

Abstract
The role of calcium (Ca2+) channel inactivation in the molecular mechanism of channel block by phenylalkylamines (PAAs) was analysed in a PAA-sensitive rabbit brain class A Ca2+ channel mutant (α1A-PAA). Use-dependent barium current (IBa) inhibition of α1A-PAA by (−)gallopamil and Ca2+ channel recovery from inactivation and block were studied with two-microlectrode voltage clamp after expression of α1A-PAA and auxiliary α2-δ- and β1a- or β2a-subunits in Xenopus oocytes.
Mutation Arg387Glu (α1A numbering) in the intracellular loop connecting domains I and II of α1A-PAA slowed the inactivation kinetics and reduced use-dependent inhibition (100 ms test pulses at 0.2 Hz from -80 to 20 mV) of the resulting mutant α1A-PAA/R-E/β1a channels by 100 μm (−)gallopamil (53 ± 2 %, α1A-PAA/β1avs. 31 ± 2 %, α1A-PAA/R-E/β1a, n ≥ 4). This amino acid substitution simultaneously accelerated the recovery of channels from inactivation and from block by (−)gallopamil.
Coexpression of α1A-PAA with the β2a-subunit reduced fast IBa inactivation and induced a substantial reduction in use-dependent IBa inhibition by (−)gallopamil (25 ± 4 %, α1A-PAA/β2a; 13 ± 1 %, α1A-PAA/R-E/β2a). The time constant of recovery from block at rest was not significantly affected.
These results demonstrate that changes in channel inactivation induced by Arg387Glu or β2a-α1-subunit interaction affect the drug-channel interaction.
Calcium (Ca2+) channel inhibition by drugs such as phenylalkylamines (PAAs), benzothiazepines (BTZs) and mibefradil increases during repetitive depolarisation of the membrane (Lee & Tsien 1983; McDonald et al. 1984; Bezprozvanny & Tsien 1995; Aczél et al. 1998). Such a ‘use-dependent’ channel inhibition reflects distinct drug interactions with the resting, open and inactivated channel states. It is believed that state-dependent Ca2+ channel block plays an important role in the therapeutic action of PAAs and BTZs as antiarrhythmics (Hondeghem & Katzung, 1984).
Functional studies on mutant Ca2+ channels enabled the first insights into the molecular architecture of the Ca2+ channel drug-binding domains (see Hockerman et al. 1997b and Striessnig et al. 1998 for review). The available data suggest that three different classes of Ca2+ channel antagonists (PAAs, BTZs and 1,4-dihydropyridines) bind in close proximity within the pore region of L-type Ca2+ channel α1-subunits (Striessnig et al. 1998). Three amino acids in segment IVS6 (Tyr1463, Ala1467, Ile1470) and four residues in transmembrane segment IIIS6 (Tyr1152, Ile1153, Phe1164 and Val1165) have been identified as crucial L-type determinants of the PAA sensitivity (Hockerman et al. 1995, 1997a). Insertion of three ‘L-type-specific’ residues (Tyr1463, Ala1467 and Ile1470) into segment IVS6 of the only weakly PAA sensitive class A (α1A) Ca2+ channel transferred PAA sensitivity to the corresponding α1A mutant (here called α1A-PAA; Hering et al. 1996).
An unequivocal identification of the PAA binding determinants by mutational analysis of α1 Ca2+ channel subunits is, however, complicated by an apparent interdependence between Ca2+ channel block and inactivation gating (see Hering et al. 1998 for review). In particular, transfer of the IVS6 L-type determinants of PAA sensitivity (Tyr1463, Ala1467 and Ile1470) to class A Ca2+ channels accelerated inactivation (Hering et al. 1996; Degtiar et al. 1997). Moreover, introduction of an additional L-type amino acid (Met1464 into IVS6 of α1A-PAA) facilitated channel inactivation and enhanced use-dependent channel block by (−)gallopamil (Hering et al. 1996). Accordingly, substitution of a PAA determinant in α1A-PAA (Ile1470 by the corresponding class A channel Met, α1C-a numbering) which substantially reduced channel inactivation induced an about 30-fold decrease of the apparent association rate for (−)devapamil (Degtiar et al. 1997) and alanine substitutions of three L-type amino acids localised close to the inner channel mouth on segment IIIS6 and IVS6 reduced Ca2+ channel inactivation and simultaneously BTZ and PAA sensitivity (Hering et al. 1997; Berjukow et al. 1999).
In our previous studies on the role of Ca2+ channel inactivation in channel block by PAAs and BTZs we have focused on residues that are located on segments IIIS6 and IVS6 (Degtiar et al. 1997; Hering et al. 1997; Berjukow et al. 1999). Here we analyse in a PAA-sensitive class A Ca2+ channel mutant (α1A-PAA) expressed in Xenopus oocytes if inactivation determinants localised outside the channel pore of an α1-subunit influence Ca2+ channel block by (−)gallopamil.
