

Abstract
The activity-dependent regulation of presynaptic K+ currents at the CA3-CA1 synapse in the rat hippocampus was investigated during a train of evoked afferent action potentials. The waveforms of presynaptic compound action potentials (cAPs) and presynaptic Ca2+ transients ([Ca2+]pre,t) were measured with fluorescent voltage-sensitive and Ca2+-sensitive indicators in rat brain slices.
Under control conditions, presynaptic cAPs and the accompanying [Ca2+]pre,t displayed similar amplitudes for each stimulus, suggesting that there was no cumulative change of K+ and Ca2+ currents during the test train. However, when a subgroup of presynaptic K+ channels was blocked by a low concentration of 4-aminopyridine (4-AP, 40 μm), a significant facilitation of the [Ca2+]pre,t was observed.
This phenomenon was not due to a direct action of 4-AP on presynaptic Ca2+ channels, but to cumulative suppression of the K+ conductance as indicated by the corresponding change in waveforms of the cAP and presynaptic fibre volley. The observed facilitation was not an artifact by virtue of increased fibre recruitment, nor was it related to the accumulation of extracellular K+; rather, it was dependent on Ca2+ influx and stimulation frequency. The time course of recovery from facilitation was closely related to the decay of the intracellular Ca2+ concentration.
The facilitation was not blocked by a saturating concentration of 4-AP (8 mM) but was reduced during the application of the K+ channel blocker tetraethylammonium (TEA, 10 mM), implicating the involvement of TEA-sensitive K+ channels. Such activity-dependent suppression of presynaptic K+ conductance could lead to excessive transmitter release and might explain the hippocampal epileptiform activity that can be induced by application of 4-AP.
Presynaptic Ca2+ influx through voltage-gated Ca2+ channels plays an important role in the release of neurotransmitters. In addition to the activity of presynaptic Ca2+ channels, the time course of presynaptic action potential repolarization, which is largely controlled by the activity of presynaptic K+ channels, determines the amount of Ca2+ influx. Modulation of neurotransmitter release by the waveform of action potentials has been shown in various preparations (Augustine, 1990; Wheeler et al. 1996; Sabatini & Regehr, 1997; Qian & Saggau, 1999). Since activation and inactivation of most presynaptic K+ channels is voltage dependent, any change in the membrane potential as the result of invading action potentials could potentially alter the activity of these channels and thereby modulate the presynaptic Ca2+ influx in response to a subsequent action potential. Furthermore, intracellular Ca2+ levels may also regulate K+ channels through Ca2+-dependent protein phosphorylation (Roeper et al. 1997). Thus, activity-dependent modulation of presynaptic K+ channels could provide neurons with a potential means for positive or negative feedback regulation of presynaptic Ca2+ influx.
Activity-dependent modulation of K+ currents has been observed at both presynaptic and postsynaptic sites. At the soma of molluscan neurons, repetitive firing broadens the action potential by cumulatively reducing repolarizing K+ currents (Aldrich et al. 1979). In hippocampal pyramidal neurons, voltage-dependent inactivation of fast inactivating K+ currents was shown to determine the frequency of repetitive somatic action potential firing (Storm, 1988). The same mechanism is believed to modulate signal propagation in dendrites of hippocampal CA1 pyramidal neurons (Hoffman et al. 1997). In presynaptic terminals of rat pituitary, voltage-dependent inactivation of K+ channels has been proposed to explain the frequency-dependent broadening of presynaptic action potentials and facilitation of Ca2+ influx (Jackson et al. 1991). The inactivation of a fast inactivating K+ channel plays a significant role in determining the reliability of action potential propagation along Schaffer collateral axons in the hippocampus (Debanne et al. 1997).
In the central nervous system, it is extremely difficult to directly assay presynaptic K+ channels due to the small structure of most presynaptic terminals and limitations of current electrophysiological techniques. Nevertheless, the duration of the presynaptic action potential, which is mediated to a great extent by voltage-gated K+ channels, determines the presynaptic Ca2+ influx. By measuring Ca2+ entry and the waveform of the presynaptic action potential with fluorescent Ca2+- and voltage-sensitive dyes, we were able to investigate the activity-dependent regulation of presynaptic K+ currents at the rat hippocampal CA3-CA1 synapse.
METHODS
Measurement of presynaptic Ca2+ transients and action potential waveform
Sprague-Dawley rats (4 weeks of age) were anaesthetized (methoxyflurane) and rapidly decapitated in accordance with the guidelines of the Baylor College of Medicine Animal Protocol Review Committee. Transverse hippocampal slices (350 μm) were prepared and incubated at 30°C in artificial cerebrospinal fluid (ACSF) comprising (mM): 124 NaCl, 3 KCl, 2.5 CaCl2, 2 MgCl2, 22 NaHCO3, 1.25 NaH2PO4 and 10 D-glucose, gassed with 95 % O2-5 % CO2 to maintain a constant pH of 7.4. The dentate gyrus and part of CA3 were routinely removed to prevent epileptiform activity during application of K+ channel blockers. A bipolar tungsten electrode was positioned in striatum radiatum (SR) of area CA1 to stimulate afferent inputs to CA1 neurons. A burst of four stimuli (0.1-0.2 ms pulse each, separated by 30 ms) was given at an interval of 30 s. Glass microelectrodes (1-5 MΩ, filled with 2 M NaCl) were used to record field excitatory postsynaptic potentials (fEPSPs) in SR of area CA1.
Presynaptic Ca2+ transients ([Ca2+]pre,t) were measured by the method described by Wu & Saggau (1994). Since we were interested in the investigation of [Ca2+]pre,t during a burst of presynaptic action potentials, the low-affinity calcium-sensitive dye (CaSD) furaptra (AM form, Molecular Probes) was employed to avoid possible saturation of the indicator during application of consecutive stimuli.
