () (C). Perfusate switched at time zero (arrow; see Methods).
) (B); in the presence of 3–5 μM TTX and 50 μM AP5, with (−) and without 10 μM CNQX () (C). Perfusate switched at time zero (arrow; see Methods).
However, there are many ways, other than via TTX-sensitive Na+ channels, that Na+ can enter the cytosol and effect depolarization. Experiments were attempted in which [Na+]o was reduced to 11.25 mM by isosmotically replacing NaCl with NMDG and replacing NaHCO3 with Hepes. This treatment alone usually caused depolarization and loss of the intracellular recording. Ultimately, reduction in [Na+]o to 11.25 mM (n = 1) or 18 mM (n = 5), or 25 mM (n = 4) was achieved without loss of recording. With 11.25 and 18 mM [Na+]o the fast action potential was completely abolished whereas in 25 mM [Na+]o the action potential could be elicited, albeit significantly reduced in amplitude.
When in vitro ischaemia was initiated in a perfusate with a lowered [Na+]o the response was significantly slower than in controls and at such a rate that RD did not occur (mean rate of depolarization 3.1 ± 1.6 mV min−1, n = 10, Fig 5B and Fig 6). When Na+ was replaced by NMDG the [Cl−]o was reduced from normal by about 25 mM. To ensure that the elimination of RD was not due to this decrement in [Cl−]o, two experiments were undertaken in which extracellular Cl− was replaced with gluconate. RD still occurred (data not shown). The ischaemia-induced rise in aKo was similar in low [Na+]o to that observed in the standard ischaemic perfusate.
These results indicate that influx of Na+ is an important contributor to RD and therefore blockade of the principal routes of Na+ entry should mimic the effects of reducing [Na+]o. It has been proposed that during ischaemia glutamate release may occur via reverse operation of the glutamate transporter (Madl & Burgesser, 1993) or even as a result of an ischaemia-induced membrane damage (Phillis et al. 1994). Such release would not be blocked by TTX but postsynaptic receptors would be activated. DVMs are known to receive a significant glutamatergic synaptic input, which results in activation of both non-NMDA and NMDA receptors (Travagli et al. 1991; Willis et al. 1996). When AMPA/kainate and NMDA subtypes of glutamate receptors were blocked with a combination of 10 μM CNQX and 50 μM AP5 (in the presence of 3–5 μM TTX, n = 4) or with only TTX and AP5 (n = 4) the mean rates of depolarization were 5.5 ± 1.4 and 6.5 ± 1.1 mV min−1, respectively (Fig 5C and Fig 6). Although not statistically significant, these rates of depolarization were about half those observed in controls (Fig. 6). EPSPs evoked prior to in vitro ischaemia by stimulation ventral to the DVMN were abolished by 10 μM CNQX and 50 μM AP5.
RD is slowed by high Mg2+-low Ca2+
Besides glutamate receptors, DVMs possess postsynaptic nicotinic cholinergic receptors, P2X2 receptors and 5HT3 receptors, all of which may permit Na+ entry during in vitro ischaemia if appropriate neurotransmitter release occurs. Thus, the effects of low [Na+]o on RD may be mimicked by blocking synaptic transmission through simultaneously raising [Mg2+]o to 10 mM and lowering [Ca2+]o to 0.5 mM. This treatment did reduce the rate of ischaemia-induced depolarization, and at 4.8 ± 2.5 mV min−1 (n = 4, Fig 6 and Fig 7A) the mean rate was similar to that observed in the presence of glutamate receptor antagonists.
Figure 7. Treatments which act at multiple targets slow or eliminate rapid depolarization.
