

Abstract
Intracellular recordings from hippocampal CA1 pyramidal neurones revealed that EPSPs evoked by selective stimulation of the isolated afferent input to the distal third of the apical dendrites were relatively insensitive to changes in dendritic membrane potential (Vm) but amplified by depolarizations of the somatic Vm. The amplification was present at potentials depolarized from resting membrane potential (RMP) but was most marked when the EPSPs were close to threshold for action potential generation. The amplification consisted of a uniform component and a variable component which was only present when the EPSPs were threshold straddling.
The somatic amplification was caused by an intrinsic membrane current which was blocked by somatic application of tetrodotoxin (TTX, 10 μm), but was insensitive to bath application of NiCl2 (100–200 μm). We therefore suggest that the amplification of the subthreshold EPSP is due primarily to the activation of a non-inactivating Na+ current (INaP).
Injection of 4-aminopyridine (4-AP, 25–50 mM) during intradendritic recordings resulted in amplification of the EPSPs in 37 % of the dendrites, which was similar to that observed in somatic recordings. However, in the one case in which somatic application of TTX was tested, dendritic amplification was blocked, suggesting that it is a reflection of the somatic amplification.
Because the shift to variable amplification was very abrupt and it is present in only a very narrow voltage range close to threshold, we suggest that the variable component is caused by the regenerative activation of INaP. The variability itself is probably due to the simultaneous activation of different outward K+ currents.
The present results indicate that the somatic region of CA1 pyramidal neurones can function as a voltage-dependent amplifier of distally evoked EPSPs and that this is due to the activation of a somatic INaP. The presence of this amplifying mechanism will have important functional consequences for the way in which distally generated EPSPs are integrated.
The cortical pyramidal neurones constantly receive a barrage of synaptic signals, which are integrated and transformed into changes in their firing behaviour. It is now evident that synaptic integration is a very complex process influenced not only by the properties of the synaptic signals, but also by the structural and physiological properties of the pyramidal neurones themselves. Over the years it has become increasingly evident that the dendrites are actively involved in the conduction and integration of synaptic potentials. The reason for this is that several of the currents originally described in somata of cortical pyramidal neurones (Brown et al. 1990; Storm, 1990) have now been shown also to be present within the dendritic membrane (for reviews see Johnston et al. 1996; Stuart et al. 1997). Recently, evidence has emerged that some of these dendritic currents are activated by subthreshold excitatory postsynaptic potentials (EPSPs) (Johnston et al. 1996) and function as amplifiers for distally generated EPSPs (Schwindt & Crill, 1995; Lipowsky et al. 1996; Gillessen & Alzheimer, 1997). Other currents have been shown to regulate dendritic excitability by damping the expression of amplifying currents (Andreasen & Lambert, 1995; Hoffman et al. 1997). Whereas dendritic currents have an obvious functional significance in synaptic integration, the involvement of somatic currents in this process is more ambiguous. While the somatic membrane of CA1 pyramidal neurones contains a host of different voltage- and Ca2+-dependent currents (for reviews see Brown et al. 1990; Storm, 1990), their functional significance has mostly been studied in relation to the firing properties of the neurones. Evidence for the involvement of somatic currents in the integration of subthreshold synaptic potentials has, however, been found in neocortical pyramidal neurones (Stafstrom et al. 1985; Thomson et al. 1988; Deisz et al. 1991; Stuart & Sakmann, 1995) and hippocampal CA3 pyramidal neurones (Miles & Wong, 1986). Here it has been suggested that the anomalous inward rectifying behaviour of the somatic membrane is caused by a non-inactivating Na+ current (INaP) (Stafstrom et al. 1985) and possibly also a low-threshold Ca2+ current (Sutor & Zieglgänsberger, 1987). The presence of these currents makes it possible for the soma to function as a voltage-dependent amplifier of EPSPs.
Somatic recordings from CA1 pyramidal neurones have indicated that they exhibit a similar type of anomalous inward rectification to that seen in CA3 and neocortical pyramidal neurones. This rectification, which occurs in the region between resting membrane potential (RMP) and the threshold for action potential generation, is sensitive to tetrodotoxin (TTX) and Mn2+ (Hotson et al. 1979), indicating that it involves the activation of both INaP (French et al. 1990) and a low-threshold Ca2+ current (Takahashi et al. 1991). Up until now, the rectifying properties of the CA1 pyramidal neurones have been suggested to be involved in the generation of prepotentials which precede action potentials (MacVicar, 1985; Hu & Hvalby, 1992; Hu et al. 1992). At the same time, however, it has been suggested that these prepotentials are predominantly of dendritic and not somatic origin (Hu & Hvalby, 1992; Hu et al. 1992). In relation to subthreshold EPSPs, the results have so far been more ambiguous in that some groups have found that blocking the anomalous inward rectification had no effect on synaptic potentials (Hotson et al. 1979; Connors & Prince, 1982), whereas others have reported a small reduction in EPSP amplitude (Puil & Carlen, 1984). However, in light of the recent studies by Alzheimer's group (Lipowsky et al. 1996; Gillessen & Alzheimer, 1997), it is possible that this reduction in EPSP amplitude is due to the block of dendritic, and not somatic, inward rectifying currents. The functional significance of somatic rectification in relationship to subthreshold EPSPs is, therefore, still not settled.
In the course of investigating the effect of dendritic propagation on synaptic efficacy (Andreasen & Lambert, 1998), we observed that distally generated EPSPs were markedly enhanced by depolarization of the somatic membrane, while they were unaffected or even reduced by depolarization of the dendritic membrane potential (Vm). These preliminary observations point to a function of the somatic region as a voltage-dependent amplifier, similar to that which has recently been proposed for neocortical layer V pyramidal neurones (Stuart & Sakmann, 1995). The purpose of the present study has, therefore, been to clarify the nature, location and properties of the current underlying the amplification.
METHODS
Experiments were performed on hippocampal slices prepared from 37 male Wistar rats (250–300 g). After anaesthetizing with chloroform, the rat was decapitated and the brain removed quickly and placed in a standard Ringer solution (see below) at 4°C. The hippocampus was dissected free and slices (400 μm thick) were cut on a McIlwain tissue chopper. The slices were immediately transferred to the recording chamber, where they were placed on a nylon-mesh grid at the interface between warm (31–33°C) standard Ringer solution and warm humidified Carbogen (95 % O2, 5 % CO2). Perfusion flow rate was 1 ml min−1.
The afferent input to the distal third of the apical dendrites of a group of CA1 pyramidal cells was isolated by making two incisions in the stratum radiatum (SR), as described previously (Andreasen & Lambert, 1998). The cuts were placed 250–300 μm from the superior border of stratum pyramidale (SP) and left a small tissue ‘bridge’ approximately 200 μm wide and parallel to SP (Fig. 1A). The cuts were made with a custom-made knife consisting of two razor blade chips mounted together in the same plane, but separated by a gap of approximately 200 μm. This procedure always left some uncut fibres on both sides of the ‘bridge’ and it was therefore necessary to complete the incisions using a micro-dissection knife (Fine Science Tools Inc., Heidelberg, Germany). This ensured complete isolation of the distal afferent input to those pyramidal neurones whose apical dendrites extended through the ‘bridge’ into the superficial part of SR and stratum lacunosum-moleculare (L-M). Following the microdissection, the slice was allowed to rest for at least 1 h before recordings were started.
Figure 1. Experimental model for isolation of the distal synaptic inputs to CA1 pyramidal neurones.
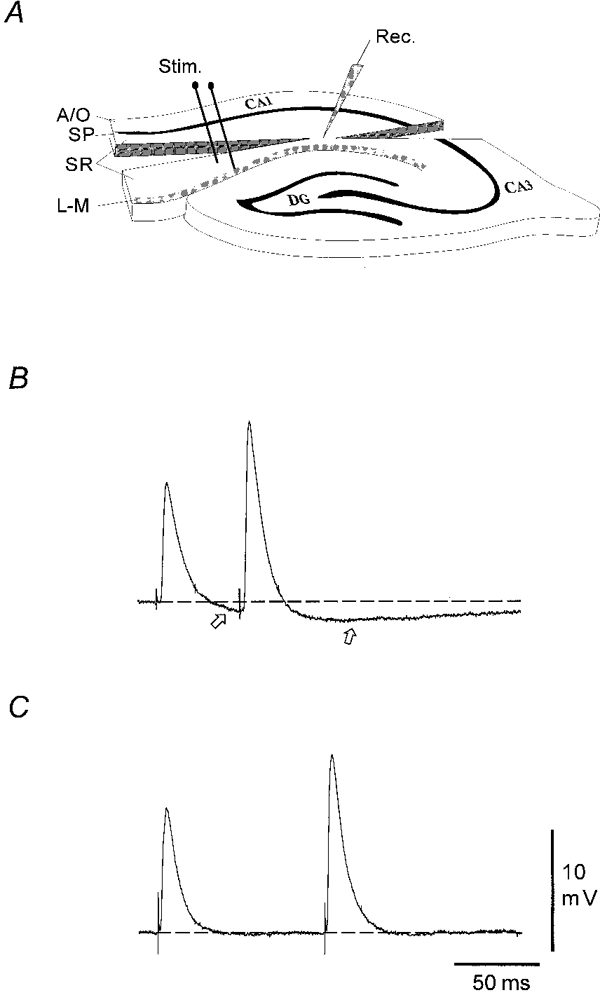
A, schematic representation of the hippocampal slice preparation and the experimental set-up for activating isolated distal synaptic inputs to CA1 pyramidal neurones. In each slice, two cuts were made which left a small tissue ‘bridge’ in SR between 250 and 300 μm from the superior border of SP. Dendritic recordings were obtained from the centre of the ‘bridge’. To activate distal afferent fibres, a bipolar stimulating electrode was placed near the subiculum close to the border between SR and L-M. A/O, alveus/oriens; SP, stratum pyramidale; SR, stratum radiatum; L-M, stratum lacunosum-moleculare; Rec, recording electrode; Stim, distal stimulation electrode. B, a typical dendritic response to distal afferent paired-pulse stimulation (450 μA) in standard Ringer solution. Note the slow IPSP (open arrows). C, a typical response of another dendrite to distal stimulation (300 μA) in the presence of Bic (10 μM), CGP 55845A (2 μM) and AP5 (50 μM). Note that the slow IPSP is now absent. In this and the following figures, the responses shown are the average of 4 to 8 individual traces unless otherwise noted. RMP: B, −68 mV; C, −71 mV.
