

Abstract
The effects of noradrenaline on neurotransmission at rat hippocampal synapses were investigated by recording autaptic currents in single neurons isolated on glial microislands. Noradrenaline reduced excitatory, but not inhibitory, autaptic currents in a pertussis toxin-sensitive manner, but the amine did not affect glutamate-evoked currents.
The inhibition of excitatory autaptic currents by noradrenaline was half-maximal at 0.11 ± 0.06 μm. The α2-adrenoceptor agonists UK 14304 and clonidine were equipotent to noradrenaline in reducing these currents, whereas the α1-adrenoceptor agonist methoxamine and the β-adrenoceptor agonist isoprenaline (isoproterenol) were ineffective. The reduction of excitatory autaptic currents by noradrenaline was not altered by the α1-adrenergic antagonist urapidil or the β-antagonist propranolol, but reduced by the α2-antagonist yohimbine. The subtype-preferring antagonists rauwolscine and phentolamine (both at 0.3 μm) caused 9-fold and 36-fold rightward shifts in the concentration-response curve for the noradrenaline-dependent reduction of excitatory autaptic currents, respectively. Prazosine (1 μm) did not affect this concentration-response curve.
Noradrenaline reduced voltage-activated Ca2+ currents in excitatory, but not in inhibitory, microisland neurons. For comparison, the GABAB agonist baclofen reduced both excitatory and inhibitory autaptic currents and diminished voltage-activated Ca2+ currents in both types of neurons. The inhibition of Ca2+ currents by noradrenaline was half-maximal at 0.17 ± 0.05 μm, and UK 14 304 and clonidine were equipotent to noradrenaline in reducing these currents. The noradrenaline-induced reduction of Ca2+ currents was antagonized by yohimbine, but not by urapidil or propranolol; the subtype-preferring α2-adrenergic antagonists displayed the following rank order of activity: phentolamine > rauwolscine > prazosine.
Noradrenaline did not affect K+ currents and failed to alter the frequency of miniature excitatory postsynaptic currents measured in mass cultures of hippocampal neurons.
These results show that noradrenaline regulates transmission at glutamatergic, but not at GABAergic, hippocampal synapses via presynaptic α2-adrenoceptors of the α2A/D subtype. This inhibitory action involves an inhibition of voltage-activated Ca2+ currents, but no modulation of spontaneous vesicle exocytosis or of voltage-activated K+ currents.
The hippocampal formation receives a dense noradrenergic innervation which originates primarily in the locus coeruleus (Loy et al. 1980), and the storage and release of noradrenaline in hippocampal preparations has been investigated in detail (Verhage et al. 1992). Moreover, neurons within the hippocampus express receptors for noradrenaline, i.e. adrenoceptors, including α1-, α2- and β-subtypes, at significant levels (Nicholas et al. 1996). Nevertheless, the actions of noradrenaline on hippocampal neurons remain controversial, ranging from inhibition to excitation. Inhibitory effects reported for noradrenaline include the following phenomena. (i) The amine may cause hyperpolarizations in hippocampal pyramidal neurons, an effect accompanied by a decrease in the frequency of spontaneous action potentials (Segal, 1981; Madison & Nicoll, 1986). (ii) Noradrenaline facilitates the presynaptic release of γ-aminobutyric acid (GABA; Pittaluga & Raiteri, 1987). (iii) The catecholamine reduces excitatory postsynaptic potentials via a presynaptic site of action (Scanziani et al. 1993) and inhibits the release of glutamate (Kamisaki et al. 1992). (iv) Endogenous noradrenaline was suggested to be involved in inhibitory postsynaptic potentials in the hippocampus (Andreasen & Lambert, 1991). On the other hand, a number of excitatory actions of noradrenaline have been detected in the hippocampal formation. Noradrenaline was found (i) to depolarize pyramidal neurons (Madison & Nicoll, 1986) and (ii) to reduce inhibitory postsynaptic potentials (Madison & Nicoll, 1988), presumably by decreasing excitatory postsynaptic potentials onto interneurons (Doze et al. 1991). In other brain regions, such as the cerebellum, noradrenaline was also found to exert contrasting actions including augmentation as well as reduction of glutamate release (Dolphin, 1982) and facilitation as well as inhibition of spontaneous and evoked inhibitory postsynaptic currents (Kondo & Marty, 1998). In contrast to these diverging results obtained in different in vitro preparations, in vivo, noradrenaline appears to be a mainly inhibitory neurotransmitter, since this biogenic amine exerts antiepileptic activity (Chauvel & Trottier, 1986).