We demonstrate that a single amino acid substitution (Arg387Glu, α1A numbering) in the intracellular loop between domains I and II slows channel inactivation and reduces sensitivity for (−)gallopamil. Furthermore, a reduced inactivation caused by coexpression of α1A-PAA with β2a- instead of the β1a-subunit reduced Ca2+ channel block by (−)gallopamil even more dramatically. Our study clearly demonstrates that inactivation determinants that are localised outside the putative drug binding regions in the channel pore affect the molecular mechanism of use-dependent Ca2+ channel block by (−)gallopamil.
METHODS
Generation of α1A-constructs
The construction of the PAA-sensitive triple rabbit brain class A Ca2+ channel mutant AL25 (named herein α1A-PAA) was previously described (Hering et al. 1996). The derived mutant α1A-PAA/R-E was constructed by introducing a single point mutation (R387E, α1A numbering) into α1A-PAA cDNA by the ‘gene SOEing’ technique (Horton et al. 1989). The point mutation was verified by sequence analysis. All constructs were inserted into the polyadenylating transcription plasmids pSPCBI 2 (a kind gift of Dr O. Pongs, University of Hamburg).
Electrophysiology
Female Xenopus laevis (NASCO, Fort Atkinson, WI, USA) were anaesthetised by exposing them for 15 min to a 0.2 % MS-222 (methane sulfonate salt of 3-aminobenzoic acid ethyl ester; Sandoz) solution before surgically removing parts of the ovaries. The frogs were then allowed to recover and returned to their tank. Each frog was reused up to two times and subsequently killed by decapitation under anaesthesia. The interval between the operations was longer than 4 months. Follicle membranes from isolated oocytes were enzymatically digested with 2 mg ml−1 collagenase (Type 1A, Sigma). Calcium channel currents (IBa) were studied 2 to 7 days after microinjection of approximately equimolar cRNA mixtures of α1 (0.3 ng per 50 nl)-β1a(β2a) (0.1 ng per 50 nl)-α2δ (0.2 ng per 50 nl) with two-microelectrode voltage clamp of Xenopus oocytes with 40 mM Ba2+ as charge carrier in a bath solution containing (mM): 40 Ba(OH)2, 40 N-methyl-D-glucamine, 10 Hepes, 10 glucose, adjusted to pH 7.4 with methanesulfonic acid as previously described (Grabner et al. 1996). Endogenous chloride currents of the oocytes were suppressed by injecting 20-40 nl of a 0.1 M BAPTA solution 30-240 min before the voltage clamp measurements. Voltage-recording and current-injecting microelectrodes were filled with 2.8 M CsCl, 0.2 M CsOH, 10 mM EGTA, 10 mM Hepes (pH 7.4) and had resistances of 0.3-2 MΩ.
Drug sensitivity was estimated as use-dependent Ca2+ channel block during 20 test pulses (100 ms) applied at 0.2 Hz from -80 mV to 20 mV corresponding to the peak current voltage of the current-voltage relationships of all studied Ca2+ channel mutants. Use-dependent block was measured after a 3 min equilibration of the oocytes in drug-containing solution. To estimate the accumulation of Ca2+ channels in inactivation under control conditions similar pulse trains were applied in the absence of drug. Resting channel block was measured in an individual set of experiments as peak IBa inhibition during 100 ms test pulses from -80 to 20 mV after a 5 min equilibration in drug-containing solution.
Recovery from inactivation was studied at a holding potential of -80 mV after depolarising Ca2+channels during a 3 s prepulse to 20 mV by applying 30 ms test pulses to 20 mV at various time intervals after the conditioning prepulse. Peak IBa values were normalised to the peak current measured during the prepulse. The time course of IBa recovery from inactivation was fitted to a biexponential function:
Initial rates of IBa decay (see Fig. 1B) were estimated by calculating the maximum derivative of mono- (α1A-PAA/β2a, α1A-PAA/R-E/β2a) or biexponential (α1A-PAA/β1a, α1A-PAA/R-E/β1a) fits to current inactivation during a 3 s depolarisation from -80 to 20 mV.
Figure 1. Mutation Arg387Glu and α1-β2a -subunit interaction reduce channel block by (−)gallopamil.