Intracellular recordings from CA1 pyramidal neurones were made using borosilicate glass microelectrodes (1.2 mm o.d., Clark Electromedical, Pangbourne, UK) filled with 4 M potassium acetate (tip resistances: 60–80 MΩ). In some experiments, 4-aminopyridine (4-AP, 25–50 mM) was included in the electrode solution. Penetrations of the distal apical dendrites were made at the centre of the ‘bridge’, as indicated in Fig. 1A. Intradendritic recordings were identified on the basis of their similarity to those reported previously from histochemically verified dendritic recordings (Andreasen & Lambert, 1995). The criteria for accepting intradendritic and intrasomatic recordings were the same as those used earlier (Andreasen & Lambert, 1995).
Teflon-insulated platinum electrodes (50 μm thick) were used for orthodromic and antidromic stimulation of the CA1 pyramidal neurones with constant current (50–500 μA; 50 μs duration; 0.1 Hz) pulses. In order to activate the distal afferent fibres, a bipolar stimulation electrode was placed on the slice close to the border between SR and L-M on the subicular side of the ‘bridge’ (Fig. 1A). For antidromic stimulation, the stimulating electrode was placed on the alveus/oriens (A/O) border. In some experiments, a paired-pulse stimulation protocol was used with an interpulse interval between 50 and 100 ms. The two responses to paired-pulse stimulation are termed the conditioning (c) and test (t) response.
Conventional recording techniques were employed, using a high input impedance amplifier (Axoclamp-2A, Axon Instruments) with bridge-balance and current injection facilities. Results were digitized on-line using a Labmaster A/D converter and pCLAMP acquisition software (Axon Instruments) on a 486 PC and recorded for off-line analysis using a modified digital audio processor (Sony PCM-701es) and a video recorder. EPSPs were quantified by their amplitude and integrated area, both measured with respect to the prestimulus baseline. All analyses were performed using pCLAMP analysis software. Values are given as means ± s.e.m. unless otherwise noted.
Drugs and solutions
The composition of the standard Ringer solution was (mM): NaCl, 124; KCl, 3.25; NaH2PO4, 1.25; NaHCO3, 20; CaCl2, 2; MgSO4, 2; D-glucose, 10; bubbled with Carbogen (pH 7.3). In some experiments, NiCl2 was added to the standard Ringer solution to give a final concentration of 100–200 μM. Unless otherwise noted, the experiments were all performed in the presence of DL-2-amino-5-phosphonovaleric acid (AP5, 50 μM), bicuculline methobromide (Bic, 10 μM) and CGP 55845A (2 μM) in order to block N-methyl-D-aspartate (NMDA), γ-aminobutyric acid (GABA)A and GABAB receptors, respectively.
For local application of TTX (10 μM), a glass pipette (o.d. 10 μm) was filled with standard Ringer solution in which TTX was dissolved and connected to a pressure injection system (PV830 Pneumatic PicoPump, WPI). The tip of the pipette was placed on the surface of the slice close to and upstream from the site of interest so that most of the TTX-containing solution was washed away by the continuous flow of standard Ringer solution, thereby limiting the lateral spread of TTX. TTX was applied by a short pressure pulse (10–20 p.s.i., for 100–300 ms), after which the TTX-containing pipette was immediately withdrawn.
All pharmacological compounds were made up in aqueous stock solutions of 100–1000 times the required final concentration and diluted in the standard Ringer solution as appropriate. TTX, 4-AP and Bic were purchased from Sigma, AP5 from Tocris Cookson (Bristol, UK) and CGP 55845A was provided by Novartis.
RESULTS
The passive membrane properties of CA1 pyramidal neurones
Intracellular recordings were obtained from 43 apical dendrites with an average RMP of −69.0 ± 0.3 mV (n = 43, range −74 to −64 mV) and an average input resistance (Rin), measured from the voltage response to small hyperpolarizing current pulses, of 19 ± 0.8 MΩ (n = 38, range 12 to 36 MΩ). The time course of the initial potential change induced by a small hyperpolarizing current pulse could be well fitted by a single exponential function giving a mean membrane time constant (τm) of 5.9 ± 0.4 ms (n = 25, range 3.2 to 9.8 ms). All of the basic membrane parameters of the apical dendrites were similar to those reported earlier (Andreasen & Lambert, 1995, 1998).
Intrasomatic recordings were obtained from 44 pyramidal neurones, with an average RMP of −69.4 ± 0.5 mV (n = 44, range −75 to −60 mV) and Rin of 32.6 ± 1.6 MΩ (range 16.5 to 55 MΩ). The decay of the somatic potential was also well described by a single exponential function giving a mean value for τm of 14.3 ± 0.9 ms (n = 17, range 8.1 to 21.5 ms), which was nearly threefold slower than the dendritic τm.
The difference in Rin between the distal apical dendrites and somata was similar to that seen in our earlier studies (Andreasen & Lambert, 1995, 1998) and could reflect either a difference in the amount of ‘leak’ introduced by the recording electrodes or a real difference in resting membrane conductances. Recently, Magee (1998) reported the same relative difference in Rin between somata and dendrites using dual patch clamp recordings. Furthermore, Magee found a six- to sevenfold higher density of hyperpolarizing-activated channels (Ih channels) in the distal apical dendrites compared with the soma. These were blocked by Cs+, which equalized the difference in Rin. This suggests that the difference in Rin observed here reflects the resting membrane conductances and is not caused by ‘leak’.
Distally generated EPSPs are differentially affected by changes in dendritic and somatic membrane potential
In standard Ringer solution, paired orthodromic stimulation of the distal afferent fibres evoked a dendritic response consisting of two fast EPSPs showing pronounced paired-pulse facilitation (PPF), followed by an inhibitory postsynaptic potential (IPSP) (Fig. 1B). At RMP the EPSPs are primarily mediated by non-NMDA receptors with a small contribution from NMDA receptors (Andreasen & Lambert, 1998) and the IPSP is blocked by CGP 55845A, indicating that it is a GABAB receptor-mediated slow IPSP. Even though there is no apparent sign of a fast IPSP, our earlier study indicated that there is nevertheless some activation of GABAA receptors which affects the time course of the EPSPs (Andreasen & Lambert, 1998). In order to avoid contamination from NMDA and GABA receptor activation, all future experiments were performed in the presence of Bic (10 μM), CGP 55845A (2 μM) and AP5 (50 μM) in the perfusion medium. The EPSPs evoked by distal paired-pulse stimulation under these conditions had time courses similar to those of EPSPs recorded without the antagonists (Fig. 1C).
Preliminary experiments indicated that the distally evoked non-NMDA receptor-mediated EPSP was differentially affected by changes in dendritic and somatic Vm. To investigate this in more detail, we compared the voltage dependency of distally generated EPSPs in the distal apical dendrites and somata. The dendritic or somatic Vm was changed by injecting long (400 ms) hyperpolarizing or depolarizing current pulses through the recording electrode. In the majority of recordings, the distally generated EPSPs were found to be strikingly insensitive to changes in dendritic Vm. In a few cases, however, the amplitude of the test EPSP (tEPSP) in particular showed the voltage dependency expected for a non-NMDA receptor-mediated EPSP (Fig. 2A) (Hestrin et al. 1990). One reason for this unexpected insensitivity to changes in Vm could be that this region of the apical dendrites has a small electrotonic length constant (Andreasen & Nedergaard, 1996). The implication of this is that current spread from the injection site at the level of the ‘bridge’ will be restricted in the distal direction, resulting in a reduced control of Vm near the active synapses.
Figure 2. The voltage dependency of distally evoked EPSPs at the dendritic and somatic level.
