

Abstract
Systemic infusion of angiotensin II (AII) increased papillary blood perfusion (PBP) measured by laser-Doppler flowmetry in rats, aged about 5 weeks.
The mechanisms involved in this response were determined by infusion of AII in the presence of systemic doses of losartan (a type 1 AII receptor antagonist), HOE-140 (a bradykinin B2 receptor antagonist), and an inhibitor of NO production - Nω -nitro-L-arginine (NOLA).
Mean arterial blood pressure (MAP) and PBP increased in a dose-dependent manner in response to intravenous infusions of AII. Infusion of losartan abolished these responses to AII but HOE-140 was without effect. Infusion of NOLA abolished the increase in PBP but did not affect the pressor response to AII. Systemic infusion of sodium nitroprusside restored the response to AII in experiments with NOLA infusion.
The results indicate that the increase in PBP caused by AII is mediated via angiotensin AT1 receptors and does not involve bradykinin B2 receptors. The AII-induced increase in PBP is dependent upon the presence of NO, thus providing a mechanism for maintenance of papillary perfusion in the face of generalized renal vasoconstriction due to AII.
Regional variations in blood flow in the kidney are related to zonal differences in function, ranging from glomerular filtration with solute and water reabsorption in the cortex to maximal osmotic concentration of urine at the papilla. The wide distribution of angiotensin II (AII) binding sites in the kidney (Mendelsohn et al. 1986) is consistent with a number of specific influences on these renal functions through effects on zonal blood flow, inter alia.
Intense interest has been shown in studying mechanisms of regulation of renal medullary and papillary blood flow in view of their role in the maintenance of countercurrent exchange and the corticomedullary osmotic gradient, and thus in sodium and water metabolism (Jamison & Kriz, 1982). The contribution of AII to the regulation of papillary blood flow and the role that renomedullary interstitial cells (RMICs) may play in the mediating mechanism are not yet clear. However, experiments by colleagues (Zhou et al. 1992, 1998) have shown that the primary AII binding sites in the renal medulla of rats were found not in the inner medulla but in the inner stripe of the outer medulla and specifically in the RMICs, which may react to peptides and so exert paracrine influences on the medullary microcirculation.
We have reported that systemic infusion of AII increases the rate of papillary blood perfusion (PBP) in young Sprague- Dawley rats, as measured by laser-Doppler flowmetry (LDF) (Nobes et al. 1991). The result differs from others (Faubert et al. 1987; Huang et al. 1991; Lu et al. 1993), which may be due to differences in species, age of rats, dose of AII, state of consciousness, or location of LDF probe in the inner medulla. The increase in PBP induced by systemic infusion of AII with our experimental procedure was shown to be independent of the increase in renal perfusion pressure by using an aortic clamp to maintain constant renal perfusion pressure (Nobes et al. 1991). In addition, equimolar infusion of angiotensin III (AIII) and AII caused similar increases in PBP despite the less potent systemic pressor effect of AIII (Nobes et al. 1991). A very recent study (Ortiz et al. 1998) concluded that pressor doses of AII that reduced renal blood flow increased PBP in a modest way.
There is considerable evidence that the renal inner medulla, and probably the RMICs located there (Tobian et al. 1969), contain and release a number of vasodilator substances such as prostaglandins, kinins, and lipids including medullipin (Muirhead et al. 1970, 1972; Muirhead & Pitcock, 1985). Later studies suggest that endogenously formed kinins influence PBP (Roman et al. 1988) and that the PBP increase due to systemic AII may be mediated by kinins (Nobes et al. 1991). A more recent study (Mattson & Cowley, 1993) showed that the increase in PBP caused by renal medullary interstitial infusion of bradykinin was mediated by a mechanism involving nitric oxide. The involvement of prostaglandins was raised again by the finding that intravenous infusion of AII did not influence medullary blood flow (Parekh et al. 1996) because prostaglandin production in the medulla antagonized the effect of some pressor hormones including AII (Parekh & Zou, 1996). Finally, a study (Ortiz et al. 1998) published since the brief reports of our results (Walker et al. 1996; Blair-West et al. 1997) indicated that the elevation of papillary blood flow caused by AII was greatly dependent on an increased production of NO, perhaps via the local release of kinins.