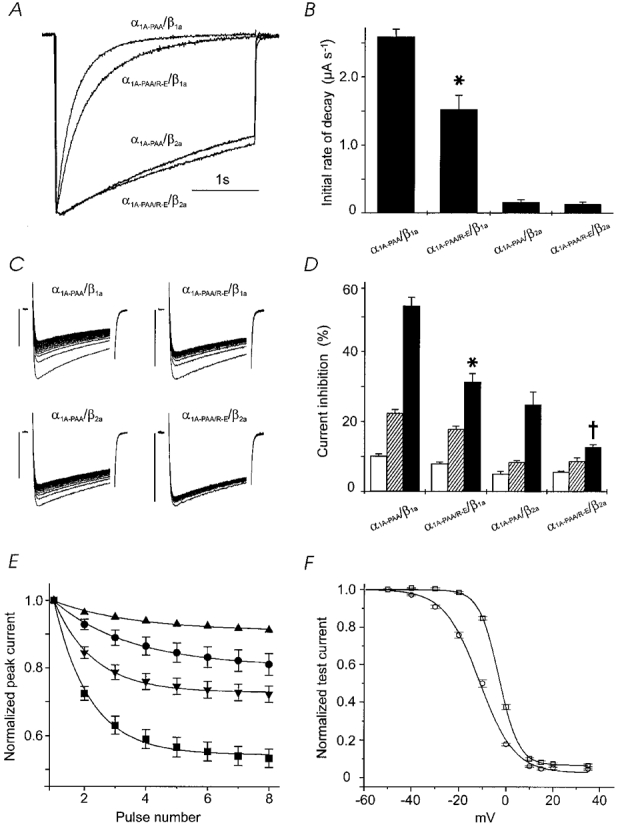
A, normalised IBa of mutants α1A-PAA/β1a, α1A-PAA/R-E/β1a, α1A-PAA/β2a and α1A-PAA/R-E/β2a during 3 s depolarising test pulses applied from a holding potential of -80 mV to 20 mV. B, comparison of the time course of IBa inactivation of mutants α1A-PAA/β1a, α1A-PAA/R-E/β1a, α1A-PAA/β2a and α1A-PAA/R-E/β2a (in control) measured as initial rate for IBa decay during a 3 s pulse from -80 mV to 20 mV (n ≥ 7, see Methods). Statistically significant reduction in the rate of current inactivation compared with α1A-PAA/β1a is indicated by the asterisk (* P < 0.05). C, use-dependent IBa inhibition of α1A-PAA/β1a, α1A-PAA/R-E/β1a, α1A-PAA/β2a and α1A-PAA/R-E/β2a by 100 μm (−)gallopamil during trains of 20 pulses (100 ms) applied at 0.2 Hz from a holding potential of -80 mV to 20 mV. Vertical bars, 0.5 μA. D, comparison of the use-dependent IBa block of α1A-PAA and α1A-PAA/R-E expressed with either the β1a- or β2a-subunit by 10 and 100 μm (−)gallopamil. The block of IBa was measured as cumulative peak current inhibition (as a percentage) during 20 depolarising pulses (100 ms, 0.2 Hz) in control (□) or in the presence of 10 μm ( ) and 100 μm (−)gallopamil (▪). Bars represent the means ±s.e.m. (n = 5-8). * Significantly different from IBa block of α1A-PAA/β1a; † Significantly different from α1A-PAA/β2a. E, initial rate of peak IBa inhibition by 100 μm (−)gallopamil (see C for details of the pulse protocol). Smooth lines are single-exponential fits to averaged peak current decays (n≥ 5) with time constants of 13.0 ± 2 s (▴, α1A-PAA/R-E/β2a), 12.5 ± 1 s (•, α1A-PAA/β2a), 6.5 ± 0.5 s (▾, α1A-PAA/R-E/β1a) and 6.0 ± 0.5 s (▪, α1A-PAA/β1a). F, inactivation curves of α1A-PAA/β1a (○) and α1A-PAA/R-E/β1a channels (□) in control. Curves are drawn according to the equation I/Imax=Iss+ (1 - Iss)/(1 + exp[(V - V0.5)/k]) (see Methods) with V0.5= -11.3 ± 0.5 mV, k = 7.4 ± 0.4 mV, Iss= 0.03 ± 0.01, n = 5 for α1A-PAA/β1a and V0.5= -3.1 ± 0.1 mV, k = 4.20 ± 0.08 mV, Iss= 0.07 ± 0.01, n = 4 for α1A-PAA/R-E/β1a.
) and 100 μm (−)gallopamil (▪). Bars represent the means ±s.e.m. (n = 5-8). * Significantly different from IBa block of α1A-PAA/β1a; † Significantly different from α1A-PAA/β2a. E, initial rate of peak IBa inhibition by 100 μm (−)gallopamil (see C for details of the pulse protocol). Smooth lines are single-exponential fits to averaged peak current decays (n≥ 5) with time constants of 13.0 ± 2 s (▴, α1A-PAA/R-E/β2a), 12.5 ± 1 s (•, α1A-PAA/β2a), 6.5 ± 0.5 s (▾, α1A-PAA/R-E/β1a) and 6.0 ± 0.5 s (▪, α1A-PAA/β1a). F, inactivation curves of α1A-PAA/β1a (○) and α1A-PAA/R-E/β1a channels (□) in control. Curves are drawn according to the equation I/Imax=Iss+ (1 - Iss)/(1 + exp[(V - V0.5)/k]) (see Methods) with V0.5= -11.3 ± 0.5 mV, k = 7.4 ± 0.4 mV, Iss= 0.03 ± 0.01, n = 5 for α1A-PAA/β1a and V0.5= -3.1 ± 0.1 mV, k = 4.20 ± 0.08 mV, Iss= 0.07 ± 0.01, n = 4 for α1A-PAA/R-E/β1a.