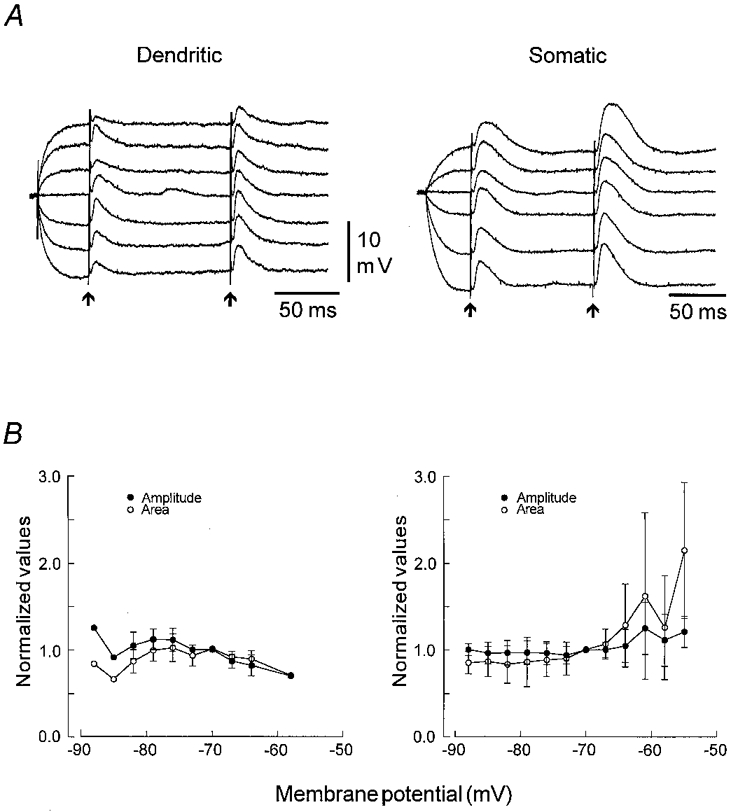
A, potential dependency of distally evoked EPSPs recorded in an apical dendrite (left) and in a soma (right) in the presence of Bic (10 μM), CGP 55845A (2 μM) and AP5 (50 μM). Vm was changed by injecting 400 ms duration depolarizing or hyperpolarizing current pulses. The dendritic cEPSPs varied in amplitude independently of Vm, while the tEPSP decreased slightly with depolarization, but was unchanged by hyperpolarization. The somatic cEPSP was also relatively insensitive to changes in Vm, while the tEPSP increased slightly in amplitude upon hyperpolarization, but was markedly increased in both amplitude and duration by depolarizations which brought the tEPSP close to action potential threshold. B, a plot of the voltage dependency of the peak amplitude (•) and area (^) of dendritically (left, n = 8) and somatically recorded tEPSPs (right, n = 17). The data were normalized with respect to responses evoked at −70 ± 1 mV and grouped according to the prestimulus Vm with a bin width of 3 mV. The mean ± s.d. area for each group was then plotted as a function of the prestimulus Vm. Apart from a slight decrease upon depolarization, both the peak amplitude and area of the dendritically recorded EPSPs show little voltage dependency. For the somatic EPSPs, the amplitude was unaffected by hyperpolarization below −70 mV whereas the area decreased slightly. At potentials more depolarized than −70 mV, both the amplitude and particularly the area increased progressively, though there was a large variability in response. RMP in A: dendrite, −71.5 mV; soma, −69.5 mV.
In contrast to the relative insensitivity of the EPSPs to changes in dendritic Vm, the distally generated EPSPs were markedly affected by changes in somatic Vm (Fig. 2A). This was an unexpected finding, considering that the active synapses are located at least 250 μm from the soma. Apart from a slight shortening in duration, the EPSPs were generally not affected by hyperpolarization of the somatic membrane. However, when the somatic membrane was depolarized there was a progressive increase in peak amplitude and duration of the EPSP which attained a maximum at potentials where the EPSPs were close to threshold for firing. At a given depolarized potential, the enhancement was most pronounced for the tEPSP (Fig. 2A). At Vm values between −56 and −54 mV, the area of the conditioning EPSP (cEPSP) was reduced to 79 ± 18 % (mean ± s.d., n = 3) of that measured at −70 mV, whereas the area of the tEPSP was increased to 215 ± 8 %. In some cases, no change was seen until Vm reached a level at which the EPSPs were threshold straddling, at which stage there was a large increase in both amplitude and duration (Fig. 2A).
In order to quantify the voltage-dependent changes, we measured the peak amplitude and area of the dendritic (n = 8) and somatic (n = 17) EPSPs evoked at different levels of Vm. Because of their more pronounced voltage dependency, tEPSPs were used for this quantification. Because of the variation in EPSP size, Rin and prestimulus Vm, the peak amplitude and area were normalized with respect to the values measured at −70 ± 1 mV and grouped over a bin width of 3 mV. The averaged values for the tEPSP were then plotted as a function of prestimulus Vm (Fig. 2B). Neither the peak amplitude nor the area of the dendritic EPSPs showed marked voltage dependency, except for a slight reduction in peak amplitude when the membrane was depolarized and a reduction in area with hyperpolarization. In the somata, the amplitude was unaffected by hyperpolarization beyond −70 mV, whereas the area was reduced slightly to a relatively constant level. At potentials more depolarized than −70 mV, both the peak amplitude and area increased in a voltage-dependent manner, although there was a great deal of variability. The reason for the decrease in the area at −58 mV could be that this point represents the average of only four recordings, in one of which the area was depressed to 60 % of that measured at −70 mV. The changes were much more pronounced for the area, which reflects a marked increase in duration of the tEPSP. The plot also indicates that the threshold for the voltage-dependent change in the distally generated EPSPs is between −70 and −65 mV, which is in the range of the RMP of CA1 pyramidal neurones.
The voltage-dependent amplification of the EPSP is caused by an intrinsic mechanism
The voltage-dependent amplification of the distally evoked EPSP can be divided into two components: a uniform component and a variable component. The uniform component was evident at RMP and increased nearly linearly with depolarization until the EPSPs reached threshold. At a given Vm, it provided a reliable and consistent amplification of the EPSP (Fig. 3A). When Vm was depolarized to a level at which the EPSP was close to threshold, another component became evident in addition to the uniform component. This component was characterized by being extremely variable in amplitude, duration and activation behaviour. Even so, it provided an additional and often very robust amplification of the EPSPs (Fig. 3A). Whenever action potentials were initiated on the variable component, the following post-spike after-hyperpolarization curtailed the response to an extent which depended on the latency of the action potential (Figs 3C, 6A and 10A). Another characteristic was that a large variable component was usually followed by an undershoot of a few millivolts (Fig. 3B). The initial rising phases of the EPSPs were identical whether or not either of the components was present (Fig. 3A). The fact that the peak amplitude often increased indicates that the amplifying mechanism is activated during the rising phase of the EPSP.
Figure 3. The amplification of distally generated EPSPs involves a uniform and a variable component.
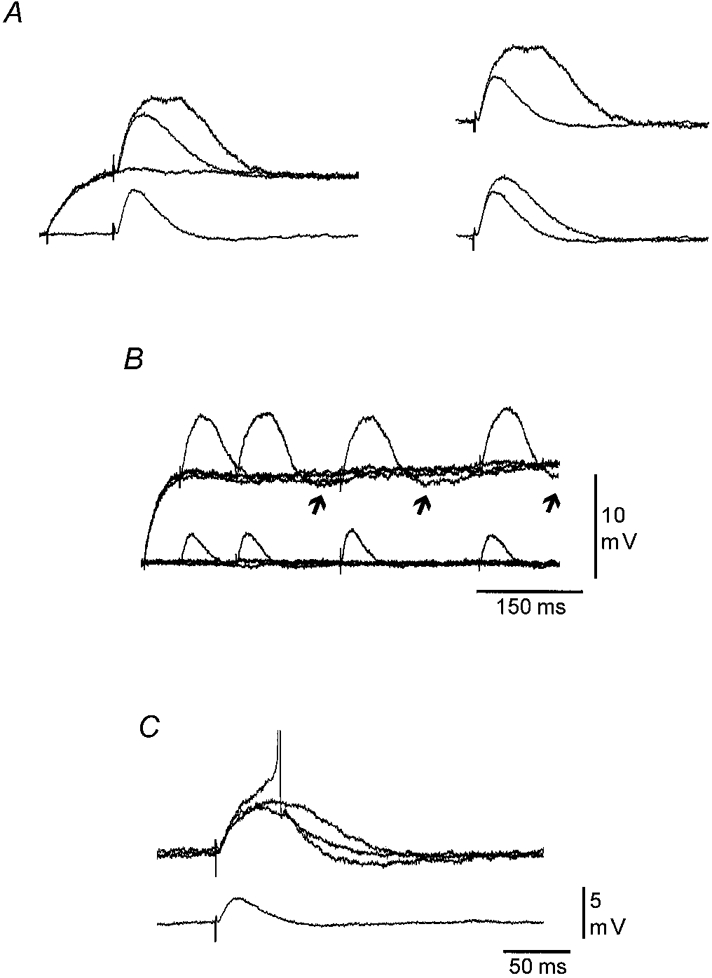
A, left, a distally evoked EPSP recorded in the soma at RMP and during a depolarizing current pulse (+0.26 nA), which depolarized Vm to −65 mV. The smallest response shown at the depolarized level is the average of four EPSPs with the lowest peak amplitude and shortest duration seen at this Vm. The largest response shown at depolarized Vm is a single trace and represents the largest amplification observed. Right, the EPSP evoked at RMP superimposed on the averaged (lower) and single (upper) EPSPs evoked during the depolarizing current pulse to exemplify the degree of amplification obtained by the uniform and the additional variable component, respectively. B, superimposed single traces of EPSPs evoked by distal stimulation at RMP and at different latencies during a long depolarizing current pulse (+0.4 nA). Note that, except for the first response, the amplified EPSPs are all followed by an undershoot (arrow). C, single traces showing that constant depolarization of the somatic Vm to a level at which the EPSP was threshold straddling still resulted in a very marked and variable amplification of the EPSP. Lower trace is the average of four EPSPs evoked at RMP. The action potentials in this and the following figures are shown truncated. Calibration bars in C also apply to A. RMP, −71 mV.
Figure 6. The amplification of the distal EPSP is insensitive to NiCl2.