We have examined further the mechanisms involved in the increase in PBP caused by systemic infusion of AII in young rats (Nobes et al. 1991). The procedures used here were the same as those used in that previous study. AII was infused in the presence of (i) losartan, the type 1 AII receptor antagonist; (ii) HOE-140, the bradykinin B2 receptor antagonist; and (iii) an inhibitor of NO production.
METHODS
Experiments were performed on Sprague-Dawley rats (body weight, 95–105 g), approximately 5 weeks of age. Animals were allowed food (GR2 Rat and Mouse Breeder Cubes, Barastoc, Australia; 0.25 % sodium) and water ad libitum before experimentation. The rats were anaesthetized with Inactin (110 mg (kg body wt)−1, i.p.) and placed on a heated table controlled via feedback from a rectal thermistor to maintain body temperature at 38°C. A tracheostomy was performed and polyethylene catheters were inserted into the jugular and femoral veins for intravenous infusions, and into the carotid artery for the recording of continuous pulsatile blood pressure. The left kidney was exposed by a flank incision and separated from the perineal attachments; the kidney capsule was left intact. The kidney was then immobilized in a stainless steel cup. The papilla was exposed by opening up the ureter to the cortical margin. Papillary blood perfusion (PBP) was recorded using a 1 mm diameter fibre-optic probe attached to a PeriFlux PF3 laser-Doppler perfusion monitor. The laser output was directed through a fibre-optic bundle to the probe, which was placed on the tip of the papilla of the left kidney.
Arterial blood pressure was measured and recorded continuously on a Grass 7D polygraph connected to a Gould P23 ID pressure transducer. Blood pressure measurements are presented as mean arterial pressure (MAP). PBP was recorded continuously through a second channel. The value of PBP was calculated as perfusion units (PU) from the trace, after calibrating the scale of the trace so that the Periflux PF3 internal standard (100 PU) was set at 100 divisions, and the external PF 1001 motility standard (250 PU) was set at 250 divisions. By this arrangement, the scale for PBP values was direct reading.
After surgical preparation, baseline arterial blood pressure and PBP were recorded for at least 10 min. AII (Auspep, Melbourne, Australia) was then infused, with a 10 min interval between doses. Specific antagonists were then administered by intravenous injection/infusion into a femoral vein. After suitable delays (see experiments 1–4, below), the AII infusions were repeated during ongoing infusion or after bolus injections of antagonists. Antagonists used were losartan (Merck Research Laboratories, Rahway, NJ, USA), HOE-140 (Hoechst, Germany), Nω-nitro-L-arginine (NOLA; Sigma) and sodium nitroprusside (SNP; Sigma). All infusion solutions were prepared in 0.9 % NaCl.
After completion of the experiment the rat was killed by an intravenous injection of saturated potassium chloride.
All experimental procedures were approved by the Institutional Animal Ethics Committee according to guidelines produced by the National Health and Medical Research Council of Australia and adopted by State Legislature.
Experiment 1 (n = 8)
In these initial experiments, a solution of AII was infused into the jugular vein for 4 min periods at three doses, 150, 300 and 600 ng kg−1 min−1, by increasing the rate of infusion from 0.75 to 1.5 and 3.0 ml h−1, respectively. The intermediate dose of 300 ng kg−1 min−1 was given routinely in all experiments and was used as the basis of comparisons in Results.
Experiment 2 (n = 7)
Losartan was injected into the femoral vein as a bolus dose of 1 mg kg−1 in 0.05 ml saline followed by an infusion of 50 ng kg−1 min−1 at 1.5 ml h−1. After waiting 45 min the AII infusions were repeated (Zhou et al. 1992).
Experiment 3 (n = 8)
HOE-140 was infused at 120 mg kg−1 min−1 into a femoral vein at 1.5 ml h−1. After 10 min the AII infusions were restarted. The effectiveness of the HOE-140 dose was confirmed by blockade of the reduction in MAP and increase in PBP caused by intravenous bradykinin (1250 ng kg−1).
Experiment 4 (n = 8)
NOLA was administered as two doses of 10 mg kg−1 in 0.05 ml saline at 5 min intervals. After 10 min the AII infusions were recommenced. SNP was then infused at 1 mg kg−1 min−1 into a femoral vein at 1.5 ml h−1 for a period of 10 min and then the AII infusions were repeated.
Statistical analysis
Results are quoted as means ± s.e.m. Effects of treatments were evaluated by the application of Student's paired t tests to the control and treatment results in individual preparations. The effects on MAP and PBP were tested for dose dependency by one-way analysis of variance.