The waveform of presynaptic action potentials was measured separately with a voltage-sensitive dye (VSD). Due to the small structure of the axon fibres, it was impossible to monitor the waveform of presynaptic action potentials at the individual terminals with current electrophysiological techniques. Instead, the VSD signals were contributed by a population of activated presynaptic fibres. To measure such a compound presynaptic action potential (cAP), thin brain slices (200 μm) were incubated for 20 min with the VSD RH 414 (100 μm, Molecular Probes). To isolate the presynaptic activity, postsynaptic responses were blocked by application of the glutamate antagonists 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 10 μm) and D-aminophosphonovalerate (D-APV, 25 μm). An area in SR with a diameter of 100 μm was illuminated at a wavelength of 520 ± 15 nm and emitted fluorescence of the VSD at wavelengths longer than 610 nm was converted to electrical signals with a single photodiode. A bipolar tungsten electrode was positioned in SR of area CA1 about 500 μm away from the recording area to stimulate Schaffer collateral axons. The presynaptic fibre volley was simultaneously monitored with extracellular field recording. Short-term bleaching of the VSD was corrected by measuring a bleaching time course without stimulation and subtracting it from traces with stimulation. Long-term bleaching of the VSD was corrected by dividing the fluorescence signal by the resting fluorescence (ΔF/F). The presented VSD traces are the averages of 15 to 20 sweeps.
Simultaneous measurement of extracellular K+ and Ca2+ transients
In some experiments, extracellular K+ concentration ([K+]o) was also measured. These experiments were conducted under similar experimental conditions to those for measuring [Ca2+]pre,t, except for the additional placement of a K+-sensitive electrode in the recording area. Briefly, double-barrelled capillary glass was used to produce ion-sensitive microelectrodes as described by Lothman & Somjen (1975). The tip of one barrel was filled with a potassium exchanger resin for K+-sensitive electrodes. The reference barrel was filled with 2 M NaCl. The electrode was calibrated before each experiment in ACSF solutions with different K+ concentrations.
Data analysis
To quantify the data, amplitudes of [Ca2+]pre,t and cAPs were measured as the difference between the stimulus-evoked peak response and the resting fluorescence prior to stimulation. Data were normalized to control conditions without application of any potassium channel blockers and expressed as the means ±s.d.
Drugs
4-AP and TEA were from Sigma, CNQX and D-APV from Tocris Cookson, ω-CgTX GVIA from Bachem and adenosine from RBI.
RESULTS
Presynaptic action potentials and Ca2+ influx during a burst of stimuli
Presynaptic action potentials and Ca2+ influx in response to a short burst of stimuli (4 pulses, each separated by 30 ms) were first examined under control conditions. The waveform of the presynaptic action potentials was measured with a fluorescent voltage-sensitive dye (VSD). Figure 1A shows a sample trace of VSD signal, which contained two components: a fast change of membrane potential that temporally coincided with the presynaptic fibre volley (Fig. 1B), which represents the presynaptic compound action potential (cAP); and a slow component, lasting for several tens of milliseconds, which was probably due to the depolarization of glial cells as a result of accumulated extracellular K+ (Konnerth & Orkand, 1986). Figure 1C shows a sample trace of the presynaptic Ca2+ transient ([Ca2+]pre,t), which was separately measured with a low-affinity Ca2+-sensitive dye (CaSD). In general, the amplitudes of the cAP, fibre volley and [Ca2+]pre,t remained similar for each stimulus within the test train. On average, there was no significant difference in the amplitudes of the fibre volley and [Ca2+]pre,t within the test train, as shown in Fig. 1D. While the cAP in response to the first stimulus exhibited a slightly larger amplitude and half-width, there was no substantial difference during the next three responses. This larger amplitude and duration of the first cAP was most probably an artifact caused by the onset of the above-mentioned slow component. These results suggest that there was no detectable cumulative change of either Ca2+ or K+ currents during a short burst of stimulation under control conditions.
Figure 1. Presynaptic signals under control conditions.

Sample traces of presynaptic compound action potentials (cAPs), fibre volleys (FVs) and Ca2+ transients ([Ca2+]pre,t) in response to a train of four stimuli at an interpulse interval of 30 ms under control conditions. A, cAPs measured with the VSD RH 414. In addition to the rapid change of membrane potential that corresponds to the presynaptic action potential, the VSD signal also contains a slow component. B, corresponding presynaptic FVs measured by extracellular field recording. C, [Ca2+]pre,t, measured separately with the Ca2+ indicator furaptra. D, summary data for cAP, FV (n = 12) and [Ca2+]pre,t (n = 18). The responses to each stimulus within the train were normalized to the mean of the last three. There was no significant difference in the amplitudes of FV and [Ca2+]pre,t between stimuli. Amplitude and half-width of the uncorrected cAP in response to the first stimulus were larger than to the rest of the stimuli. This was most probably due to the artifact of the slow component.
Facilitation of [Ca2+]pre,t in the presence of 4-AP
In contrast to the unchanged amplitudes under control conditions, a facilitation of [Ca2+]pre,t was observed when a portion of the presynaptic K+ channels were blocked by application of 40 μm 4-AP, as shown in Fig. 2A. 4-AP not only increased the [Ca2+]pre,t evoked by the first stimulus but also facilitated those for subsequent stimuli within the test train. The [Ca2+]pre,t evoked by the last stimulus was about 100 % larger in amplitude than the first. This facilitation of [Ca2+]pre,t was also accompanied by a change in the waveform of the presynaptic fibre volley (Fig. 2B). The duration of the fibre volley was progressively prolonged, but not the amplitude. Therefore, the observed progressive increase in CaSD signals was not due to the recruitment of fibres by the subsequent stimuli, otherwise the amplitude of the fibre volley would have changed accordingly. The inset in Fig. 2B shows superimposed sample traces of fibre volleys: 4-AP not only broadened the duration of the action potential compared with control, but also significantly increased the duration of the subsequent action potentials within the test train. The VSD signal exhibited a similar change, as shown in Fig. 2C. Unfortunately, during application of 40 μm 4-AP, the slow component of the VSD signal became more prominent, making it difficult to precisely measure the waveform of the cAP. Therefore, the slow component was fitted by exponential functions (thin trace in Fig. 2C) and subtracted from the raw traces. The inset shows summary data for amplitude and half-width of the cAP measured from both raw traces and corrected traces. 4-AP significantly increased both the amplitude and half-width of the cAP not only compared with control but also within the test train.