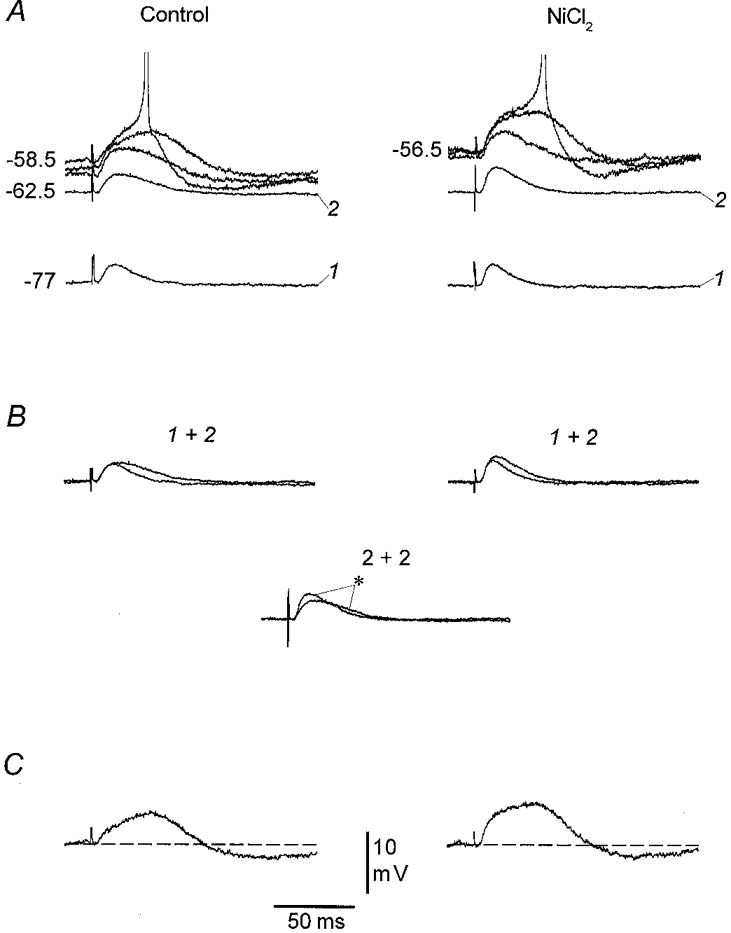
A, somatic recordings of EPSPs evoked by distal stimulation at different levels of Vm before (left) and after (right) 35 min of perfusion with 200 μM NiCl2. In the control situation, there was a characteristic amplification of the EPSP which was most pronounced close to threshold. NiCl2 had little effect on the overall amplification or the variability at Vm levels close to threshold: if anything, the variable component was larger in the presence of NiCl2. NiCl2 did, however, appear to cause a depolarizing shift in the level at which the variable component was evident and in the threshold for action potentials. B, superimpositions of the EPSPs marked 1 and 2 to show the size of the uniform amplification in the two recording conditions and to compare the size of the uniform component in control and in the presence of NiCl2, respectively (asterisk indicates the response in NiCl2). C, the undershoot seen after large EPSPs was insensitive to NiCl2. RMP, −68 mV.
Figure 10. The variable component of the somatic amplification is only activated in a very narrow voltage range.
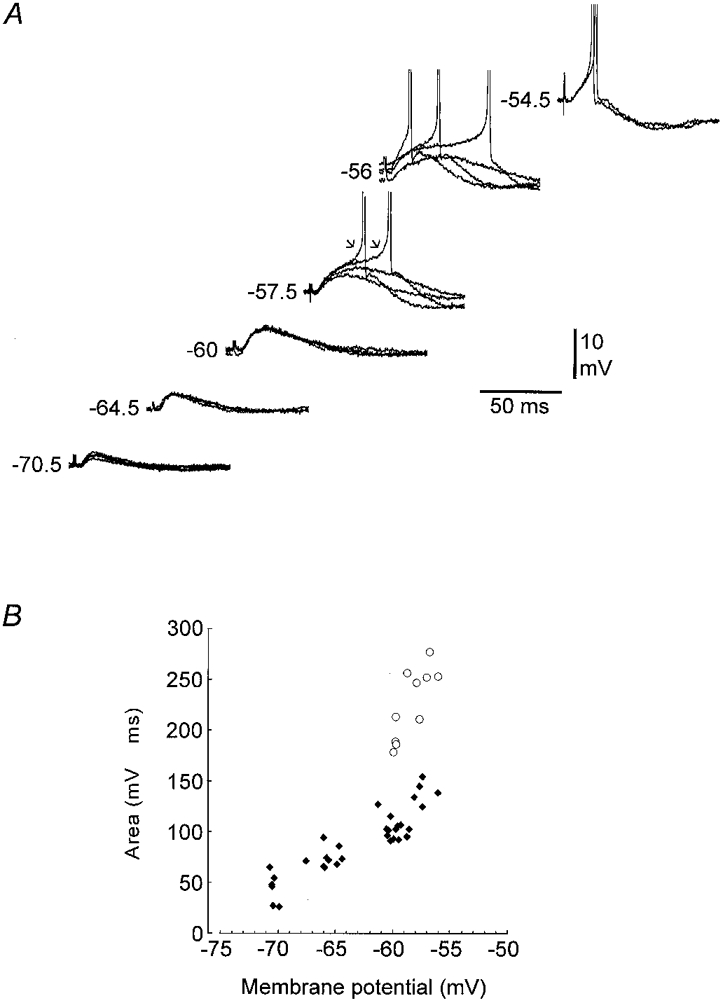
A, distally evoked individual EPSPs recorded at different levels of somatic Vm (indicated by the numbers to the left of each set of records). When Vm is depolarized from −70.5 to −60 mV, there is a progressive and uniform amplification of the EPSP. Between −60 and −56 mV, the amplification increases, but becomes more variable and action potentials are now initiated by a prepotential (arrows) at different latencies. At −54.5 mV, action potentials are reliably initiated at a short and constant latency. B, a plot of the area of the individual EPSPs in relation to the prestimulus Vm. At hyperpolarized levels up to about −60 mV, there is a progressive increase in the area of the EPSP which shows only a slight variability. However, the variability increases dramatically in a narrow range of potentials more depolarized than −60 mV. The filled symbols represent EPSPs amplified by a uniform component, whereas the open symbols represent EPSPs amplified by both a uniform and a variable component. RMP, −69 mV.
In neocortical layer II/III pyramidal neurones, subthreshold EPSPs are amplified in a voltage-dependent manner similar to that described here. However, part of this amplification was found to have a time dependent component (Deisz et al. 1991). To investigate whether the amplification described here also showed time dependency, we conducted experiments in which the distal afferent fibres were stimulated at different latencies from the onset of the depolarizing current pulse. In Fig. 3B, the somatic Vm has been depolarized to a level at which a pronounced amplification of the EPSP occurred which was mostly due to the activation of a large variable component. As can be seen, the activation and size of this component were independent of the latency of the afferent stimulation. At more hyperpolarized levels of Vm where the uniform component was evoked in isolation, it too showed no time dependency. In fact, even when the membrane was depolarized by constant current injection, a pronounced amplification of the EPSPs was still observed (Fig. 3C). These experiments therefore strongly suggest that the mechanism underlying the amplification is voltage dependent, but time independent.
The voltage-dependent amplification of EPSPs in neocortical and hippocampal CA3 pyramidal neurones has previously been attributed to the activation of intrinsic membrane currents (Stafstrom et al. 1985; Miles & Wong, 1986; Thomson et al. 1988; Deisz et al. 1991). In order to investigate this possibility, we injected small depolarizing current pulses tailored to give a depolarization of similar amplitude and duration to the distally generated EPSPs recorded at the somatic level (cf. Miles & Wong, 1986; Deisz et al. 1991) (Fig. 4A). The voltage response to these current pulses was amplified by somatic depolarization in a similar manner to that of the synaptic potentials (compare Figs 3A and 4A). The amplification contained both a uniform component, which activated around RMP, and a variable component, which appeared when the responses were close to threshold. The variable component accompanying current injection was just as labile as with EPSPs, and also attained similar magnitudes. Furthermore, the amplification of the current-induced response and the EPSPs had the same threshold and voltage dependency (Fig. 4B). Figure 4B also shows that the uniform component was of similar size for the EPSP and current-evoked response. These results strongly suggest that the amplification of the distally evoked EPSPs is indeed caused by the activation of one or more intrinsic membrane currents and has nothing to do with the EPSP generating mechanism per se.
Figure 4. The amplification of the EPSPs is due to intrinsic membrane properties.
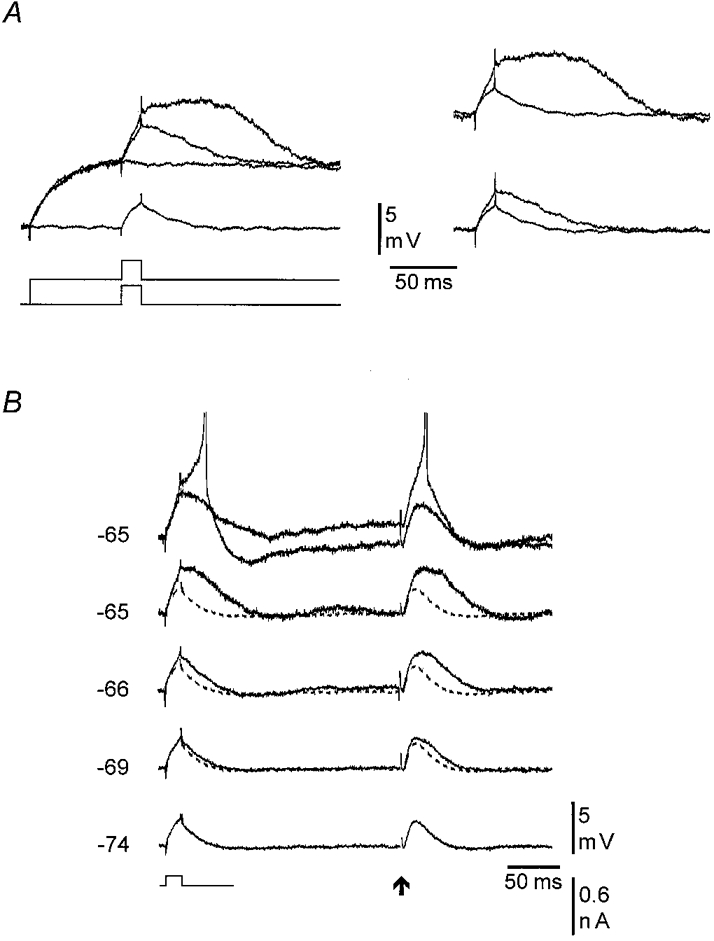
All records are from the same neurone as in Fig. 3. A, left, similar to Fig. 3A, except that here a small depolarizing current pulse (+0.2 nA, 15 ms) was used instead of distal afferent stimulation. B, the response to a small depolarizing current (+0.1 nA, 15 ms) followed after 225 ms by a single distal afferent stimulation (200 μA). The current and stimulating intensities were adjusted to give equally sized responses at −74 mV. Holding Vm at potentials between −74 and −65 mV indicated that the amplification of the current-induced response and the EPSP had similar voltage dependency and magnitude. Responses at −65 mV are single traces to show that both responses were threshold straddling at the same Vm. For comparison, the responses evoked at −74 mV (dashed line) have been superimposed on the responses evoked at more depolarized potentials.