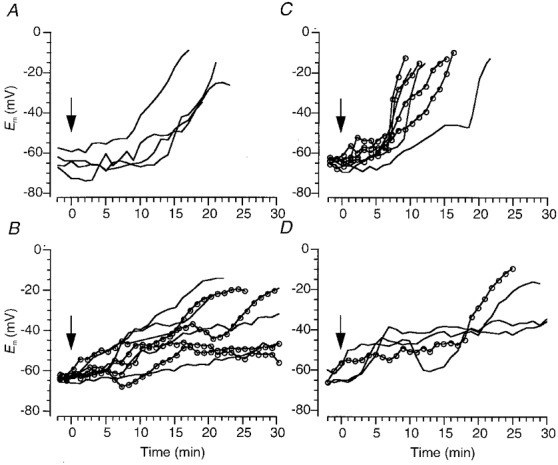
Ischaemia-induced changes in membrane potential of DVMs in the presence of: 10 mM Mg2+ and 0.5 mM Ca2+ (A); 0.6 mM Ni2+ () or 1.33 mM Ni2+ (−) (B); 0.3 mM Ni2+ () or 0.6 mM Cd2+ (−) (C); 0.2 mM Cd2+, 0.3 mM Ni2+, 50 μM AP5 and 10 μM bepridil, with (−) and without () 3–5 μM TTX (D). Perfusate switched at time zero (arrow; see Methods).
RD is blocked by high concentrations of Ni2+
Studies on hippocampal CA1 neurones have been discordant in relation to the effects of 2 mM Ni2+ on RD, but in DVMs both 0.6 mM and 1.33 mM Ni2+ consistently prevented RD. The mean rates of depolarization were 1.5 ± 2.5 and 1.8 ± 2.5 mV min−1, respectively, and these were significantly different from controls (Fig 6 and Fig 7B). In several cells there was an initial early depolarization from rest to about −50 mV, but membrane potential barely changed thereafter. Addition of TTX (5 μM) and Cd2+ (0.2 mM), or TTX (5 μM), CNQX (10 μM) and AP5 (50 μM) did not further alter the response (n = 2, data not shown). In contrast, neither 0.3 mM Ni2+ (n = 4; Fig. 7C) nor 0.6 mM Cd2+ (n = 3; Fig. 7C) slowed RD (mean rates of depolarization 9.1 ± 2.5 and 14.2 ± 2.9 mV min−1, respectively). The effects of 0.6 mM Ni2+ on the action potential were as would be expected of Ca2+ channel block (Zamponi et al. 1996) and a secondary effect on Ca2+-dependent K+ channels, i.e. a slight increase in amplitude, decrease in half-width, and decreases in both the amplitude and duration of the after-hyperpolarization. Furthermore, EPSPs induced by perivagal stimulus were blocked, as they were by 0.6 mM Cd2+.
In addition to its action on voltage-dependent Ca2+ channels, 0.6 mM Ni2+ would be expected to block NMDA receptors (Mayer & Westbrook, 1985) and inhibit the Na+-Ca2+ exchanger (Kimura et al. 1987). Therefore the effects of a combination of 50 μM AP5, 0.2 mM Cd2+, 0.3 mM Ni2+and 10 μM bepridil on RD were tested (usually in the presence of 3–5 μM TTX). As shown in Fig. 7D, RD did not occur within the 30 min time frame in two cells, and in the other two cells a late, slow depolarization from a relatively positive membrane potential occurred (mean rate of depolarization 2.4 ± 2.5 mV min−1, n = 4). Despite the small sample size this result was significantly different from controls (Fig. 6).
DISCUSSION
The results presented in this paper clearly indicate that the rapid depolarization induced by in vitro ischaemia (RD) in DVMs is not due to development of non-specific membrane leakiness. The idea that RD occurs because the membrane develops a general ‘leakiness’ has arisen in the literature because many earlier studies failed to find drugs that could block RD. But here application of Ni2+ in concentrations as low as 0.6 mM, or a combination of TTX, voltage-dependent Ca2+ channel blockers, an NMDA receptor antagonist and an inhibitor of Na+-Ca2+ exchange eliminated the rapid phase of ischaemia-induced depolarization. Despite the very different rates of ischaemia-induced depolarization of DVMs and CA1 neurones, the mechanisms contributing to RD seem very similar. In CA1 neurones, 2 mM Ni2+ in combination with TTX and the glutamate receptor antagonists DNQX and AP5 blocks RD (Müller & Somjen, 1998). Unfortunately, Müller & Somjen did not explore whether the inclusion of the AMPA/kainate receptor antagonist DNQX was absolutely necessary to prevent RD.