Figure 2. 4-AP increases and facilitates both [Ca2+]pre,t and cAP.
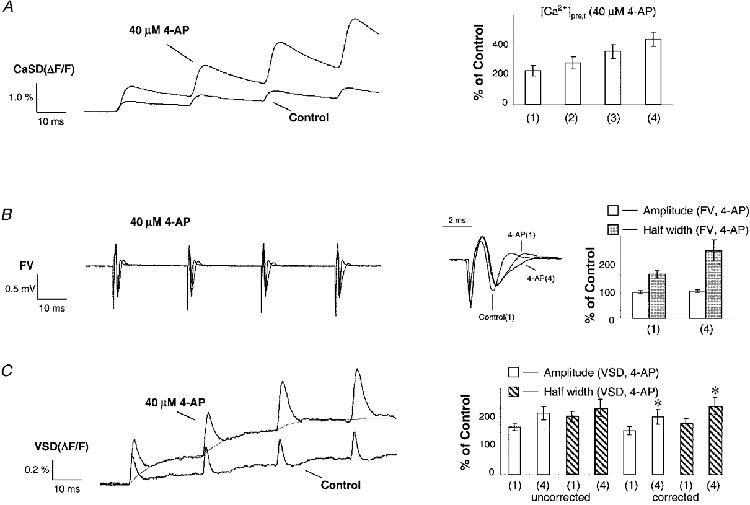
Sample traces of [Ca2+]pre,t, FVs and cAPs in response to a train of stimuli under control conditions and in 4-AP. Application of 40 μm 4-AP broadened the FV, increased the [Ca2+]pre,t and prolonged repolarization of the VSD signals. In contrast to a similar duration of FVs for each stimulus in control, FVs were progressively broadened for the subsequent stimuli in 4-AP. Consistently, the amplitudes of [Ca2+]pre,t were facilitated in 4-AP. A, on average, the [Ca2+]pre,t was increased to 226 ± 36, 278 ± 40, 356 ± 47 and 437 ± 47 % of control (n = 8) during application of 4-AP for successive stimuli within the train. B, the half-width of FV in response to the first and last stimulus was 162 ± 13 and 248 ± 38 % of control (n = 12) during application of 4-AP, respectively. The amplitude of the FV was not significantly changed (see inset). This argues against a possible recruitment of presynaptic fibres by the subsequent stimuli. C, the slow component of the VSD signal during application of 4-AP was corrected for by subtracting fitted exponential functions (thin line). The chart summarizes amplitude and half-width of VSD signals for the first and last stimulus in the presence of 4-AP. The left-hand side shows raw data of the first and last cAP (n = 12; amplitude, 164 ± 11 and 213 ± 23 %; half-width, 201 ± 18 and 229 ± 30 %); the right-hand side shows corrected data (n = 12; amplitude, 152 ± 14 and 200 ± 24 %; half-width, 179 ± 15 and 236 ± 31 %). There was a significant increase in both amplitude and half-width of the cAP waveform after correction for the slow component (paired two-tailed t test, * P < 0.001).
Facilitation of Ca2+ influx is both Ca2+ and frequency dependent
We found that the facilitation of [Ca2+]pre,t was dependent on Ca2+ influx. Figure 3A shows a typical time course of facilitated Ca2+ influx and the effect of manipulating Ca2+ influx by application of the neuromodulator adenosine, the non-specific Ca2+ channel blocker Cd2+ and the specific N-type Ca2+ channel blocker ω-CgTX GVIA. Application of these agents not only decreased [Ca2+]pre,t but also reduced their facilitation. As shown by the sample traces in Fig. 3B, 50 μm Cd2+ almost completely abolished the facilitation. Figure 3D summarizes the results from experiments in which adenosine (100 μm), Cd2+ (30 μm) or ω-CgTX GVIA (1 μm) was applied in the presence of 4-AP (40 μm). Clearly, the facilitation of the [Ca2+]pre,t was dependent on the amplitude: the more the [Ca2+]pre,t decreased, the less facilitation was observed.
Figure 3. Facilitation of [Ca2+]pre,t is dependent on Ca2+ influx.
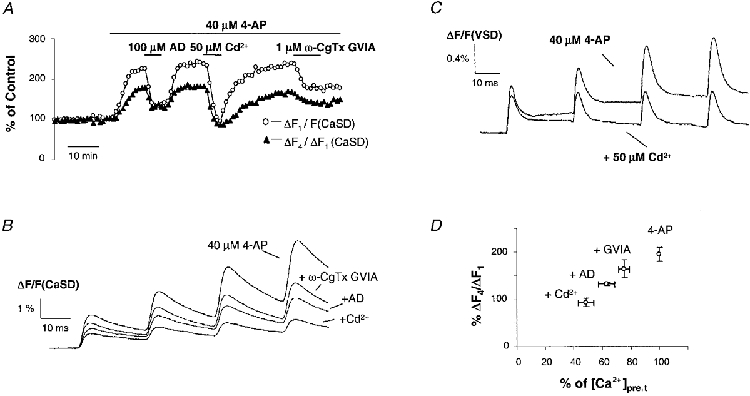
A, time course of normalized [Ca2+]pre,t and its facilitation from a typical experiment. The amount of facilitation is presented as the amplitude ratio of the last to the first Ca2+ influx (ΔF4/ΔF1). In the presence of 4-AP, application of adenosine (AD), Cd2+ and ω-CgTX GVIA inhibited the [Ca2+]pre,t and reduced the facilitation. B, sample traces of the CaSD signals shown in A. C, VSD signals in the presence of 4-AP and during application of Cd2+. This non-specific blocker of voltage-dependent Ca2+ channels eliminated the progressive increase of cAP waveform, consistent with the Ca2+ dependency observed in the CaSD signal. D, summary data illustrating that the facilitation of [Ca2+]pre,t was dependent on presynaptic Ca2+ influx. In the presence of 4-AP (40 μm), application of ω-CgTX GVIA (1 μm), AD (100 μm) and Cd2+ (30 μm) inhibited [Ca2+]pre,t by 25 ± 4 % (n = 8), 37 ± 6 % (n = 4) and 51 ± 6 % (n = 7), respectively. The corresponding response to the fourth stimulus was reduced to 164 ± 19, 133 ± 3 and 93 ± 9 % of the first.