Voltage dependence of IBa inactivation (‘inactivation curve’) was measured as normalised peak current during a 30 ms test pulse that was applied 3 ms after a 3 s conditioning prepulse to a given voltage. Conditioning and test pulses were applied every 60 s from a holding potential of -100 mV. Inactivation curves were fitted to the equation:
Data are given as means ±s.e.m. Statistical significance was calculated according to Student's unpaired t test (P < 0.05).
RESULTS
Mutation Arg387Glu in the domain I-II loop of the PAA-sensitive α1A-PAA-subunit and β2a-subunit interaction affect channel inactivation and sensitivity for (−)gallopamil
We have previously reported that amino acid substitutions in transmembrane segments IIIS6 and IVS6 of class A and class C α1-subunits affect Ca2+ channel inactivation kinetics and simultaneously sensitivity for PAA and BTZ (Hering et al. 1998). In order to further characterise the role of channel inactivation in Ca2+ channel block by PAAs we substituted arginine by glutamine in position 387 of the intracellular loop between domains I and II of the PAA-sensitive class A channel mutant α1A-PAA (see Herlitze et al. 1997).
As expected from previous studies on wild-type class A channels (Herlitze et al. 1997), the I-II loop mutation Arg387Glu reduced the rate of current decay of the resulting quadruple mutant α1A-PAA/R-E and shifted the mid-point of the inactivation curve to more positive potentials (Fig. 1A, B and F). IBa of α1A-PAA/R-E/β1a displayed less use-dependent IBa inhibition by (−)gallopamil (10 and 100 μm) compared with α1A-PAA/β1a (Fig. 1C andD).
Next we determined if a modulation of the current decay by different β-subunits would affect Ca2+ channel block by (−)gallopamil. It is well established that coexpression of the β2a-isoform induces a slow rate of voltage-dependent inactivation of Ca2+ channels (Stea et al. 1994). Accordingly, coexpression of the mutants α1A-PAA and α1A-PAA/R-E with the β2a-subunit dramatically decreased IBa inactivation (Fig. 1A and B). Again, slower inactivation kinetics induced significantly less use-dependent block of α1A-PAA/β2a and α1A-PAA/R-E/β2a compared with α1A-PAA/β1a and α1A-PAA/R-E/β1a channels (Fig. 1C and D). Interestingly, peak IBa inhibition by (−)gallopamil occurred at a faster rate if Ca2+ channels were coexpressed with the β1a-subunit (Fig. 1E). There was little additional effect of the point mutation on inactivation rate once the β2a-subunit was coexpressed (Fig. 1A); however, there was still a substantial effect on drug sensitivity (Fig. 1D).
We did not observe significant resting channel block (<5 %) by 10 μm (−)gallopamil. Resting channel inhibition induced by 100 μm (−)gallopamil was 15.0 ± 1.7 % (α1A-PAA/β1a), 6.7 ± 1.1 % (α1A-PAA/R-E/β1a, P < 0.01 compared with α1A-PAA/β1a), 20.1 ± 1 % (α1A-PAA/β2a) and 18.2 ± 1 % (α1A-PAA/R-E/β2a) (n≥ 4).
Mutation Arg387Glu and α1-β2a-subunit interaction differently affect recovery of Ca2+ channels from block
To elucidate the molecular basis of the different (−)gallopamil sensitivities of α1A-PAA and α1A-PAA/R-E coexpressed either with the β1a- or β2a-subunit, we analysed the drug-induced changes in IBa recovery from inactivation and block. The time courses of IBa recovery were fitted to a double-exponential function (see Methods). Under control conditions the slow component in Ca2+ channel repriming reflects recovery of Ca2+ channels from a slow or ultra-slow inactivated state (Boyett et al. 1994). (−)Gallopamil dose-dependently slowed recovery in all mutants (Fig. 2) whereas the time constant of recovery from fast inactivation was not significantly affected by the drug. The latter finding suggests that slow recovery reflects the fraction of drug-modified Ca2+ channels.
Figure 2. Recovery of Ca2+ channels from inactivation and block by (−)gallopamil.
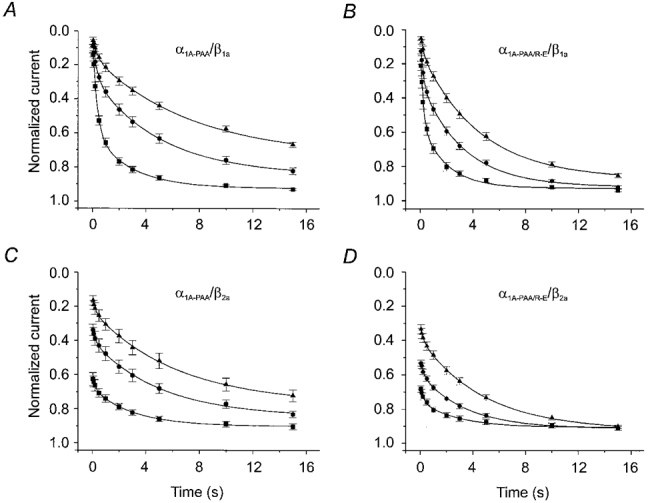
IBa recovery of α1A-PAA/β1a (A), α1A-PAA/R-E/β1a (B), α1A-PAA/β2a (C) and α1A-PAA/R-E/β2a (D) was measured by a two-pulse protocol in the absence (▪) and presence of 10 (•) and 100 μm (−)gallopamil (▴). Test pulses were applied at various times (between 50 ms and 15 s) after a conditioning prepulse (see Methods). IBa values were normalised to peak IBa of the conditioning prepulse and plotted as a function of time. Data points are fitted by a double-exponential function (see Table 1 for mean values).