RESULTS
Effect of angiotensin II (Fig. 1)
Figure 1. Efect of i.v. infusion of AII at doses of 150, 300 and 600 ng kg−1 min−1.
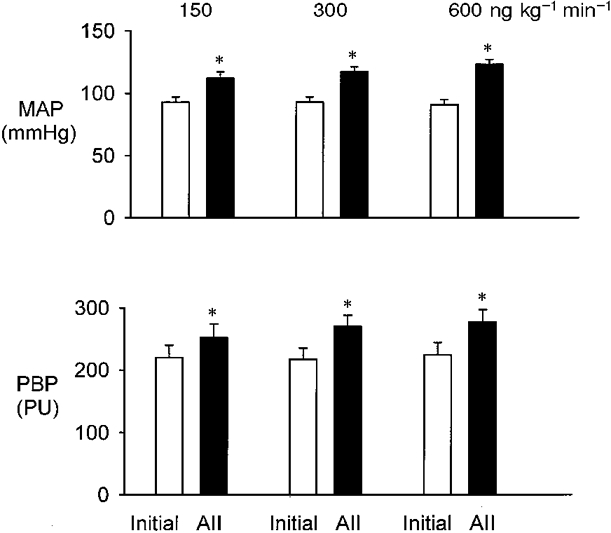
Top, mean arterial pressure (MAP). Bottom, renal papillary blood perfusion (PBP) expressed in perfusion units (PU). Results are means ± s.e.m., n = 8. * P < 0.001 compared with initial control value.
The infusions of AII caused dose-dependent increases in MAP and PBP. Doses of 150, 300 and 600 ng kg−1 min−1 increased MAP from an initial value of 93 ± 4 mmHg to 112 ± 5 (P < 0.001), 117 ± 4 (P < 0.001) and 123 ± 4 mmHg (P < 0.001), respectively (comparison of individual increases by ANOVA; P < 0.001). PBP increased from 220 ± 20 to 252 ± 22 PU with 150 ng kg−1 min−1 AII (P < 0.001), 269 ± 18 PU with 300 ng kg−1 min−1 AII (P < 0.001) and 277 ± 20 PU with 600 ng kg−1 min−1 AII (P < 0.001). The increase in PBP with 300 ng kg−1 min−1 was significantly greater than with the lower dose (P < 0.01, paired t test). Comparison of the increases in PBP for the two higher doses of AII showed that these were not significantly different. These data indicate that the change in PBP during graded AII infusions was dose dependent, tending to plateau at around 300 ng kg−1 min−1. The dose of 300 ng kg−1 min−1 was therefore chosen as the routine dose in all subsequent experiments.
Effect of losartan (Fig. 2)
Figure 2. Effect of i.v. infusion of losartan (1 mg kg−1 bolus, 50 ng kg−1 min−1) on responses to i.v. infusion of AII at 300 ng kg−1 min−1.
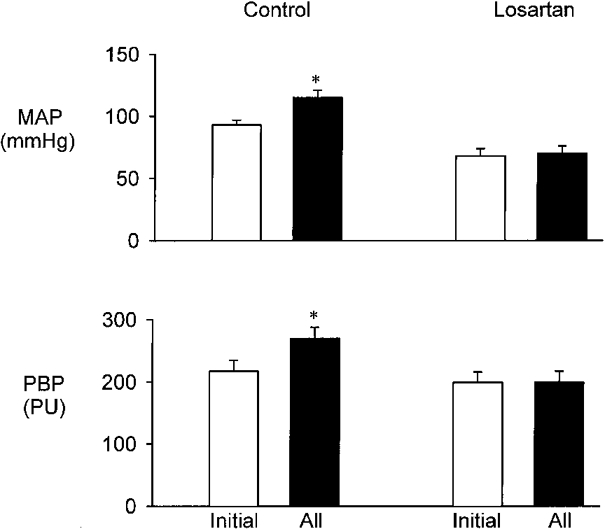
Top, MAP. Bottom, renal PBP. Results are means ± s.e.m., n = 7. * P < 0.001 compared with initial control value.
The initial values for MAP and PBP were 93 ± 4 mmHg and 217 ± 18 PU. Infusion of AII increased MAP to 115 ± 6 mmHg and PBP to 270 ± 18 PU (both P < 0.001). Infusion of losartan reduced MAP to 68 ± 6 mmHg (P < 0.001) but PBP was unchanged. Subsequent infusion of AII did not alter either MAP or PBP.