Presynaptic cAPs measured during application of 4-AP displayed a similar dependence on Ca2+ influx to that of [Ca2+]pre,t. In the presence of 40 μm 4-AP, application of 50 μm Cd2+ substantially reduced the facilitation in amplitude of cAPs as shown in Fig. 3C. This dependence of facilitation on Ca2+ influx is inconsistent with a scenario in which 4-AP molecules that are blocking open K+ channels are slowly released and cause fewer channels to be available when the next action potential invades the presynaptic terminals. To date, there is no evidence indicating that Ca2+ interferes with the interaction between 4-AP and K+ channels. Taken together, this strongly suggests that the observed facilitation was not an artifact due to the kinetics of the interaction between 4-AP and K+ channels.
Furthermore, the observed facilitation was also found to depend on the frequency of stimulation. Figure 4A shows a sample trace of VSD signals in the presence of 40 μm 4-AP. When a train of four stimuli was delivered at an interval of 100 ms instead of 30 ms, there was no significant facilitation in the waveform of the cAPs (n = 3). Consistent with this frequency-dependent facilitation of cAPs, no facilitation of [Ca2+]pre,t was observed when the CA1 afferent pathway was stimulated at the same frequency (Fig. 4B, n = 3).
Figure 4. Facilitation of [Ca2+]pre,t depends on the frequency of stimulation.
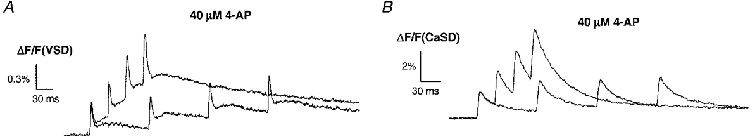
Sample traces of cAPs (A) and [Ca2+]pre,t (B) evoked by stimulation with different interstimulus intervals in the presence of 4-AP. Facilitation of cAPs was dependent on the stimulation frequency. When stimuli were delivered at an interval of 100 ms, there was no progressive increase in the amplitude of the cAP. The facilitation of the [Ca2+]pre,t shows a similar frequency dependence to the cAP.
Time course of [Ca2+]pre,t facilitation
We also tested the time course of recovery from facilitation of the [Ca2+]pre,t. The observed Ca2+ dependency and frequency dependency of [Ca2+]pre,t facilitation suggests that the mechanism for suppression of K+ currents during the test train may be related to the residual Ca2+ concentration ([Ca2+]res). To examine this hypothesis, the time course of facilitation was compared with that of [Ca2+]res. As illustrated in Fig. 5A, a burst of conditioning stimuli was used to raise [Ca2+]res, then a test stimulus was applied at various intervals after the last conditioning stimulus. In the presence of 4-AP, the [Ca2+]res was greatly increased by the conditioning stimulation and quickly decayed. Figure 5B summarizes the results from eight experiments. By visual inspection, the recovery from facilitation of [Ca2+]pre,t had a similar time course to the decay of [Ca2+]res. During conditioning stimulation, the facilitation of [Ca2+]pre,t had a relationship with [Ca2+]res slightly different from that during testing (Fig. 5C). This difference may reflect a small contribution of cumulative voltage-dependent modulation as the interval between the conditioning stimulation and the test stimulus increased during the decay phase. Therefore, comparison of the time courses during the decay phase of [Ca2+]res would minimize the contribution of such a cumulative voltage-dependent modulation induced by the conditioning stimulation. The observed similarity of the decay phase time courses suggests that the suppression of K+ currents is related to the [Ca2+]res.
Figure 5. Recovery time course of [Ca2+]pre,t facilitation.
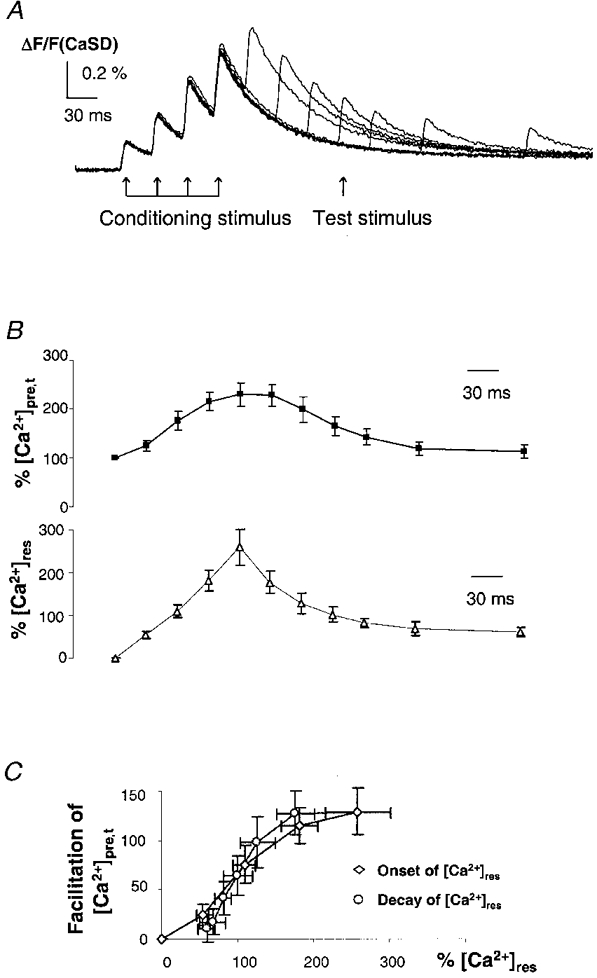
A, [Ca2+]pre,t from a typical experiment to measure the time course of recovery from facilitation of [Ca2+]pre,t. Following the conditioning stimulation train, a single test stimulus was applied at various intervals. B, summary data to show the time course of [Ca2+]pre,t facilitation and [Ca2+]res (n = 8). The recovery is represented by the [Ca2+]pre,t in response to the test stimulus after the conditioning stimulation, which was measured after subtracting the decay time course of residual Ca2+ traces ([Ca2+]res). By visual inspection, the recovery time course for the facilitation appears similar to the decay of the [Ca2+]res. This suggests that the suppression of K+ currents is related to the [Ca2+]res. C, relationship between facilitation of [Ca2+]pre,t and normalized [Ca2+]res during conditioning stimulation and testing. The onset and decay phases of [Ca2+]res exhibit a slightly different relationship with [Ca2+]pre,t. This difference may reflect a small amount of cumulative voltage-dependent modulation during the conditioning stimulation.