The voltage-dependent enhancement of the EPSPs is caused by a TTX-sensitive current
There are several types of membrane currents which could give rise to a voltage-dependent enhancement of EPSPs at the subthreshold level. However, we will focus on the involvement of the non-inactivating Na+ current (INaP) and the low threshold T-type Ca2+ current (IT(Ca)) because both of these have been shown to be involved in voltage-dependent amplification of subthreshold EPSPs (Deisz et al. 1991; Stuart & Sakmann, 1995; Lipowsky et al. 1996; Gillessen & Alzheimer, 1997). We initially tested whether the enhancement was sensitive to TTX, which blocks INaP. TTX (10 μM) was applied locally to the somatic region close to and upstream from the recording electrode, so as to limit the spread of TTX into SR (Fig. 5A). The voltage dependency of the EPSP was recorded before TTX was applied. A depolarization was then chosen which gave the largest enhancement of the EPSPs, and six to eight control responses were collected. A short application of TTX was then made and the TTX pipette was immediately retracted from the surface of the slice. Stimulation was continued unchanged until the effect of TTX had reach a stable level, at which point examination of the voltage dependency was repeated. As seen in Fig. 5B, TTX completely blocked the voltage-dependent amplification of the distally evoked EPSPs without affecting EPSPs evoked at hyperpolarized potentials. The voltage dependency of the blocking action of TTX is clearly evident in Fig. 5C, which shows a plot of the area of the tEPSPs measured before and in the presence of TTX in relation to prestimulus Vm. In the control situation, there was an abrupt increase in the area of the tEPSPs at potentials more depolarized than −75 mV. This increase was completely blocked by TTX and the area now decreased slightly with depolarization. That TTX had no effect on the EPSPs at hyperpolarized potentials indicates that it had not diffused into the region of active synapses and blocked the terminals of the distal afferent fibres. The relative effect of TTX on the amplitude and area of the c- and tEPSPs from a total of five experiments is shown in Fig. 5D. The effect on the tEPSPs was measured on averaged responses recorded at −64.1 ± 1.6 mV and −84.8 ± 1.7 mV. TTX reduced the tEPSP amplitude by 38 ± 7 % and the area by 55 ± 8 % at depolarized Vm, whereas there was no effect on the amplitude (3 ± 8 %) and only a slight effect on the area (13 ± 6 %) at hyperpolarized Vm. To control for possible direct effects of TTX on the distal synaptic transmission, we also measured its effect on cEPSPs recorded at −84.1 ± 2.3 mV. As seen in Fig. 5D, the amplitude of the cEPSP was unchanged (4 ± 8 %), whereas there was a small increase (13 ± 11 %) in area, indicating that the effect of TTX on the tEPSP cannot be explained by a direct action on the distal afferent fibres. These results therefore provide strong evidence that the amplification of the EPSPs is due to the activation of INaP.
Figure 5. The amplification of the distal EPSP is sensitive to somatic application of TTX.

A, the experimental set-up for local application of TTX (10 μM) to the somatic region. TTX was applied upstream from the recorded pyramidal neurone, and was washed away by the flow of standard Ringer solution (shaded area). B, the somatic responses to paired distal stimulation evoked at different levels of Vm before (left) and after TTX application (right). Note that the marked enhancement of the EPSPs at depolarized levels of Vm is blocked by TTX, whereas the EPSPs at hyperpolarized levels of Vm are unaffected. C, a plot of the effect of TTX on the tEPSP area as a function of prestimulus Vm. The data are from the experiment shown in B. D, histogram showing the relative effect of TTX on the amplitude and area of the c- and tEPSPs (mean ± s.e.m., n = 5). The cEPSP was measured at −84.1 ± 2.3 mV and the tEPSP at −64.1 ± 1.6 mV (tEPSP (Dep.)) and −84.8 ± 1.7 mV (tEPSP (Hyp.)). RMP in B, −72 mV.
Because of its voltage dependency, the activation of IT(Ca) could be partly dependent on the activation of INaP. Therefore, the complete block of the amplification by TTX does not necessarily exclude a contribution by IT(Ca). We therefore examined the effect of Ni2+, which in low concentrations is a relatively selective blocker of IT(Ca) (Magee & Johnston, 1995a). In the presence of NiCl2 (100–200 μM), the distally evoked EPSPs were amplified to a similar extent by somatic depolarization as under control conditions (Fig. 6, n = 7). In two experiments, we were able to compare responses before and during perfusion of NiCl2. In both cases, the uniform component was not reduced by NiCl2. In the example shown in Fig. 6, the EPSP in fact had a larger peak amplitude and a faster rate of rise in the presence of NiCl2 (Fig. 6B). In both cases, the variable component was also unchanged by NiCl2 with respect to variability (Fig. 6A) and size (Fig. 6C). However, the Vm level at which the variable component appeared was moved about 2 mV in the depolarizing direction, as was the action potential threshold. Together, these results indicate that IT(Ca) is not involved in the voltage-dependent amplification of the distally generated EPSP to any great extent. Interestingly, the undershoot which follows particularly large variable components was still seen in the presence of NiCl2 (Fig. 6C).
The voltage-dependent amplification of the distally evoked EPSP is a somatic phenomenon
There are two indications that the amplification described here is a somatic phenomenon: the amplification is blocked by local application of TTX to the somatic region and dendritic depolarization had virtually no effect on the distally evoked EPSP. Even though precautions were taken to limit the lateral spread of TTX, we cannot exclude the possibility that TTX had not diffused into the deep parts of SR. It is therefore possible that at least part of the amplification is a result of activation of INaP residing in the proximal apical dendrites. Recently, Na+ channels in the proximal apical dendrites of CA1 pyramidal neurones (Lipowsky et al. 1996) and neocortical pyramidal neurones (Schwindt & Crill, 1995) have been suggested to be involved in the amplification of distally evoked EPSPs. To gain more information about the location of the current involved, we made intradendritic recordings (n = 7) from the proximal apical dendrites at a distance of 100–125 μm from the superficial border of SP (Fig. 7A). One dendrite exhibited compound spiking (Andreasen & Lambert, 1995), which made it impossible to investigate the voltage dependency of the EPSP and was therefore rejected. In five dendrites, the EPSP amplitude and duration decreased with depolarization (Fig. 7B) or was insensitive to changes in Vm. In the remaining dendrite, the EPSP was amplified by dendritic depolarization. These experiments therefore support the notion that the voltage-dependent amplification of the distally generated EPSP is primarily due to a somatically located INaP.
Figure 7. Distally evoked EPSPs are insensitive to depolarization of the proximal apical dendrites.
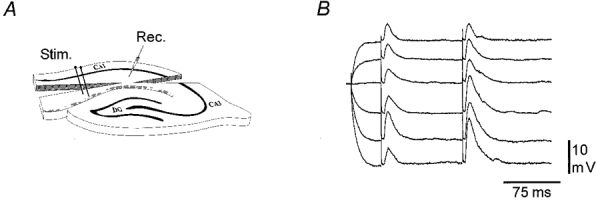
A, experimental set-up for intracellular recordings from proximal apical dendrites located about 100–125 μm from the superficial border of SP. B, the voltage dependency of dendritic responses to paired stimulation of the distal afferent fibres. Vm was changed by intradendritic injection of long depolarizing or hyperpolarizing current pulses. At each membrane potential four responses were collected and averaged. Both the c- and the tEPSP decreased with depolarization without any evidence of a voltage-dependent enhancement of the EPSP peak amplitude and area. Although the voltage-dependent reduction in PPF could suggest the involvement of a postsynaptic component in PPF, this was not a consistent finding. RMP, −66.7 mV.
Recently, Lipowsky et al. (1996) have suggested that the dendritic A-type K+ current, IA, (Andreasen & Lambert, 1995; Hoffman et al. 1997) could suppress the dendritic INaP. We therefore investigated the effect of 4-AP, which blocks IA (Storm, 1990). To avoid changes in presynaptic release of transmitter associated with extracellular application of 4-AP, 4-AP (25–50 mM) was dissolved in the electrode solution and applied intradendritically by way of diffusion. As expected from our earlier work (Andreasen & Lambert, 1995), application of 4-AP had a marked effect on the firing properties of the distal apical dendrites. During the course of the recording, the dendritic excitability increased and both depolarizing current pulses and synaptic stimulation induced compound spiking (see inset in Fig. 9A). However, the effect of blocking IA on the voltage dependency of the subthreshold EPSPs was very inconsistent. In 16 % (3/19) of dendrites, the EPSPs were still unaffected by dendritic depolarization. In 47 % (9/19) of dendrites, depolarization induced a slight increase in the duration of the EPSPs but at the same time the amplitude was reduced. In the remaining 37 % (7/19), there was a voltage-dependent amplification of the EPSP, primarily evident as a prolongation of the decaying phase, in some cases to the point where the EPSPs had a plateau-like appearance (Figs 8 and 9A). A similar amplification was seen when small depolarizing current pulses were injected to simulate EPSPs (Fig. 8), verifying that the amplification of the EPSPs was not due to a change in presynaptic glutamate release. When the EPSPs were close to threshold, part of the response had a variable behaviour similar to that observed in somatic recordings. In other respects, however, the amplification was different from that seen in somatic recordings. Firstly, it was not apparent until the EPSPs were threshold straddling, and then it mostly resembled the variable component of the somatic amplification. Secondly, only small (< 5 mV) EPSPs were amplified. These observations, together with the fact that threshold spikes evoked by distally generated EPSPs are primarily triggered at the initial segment (Andreasen & Lambert, 1998), could indicate that the dendritic amplification is, in fact, a reflection of what is occurring in the somatic region (Stuart & Sakmann, 1995). Because a larger depolarization of the dendritic membrane is needed to depolarize the somatic membrane sufficiently, this would also explain why only small EPSPs are amplified. In one experiment we were able to test this hypothesis by local application of TTX (10 μM) to the somatic region while recording from a distal apical dendrite. Antidromic stimulation of A/O was used as a control for the effect of TTX. As shown in Fig. 9B, somatic application of TTX rapidly blocked the A/O evoked response. This was followed by a progressive reduction in the distally evoked EPSP, until it reached a stable value after 180 s. At this time the firing induced by distal afferent stimulation and the amplification of the EPSP were blocked and the EPSP was now reduced by dendritic depolarization. At the same time, the response recorded at RMP was unaffected by TTX, indicating that the effects at depolarized potentials were not due to TTX having diffused into the superficial part of SR and L-M and reduced synaptic transmission directly. These effects were partly reversed 15 min after the TTX application (Fig. 9C). This experiment indicates that the dendritic amplification following intracellular application of 4-AP is of somatic and not dendritic origin.