Effect of HOE-140 (Fig. 3)
Figure 3. Effect of i.v. infusion of HOE-140 (120 mg kg−1 min−1) on responses to i.v. infusion of AII at 300 ng kg−1 min−1.
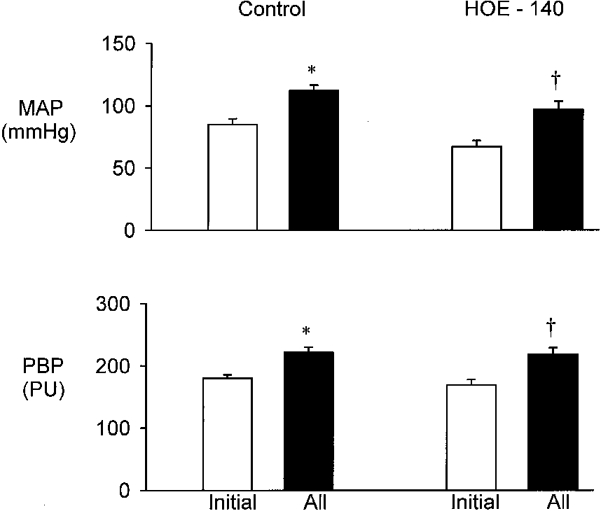
Top, MAP. Bottom, renal PBP. Results are means ± s.e.m., n = 8. * P < 0.001 compared with initial control value; † P < 0.001 compared with initial HOE-140 value.
Treatment with HOE-140 reduced MAP from its initial value of 85 ± 5 to 68 ± 5 mmHg (P < 0.001) whereas PBP was not significantly altered. Infusion of AII increased MAP and PBP both before and after treatment with HOE-140. PBP increased from 180 ± 5 to 223 ± 8 PU in the control experiment and from 169 ± 9 to 219 ± 11 PU after HOE-140 infusion (both P < 0.001).
Effect of NOLA and SNP (Fig. 4)
Figure 4. Effect of i.v. infusion of NOLA (2 doses of 10 mg kg−1 with 5 min interval) and SNP on responses to i.v. infusion of AII at 300 ng kg−1 min−1.
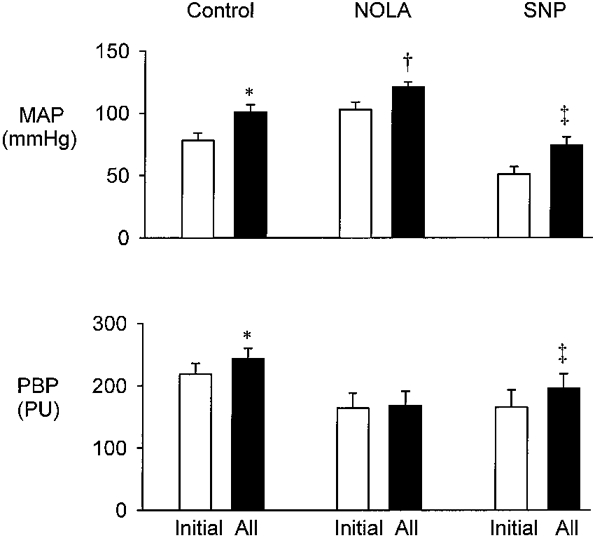
Top, MAP. Bottom, renal PBP. Results are means ± s.e.m., n = 8. * P < 0.001 compared with initial control value; † P < 0.01 compared with initial NOLA value; ‡ P < 0.01 compared with initial SNP value.
Infusion of AII increased MAP and PBP as in previous control experiments (P < 0.001). Treatment with NOLA increased MAP from 78 ± 6 to 103 ± 6 mmHg (P < 0.001) and PBP was reduced from 219 ± 17 to 164 ± 24 PU (P < 0.001). Subsequent AII infusion increased MAP to 121 ± 4 mmHg (P < 0.01) but PBP was steady at 168 ± 23 PU. Further treatment with SNP reduced initial MAP to 51 ± 6 mmHg (P < 0.001) but initial PBP was unchanged at 166 ± 27 PU. Subsequent infusion of AII increased MAP to 74 ± 7 mmHg (P < 0.01) and PBP to 196 ± 23 PU (P < 0.01).