The present study was performed to re-evaluate the actions of noradrenaline in hippocampal neurons and focused particularly on presynaptic actions of the amine. To identify effects of noradrenaline specific for either GABAergic or glutamatergic neurons, single hippocampal neurons isolated on microislands of glial cells were used; these neurons form synapses exclusively onto themselves, i.e. autapses (Bekkers & Stevens, 1991). Such cultures are ideally suited to investigate the function of neurotransmitter receptors in either glutamatergic or GABAergic neurons separated from each other (Boehm & Betz, 1997).
METHODS
Cell culture
Microisland as well as mass cultures of rat hippocampal neurons were prepared as described before (Boehm & Betz, 1997). Briefly, hippocampi were dissected from neonatal Sprague-Dawley rats which had been killed by decapitation in accordance with the rules of the university animal welfare committee. The tissue was cut into small pieces which were incubated in papain (1 mg ml−1 in L-15 Leibovitz Medium; Worthington) for 30 min at 36°C, and dissociated by trituration in Dulbecco's modified Eagle's medium (Gibco) containing 10 % fetal celf serum and 5 μg ml−1 insulin, 5 μg ml−1 transferrin, 5 ng ml−1 sodium selenite (Boehringer, Mannheim, Germany), 10 nM progesterone, 2 mM MgSO4, 25 000 i.u. l−1 penicillin, and 25 mg l−1 streptomycin (Sigma, Vienna, Austria). To obtain mass cultures, approximately 50 000 cells were seeded into microchambers created by glass rings (i.d., 10 mm) placed in the centre of 35 mm culture dishes (Nunc no. 150350; Rosklide, Denmark) coated with poly-D-lysine (Sigma no. 1149; 1 mg ml−1). To obtain microisland cultures, about 50 000 cells were plated in 35 mm culture dishes prepared as follows. The dishes were first coated with 0.15 % agarose, sterilized by UV irradiation, and then a mixture of poly-D-ornithine (1 mg ml−1) and collagen (4 mg ml−1) was sprayed onto the dishes under sterile conditions using a chromatography microatomizer (NeoLab, Heidelberg, Germany). After 3-5 days, 3 μm cytosine arabinoside was added to the culture medium to reduce the proliferation of non-neural cells. Mass cultures remained unaltered until experiments were performed. After 6 days in vitro, microisland cultures were treated with 100 μm glutamate in recording solution (see below) containing no Mg2+, but 2 mM Ca2+, for 60 min at room temperature; this procedure removed virtually all neurons and left pure glial microislands behind. On day 7, approximately 40 000 freshly dissociated hippocampal cells were seeded onto these microislands. Cytosine (3 μm) arabinoside was added again after 3-5 days.
Electrophysiological recordings of autaptic, glutamate-evoked, and voltage-activated Ca2+ and K+ currents in microculture neurons
The electrophysiological procedures employed with hippocampal microculture neurons have previously been described in detail (Boehm & Betz, 1997). Microcultures were used for whole-cell patch-clamp (Hamill et al. 1981) experiments after 6-18 days in vitro. To record autaptic currents, neurons were clamped at a holding potential of -70 mV and depolarized for 1 ms to voltages between 0 and 30 mV. This stimulation protocol was repeated once per 20 s. Currents through glutamate receptors were elicited by the direct application of 100 μm glutamate to neurons clamped at -70 mV in either the absence or the continuous presence of noradrenaline. Ca2+ currents were elicited by 30 ms depolarizations from a holding potential of -80 mV to 0 mV once every 20 s. K+ currents were evoked by 200 ms ramp depolarizations from -70 to +50 mV, again once every 20 s. When Ca2+ or K+ currents were recorded, the bathing solution contained 1 μm tetrodotoxin (TTX). All current recordings were performed with an EPC-7 (List Medical, Darmstadt, Germany) amplifier linked to an Olivetti personal computer controlled by pCLAMP software (version 6.0; Axon Instruments, Foster City, CA, USA). Electrodes were pulled from borosilicate glass capillaries (Science Products, Frankfurt/Main, Germany) with a Flaming-Brown puller (Sutter Instruments, Novato, CA, USA) to yield tip resistances of 2-5 MΩ.