Mutation Arg387Glu did more than just slow the rate of current inactivation (see Fig. 1A). As shown in Fig. 2A and B, α1A-PAA/R-E/β1a channels recovered significantly faster from inactivation (τfast= 0.20 ± 0.04 s, τslow= 1.9 ± 0.3 s, n = 7) compared with α1A-PAA/β1a (τfast= 0.34 ± 0.04 s, τslow= 2.8 ± 0.4 s, n = 7, P < 0.05, Table 1). Faster IBa recovery from inactivation was accompanied by significantly faster recovery of α1A-PAA/R-E/β1a channels from block by (−)gallopamil (α1A-PAA/R-E/β1a: τslow, 100 μm= 4.4 ± 0.2 s, n = 6; α1A-PAA/β1a: τslow, 100 μm= 7.3 ± 0.6 s, n = 6, P < 0.05, Table 1).
Table 1.
Time constants (τ, s) and corresponding amplitude coefficients (A) of double-exponential IBa recovery from inactivation (see Fig. 2)
| Construct | Conditions | τfast | Afast | τslow | Aslow | n |
|---|---|---|---|---|---|---|
| α1A-PAA/β1A | Control | 0.34 ± 0.04 | 0.54 ± 0.03 | 2.8 ± 0.4 | 0.33 ± 0.03 | 7 |
| 10 μm | 0.34 ± 0.06 | 0.21 ± 0.02 | 5.0 ± 0.3 | 0.57 ± 0.01 | 5 | |
| 100 ± M | 0.34* | 0.11 ± 0.02 | 7.3 ± 0.6 | 0.59 ± 0.01 | 6 | |
| α1A-PAA/R-E/β1A | Control | 0.20 ± 0.04 | 0.43 ± 0.04 | 1.9 ± 0.3 | 0.39 ± 0.04 | 7 |
| 10 μm | 0.23 ± 0.04 | 0.24 ± 0.02 | 3.3 ± 0.2 | 0.61 ± 0.02 | 4 | |
| 100 μm | 0.2* | 0.10 ± 0.01 | 4.4 ± 0.2 | 0.75 ± 0.01 | 6 | |
| α1A-PAA/β2A | Control | 0.23 ± 0.05 | 0.073 ± 0.007 | 3.0 ± 0.2 | 0.221 ± 0.007 | 4 |
| 10 μm | 0.4 ± 0.2 | 0.09 ± 0.02 | 5.1 ± 0.4 | 0.43 ± 0.02 | 4 | |
| 100 μm | 0.23* | 0.08 ± 0.01 | 6.3 ± 0.4 | 0.54 ± 0.01 | 4 | |
| α1A-PAA/R-E/β2A | Control | 0.24 ± 0.07 | 0.09 ± 0.01 | 2.8 ± 0.4 | 0.15 ± 0.01 | 5 |
| 10 μm | 0.28 ± 0.09 | 0.08 ± 0.01 | 3.5 ± 0.2 | 0.32 ± 0.01 | 5 | |
| 100 μm | 0.24* | 0.071 ± 0.008 | 4.9 ± 0.2 | 0.53 ± 0.01 | 5 |
Data were obtained by fitting the kinetics of IBa recovery to a biexponential function (Fig. 2, see Methods).
In the presence of 100 μm (−)gallopamil τfast was fixed to τfast,control.
As shown in Fig. 1A, coexpressing α1A-PAA and α1A-PAA/R-E with the β2a-subunit almost completely diminished fast IBa inactivation. This finding was confirmed by the corresponding recovery experiments. During the 3 s conditioning pulse less than 10 % of α1A-PAA/β2a or α1A-PAA/R-E/β2a channels accumulated in fast inactivation compared with more than 50 % of α1A-PAA/β1a and α1A-PAA/R-E/β1a (Fig. 2). Subsequent application of 10 or 100 μm (−)gallopamil substantially accelerated the IBa decay of all channel constructs (Fig. 3) and attenuated the slow component in IBa recovery (Fig. 2). Furthermore, drug-induced acceleration of the current decay was more prominent in α1A-PAA/β2a and α1A-PAA/R-E/β2a channels. As previously observed for α1A-PAA/R-E/β1a, recovery of α1A-PAA/R-E/β2a channels from block was more rapid than recovery of α1A-PAA/β2a (Fig. 2 and Table 1).