Previously, Haddad & Jiang (1993) showed that in brainstem neurones a reduction of [Na+]o to 5 mM limited the size and speed of RD but the reasons for this were not studied. In CA1 hippocampal neurones, Tanaka et al. (1997) and Yamamoto et al. (1997) reported that neither a reduction of [Na+]o to 28.6 mM nor application of 2 mM Ni2+ prevented RD. However, there was a significant reduction in the rate of depolarization with these treatments. High concentrations (0.25 mM) of AP5 slowed RD but the reduction of [Ca2+]o to 0.25 mM had the most dramatic effects of any treatment (Tanaka et al. 1997). However, it is only when drugs are used in combinations, as in the present study, and as reported by Somjen and colleagues (Jing et al. 1993; Müller & Somjen, 1998) that RD is prevented.
Block of action potential and exocytosis
The ability of low [Na+]o to prevent RD in DVMs suggests that blocking pathways that permit Na+ entry should produce a similar result. In keeping with results reported in other studies (see Introduction), TTX did not block RD, which indicates that activation of TTX-sensitive Na+ channels or any process which depends upon their action, e.g. physiological exocytotic neurotransmitter release, does not contribute significantly to RD. Nevertheless combined TTX, CNQX and AP5, or TTX and AP5 clearly reduced the rate of depolarization. This suggests that glutamate release by non-exocytotic pathways occurs during ischaemia and is consistent with slice studies showing glutamate release by reversal of the glutamate transporter when ATP is reduced (Madl & Burgesser, 1993) or during oxygen and glucose deprivation (Roettger & Lipton, 1996), and with studies showing that ischaemia-induced glutamate release is largely Ca2+ independent (Ikeda et al. 1989; Rubio et al. 1991; Lobner & Lipton, 1993). Activation of AMPA/kainate receptors appears to contribute little to RD in DVMs, possibly because of their rapid desensitization.
When transmission at all synapses was blocked using a high Mg2+-low Ca2+ perfusate, RD was also slowed. This result is inconsistent with the effects of TTX and low concentrations of Cd2+ and Ni2+, which also block synaptic transmission. Thus, high Mg2+-low Ca2+ probably slows RD through inhibition of NMDA receptors (Mayer & Westbrook, 1987), although high concentrations of Mg2+ have been reported to inhibit the plasmalemmal Ca2+-ATPase in red blood cells (Enyedi et al. 1982) and the Na+-Ca2+exchanger in renal epithelial cells (Lyu et al. 1991).
Actions of Ni2+
Interestingly, concentrations of Ni2+ as low as 0.6 mM were able to prevent RD. Nickel may protect against RD by four possible modes of action: block of voltage-dependent Ca2+ channels, block of the NMDA receptor (Mayer & Westbrook, 1985), inhibition of the Ca2+ pump (Enyedi et al. 1982) or block of the Na+-Ca2+ exchanger (Kimura et al. 1987). Preliminary observations in this laboratory indicate that the combination of 0.2 mM Cd2+ and 0.3 mM Ni2+provides nearly complete block of all voltage-dependent Ca2+ channels of DVMs, a result which is consistent with those obtained in CA3 hippocampal neurones (Avery & Johnston, 1996) and neurones from the central amygdala (Yu & Shinnick-Gallagher, 1997). Since neither these concentrations of divalent ions nor 10 mM Mg2+ and 0.5 mM Ca2+ were able to block RD as effectively as high Ni2+, it is highly unlikely that protection by higher concentrations of Ni2+ is solely through blockade of voltage-dependent Ca2+ channels. It is also unlikely that the action of Ni2+ is mediated solely by block of the NMDA receptor because the effect of Ni2+ on RD was also more dramatic than that of TTX and AP5.