Figure 9. The dendritic amplification is blocked by somatic application of TTX.
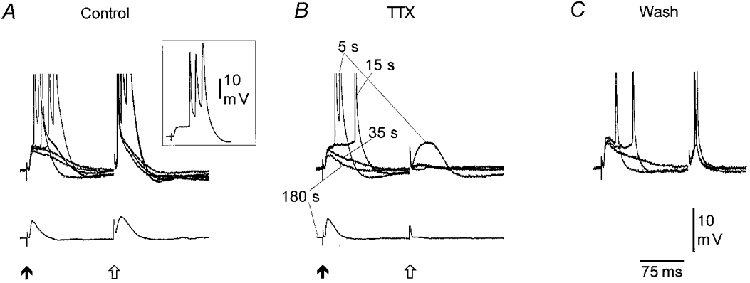
A, recordings from a distal apical dendrite with an electrode containing 4 M potassium acetate and 25 mM 4-AP. Responses were evoked by distal afferent stimulation (filled arrow) followed after 150 ms by A/O stimulation (open arrow). The stimulating intensity was adjusted to evoke EPSPs of similar size at RMP (−65 mV, lower records). On depolarization to −52 mV (upper records, five superimposed traces), A/O stimulation evoked single and compound spiking, whereas distal stimulation evoked compound spiking (see single response in inset) or EPSPs with a prolonged decaying phase. Note that firing was often triggered on a plateau-like potential (see inset). B, local application of TTX (10 μM) to the somatic region blocked the A/O evoked response after 15 s whereas there was a progressive reduction in the distally evoked response which attained a stable level after 180 s. The numbers indicate the time in seconds following the TTX application. Note that, whereas the A/O response at RMP is completely blocked by TTX the distally evoked EPSP is unchanged. C, 15 min after the TTX application both the distally and A/O evoked responses have partly recovered.
Figure 8. Intracellular injection of 4-AP causes dendritic amplification of the distally generated EPSP.
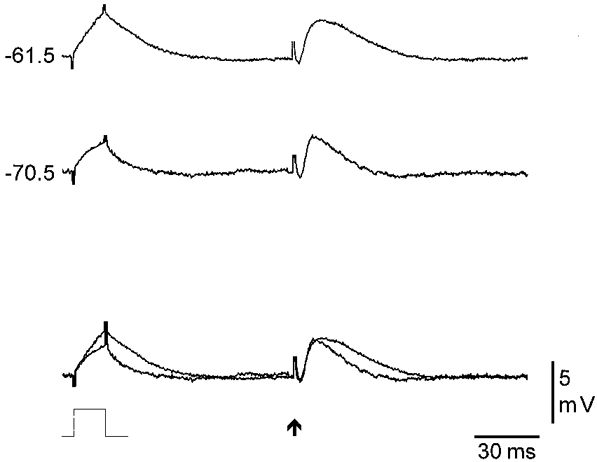
The response to a small depolarizing current pulse (+0.2 nA, 15 ms) and a single distal afferent stimulation (38 μA) recorded from a distal apical dendrite with a 4-AP (50 mM)-containing electrode. The current and stimulating intensities were adjusted to give similar sized responses at −70.5 mV. Depolarization of the dendritic membrane to −61.5 mV resulted in an amplification of both responses. For comparison, the responses evoked at both potentials have been superimposed below.
The voltage dependency of the individual components of the somatic amplification
To investigate the voltage dependence of the uniform and variable amplification in greater detail, we used constant current injection to change the somatic Vm to various potentials during which the distal afferent fibres were stimulated. One of these experiments is shown in Fig. 10A, where the change in the amplification is clearly evident. From −70.5 to −60 mV, there was a progressive increase in EPSP amplitude and duration which, at a given Vm, changed very little. However, when the somatic membrane was further depolarized to −56 mV, the variability in EPSP amplitude and duration increased greatly. Furthermore, action potentials were occasionally evoked at variable latencies and usually rode on prepotentials (Hu et al. 1992) of variable duration (Fig. 10A). With further depolarization to −54.5 mV, action potentials were consistently generated at a relatively short but constant latency. When plotting the area of each individual subthreshold EPSP in relation to the prestimulus Vm, two interesting properties emerged (Fig. 10B). Firstly, the shift from a relatively constant to a highly variable amplification was very abrupt and occurred around −59 mV. Secondly, the voltage range in which variable amplification occurred was very narrow. In the neurone shown, the range was between −59 mV (determined by the threshold for activation of the variable component) and −56 mV (determined by the threshold for action potential generation) as the post-spike after-hyperpolarization curtails the expression of the variable amplification (Fig. 10A). In Fig. 10B, the filled symbols represent EPSPs in which only the uniform component of the amplification was present, whereas the open symbols represent EPSPs in which the variable component was also present. An identical picture was found in all 16 pyramidal neurones in which a total of 28 EPSPs were examined. The main difference between the individual neurones was the level of membrane potential at which the shift to the variable component occurred. For EPSPs with amplitudes < 5 mV (range: 2 to 4.5 mV), the shift occurred in 25 % (5/20) of cases at a Vm below −65 mV, in 65 % (13/20) of cases between −65 and −60 mV (as in Fig. 10B) and in the remaining 10 % (2/20) above −60 mV. No correlation was found between the peak amplitude of the EPSP and the level at which the abrupt shift occurred. For EPSPs > 5 mV, the shift usually occurred at potentials between −70 and −65 mV.
DISCUSSION
A somatic non-inactivating Na+ current functions as a voltage-dependent amplifier for distally generated EPSPs
The present study shows that distally generated non-NMDA receptor-mediated EPSPs are amplified in a voltage-dependent manner by somatic membrane depolarization resulting in an enhancement of both the peak amplitude and the duration. The amplification was most marked at membrane potentials at which the EPSP was close to threshold for action potential initiation, but was already evident at membrane potentials around −70 mV, which is close to the averaged RMP of the CA1 pyramidal neurones. At more hyperpolarized potentials, the distal EPSPs were little affected by changes in somatic Vm, which is to be expected considering that the active synapses are located more than 250 μm from the soma in a part of the apical dendrite that has a small electrotonic length constant (Andreasen & Nedergaard, 1996). A similar type of voltage dependency of EPSPs has been described in neocortical pyramidal neurones (Stafstrom et al. 1985; Thomson et al. 1988; Deisz et al. 1991; Stuart & Sakmann, 1995) and in CA3 pyramidal neurones (Miles & Wong, 1986).
The presence of AP5 in the perfusion medium excludes the possibility that the amplification is caused by a voltage-dependent unblocking of the NMDA receptor-ionophore complex (Nowak et al. 1984). Furthermore, a similar type of voltage-dependent amplification could be obtained by using small depolarizing current pulses to simulate EPSPs, indicating that the amplification is caused by the activation of an intrinsic membrane current.