Figure 3. (−)Gallopamil-induced acceleration of IBa decay.
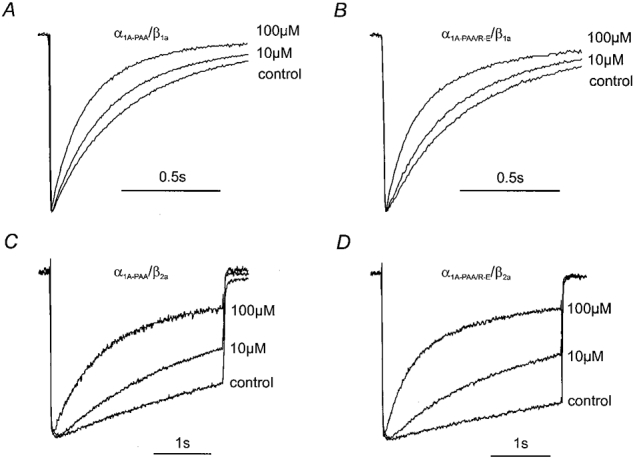
Normalised IBa of α1A-PAA/β1a, α1A-PAA/R-E/β1a, α1A-PAA/β2a and α1A-PAA/R-E/β2a in control and in the presence of 10 and 100 μm (−)gallopamil are superimposed to illustrate channel block during 1 s (α1A-PAA/β1a, α1A-PAA/R-E/β1a, A and B) or 3 s (α1A-PAA/β2a, α1A-PAA/R-E/β2a, C and D) test pulses from -80 mV to 20 mV.
DISCUSSION
Photoaffinity labelling experiments, radioligand binding studies and alanine scanning mutagenesis of L-type transmembrane segments IIIS6 and IVS6 suggest that the drug binding pockets for PAA, BTZ and 1,4-dihydropyridines are located near the Ca2+ channel pore (see Striessnig et al. 1998 for review). However, substitutions of inactivation determinants that have been identified in close proximity to the putative drug binding determinants on pore forming S6 segments significantly modulate sensitivity for PAA (see Hering et al. 1997) and BTZ (Berjukow et al. 1999). The latter findings suggest that those amino acids form either part of the drug-binding site or, alternatively, affect PAA and BTZ sensitivity in an indirect manner (i.e. via conformational changes modulating drug access, drug trapping or the steric orientation of the receptor determinants in the pore region, see Hering et al. 1998 for review). We have, therefore, investigated if inactivation determinants that are localised outside the putative drug-binding region have similar modulatory effects on sensitivity for the phenylalkylamine (−)gallopamil.
Here we demonstrate that an amino acid substitution (Arg387Glu) in the intracellular loop between domains I and II of the PAA-sensitive class A Ca2+ channel mutant α1A-PAA and coexpression of α1A-PAA with the β2a-subunit slow the rate of IBa inactivation and, simultaneously, reduce use-dependent Ca2+ channel block by (−)gallopamil (Fig. 1).
It is highly unlikely that Arg387 forms part of the PAA-binding site. Hence, arginine in position 387 of the α1A-PAA-subunit was mutated to the corresponding glutamate of the L-type α1C-a-subunit. Since L-type channels carry the high-affinity PAA-binding site, transfer of a L-type amino acid to α1A-PAA would be expected to increase and not, as shown here, to decrease PAA sensitivity (Fig. 1C, D and E).
It appears more likely that mutation Arg387Glu and the association of the β2a-subunit with a motif on the I-II loop known as the ‘alpha interaction domain’ (AID; Pragnell et al. 1994; see also Walker et al. 1998 for a second β-interaction motif located on the carboxyl terminus of α1A) affect drug sensitivity indirectly by modulating channel inactivation. In other words, our data are consistent with the hypothesis that the observed effects on gallopamil sensitivity are secondary to the change in inactivation and recovery rates. It appears, therefore, likely that the different inactivation rates of naturally occurring Ca2+ channel splice variants (Zuhlke et al. 1998; Bourinet et al. 1999) affect their pharmacological properties.
Such a mechanism clearly differs from observations of Zamponi et al. (1996) indicating that the I-II loop of class A channels forms part of the piperidine receptor.
As shown in Fig. 2B, α1A-PAA/R-E/β1a channels display a faster recovery from inactivation than α1A-PAA/β1a (see also Table 1). α1A-PAA/R-E/β1a also recovered more rapidly from block by (−)gallopamil suggesting that mutation Arg387Glu affects sensitivity for (−)gallopamil by accelerating channel unblock at rest (Table 1).
The β2a-subunit-induced changes in α1A-PAA inactivation (Fig. 1) and the consequences for use-dependent channel block by (−)gallopamil were even more dramatic (Fig. 2). Coexpression of the β2a-subunit almost completely diminished fast inactivation during a depolarising test pulse (Fig. 1A). However, the strong time-dependent block of α1A-PAA/β2a channels indicates that nearly complete lack of fast inactivation does not prevent Ca2+ channel block (Fig. 3C and D). This finding is in line with the comparable resting channel inhibition by 100 μm (−)gallopamil observed for α1A-PAA/β2a, α1A-PAA/R-E/β2a and α1A-PAA/β1a.