DISCUSSION
The major finding from these experiments was that the increase in PBP caused by intravenous infusion of AII was mediated through type 1 AII (AT1) receptors via a vasodilatory mechanism that probably involves the production of NO. Treatment with an inhibitor of NO production, NOLA, did not significantly affect the pressor response to intravenous infusion of AII, but it did reduce PBP and it abolished the PBP increase evoked by AII infusion. The ability of the NO donor SNP to reinstate the PBP response to AII provides further evidence that the vasodilatory mechanism evoked by intravenous AII depends on the presence of NO.
This result and proposal are in agreement with the results of Ortiz et al. (1998). These authors observed that chronic NO deficiency in rats caused arterial hypertension, renal vasoconstriction and reduced papillary blood flow partly due to the effects of AII, acting via AT1 receptors. They suggested that the reduction in papillary blood flow was due to the disappearance of the tonic release of NO into the medullary environment. They also showed that AII infusion elevated papillary blood flow dependent on increased production of NO and suggested that NO may be an important antagonist of AII in the papillary circulation where it may maintain blood flow despite a reduction in cortical blood flow.
Another important finding was that treatment with the bradykinin B2 receptor blocker HOE-140, which completely inhibited responses to a large intravenous dose (1250 ng kg−1) of bradykinin, did not significantly affect the pressor response to AII, nor did it modulate the associated increase in PBP. This result is important because previous work with the same preparation (Nobes et al. 1991) found that PBP did not increase when AII was infused during aprotinin administration, indicating that kinin release may be the cause of the increase of PBP by AII. That study (Nobes et al. 1991) also showed that the increase of PBP by AII did not appear to be mediated by prostaglandins, although such a mechanism has been indicated by other experiments in vivo (Parekh et al. 1996) and in vitro (Grenier et al. 1981). Our results raise the possibility that a kinin other than bradykinin is implicated or that the aprotinin-induced inhibition of the AII response (Nobes et al. 1991) was due to an action other than its inhibition of kallikrein. A possible explanation for the decrease in MAP during HOE-140 experiments in this preparation could be the longer than usual period of time before returning to AII infusion. During this period an effective dose of bradykinin was established and the effectiveness of the HOE-140 blockade was proved.
It has been established that endogenously formed kinins may influence renal inner medullary blood flow in the rat. For example, intravenous infusion of an inhibitor of neutral endopeptidase, the major kininase in the rat kidney, increased inner medullary blood flow and this effect was blocked by a kinin receptor antagonist (Roman et al. 1988). Also, blockade of endogenous kinin degradation by intravenous enalaprilat (Roman et al. 1988) or by interstitial infusion of captopril (Mattson & Cowley, 1993) increased papillary blood flow. In addition, pretreatment with NOLA abolished the increases in papillary blood flow due to interstitial administration of either bradykinin or captopril (Mattson & Cowley, 1993). These data and our present findings support the proposition that the increase in PBP observed with intravenous AII may be NO dependent.
The evidence presented in this paper indicates that the graded PBP response to AII might be dependent on the presence, but not the graded production, of NO and raises the possibility that some other regulatory pathway may be involved. Taken together, our earlier experiments (Nobes et al. 1991), the results of the HOE-140 experiments from the present study, and the studies mentioned above suggest that the release of a kinin other than bradykinin could be a contributory factor in the graded modulation of the response to AII.
Our proposal that the increase of PBP caused by intravenous AII is independent of the systemic vascular response to AII is based on evidence (Nobes et al. 1991) that similar PBP responses were observed with intravenous angiotensin III (AIII) infusion and that PBP increased despite aortic clamping to maintain constant renal perfusion pressure during AII infusion. These observations suggest that the changes in ‘initial’ MAP caused by various treatments in the present experiments, e.g. HOE-140, NOLA and SNP, none of which modified the MAP response to AII, did not contribute to the associated response of PBP to AII. Furthermore, treatment with NOLA increased ‘initial’ MAP and lowered ‘initial’ PBP with abolition of the PBP response to AII. Subsequent treatment with SNP (Fig. 4) did not restore the ‘initial’ PBP to control levels but it did restore the increase in PBP caused by infusion of AII. In this context it is pertinent that MAP increased in a graded manner with increasing infusion of AII. In contrast, PBP increased significantly when the effects of the two lower rates of infusion were compared, but reached a plateau with the intermediate dose. This provides evidence for dissociation of PBP and arterial blood pressure and indicates autoregulation of PBP at higher perfusion pressures.