Effects of TEA on [Ca2+]pre,t and cAP
Facilitation of [Ca2+]pre,t and progressive broadening of action potentials were observed during application of 4-AP. Thus, 4-AP unmasked the activity-dependent modulation of K+ currents by blocking ‘non-modulated’ K+ currents. Therefore, K+ channels sensitive to low concentrations of 4-AP apparently were not those exhibiting activity-dependent modulation. In order to confirm this conclusion, another K+ channel blocker, TEA, was applied to block TEA-sensitive currents while sparing 4-AP-sensitive K+ currents. Application of TEA has been shown to broaden the presynaptic fibre volley in both cerebellum and hippocampus (Laerum & Storm, 1994; Sabatini & Regehr, 1997). Figure 6A and B shows sample traces of cAPs and [Ca2+]pre,t under control conditions and during application of 10 mM TEA. As expected, TEA broadened the presynaptic fibre volley (inset in Fig. 6A). Consistently, the cAP exhibited a pattern of increased amplitude and half-width compared with control, and an increased [Ca2+]pre,t was observed. Figure 6C summarizes the effects of TEA on the [Ca2+]pre,t. In addition to broadening the fibre volley, 10 mM TEA also reduced the amplitude of the fibre volley by 15 ± 3 % (n = 5), suggesting a decrease in the fibre excitability during application of a high concentration of TEA. This indicates that the amount of increase in the Ca2+ influx caused by 10 mM TEA was probably underestimated by the measured [Ca2+]pre,t. In contrast to the facilitation of both [Ca2+]pre,t and cAPs observed in the presence of 4-AP, during application of TEA there was much less facilitation of [Ca2+]pre,t and no difference was detected in the waveform of the fibre volley for subsequent stimuli. We also measured the [Ca2+]pre,t during application of 10 μm 4-AP, at which concentration a significant facilitation was observed (n = 3; 177 ± 2, 203 ± 4, 237 ± 6 and 270 ± 9 %, for successive stimuli within the test train) even though the [Ca2+]pre,t was only increased to a level similar to application of 10 mM TEA. This result suggests that the presence of K+ channels sensitive to TEA is essential to the observed phenomena, implying that the TEA-sensitive K+ currents undergo the activity-dependent modulation.
Figure 6. Effects of TEA on the cAP and [Ca2+]pre,t.
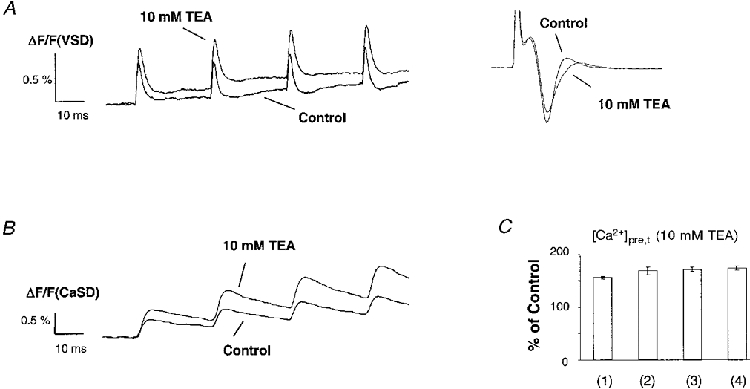
Sample traces of cAPs and [Ca2+]pre,t under control conditions and in the presence of 10 mM TEA. A, in contrast to application of 4-AP, TEA did not evoke a progressive increase in the amplitude of VSD signals. No difference was detected in the waveform of the FV among stimuli within the test train. The inset shows superimposed presynaptic FVs. TEA not only increased the duration but also reduced the amplitude of the FV compared with control, suggesting a decreased excitability of fibres in the presence of high concentrations of TEA. B, [Ca2+]pre,t in control and in the presence of 10 mM TEA. Consistent with the observed cAP, there was much less facilitation of the [Ca2+]pre,t for subsequent stimuli within the test train. C, summary data for the [Ca2+]pre,t in the presence of TEA. On average, 10 mM TEA increased the [Ca2+]pre,t to 156 ± 3, 168 ± 7, 171 ± 5 and 174 ± 4 % of control (n = 4) for successive stimuli within the test train. The amount of [Ca2+]pre,t induced by 10 mM TEA is probably an underestimate, due to the reduced amount of fibres activated. There was no significant facilitation of [Ca2+]pre,t as compared with application of 4-AP.
Facilitation of [Ca2+]pre,t is not related to [K+]o
Even though recruitment of axons has been ruled out as a reason for the observed facilitation of [Ca2+]pre,t, a possible elevation of [K+]o could potentially reduce the driving force for presynaptic K+ currents and thereby prolong action potentials. To test this possibility, [K+]o was simultaneously measured with [Ca2+]pre,t. A K+-sensitive electrode was positioned in the SR of area CA1 to measure the change in [K+]o during a burst of stimuli. Figure 7 shows the time course of [K+]o and [Ca2+]pre,t evoked by such a burst in the presence of 40 μm 4-AP. The [K+]o reached a peak of about 7 mM above resting concentration (3 mM) and then slowly recovered. Application of the glutamate receptor antagonists CNQX (10 μm) and D-APV (25 μm), which completely eliminated the fEPSP as monitored by extracellular field recording (not shown here), almost abolished the K+ response. In the presence of CNQX and D-APV, the increase of [K+]o was less than 1 mM in this experiment. However, [Ca2+]pre,t still displayed the typical pattern of facilitation, indicating that an accumulation of [K+]o was not responsible for the observed facilitation of [Ca2+]pre,t.