There are several intrinsic membrane currents which could be involved in the observed amplification of subthreshold EPSPs. One is INaP, which has been described in cat neocortical neurones (Stafstrom et al. 1985), cerebellar Purkinje cells (Llinás & Sugimori, 1980) and hippocampal CA1 pyramidal neurones (French et al. 1990). Another is IT(Ca), which also has been described in a variety of central neurones (for review see Huguenard, 1996), including hippocampal CA1 pyramidal neurones (Takahashi et al. 1991). Amplification of the distal EPSPs at around −70 mV coincides well with the activation threshold for both INaP (French et al. 1990) and IT(Ca) (Takahashi et al. 1991). Indeed, there is already evidence that EPSPs activate dendritic INaP and T-type Ca2+ channels in CA1 pyramidal neurones (Magee & Johnston, 1995b; Lipowsky et al. 1996; Gillessen & Alzheimer, 1997). Furthermore, the area of the EPSP usually increased to a greater extent than the peak amplitude (Fig. 2B), indicating that the modulation primarily affects the duration of the distal EPSPs. This is what would be expected considering the kinetic properties of these two currents. INaP has a fast activation and reaches its non-inactivating steady-state level 2–4 ms after a step change in Vm (Stafstrom et al. 1985). The T-type Ca2+ current is generally referred to as a transient current by virtue of its fast inactivation. However, the time courses of both activation and inactivation are voltage dependent and considerably slower at subthreshold Vm than at more depolarized levels (Takahashi et al. 1991). Both INaP and IT(Ca) have sigmoidal activation curves, which would explain the progressive increase in amplification with somatic depolarization. The fact that the amplification also included an increase in peak amplitude (Fig. 3A) means that the current must have been activated during the rising phase of the EPSPs. Even though the distally evoked EPSPs recorded at the somatic level have a relatively slow rise time of about 7 ms (Andreasen & Lambert, 1998), the increase in peak amplitude seems to be more in accordance with the fast activation kinetics of INaP (Stafstrom et al. 1985) than the slow activation kinetics of IT(Ca) at subthreshold potentials (Takahashi et al. 1991; Huguenard, 1996). The enhancement of the distal EPSP was also blocked by TTX (Fig. 5), whereas it was unaffected by NiCl2 (Fig. 6) at concentrations which are known to block or substantially reduce IT(Ca) (Magee & Johnston, 1995a). This indicates that the amplification is primarily due to the activation of INaP and that there is no significant interaction between INaP and IT(Ca). This is similar to what has been reported for neocortical layer V neurones (Stuart & Sakmann, 1995), but in contrast to neocortical layer II/III neurones, in which both currents where found to be involved (Deisz et al. 1991). The time-dependent component of the depolarization-induced amplification in neocortical layer II/III neurones was attributed to the activation of IT(Ca) (Deisz et al. 1991) and results in the amplification being larger within the initial 80–100 ms of a depolarizing step. This is in good agreement with the activation of T-type Ca2+ channels in that these will enter an inactivated state within 100 ms after a step depolarization (Takahashi et al. 1991). The lack of any time dependency in the present study therefore further strengthens the case for the involvement of INaP, which is very resistant to steady-state inactivation (French et al. 1990). Dihydropyridine-sensitive L-type Ca2+ channels have been shown to open at subthreshold potentials in both CA3 (Avery & Johnston, 1996) and CA1 (Fisher et al. 1990) pyramidal neurones and could therefore be involved in the amplification of subthreshold EPSPs. However, there are indications that subthreshold openings of L-type channels only occur if preceded by a long depolarization, such as a burst of action potentials (Fisher et al. 1990; Kavalali & Plummer, 1994). Furthermore, the concentrations of Ni2+ used here have been shown to reduce subthreshold L-type Ca2+ currents by 30–40 % (Avery & Johnston, 1996). The lack of change in amplification in the presence of Ni2+ would argue against any major involvement of L-type Ca2+ channels in the voltage-dependent amplification of subthreshold EPSPs. Another possibility is that a depolarization-induced deactivation of Ih is involved in the uniform component of the amplification. It has recently been shown that this current contributes to the resting membrane conductance of CA1 pyramidal neurones and that Cs+ leads to an increase in both amplitude and duration of subthreshold EPSPs (Magee, 1998). Depolarization of Vm would result in a progressive deactivation of Ih until this was complete at about −50 mV. However, the complete block of the amplification by TTX argues against a contribution by deactivation of Ih, since this would not be expected to be affected by TTX.
When the amplification was pronounced, the EPSPs were often followed by an undershoot (Figs 3 and 6). In neocortical layer II/III pyramidal neurones, there was a close correlation between the time-dependent part of the amplification and the appearance of a similar undershoot, which is therefore likely to be a Ca2+-dependent K+ current. However, no such time dependency was found in the present study (Fig. 3) and the undershoot observed here was still present following the application of NiCl2 (Fig. 6C). In pyramidal neurones from the sensorimotor cortex (Thomson et al. 1988; Stuart & Sakmann, 1995) and in CA3 pyramidal neurones (Miles & Wong, 1986), an undershoot similar to that described here has been reported and was found to be blocked by intracellular application of tetraethylammonium and Cs+, indicating that it is due to the activation of one or more voltage-dependent K+ currents.
The voltage-dependent amplification of the distally generated EPSPs was seen in somatic recordings but not in recordings from the apical dendrites 100–150 μm or 250–300 μm from the superficial border of SP. This indicates that the amplification is due to the activation of a somatic INaP. Furthermore, local application of TTX to the somatic region completely blocked the amplification without affecting the EPSPs evoked at potentials negative to RMP, indicating that the effect of TTX was not due to a reduction in the number of activated synapses. These findings are somewhat in contrast to those recently reported by Lipowsky et al. (1996), who found that the amplification of distal EPSPs in CA1 pyramidal neurones was mainly due to the activation of dendritic Na+ currents with only a small contribution from axosomatic Na+ currents. One explanation for this apparent discrepancy could be the presence of different dendritic K+ currents and in particular the A-type K+ current, IA, which exerts a significant influence on dendritic excitability (Andreasen & Lambert, 1995; Hoffman et al. 1997). On the basis of computer simulations, Lipowsky et al. (1996) predicted that the presence of a dendritic IA could partly counteract the activation of a dendritic Na+ current and later Hoffman et al. (1997) reported that TTX blocks the 4-AP-induced enhancement of the response to small dendritic current injections. Even though the blockade of the dendritic IA yielded very inconsistent results in the present study, in about 37 % of the dendritic recordings the distally evoked EPSP was amplified by depolarization somewhat similar to that seen in somatic recordings. The characteristics of this dendritic amplification seem to indicate that it was not generated locally, but was a reflection of the somatic amplification (Stuart & Sakmann, 1995). This was supported by the one case in which we were able to show that dendritic amplification was blocked by local application of TTX to the somatic region (Fig. 9).
Although the A-type K+ current is also found in the somatic membrane, its density is substantially lower than in the distal apical dendrites (Hoffman et al. 1997) and the damping effect on the activation of the somatic INaP would therefore be less. The resolution of the present experiments is, however, not high enough to exclude the possibility that part of the amplification induced by somatic depolarization is caused by activation of INaP in the proximal dendrites. Because the recordings from the mid-dendritic region are likely to be from large diameter dendrites (Bannister & Larkman, 1995), current injected here will spread in both directions and thereby change the dendritic Vm for some distance from the recording site. This means that if INaP is activated in the proximal dendrites, this should be reflected as a voltage-dependent amplification of the distally evoked EPSP. In most (5/6) experiments, no voltage-dependent amplification was observed, suggesting that the proximal apical dendrites are not involved to any significant extent. The present results are therefore in agreement with a recent study by Stuart & Sakmann (1995), and strongly suggest that the amplification of the distally evoked EPSPs is primarily due to the activation of a somatic INaP, with a negligible contribution from dendritic Na+ currents.
In addition to the voltage dependency of the somatic amplification, there was a shift in its character from being very consistent to highly variable. The shift occurred very abruptly around the point at which the EPSP was close to threshold for firing and was therefore less negative for small EPSPs (< 5 mV) than for larger EPSPs. To the best of our knowledge, this change in the voltage-dependent amplification has not been reported before in detail. However, Miles & Wong (1986) did observe that subthreshold EPSPs could be dramatically prolonged in an all-or-none fashion when Vm was 10–15 mV below threshold. The abrupt shift in the type of amplification can be explained from the sigmoidal activation curve of the hippocampal INaP (French et al. 1990). At Vm levels around RMP, the activation curve is very shallow, which explains the near linear increase in amplification with small depolarization. However, as Vm is depolarized further, the peak of the EPSPs brings the membrane into a region in which the curve is very steep and the activation of INaP becomes regenerative, giving rise to the highly variable type of amplification seen at potentials close to action potential threshold. On the other hand, regenerative activation of INaP seems to be inconsistent with the high degree of variability since a near maximal activation of available channels, and therefore a more consistent response, would be expected. One explanation for the observed variability could be that simultaneous activation of voltage-dependent outward currents dampens the expression of INaP. There is abundant evidence that simultaneous activation of a variety of outward K+ currents curtails the full expression of INaP (Stafstrom et al. 1985; Miles & Wong, 1986; French et al. 1990; Lipowsky et al. 1996; Hoffman et al. 1997). The undershoot observed following the activation of a large variable component is, as stated above, likely to be caused by the simultaneous activation of outward K+ currents. At least part of the variability could therefore be due to alternating activation of these voltage-dependent K+ currents.
The functional implications of a voltage-dependent somatic amplification
In CA1 pyramidal neurones, INaP has been suggested to mediate prepotentials that precede action potentials (MacVicar, 1985; Hu & Hvalby, 1992; Hu et al. 1992) or to be involved in burst generation (Azouz et al. 1996). To the best of our knowledge, the present work is the first account of a somatic INaP that functions as a voltage-dependent amplifier of subthreshold EPSPs in CA1 pyramidal neurones. The presence of a voltage-dependent somatic amplifier can have several important implication for the integration of distally evoked EPSPs. (1) Because amplification is evident already at RMP, the somatic INaP will act in concert with dendritic Na+ and Ca2+ currents to compensate for the electrotonic attenuation of distally generated EPSPs (Andreasen & Lambert, 1998). (2) Because of the voltage dependency of INaP, large EPSPs will be amplified to a greater extent than small EPSPs, at a given Vm as is evident here by the difference in amplification of the cEPSP and tEPSP. The presence of INaP will therefore provide the pyramidal neurones with a mechanism for increasing the signal-to-noise ratio and a postsynaptic mechanism for enhancing PPF. Furthermore, the somatic INaP will also enhance the pyramidal neurone's ability to differentiate between low- and high-frequency synaptic inputs. This is because high-frequency synaptic inputs are more likely to summate, resulting in an increase in amplitude and thereby increased activation of INaP at the somatic level. This is in contrast to low-frequency synaptic inputs, where summation is less likely to occur. (3) Because the amplification was primarily associated with an increase in the duration of the EPSPs, it increased the frequency range at which temporal summation can occur. (4) Because the activation kinetics of INaP are relatively fast and it is very resistant to inactivation (Stafstrom et al. 1985; French et al. 1990), it will be activated not only by a short depolarization, as occurs during an EPSP or a short train of EPSPs, but also when the somatic membrane is tonically depolarized. This means that neurotransmitters such as acetylcholine and noradrenaline, which cause prolonged depolarizations of the CA1 pyramidal neurones (Benardo & Prince, 1982; Madison & Nicoll, 1986) can upregulate the amplification of incoming EPSPs. This could be of significant importance for distally evoked EPSPs, which are electrotonically attenuated and therefore of small amplitude when arriving at the soma (Stuart & Sakmann, 1995; Andreasen & Lambert, 1998). A depolarization of the somatic Vm by acetylcholine or noradrenaline would increase the amplification of these EPSPs and thereby increase the probability that the distal inputs induce action potential firing. On the other hand, transmitters like serotonin, acting on 5-HT1 receptors, and GABA, acting on GABAB receptors (Andrade et al. 1986), give rise to prolonged hyperpolarizations, which would downregulate the amplifying current.