The time constants of α1A-PAA/β2a recovery from fast and slow inactivation did not significantly differ from α1A-PAA/β1a. Accordingly, drug bound α1A-PAA/β2a channels recovered at nearly the same rate as the ‘higher sensitive’α1A-PAA/β1a channels (Fig. 2 and Table 1), and the reduced use-dependent inhibition of α1A-PAA/β2a channels is, in line with the slower rate of IBa inhibition (Fig. 1E), caused by a reduced block development during membrane depolarisation.
Arg387Glu also diminished use-dependent channel block by (−)gallopamil (Fig. 1D). However, contrary to the effect of the β2a-subunit, this amino acid substitution affected not only the rate of block development but also the rate of recovery from block at rest. ‘Lower PAA sensitivity’ of α1A-PAA/R-E/β1a (see Fig. 1) is, therefore, mainly caused by a faster channel unblock between individual pulses (Fig. 2A and B). This hypothesis fits nicely with Arg387Glu-induced changes in channel inactivation (Fig. 1B and F) that would reduce drug trapping in inactivation and facilitate recovery from block at rest (Fig. 2A and B).
Under control conditions, the effect of the Arg387Glu substitution on IBa recovery of α1A-PAA/R-E/β2a was less evident than in α1A-PAA/R-E/β1a channels. However, a crucial role of Arg387Glu for channel unblock was confirmed by significantly faster recovery of α1A-PAA/R-E/β2a (compared with α1A-PAA/β2a) in the presence of 10 and 100 μm (−)gallopamil (Fig. 2 and Table 1). In other words, reduced use-dependent block of α1A-PAA/R-E/β2a (compared with α1A-PAA/β2a, Fig. 1D) is also due to facilitated channel unblock between individual test pulses of a train (Table 1).
Taken together, use-dependent block was reduced either by slowing channel inactivation (caused by β2a-interaction) or by speeding recovery (mutation Arg387Glu). The additive and kinetically different effects of the β2a-subunit interaction and mutation Arg387Glu on channel block (Fig. 1D) and recovery (Fig. 2) indicate that the corresponding conformational changes in the α1-subunit are distinct and independent. Interestingly, only α1A-PAA/R-E/β1a, but not α1A-PAA/β2a or α1A-PAA/R-E/β2a, displayed significantly different resting channel block compared with α1A-PAA/β1a channels.
It is tempting to speculate that reduced use-dependent block of α1A-PAA/β2a channels during a train (Fig. 1D) reflects β2a-induced changes in the steric orientation of the putative PAA binding determinants on pore forming S6-segments during a membrane depolarisation whereas Arg387Glu-induced changes in inactivation promote accelerated channel unblock. A detailed characterisation of the functional consequences of an amino acid substitution (i.e. possible modulation of fast and/or slow inactivation gating) is, therefore, a key requirement for future mutational studies directed towards the identification of drug-binding domains.
Acknowledgments
We thank Professor H. Glossmann for continuous support of our work and Dr Stanislav Berjukow for comments on the manuscript. We also thank Drs Y. Mori and K. Imoto for the gift of the α1A cDNA, Dr A. Schwartz for providing the α2/δ cDNA, Dr L. Birnbaumer for providing the β2a cDNA and Dr Traut (Knoll AG, Ludwigshafen, Germany) for providing (−)gallopamil. This work was supported by grants from the Fonds zur Förderung der Wissenschaftlichen Forschung P12649 (S.H.), a grant from the Else Kröner Fresenius Stiftung (S.H.), and a grant from the Österreichische Nationalbank (S.H.), and is part of the thesis of S.S.