The consistent and dose-dependent PBP responses to doses of intravenous AII in the range 150–600 ng kg−1 min−1 confirm our earlier report of an enhancement of PBP by AII in the systemic circulation (Nobes et al. 1991). This observation appears to conflict with various other findings (Faubert et al. 1987; Huang et al. 1991; Lu et al. 1993) but there are a number of possible reasons for this including differences in methods, doses of AII and species. The data in dogs (Faubert et al. 1987) involve (i) a different method and site of measurement of PBP, (ii) AII application by renal arterial infusion at 0.5 ng kg−1 min−1 and (iii) a possible species difference in responsiveness. The experiments of Huang et al. (1991) involved young Wistar rats anaesthetized with sodium pentobarbitone. Those workers also used LDF to measure PBP but their intravenous doses of AII were without effect on PBP. Their highest dose, 150 ng kg−1 min−1, was equal to the smallest dose used in the present experiments, and in our hands this dose often produced small or even doubtful responses so that higher doses were routinely used. The study of Lu et al. (1993) was performed with adult Sprague- Dawley rats, and measured cortical and medullary blood flow using chronically implanted fibre-optic probes in conscious animals. Bolus intravenous injections of 12.5 ng of AII reduced blood flow in both regions. Amongst other differences, that study did not relate specifically to blood perfusion in the renal papilla.
An important issue here is the siting of the fibre-optic probe for optimal measurement of PBP. The young (100 g) Sprague-Dawley rat has a prominent renal papilla that extends into the ureter. In our experiments, the fibre-optic probe was positioned towards the tip of the papilla so that the 1 mm tip of the probe was still in full and intimate contact with the tissue surface. This position was therefore the last possible position before the probe tip lost full contact with the papilla. Moving the tip of the probe towards the outer medulla offered a number of options for positioning. This lack of certainty was always avoided in our experiments as our method provided assurance that the probe was placed in a unique, reproducible position. This precise procedure for placement of the probe has not been used by other workers reporting measurements of PBP or medullary blood flow.
It should be noted that the initial MAP in these young rats tended to be lower than would be expected for adult rats with similar anaesthesia and surgical preparation. The lowest initial MAP was in the NOLA experiments, but this value was not significantly less than the values in the other sets of experiments.
In summary, intravenous infusion of AII causes an increase in PBP. This response is mediated through AT1 receptors but does not involve bradykinin B2 receptors. Further, it is concluded that the AII-induced increase in PBP depends on the presence of endogenous NO. This mechanism would provide a basis for the maintenance of papillary perfusion during renal vasoconstriction as a consequence of elevated systemic or intrarenal levels of AII.
Acknowledgments
This project was funded by a grant from the National Health and Medical Research Council of Australia. A. A. J. Rajaratne received a travel grant from The University of Melbourne.
References
- Blair-West JR, Walker LL, Rajaratne AAJ, Harris PJ. Nitric oxide modulates the effects of angiotensin II on blood perfusion in the rat renal papilla. Proceedings of the XXXIII International Congress of Physiological Sciences. 1997 St Petersburg, P004.08. [Google Scholar]
- Faubert PF, Chou SY, Porush JG. Regulation of papillary plasma flow by angiotensin II. Kidney International. 1987;32:472–478. doi: 10.1038/ki.1987.234. [DOI] [PubMed] [Google Scholar]
- Grenier FC, Rollins TE, Smith WL. Kinin-induced prostaglandin synthesis by renal papillary collecting tubule cells in culture. American Journal of Physiology. 1981;241:F 94–104. doi: 10.1152/ajprenal.1981.241.1.F94. [DOI] [PubMed] [Google Scholar]