Typically, 5 mM Ni2+ is used to block the Na+-Ca2+ exchanger, but in ventricular myocytes 1 mM appears to block its activity by about 60 % (Kimura et al. 1987). However, results pertaining to the sensitivity of the cardiac splice variant to Ni2+ cannot necessarily be applied to the neuronal Na+-Ca2+ exchanger because it has been shown that although all three isoforms (NCX1–3) of the Na+-Ca2+ exchanger exist in the brain, only three splice variants of NCX1 (NCX1.4–1.6) are found there and these differ from the cardiac splice variant (NCX1.1) (Quednau et al. 1997).
Ni2+ in concentrations greater than 0.2 mM has been shown to inhibit the Ca2+-ATPase of red cells by interacting non-specifically with an internal site on the pump (Enyedi et al. 1982). Although Ni2+ may have crossed the plasma membrane it probably did not attain sufficiently high intracellular concentrations to significantly affect the Ca2+ pump, especially when an effective external concentration was as low as 0.6 mM.
This analysis of the potential actions of Ni2+ points to the strong likelihood that it prevents RD not by a single action but most probably by partly limiting Ca2+ influx via voltage-gated Ca2+ channels and partly limiting both Na+ and Ca2+ influx through the NMDA receptor ionophore. Additionally, partial inhibition of the Na+-Ca2+ exchanger could be important. If this is the case then a similar result should be produced by use of 0.2 mM Cd2+, 0.3 mM Ni2+, AP5 and bepridil. In general this proved to be the case. A residual, late depolarization from very depolarized potentials in some cells treated with these drugs may reflect Na+ entry via AMPA/kainate receptors.
Na+-Ca2+ exchanger
Because of its electrogenic nature, the Na+-Ca2+ exchanger could contribute to membrane depolarization during ischaemia if it operated in the forward mode. Bepridil was chosen to block this exchanger because it has proved to be neuroprotective in white matter exposed to oxygen and glucose deprivation (reviewed by Stys, 1998). At low concentrations (10 μm) it targets the Na+-Ca2+ exchanger and the NMDA receptor (Sobolevsky et al. 1997) but at higher concentrations (30 μM or more) it has also been shown to depress the sodium current, INa, in ventricular cells. In the presence of TTX no obvious effects of bepridil were observed, suggesting little contribution of electrogenic Na+-Ca2+ exchange to RD. This would be consistent with a report that the NCX1 and 2 isoforms of the exchanger are inhibited by depletion of cellular ATP (Linck et al. 1998). Nevertheless, further experiments are required to clearly establish a role, if any, of the exchanger in RD.
Residual slow depolarization
In a number of cells a slow depolarization persisted after block of RD. This is consistent with failure of the Na+-K+-ATPase. Perhaps the more intriguing observation is why some cells were able to maintain a membrane potential between −40 and −50 mV for 30 or more minutes. The most likely explanation is that the metabolic demands of these cells have been sufficiently slowed by depletion of [Na+]o that any residual oxygen available to them is sufficient to fuel the Na+-K+-ATPase.
Summary
The results presented in this paper suggest that ischaemia-induced rapid depolarization is due to a combination of Na+ and Ca2+ entry, particularly through voltage-dependent Ca2+ channels and the NMDA receptor ionophore. These results are consistent with studies indicating that at the time of RD there is an abrupt reduction in [Na+]o and [Ca2+]o (Hansen, 1985; Silver & Erecinska, 1990) and with studies showing that in the early phase of ischaemia some of the rise in [Ca2+]i can be reduced by blocking NMDA receptors and voltage-dependent Ca2+ channels (Lobner & Lipton, 1993).
Acknowledgments
This study was supported by the Australian Research Council. I wish to thank Gerlinde Lenz for technical assistance, especially for her contribution to construction of K+-selective microelectrodes.