Figure 7. Modulation of K+ conductance is not related to [K+]o.
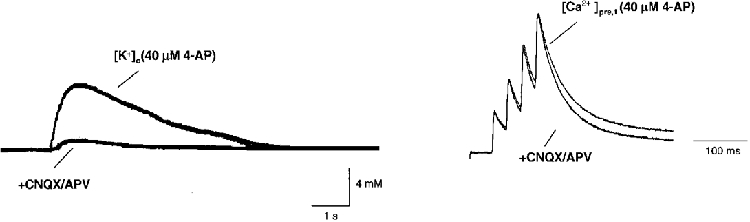
[K+]o and [Ca2+]pre,t in the presence of 4-AP and during application of the glutamate receptor antagonists CNQX and D-APV. Blockade of postsynaptic responses with 10 μm CNQX and 25 μm D-APV did not eliminate the facilitation of [Ca2+]pre,t while this manipulation greatly reduced the stimulation-evoked increase of [K+]o. This suggests that the observed activity-dependent modulation of K+ conductance was not related to [K+]o.
Facilitation of [Ca2+]pre,t is insensitive to high concentrations of 4-AP
The slowly inactivating delayed K+ D-type current, which is sensitive to low concentrations of 4-AP, is unlikely to be responsible for the observed facilitation of [Ca2+]pre,t because this facilitation was found to be more prominent after blocking the K+ currents with 40-100 μm 4-AP. The fast inactivating K+ (A-type) current, which is also sensitive to TEA at a concentration of 10 mM, is a potential candidate for activity-dependent modulation. Indeed, voltage-dependent transient K+ channels have been found to exhibit Ca2+-dependent inactivation (Chen & Wong, 1991; Roeper et al. 1997). Thus, high concentrations of 4-AP were applied to test whether A-type-like channels are involved in the facilitation of [Ca2+]pre,t. Figure 8A compares the measured [Ca2+]pre,t during application of 40 μm and 8 mM 4-AP. In general, high concentrations of 4-AP did not block the facilitation, but rather enhanced the facilitation as indicated by an increase in the relative amplitude of [Ca2+]pre,t for the third stimulus. It should be noted that 8 mM 4-AP also reduced the size of the fibre volley, similar to high concentrations of TEA. Therefore, the fluorescence signal observed in the presence of high concentrations of 4-AP led to an underestimation of presynaptic Ca2+ influx at the terminals as compared with the Ca2+ influx under application of low concentrations of 4-AP.
Figure 8. Facilitation of [Ca2+]pre,t is insensitive to high concentrations of 4-AP.
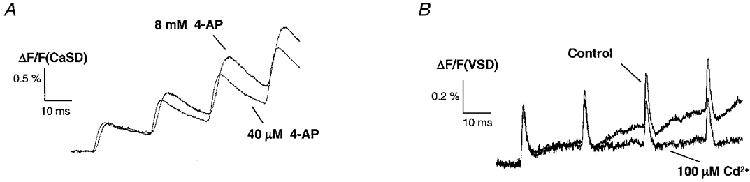
A, [Ca2+]pre,t in the presence of 8 mM 4-AP. This concentration is supposed to block fast inactivating (A-type) K+ currents; however, it did not eliminate the facilitation of [Ca2+]pre,t, suggesting that A-type K+ channels were not involved. High concentrations of 4-AP reduced the amplitude of FVs (not shown). Therefore, the trace of [Ca2+]pre,t shown here is an underestimate of presynaptic Ca2+ influx as compared with application of 40 μm 4-AP. B, application of Cd2+ (100 μm), abolished the afterhyperpolarization, suggesting the existence of presynaptic Ca2+-sensitive K+ currents.
Involvement of Ca2+-dependent K+ channels in the repolarization of action potentials
In addition to K+ channels sensitive to TEA or 4-AP, the contribution of action potential repolarization by Ca2+-dependent K+ currents was also examined. Figure 8B shows cAPs evoked by a burst of stimuli under control conditions and during application of 100 μm Cd2+. Blocking of Ca2+ influx with Cd2+ clearly abolished the after-hyperpolarization (AHP) for subsequent stimuli. This suggests that Ca2+-dependent K+ channels are also present at presynaptic terminals of the rat hippocampal CA3-CA1 synapse. The AHP of the first stimulus was probably masked by the onset of the slow component of VSD signals.
DISCUSSION
Types of presynaptic K+ channels that contribute to the repolarization of action potentials
At the hippocampal CA3-CA1 synapse, a K+ current sensitive to low concentrations of 4-AP is the most prominent component of the outward K+ current during the repolarization of presynaptic action potentials. Block of this type of K+ channel by low concentrations of 4-AP greatly broadened the fibre volley and enhanced [Ca2+]pre,t as well. This K+ current is also present at other presynaptic terminals such as the squid giant synapse (Augustine, 1990), the mouse neuromuscular junction (Brigant & Mallart, 1982), the rat posterior pituitary (Bielefeldt et al. 1992) and at the calyx of Held synapse in the rat brainstem (Forsythe, 1994).
TEA-sensitive K+ currents also play a significant role in action potential repolarization at the presynaptic terminals of the hippocampal CA3-CA1 pathway. Application of 10 mM TEA increased both cAPs and [Ca2+]pre,t. However, the amplitude of presynaptic fibre volleys was significantly reduced during application of TEA in our experiments. This makes a quantitative interpretation of cAPs and [Ca2+]pre,t difficult.