References
- Aczél S, Kurka B, Hering S. Mechanism of voltage- and use-dependent block of class A Ca2+ channels by mibefradil. British Journal of Pharmacology. 1998;125:447–454. doi: 10.1038/sj.bjp.0702092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berjukow S, Gapp F, Aczél S, Sinnegger MJ, Mitterdorfer J, Glossmann H, Hering S. Sequence differences between α1C and α1S Ca2+ channel subunits reveal structural determinants of a guarded and modulated benzothiazepine receptor. Journal of Biological Chemistry. 1999;274:6154–6160. doi: 10.1074/jbc.274.10.6154. [DOI] [PubMed] [Google Scholar]
- Bezprozvanny I, Tsien RW. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40–5967) Molecular Pharmacology. 1995;48:540–549. [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Sutton K, Slaymaker S, Mathews E, Monteil A, Zamponi GW, Nargeot J, Snutch TP. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nature Neuroscience. 1999;2/5:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- Boyett MR, Honjo H, Harrison SM, Zang WJ, Kirby MS. Ultra-slow voltage-dependent inactivation of the calcium current in guinea-pig and ferret ventricular myocytes. Pflügers Archiv. 1994;428:39–50. doi: 10.1007/BF00374750. [DOI] [PubMed] [Google Scholar]
- Degtiar VE, Aczél S, Doring F, Timin EN, Berjukow S, Kimball D, Mitterdorfer J, Hering S. Calcium channel block by (−)devapamil is affected by the sequence environment and composition of the phenylalkylamine receptor site. Biophysical Journal. 1997;73:157–167. doi: 10.1016/S0006-3495(97)78056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabner M, Wang Z, Hering S, Striessnig J, Glossmann H. Transfer of 1,4-dihydropyridine sensitivity from L-type to class A (BI) calcium channels. Neuron. 1996;16:207–218. doi: 10.1016/s0896-6273(00)80037-9. [DOI] [PubMed] [Google Scholar]
- Hering S, Aczél S, Grabner M, Doring F, Berjukow S, Mitterdorfer J, Sinnegger MJ, Striessnig J, Degtiar VE, Wang Z, Glossmann H. Transfer of high sensitivity for benzothiazepines from L-type to class A (BI) calcium channels. Journal of Biological Chemistry. 1996;271:24471–24475. doi: 10.1074/jbc.271.40.24471. [DOI] [PubMed] [Google Scholar]
- Hering S, Aczél S, Kraus RL, Berjukow S, Striessnig J, Timin EN. Molecular mechanism of use-dependent calcium channel block by phenylalkylamines: role of inactivation. Proceedings of the National Academy of Sciences of the USA. 1997;94:13323–13328. doi: 10.1073/pnas.94.24.13323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hering S, Berjukow S, Aczél S, Timin EN. Calcium channel block and inactivation: common molecular determinants. Trends in Pharmacological Sciences. 1998;19:439–443. doi: 10.1016/s0165-6147(98)01258-9. [DOI] [PubMed] [Google Scholar]
- Herlitze S, Hockerman HG, Scheuer T, Catterall WA. Molecular determinants of inactivation and G protein modulation in the intracellular loop connecting domains I and II of the calcium channel α1A subunit. Proceedings of the National Academy of Sciences of the USA. 1997;94:1512–1516. doi: 10.1073/pnas.94.4.1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hockerman GH, Johnson BD, Abbott MR, Scheuer T, Catterall WA. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels in transmembrane segment IIIS6 and the pore region of the alpha1 subunit. Journal of Biological Chemistry. 1997a;272:18759–18765. doi: 10.1074/jbc.272.30.18759. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Johnson BD, Scheuer T, Catterall WA. Molecular determinants of high affinity phenylalkylamine block of L-type calcium channels. Journal of Biological Chemistry. 1995;270:22119–22122. doi: 10.1074/jbc.270.38.22119. [DOI] [PubMed] [Google Scholar]
- Hockerman GH, Peterson BZ, Johnson BD, Catteral WA. Molecular determinants of drug binding and action on L-type calcium channels. Annual Review of Pharmacology and Toxicology. 1997b;37:361–396. doi: 10.1146/annurev.pharmtox.37.1.361. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annual Review in Pharmacology and Toxicology. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene. 1989;77:61–68. doi: 10.1016/0378-1119(89)90359-4. [DOI] [PubMed] [Google Scholar]
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Pelzer D, Trautwein W. Cat ventricular muscle treated with D600: characteristics of calcium channel block and unblock. The Journal of Physiology. 1984;352:217–241. doi: 10.1113/jphysiol.1984.sp015288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pragnell M, De Waard M, Mori Y, Tanabe T, Snutch TP, Campbell KP. Calcium channel β-subunit binds to a conserved motif in the I-II cytoplasmic linker of the α1-subunit. Nature. 1994;368:67–70. doi: 10.1038/368067a0. [DOI] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, Snutch TP. Localisation and functional properties of a rat brain α1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:10576–10580. doi: 10.1073/pnas.91.22.10576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinnegger MJ, Glossmann H. Structural basis of drug binding to L Ca2+ channels. Trends in Pharmacological Sciences. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- Walker D, Bichet D, Campbell KP, De Waard M. A β4 isoform-specific interaction site in the carboxyl-terminal region of the voltage-dependent Ca2+ channel α1A subunit. Journal of Biological Chemistry. 1998;273:2361–2367. doi: 10.1074/jbc.273.4.2361. [DOI] [PubMed] [Google Scholar]
- Zamponi GW, Soong TW, Bourinet E, Snutch TP. β subunit coexpression and the α1 subunit domain I-II linker affect piperidine block of neuronal calcium channels. Journal of Neuroscience. 1996;16:2430–2443. doi: 10.1523/JNEUROSCI.16-08-02430.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuhlke RD, Bouron A, Soldatov NM, Reuter H. Ca2+ channel sensitivity towards the blocker isradipine is affected by alternative splicing of the human α1C subunit gene. FEBS Letters. 1998;427:220–224. doi: 10.1016/s0014-5793(98)00425-6. [DOI] [PubMed] [Google Scholar]