The functional significance of the abrupt change in amplification is not immediately apparent. Even though the variable component often provides a marked amplification of both the EPSP amplitude and duration (Fig. 3), its variable nature make it very unreliable. Furthermore, when the EPSPs were threshold straddling as a result of the variable amplification, the latency from the arrival of the EPSP to initiation of an action potential was very inconsistent, and on occasions extremely long (Fig. 10). This will not only disrupt the time lock between the peak of the EPSP and the action potential, but also means that the efficacy of the EPSPs will be very variable. This point has already been discussed by Stuart & Sakman (1995), who also observed a great variability in action potential latency in neocortical layer V neurones. However, once the EPSP has passed this level of unstable amplification, action potentials are reliably triggered and time locked to the peak of the EPSPs. Another aspect of this region of highly unpredictable amplification is that very small changes in Vm can drastically change the functional importance of a distally evoked EPSP, which makes EPSPs evoked close to this voltage range extremely sensitive to preceding activity in the soma such as action potential firing.
Acknowledgments
We would like to thank Dr Steen Nedergaard (University of Aarhus) for helpful discussions. This work was supported by the Danish Medical Research Council and Aarhus Universitets Forskningsfond. Novartis is thanked for providing CGP 55845A.
References
- Andrade R, Malenka RC, Nicoll RA. A G protein couples serotonin and GABAB receptors to the same channels in hippocampus. Science. 1986;234:1261–1265. doi: 10.1126/science.2430334. [DOI] [PubMed] [Google Scholar]
- Andreasen M, Lambert JDC. Regenerative properties of pyramidal cell dendrites in area CA1 of the rat hippocampus. The Journal of Physiology. 1995;483:421–441. doi: 10.1113/jphysiol.1995.sp020595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Lambert JDC. Factors determining the efficacy of distal excitatory synapses in rat hippocampal CA1 pyramidal neurones. The Journal of Physiology. 1998;507:441–462. doi: 10.1111/j.1469-7793.1998.441bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreasen M, Nedergaard S. Dendritic electrogenesis in rat hippocampal CA1 pyramidal neurons: functional aspects of Na+ and Ca2+ currents in apical dendrites. Hippocampus. 1996;6:79–95. doi: 10.1002/(SICI)1098-1063(1996)6:1<79::AID-HIPO13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Avery RB, Johnston D. Multiple channel types contribute to the low-voltage-activated calcium current in hippocampal CA3 pyramidal neurons. Journal of Neuroscience. 1996;16:5567–5582. doi: 10.1523/JNEUROSCI.16-18-05567.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. The Journal of Physiology. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bannister NJ, Larkman AU. Dendritic morphology of CA1 pyramidal neurones from the rat hippocampus. I. Branching patterns. Journal of Comparative Neurology. 1995;360:150–160. doi: 10.1002/cne.903600111. [DOI] [PubMed] [Google Scholar]
- Benardo LS, Prince DA. Cholinergic excitation of mammalian hippocampal pyramidal cells. Brain Research. 1982;249:315–331. doi: 10.1016/0006-8993(82)90066-x. [DOI] [PubMed] [Google Scholar]
- Brown DA, Gähwiler BH, Griffith WH, Halliwell JV. Membrane currents in hippocampal neurons. In: Storm-Mathisen J, Zimmer J, Ottersen OP, editors. Progress in Brain Research. Vol. 83. Amsterdam: Elsevier Science Publishers B.V.; 1990. pp. 141–160. [DOI] [PubMed] [Google Scholar]
- Connors BW, Prince DA. Effects of local anesthetic QX-314 on the membrane properties of hippocampal pyramidal neurons. Journal of Pharmacology and Experimental Therapeutics. 1982;220:476–481. [PubMed] [Google Scholar]
- Deisz RA, Fortin G, Zieglgänsberger W. Voltage dependence of excitatory postsynaptic potentials of rat neocortical neurons. Journal of Neurophysiology. 1991;65:371–382. doi: 10.1152/jn.1991.65.2.371. [DOI] [PubMed] [Google Scholar]
- Fisher RE, Gray R, Johnston D. Properties and distribution of single voltage-gated calcium channels in adult hippocampal neurons. Journal of Neurophysiology. 1990;64:91–104. doi: 10.1152/jn.1990.64.1.91. [DOI] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. Journal of General Physiology. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillessen T, Alzheimer C. Amplification of EPSPs by low Ni2+- and amiloride-sensitive Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. Journal of Neurophysiology. 1997;77:1639–1643. doi: 10.1152/jn.1997.77.3.1639. [DOI] [PubMed] [Google Scholar]
- Hestrin S, Nicoll RA, Perkel DJ, Sah P. Analysis of excitatory synaptic action in pyramidal cells using whole-cell recording from rat hippocampal slices. The Journal of Physiology. 1990;422:203–225. doi: 10.1113/jphysiol.1990.sp017980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Hotson JR, Prince DA, Schwartzkroin PA. Anomalous inward rectification in hippocampal neurons. Journal of Neurophysiology. 1979;42:889–895. doi: 10.1152/jn.1979.42.3.889. [DOI] [PubMed] [Google Scholar]
- Hu G-Y, Hvalby O. Glutamate-induced action potentials are preceded by regenerative prepotentials in rat hippocampal pyramidal cells in vitro. Experimental Brain Research. 1992;88:485–494. doi: 10.1007/BF00228178. [DOI] [PubMed] [Google Scholar]
- Hu G-Y, Hvalby O, Lacaille J-C, Piercey B, Ostberg T, Andersen P. Synaptically triggered action potentials begin as a depolarizing ramp in rat hippocampal neurones in vitro. The Journal of Physiology. 1992;453:663–687. doi: 10.1113/jphysiol.1992.sp019250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annual Review of Physiology. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Johnston D, Magee JC, Colbert CM, Christie BR. Active properties of neuronal dendrites. Annual Review of Neuroscience. 1996;19:165–186. doi: 10.1146/annurev.ne.19.030196.001121. [DOI] [PubMed] [Google Scholar]
- Kavalali ET, Plummer MR. Selective potentiation of a novel calcium channel in rat hippocampal neurones. The Journal of Physiology. 1994;480:475–484. doi: 10.1113/jphysiol.1994.sp020376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipowsky R, Gillessen T, Alzheimer C. Dendritic Na+ channels amplify EPSPs in hippocampal CA1 pyramidal cells. Journal of Neurophysiology. 1996;76:2181–2191. doi: 10.1152/jn.1996.76.4.2181. [DOI] [PubMed] [Google Scholar]
- Llinás R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell somata in mammalian cerebellar slices. The Journal of Physiology. 1980;305:171–195. doi: 10.1113/jphysiol.1980.sp013357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacVicar BA. Depolarizing prepotentials are Na+ dependent in CA1 pyramidal neurons. Brain Research. 1985;333:378–381. doi: 10.1016/0006-8993(85)91597-5. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 pyramidal neurones, in vitro. The Journal of Physiology. 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC. Dendritic hyperpolarization-activated currents modify the integrative properties of hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1998;18:7613–7624. doi: 10.1523/JNEUROSCI.18-19-07613.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Characterization of single voltage-gated Na+ and Ca2+ channels in apical dendrites of rat CA1 pyramidal neurons. The Journal of Physiology. 1995a;487:67–90. doi: 10.1113/jphysiol.1995.sp020862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995b;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- Miles R, Wong RKS. Excitatory synaptic interactions between CA3 neurones in the guinea-pig hippocampus. The Journal of Physiology. 1986;373:397–418. doi: 10.1113/jphysiol.1986.sp016055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurones. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Puil E, Carlen PL. Attenuation of glutamate-action, excitatory postsynaptic potentials, and spikes by intracellular QX 222 in hippocampal neurons. Neuroscience. 1984;11:389–398. doi: 10.1016/0306-4522(84)90031-9. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. Journal of Neurophysiology. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Schwindt PC, Chubb MC, Crill WE. Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. Journal of Neurophysiology. 1985;53:153–170. doi: 10.1152/jn.1985.53.1.153. [DOI] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. In: Storm-Mathisen J, Zimmer J, Ottersen OP, editors. Progress in Brain Research. Vol. 83. Amsterdam: Elsevier Science Publishers; 1990. pp. 161–187. [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Häusser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends in Neurosciences. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- Sutor B, Zieglgänsberger W. A low-voltage activated, transient calcium current is responsible for the time-dependent depolarizing inward rectification of rat neocortical neurons in vitro. Pflügers Archiv. 1987;410:102–111. doi: 10.1007/BF00581902. [DOI] [PubMed] [Google Scholar]
- Takahashi K, Ueno S, Akaike N. Kinetic properties of T-type Ca2+ currents in isolated rat hippocampal CA1 pyramidal neurons. Journal of Neurophysiology. 1991;65:148–154. doi: 10.1152/jn.1991.65.1.148. [DOI] [PubMed] [Google Scholar]
- Thomson AM, Girdlestone D, West DC. Voltage-dependent currents prolong single-axon postsynaptic potentials in layer III pyramidal neurons in rat neocortical slices. Journal of Neurophysiology. 1988;60:1896–1907. doi: 10.1152/jn.1988.60.6.1896. [DOI] [PubMed] [Google Scholar]