The role of presynaptic Ca2+-dependent K+ channels was also examined by comparing the waveform of the cAP in control and during application of a non-specific Ca2+ channel blocker. Cd2+ (100 μm) abolished the AHP, indicating that Ca2+-activated K+ currents do substantially contribute to the repolarization of action potentials. At the neuromuscular junction, Ca2+-dependent K+ currents play a significant role in the modulation of transmitter release (Robitaille et al. 1993). In contrast, measuring the presynaptic waveform at the cerebellar parallel synapse did not reveal an involvement of this type of K+ current in the repolarization of action potentials (Sabatini & Regehr, 1997).
In addition to the types of K+ channel mentioned above, a fast-inactivating K+ current could also play a significant role in the repolarization of action potentials in the mammalian central nervous system. In the hippocampus, in situ hybridization studies revealed an abundant distribution of this type of K+ channel in axons and nerve terminals (Sheng et al. 1993). A study of the coupling between CA3 and CA1 pyramidal neurons from dual cell patch recording suggests that A-type K+ currents regulate action potential propagation along Schaffer collateral axons (Debanne et al. 1997). Due to lack of specific antagonists for this type of K+ channel, the actual contribution of fast-inactivating K+ currents was not investigated.
Activity-dependent suppression of K+ currents
The signals obtained with optical and electrical recording techniques in the present study show a facilitation of Ca2+ influx and a corresponding change in the waveform of presynaptic action potentials during application of 4-AP. Our experimental results indicate that the observed phenomena neither represent an artifact of the pharmacological interaction between 4-AP and K+ channels, nor are related to an accumulation of extracellular K+ that might occur during application of 4-AP. Rather, our results suggest that an activity-dependent suppression of TEA-sensitive presynaptic K+ currents, which is presumably mediated by accumulated intracellular Ca2+, underlies the observed progressive change in the waveform of action potentials which thereby facilitates Ca2+ influx. This facilitation of Ca2+ influx is unlikely to be due to the action of Ca2+-activated K+ channels, since this would be expected to speed up the repolarization of action potentials. Under control conditions, the effect of such a modulation of TEA-sensitive K+ channels is masked, as a 4-AP-sensitive K+ current is the most prominent component of the outward current during repolarization. In addition, under control conditions, Ca2+ influx is not large enough to substantially increase the residual Ca2+ concentration. In rat pituitary presynaptic terminals, a similar frequency-dependent facilitation of presynaptic Ca2+ influx has been observed (Jackson et al. 1991). This facilitation of Ca2+ influx correlated well with the increasing duration of action potentials. A voltage-dependent inactivation of K+ channels was suggested to explain the cumulative broadening of action potentials. A similar activity-dependent broadening of action potentials was also observed in molluscan neurons (Aldrich et al. 1979), where action potentials recorded from somata increased in duration during low frequency repetitive firing. Spike broadening was also sensitive to Ca2+ channel blockers. Although these authors had shown that TEA-sensitive K+ channels undergo voltage-dependent inactivation, inactivation of K+ channels alone could not fully account for the observed cumulative broadening of action potentials. The requirement for additional Ca2+ influx was interpreted as an increase in the Ca2+-dependent spike shoulder, which causes a sufficient inactivation of K+ channels. However, the Ca2+-dependent spike shoulder could alternatively be explained as a Ca2+-dependent reduction of K+ currents, which would in turn prolong spike duration and cause more Ca2+ influx. In the hippocampus, intracellular Ca2+ has been shown to suppress a K+ current (Chen & Wong, 1991). Recently, a frequency-dependent reduction of mammalian transient K+ channels was demonstrated to be regulated by Ca2+-calmodulin-dependent protein kinase (Roeper et al. 1997). Therefore, in addition to voltage-dependent inactivation, Ca2+-dependent suppression of K+ currents could be a potential mechanism underlying the observed facilitation of Ca2+ influx in our experiments. In the present study, the suppression of presynaptic K+ currents was shown to be dependent on Ca2+ influx. Moreover, similar time courses were observed between the recovery from facilitation of [Ca2+]pre,t and the decay of [Ca2+]res. This suggests that the suppression of the TEA-sensitive K+ current is related to the intracellular Ca2+ concentration at the investigated presynaptic terminals.
Acknowledgments
We acknowledge Dr Sinha for his critical and helpful comments on the manuscript. The computer software for data acquisition and analysis was developed by Dr S. S. Patel. We thank Dr Janet Stringer for her advice and Jason Xiong for his assistance in measuring extracellular potassium concentration. This work was supported by National Institutes of Health Grant NS-33147 to P. Saggau.
APPENDIX
Measurement of cAPs with voltage-sensitive dyes
Due to the nature of the optical recording technique, the cAP measured with the VSD represents a convolution of the waveform of the action potential with a spreading function of this wave within the optical recording area. The direct effect of this convolution is that a broadened action potential leads to an increase in both the amplitude and half-width of the optically measured cAP. Figure 9 describes the steps to test this interpretation. The spreading function was first estimated by de-convoluting the optically measured control cAP with the waveform of a corrected presynaptic fibre volley, which is the best approximation to the waveform of a true action potential that we could obtain without any bias. Once the spreading function was determined, the waveforms of cAPs for a number of action potentials with different durations were calculated by convoluting the presumed waveform of the action potentials with the estimated spreading function. Gaussian functions were used to mimic the waveform of action potential repolarization. Figure 9C shows the mean measured waveform of cAPs after correction for the slow component under control conditions and during application of 4-AP. These measured cAPs exhibit waveforms very similar to the calculated ones. Therefore, the VSD signals provide consistent evidence for our conclusion that activity-dependent progressive prolongation of action potential repolarization is responsible for the facilitation of Ca2+ influx as observed in the presence of 4-AP.
Figure 9. Interpretation of cAPs measured with voltage-sensitive dyes.