References
- Aitken PG, Balestrino M, Somjen GG. NMDA antagonists: lack of protective effect against hypoxic damage in CA1 region of hippocampal slices. Neuroscience Letters. 1988;89:187–192. doi: 10.1016/0304-3940(88)90379-5. [DOI] [PubMed] [Google Scholar]
- Aitken PG, Jing J, Young J, Somjen GG. Ion channel involvement in hypoxia-induced spreading depression in hippocampal slices. Brain Research. 1991;541:7–11. doi: 10.1016/0006-8993(91)91067-b. [DOI] [PubMed] [Google Scholar]
- Andreasen M, Lambert JD. Regenerative properties of pyramidal cell dendrites in area CA1 of the rat hippocampus. The Journal of Physiology. 1995;483:421–441. doi: 10.1113/jphysiol.1995.sp020595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery RB, Johnston D. Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. Journal of Neuroscience. 1996;16:5567–5582. doi: 10.1523/JNEUROSCI.16-18-05567.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PF, Ribeiro MA, Santos AI. Afterpotential characteristics and firing patterns in maturing rat hippocampal CA1 neurones in in vitro slices. Developmental Brain Research. 1991;62:263–272. doi: 10.1016/0165-3806(91)90174-h. [DOI] [PubMed] [Google Scholar]
- Cowan AI, Martin RL. Ionic basis of membrane potential changes induced by anoxia in rat dorsal vagal motoneurones. The Journal of Physiology. 1992;455:89–109. doi: 10.1113/jphysiol.1992.sp019292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan AI, Martin RL. Simultaneous measurement of pH and membrane potential in rat dorsal vagal motoneurons during normoxia and hypoxia: a comparison in bicarbonate and Hepes buffers. Journal of Neurophysiology. 1995;74:2713–2721. doi: 10.1152/jn.1995.74.6.2713. [DOI] [PubMed] [Google Scholar]
- Enyedi A, Sarkadi B, Nyers A, Gardos G. Effects of divalent metal ions on the calcium pump and membrane phosphorylation in human red cells. Biochimica et Biophysica Acta. 1982;690:41–49. doi: 10.1016/0005-2736(82)90236-x. [DOI] [PubMed] [Google Scholar]
- Ewart HS, Klip A. Hormonal regulation of the Na(+)-K(+)-ATPase: mechanisms underlying rapid and sustained changes in pump activity. American Journal of Physiology. 1995;269:C295–311. doi: 10.1152/ajpcell.1995.269.2.C295. [DOI] [PubMed] [Google Scholar]
- Fujiwara N, Higashi H, Shimoji K, Yoshimura M. Effects of hypoxia on rat hippocampal neurones in vitro. The Journal of Physiology. 1987;384:131–151. doi: 10.1113/jphysiol.1987.sp016447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haddad GG, Jiang C. Mechanisms of anoxia-induced depolarization in brainstem neurons: in vitro current and voltage-clamp studies in the adult rat. Brain Research. 1993;625:261–268. doi: 10.1016/0006-8993(93)91067-3. [DOI] [PubMed] [Google Scholar]
- Hansen AJ. Effect of anoxia on ion distribution in the brain. Physiological Reviews. 1985;65:101–148. doi: 10.1152/physrev.1985.65.1.101. [DOI] [PubMed] [Google Scholar]
- Hernándéz-Cáceres J, Macias-González R, Brozek G, Bures J. Systemic ketamine blocks cortical spreading depression but does not delay the onset of terminal anoxic depolarisation in rats. Brain Research. 1987;437:360–364. doi: 10.1016/0006-8993(87)91652-0. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Nakazawa T, Abe K, Kaneko T, Yamatsu K. Extracellular accumulation of glutamate in the hippocampus induced by ischemia is not calcium dependent - in vitro and in vivo evidence. Neuroscence Letters. 1989;96:202–206. doi: 10.1016/0304-3940(89)90058-x. [DOI] [PubMed] [Google Scholar]
- Jing J, Aitken PG, Somjen GG. Role of calcium channels in spreading depression in rat hippocampal slices. Brain Research. 1993;604:251–259. doi: 10.1016/0006-8993(93)90376-x. [DOI] [PubMed] [Google Scholar]