- Huang CL, Davis G, Johns EJ. A study of the action of angiotensin II on perfusion through the cortex and papilla of the rat kidney. Experimental Physiology. 1991;76:787–798. doi: 10.1113/expphysiol.1991.sp003544. [DOI] [PubMed] [Google Scholar]
- Jamison RL, Kriz W. Urinary Concentrating Mechanism. New York: Oxford University Press; 1982. pp. 79–271. [Google Scholar]
- Lu S, Mattson DL, Roman RJ, Becker CG, Cowley AW., Jr Assessment of changes in intrarenal blood flow in conscious rats using laser-Doppler flowmetry. American Journal of Physiology. 1993;264:F956–962. doi: 10.1152/ajprenal.1993.264.6.F956. [DOI] [PubMed] [Google Scholar]
- Mattson DL, Cowley AW., Jr Kinin actions on renal papillary blood flow and sodium excretion. Hypertension. 1993;21:961–965. doi: 10.1161/01.hyp.21.6.961. [DOI] [PubMed] [Google Scholar]
- Mendelsohn FAO, Dunbar M, Allen A, Chou ST, Millan MA, Aguilera G, Catt KJ. Angiotensin II receptors in the kidney. Federation Proceedings. 1986;45:1420–1425. [PubMed] [Google Scholar]
- Muirhead EE, Brown GB, Germain GS, Leach BE. The renal medulla as an antihypertensive organ. Journal of Laboratory and Clinical Medicine. 1970;76:641–651. [PubMed] [Google Scholar]
- Muirhead EE, Germain G, Leach BE, Pitcock JA, Stephenson P, Brooks B, Brosius WL, Daniels EG, Hinman JW. Production of renomedullary prostaglandins by renomedullary interstitial cells grown in tissue culture. Circulation Research. 1972;31(suppl. 2):161–172. [PubMed] [Google Scholar]
- Muirhead EE, Pitcock JA. The renal antihypertensive hormone. Journal of Hypertension. 1985;3:1–8. doi: 10.1097/00004872-198502000-00001. [DOI] [PubMed] [Google Scholar]
- Nobes MS, Harris PJ, Yamada H, Mendelsohn FAO. Effects of angiotensin on renal cortical and papillary blood flows measured by laser-Doppler flowmetry. American Journal of Physiology. 1991;261:F 998–1006. doi: 10.1152/ajprenal.1991.261.6.F998. [DOI] [PubMed] [Google Scholar]
- Ortiz MC, Fortepiani LA, Ruis-Marcos FM, Atucha NM, Garcia-Estan J. Role of AT1 receptors in the renal papillary effects of acute and chronic nitric oxide inhibition. American Journal of Physiology. 1998;274:R 760–766. doi: 10.1152/ajpregu.1998.274.3.R760. [DOI] [PubMed] [Google Scholar]
- Parekh N, Dobrowolski L, Zou AP, Steinhausen M. Nitric oxide modulates angiotensin II- and norepinephrine-dependent vasoconstriction in rat kidney. American Journal of Physiology. 1996;270:R 630–635. doi: 10.1152/ajpregu.1996.270.3.R630. [DOI] [PubMed] [Google Scholar]
- Parekh N, Zou AP. Role of prostaglandins in renal medullary circulation: response to different vasoconstrictors. American Journal of Physiology. 1996;271:F 653–658. doi: 10.1152/ajprenal.1996.271.3.F653. [DOI] [PubMed] [Google Scholar]
- Roman RJ, Kaldunski ML, Scicli AG, Carretero OA. Influence of kinins and angiotensin II on the regulation of papillary blood flow. American Journal of Physiology. 1988;255:F 690–698. doi: 10.1152/ajprenal.1988.255.4.F690. [DOI] [PubMed] [Google Scholar]
- Tobian L, Ishii M, Duke M. Relationship of cytoplasmic granules in renal papillary interstitial cells to ‘postsalt’ hypertension. Journal of Laboratory and Clinical Medicine. 1969;73:309–319. [PubMed] [Google Scholar]
- Walker LL, Rajaratne AAJ, Blair-West JR, Harris PJ. Modulation of effects of angiotensin on rat papillary blood flow by nitric oxide. Proceedings of the Australian Physiological and Pharmacological Society. 1996;27:210. P. [Google Scholar]
- Zhou J, Alcorn D, Allen AM, Mendelsohn FAO. High resolution localization of angiotensin II receptors in the rat renal medulla. Kidney International. 1992;42:1372–1380. doi: 10.1038/ki.1992.429. [DOI] [PubMed] [Google Scholar]
- Zhou J, Dean R, Maric C, Aldred PG, Harris P, Alcorn D, Mendelsohn FAO. Localization and interactions of vasoactive peptide receptors in renomedullary interstial cells of the kidney. Kidney International. 1998;54(suppl. 67):S22–S28. doi: 10.1046/j.1523-1755.1998.06705.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Thomas D, Harris PJ, Skinner SL. The role of endogenous angiotensin II in the regulation of renal haemodynamics and proximal fluid reabsorption in the rat. The Journal of Physiology. 1992;435:1–13. doi: 10.1113/jphysiol.1992.sp019214. [DOI] [PMC free article] [PubMed] [Google Scholar]