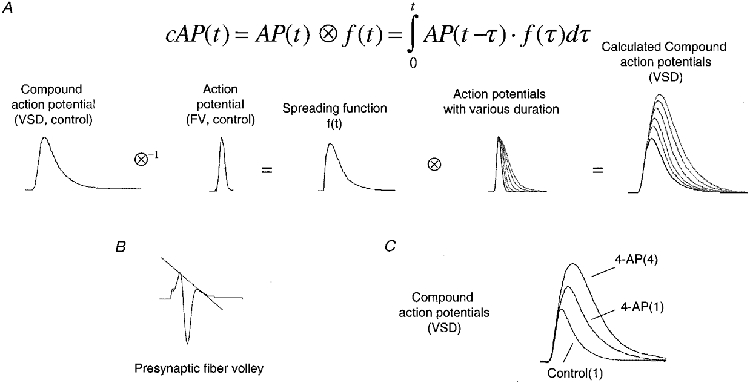
Due to the nature of the optical recording technique, a cAP measured with the VSD is actually a convolution of the waveform of the true action potential (AP) with a spreading function, f(t), of the AP within the optical recording area. A, to estimate the spreading function, a cAP measured under control conditions was de-convoluted with the waveform of an AP, obtained from a corrected presynaptic fibre volley in control (see B). Then, cAPs for APs of various durations were obtained by convoluting the waveform of APs with the estimated spreading function. Gaussian functions were used to simulate the waveform of action potential repolarization. B, mean waveform of presynaptic FVs under control conditions. The dotted line indicates the correction for the field effect. C, mean waveform of cAPs after correction for the slow component under control conditions and during application of 4-AP. These cAPs measured with the VSD show patterns similar to the calculated waveforms of cAPs.
References
- Aldrich RW, Getting PA, Thompson SH. Mechanisms of frequency-dependent broadening of molluscan neurone soma spikes. The Journal of Physiology. 1979;291:531–544. doi: 10.1113/jphysiol.1979.sp012829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ. Regulation of transmitter release at the squid giant synapse by presynaptic delayed rectifier potassium current. The Journal of Physiology. 1990;431:343–364. doi: 10.1113/jphysiol.1990.sp018333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Rotter JI, Jackson MB. Three potassium channels in rat posterior pituitary nerve endings. The Journal of Physiology. 1992;458:41–67. doi: 10.1113/jphysiol.1992.sp019405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigant DC, Mallart A. Presynaptic currents in mouse motor endings. The Journal of Physiology. 1982;333:619–639. doi: 10.1113/jphysiol.1982.sp014472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen QX, Wong RKS. Intracellular Ca2+ suppressed a transient potassium current in hippocampal neurons. Journal of Neuroscience. 1991;11:337–343. doi: 10.1523/JNEUROSCI.11-02-00337.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debanne D, Guerineau NC, Gahwller BH, Thompson SM. Action-potential propagation gated by an axonal IA-like K+ conductance in hippocampus. Nature. 1997;389:286–289. doi: 10.1038/38502. [DOI] [PubMed] [Google Scholar]
- Forsythe ID. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. The Journal of Physiology. 1994;479:381–387. doi: 10.1113/jphysiol.1994.sp020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman DA, Magee JC, Colbert CM, Johnston D. K+ channel regulation of signal propagation in dendrites of hippocampal pyramidal neurons. Nature. 1997;387:869–875. doi: 10.1038/43119. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, Augustine GA. Action potential broadening and frequency-dependent facilitation of Ca2+ signals in pituitary nerve terminals. Proceedings of the National Academy of Sciences of the USA. 1991;88:380–384. doi: 10.1073/pnas.88.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konnerth A, Orkand RK. Voltage-sensitive dyes measure potential changes in axons and glia of the frog optic nerve. Neuroscience Letters. 1986;66:49–54. doi: 10.1016/0304-3940(86)90164-3. [DOI] [PubMed] [Google Scholar]
- Laerum H, Storm JF. Hippocampal long-term potentiation is not accompanied by presynaptic spike broadening, unlike synaptic potentiation by K+ channel blockers. Brain Research. 1994;637:349–355. doi: 10.1016/0006-8993(94)91260-2. [DOI] [PubMed] [Google Scholar]
- Lothman EW, Somjen GG. Extracellular potassium activity, intracellular and extracellular potential responses in the spinal cord. The Journal of Physiology. 1975;252:115–136. doi: 10.1113/jphysiol.1975.sp011137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian J, Saggau P. Modulation of transmitter release by action potential duration at the hippocampal CA3-CA1 synapse. Journal of Neurophysiology. 1999;81:288–298. doi: 10.1152/jn.1999.81.1.288. [DOI] [PubMed] [Google Scholar]
- Robitaille R, Adler EM, Charlton MP. Calcium channels and calcium-gated potassium channels at the frog neuromuscular junction. Journal de Physiologie. 1993;87:15–24. doi: 10.1016/0928-4257(93)90020-t. [DOI] [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channels Kv1.4 regulated by Ca2+/calmodulin-dependent protein kinase. Journal of Neuroscience. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatini BL, Regehr WG. Control of neurotransmitter release by presynaptic waveform at the granule cell to Purkinje cell synapse. Journal of Neuroscience. 1997;17:3425–3435. doi: 10.1523/JNEUROSCI.17-10-03425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng M, Luao YJ, Jan YN, Jan LY. Presynaptic A-current based on heteromultimeric K+ channels detected in vivo. Science. 1993;365:72–75. doi: 10.1038/365072a0. [DOI] [PubMed] [Google Scholar]
- Storm JF. Temporal integration by a slowly inactivating K+ current in hippocampal neurons. Nature. 1988;336:379–381. doi: 10.1038/336379a0. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Changes in action potential duration alter reliance of excitatory synaptic transmission on multiple types of Ca2+ channels in rat hippocampus. Journal of Neuroscience. 1996;16:2226–2237. doi: 10.1523/JNEUROSCI.16-07-02226.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu LG, Saggau P. Presynaptic calcium is increased during normal synaptic transmission and paired-pulse facilitation, but not in long-term potentiation in area CA1 of hippocampus. Journal of Neuroscience. 1994;14:645–654. doi: 10.1523/JNEUROSCI.14-02-00645.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]