- Kimura J, Miyamae S, Noma A. Identification of sodium-calcium exchange current in single ventricular cells of guinea-pig. The Journal of Physiology. 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leão AAP. Spreading depression of activity in the cerebral cortex. Journal of Neurophysiology. 1944;7:359–390. doi: 10.1152/jn.1947.10.6.409. [DOI] [PubMed] [Google Scholar]
- Linck B, Qiu Z, He Z, Tong Q, Hilgemann DW, Philipson KD. Functional comparison of the three isoforms of the Na+/Ca2+ exchanger (NCX1, NCX2, NCX3) American Journal of Physiology. 1998;274:C415–423. doi: 10.1152/ajpcell.1998.274.2.C415. [DOI] [PubMed] [Google Scholar]
- Lobner D, Lipton P. Intracellular calcium levels and calcium fluxes in the CA1 region of the rat hippocampal slice during in vitro ischemia: relationship to electrophysiological cell damage. Journal of Neuroscience. 1993;13:4861–4871. doi: 10.1523/JNEUROSCI.13-11-04861.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyu RM, Smith L, Smith JB. Sodium-calcium exchange in renal epithelial cells: dependence on cell sodium and competitive inhibition by magnesium. Journal of Membrane Biology. 1991;124:73–83. doi: 10.1007/BF01871366. [DOI] [PubMed] [Google Scholar]
- Madl JE, Burgesser K. Adenosine triphosphate depletion reverses sodium-dependent neuronal uptake of glutamate in rat hippocampal slices. Journal of Neuroscience. 1993;13:4429–4444. doi: 10.1523/JNEUROSCI.13-10-04429.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marrannes R, De Prins E, Willems R, Wauquier A. NMDA antagonists inhibit cortical spreading depression but accelerate the onset of neuronal depolarisation induced by asphyxia. In: Somjen G, editor. Mechanisms of Cerebral Hypoxia and Stroke. New York: Plenum Press; 1988. pp. 303–304. [Google Scholar]
- Martin RL. Mechanisms underlying neuronal membrane potential changes induced by deprivation and restoration of oxygen and glucose. Society for Neuroscience Abstracts. 1997;23:544. [Google Scholar]
- Martin RL. Poster Abstracts of 7th International Symposium on Pharmacology of Cerebral Ischemia. Germany: Marburg; 1998. Nickel ions prevent rapid depolarisation induced by hypoxia/aglycaemia in rat dorsal vagal motoneurones in vitro. [Google Scholar]
- Martin RL, Lloyd HGE, Cowan AI. The early events of oxygen and glucose deprivation: setting the scene for neuronal death? Trends in Neurosciences. 1994;17:251–257. doi: 10.1016/0166-2236(94)90008-6. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. The action of N-methyl-D-aspartic acid on mouse spinal neurones in culture. The Journal of Physiology. 1985;361:65–90. doi: 10.1113/jphysiol.1985.sp015633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer ML, Westbrook GL. Permeation and block of N-methyl-D-aspartic acid receptor channels by divalent cations in mouse cultured central neurones. The Journal of Physiology. 1987;394:501–527. doi: 10.1113/jphysiol.1987.sp016883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller M, Somjen GG. Inhibition of major cationic inward currents prevents spreading depression-like hypoxic depolarization in rat hippocampal tissue slices. Brain Research. 1998;812:1–13. doi: 10.1016/s0006-8993(98)00812-9. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Smith-Barbour M, Perkins LM, O'Regan MH. Characterisation of glutamate, aspartate, and GABA release from ischemic rat cerebral cortex. Brain Research Bulletin. 1994;34:457–466. doi: 10.1016/0361-9230(94)90019-1. [DOI] [PubMed] [Google Scholar]
- Quednau BD, Nicoll DA, Philipson KD. Tissue specificity and alternative splicing of the Na+/Ca2+ exchanger isoforms NCX1, NCX2, and NCX3 in rat. American Journal of Physiology. 1997;272:C1250–1261. doi: 10.1152/ajpcell.1997.272.4.C1250. [DOI] [PubMed] [Google Scholar]
- Roettger V, Lipton P. Mechanism of glutamate release from rat hippocampal slices during in vitro ischemia. Neuroscience. 1996;75:677–685. doi: 10.1016/0306-4522(96)00314-4. [DOI] [PubMed] [Google Scholar]
- Rubio I, Torres M, Miras-Portugal MT, Sanchez-Prieto J. Ca2+-independent release of glutamate during in vitro anoxia in isolated nerve terminals. Journal of Neurochemistry. 1991;57:1159–1164. doi: 10.1111/j.1471-4159.1991.tb08274.x. [DOI] [PubMed] [Google Scholar]
- Sah P. Different calcium channels are coupled to potassium channels with distinct physiological roles in vagal neurons. Proceedings of the Royal Society. 1995;260:105–111. doi: 10.1098/rspb.1995.0066. B. [DOI] [PubMed] [Google Scholar]
- Silver IA, Erecinska M. Intracellular and extracellular changes of [Ca2+] in hypoxia and ischemia in rat brain in vivo. Journal of General Physiology. 1990;95:837–866. doi: 10.1085/jgp.95.5.837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky A, Koshelev S, Khodorov BI. Bepridil-induced blockade of NMDA channels in rat hippocampal neurones. Neuropharmacology. 1997;36:319–324. doi: 10.1016/s0028-3908(97)00003-8. [DOI] [PubMed] [Google Scholar]
- Stys PK. Anoxic and ischemic injury of myelinated axons in CNS white matter: from mechanistic concepts to therapeutics. Journal of Cerebral Blood Flow and Metabolism. 1998;18:2–25. doi: 10.1097/00004647-199801000-00002. [DOI] [PubMed] [Google Scholar]
- Tanaka E, Yamamoto S, Kudo Y, Mihara S, Higashi H. Mechanisms underlying the rapid depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology. 1997;78:891–902. doi: 10.1152/jn.1997.78.2.891. [DOI] [PubMed] [Google Scholar]
- Travagli RA, Gillis RA, Rossiter CD, Vincini S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. American Journal of Physiology. 1991;260:G531–535. doi: 10.1152/ajpgi.1991.260.3.G531. [DOI] [PubMed] [Google Scholar]
- Willis A, Mihalevich M, Neff RA, Mendelowitz D. Three types of postsynaptic glutamatergic receptors are activated in DMNX neurons upon stimulation of NTS. American Journal of Physiology. 1996;271:R1614–1619. doi: 10.1152/ajpregu.1996.271.6.R1614. [DOI] [PubMed] [Google Scholar]
- Xie Y, Dengler K, Zacharias E, Wilffert B, Tegtmeier F. Effects of the sodium channel blocker tetrodotoxin (TTX) on cellular ion homeostasis in rat brain subjected to complete ischemia. Brain Research. 1994;652:216–224. doi: 10.1016/0006-8993(94)90230-5. [DOI] [PubMed] [Google Scholar]
- Xie Y, Zacharias E, Hoff P, Tegtmeier F. Ion channel involvement in anoxic depolarization induced by cardiac arrest in rat brain. Journal of Cerebral Blood Flow and Metabolism. 1995;15:587–594. doi: 10.1038/jcbfm.1995.72. [DOI] [PubMed] [Google Scholar]
- Yamamoto S, Tanaka E, Shoji Y, Kudo Y, Inokuchi H, Higashi H. Factors that reverse the persistent depolarization produced by deprivation of oxygen and glucose in rat hippocampal CA1 neurons in vitro. Journal of Neurophysiology. 1997;78:903–911. doi: 10.1152/jn.1997.78.2.903. [DOI] [PubMed] [Google Scholar]
- Young JN, Aitken PG, Somjen GG. Calcium, magnesium, and long-term recovery from hypoxia in hippocampal tissue slices. Brain Research. 1991;548:343–345. doi: 10.1016/0006-8993(91)91146-r. [DOI] [PubMed] [Google Scholar]
- Yu B, Shinnick-Gallagher P. Dihydropyridine- and neurotoxin-sensitive and -insensitive calcium currents in acutely dissociated neurons of the rat central amygdala. Journal of Neurophysiology. 1997;77:690–701. doi: 10.1152/jn.1997.77.2.690. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Bourinet E, Snutch TP. Nickel block of a family of neuronal calcium channels: subtype- and subunit-dependent action at multiple sites. Journal of Membrane Biology. 1996;151:77–90. doi: 10.1007/s002329900059. [DOI] [PubMed] [Google Scholar]