Electrophysiological recordings of miniature excitatory postsynaptic, voltage-activated Ca2+ and afterhyperpolarization currents in mass culture neurons
Miniature excitatory postsynaptic currents (mEPSCs), voltage-activated Ca2+ currents, and afterhyperpolarization K+ currents were also recorded in mass culture neurons. Like microcultures, mass cultures were used for experiments 6-18 days after plating. The bathing solution (see below) contained 1 μm TTX, 2 mM kynurenic acid, and 30 μm bicuculline methiodide (BMI) to minimize the probability of polysynaptic events. The neuron from which mEPSCs were recorded was continuously superfused with bathing solution containing only TTX and bicuculline to permit the occurence of mEPSCs. Whole-cell afterhyperpolarization currents (IAHP) were recorded by the amphotericin B perforated patch technique in order to prevent rundown which is prominent under open-tip whole-cell recording conditions (Boehm, 1998).
Solutions and drug application
The bathing solution consisted of (mM): NaCl, 140; KCl, 3; CaCl2, 3; MgCl2, 2; glucose, 20; and Hepes, 10; adjusted to pH 7.4 with NaOH. Neurons under investigation were continuously superfused with this solution containing test drugs when appropriate. Superfusion was performed with a DAD-12 (Adams & List, Westbury, NY, USA) drug application system which permits complete exchange of solutions within less than 100 ms (Boehm et al. 1997).
Pipettes were filled with a solution containing (mM): KCl, 140; CaCl2, 1.6; EGTA, 10; Hepes, 10; Mg-ATP, 2; Li-GTP, 2; adjusted to pH 7.3 with KOH to record autaptic currents, K+ currents and mEPSCs. For the measurement of Ca2+ curents, the pipette solution contained (mM): CsCl, 120; tetraethylammonium chloride, 20; CaCl2, 0.24; glucose, 10; Hepes, 10; EGTA, 5; Mg-ATP, 2; Li-GTP, 2; adjusted to pH 7.3 with NaOH. For amphotericin B perforated patch recordings, patch pipettes were first front-filled with a solution consisting of (mM): K2SO4, 75; KCl, 55; MgCl2, 8; Hepes, 10; adjusted to pH 7.3 with KOH, and then back-filled with the same solution containing 200 μg ml−1 amphotericin B (in 0.8 % DMSO, see Boehm, 1998).
Calculations and statistics
Current amplitudes in the presence (B) of noradrenaline and other test drugs were compared with those obtained before application (A) and after washout (C); drug effects were evaluated either as a percentage of control (= 200 ×B/(A+C)) or as percentage inhibition (= 100 × (1 - 2B/(A+C))).
Results are presented as arithmetic means ±s.e.m.; n is number of cells. Differences between data points were evaluated by the Mann-Whitney U test. Concentration-response curves were fitted to experimentally obtained data by the ALLFIT program (DeLean et al. 1978) which also determines differences between single concentration-response curves by simultaneous fitting with shared parameters and subsequent calculation of the F statistic on the resulting ‘extra sum of squares’.
Materials
Noradrenaline, methoxamine, isoprenaline (isoproterenol), prazosine, phentolamine, rauwolscine, urapidil, yohimbine, bicuculline methiodide (BMI), glutamate, kynurenic acid, and pertussis toxin were obtained from Sigma (Vienna, Austria); cyano-2,3-dihydroxy-7-nitroquinoxaline (CNQX) from Tocris Cookson (Bristol, UK); tetrodotoxin (TTX) from Latoxan (Rosans, France); 5-bromo-N-(4,5-dihydro-1H-imidazol-2-yl)-6-quinoxalinamine (UK 14 304) from Research Biochemicals Inc. (Natick, MA, USA).
RESULTS
Effects of noradrenaline on autaptic and glutamate-evoked currents in hippocampal microisland neurons
Depolarization of microisland neurons for 1 ms to between 0 and +30 mV evoked either excitatory or inhibitory autaptic currents (EACs and IACs, respectively), depending on the type of neuron investigated. As reported previously (Boehm & Betz, 1997), EACs (Fig. 1A) decayed rapidly and lasted only up to 20 ms, whereas IACs (Fig. 1B) lasted more than 100 ms. Furthermore, EACs were reduced by the non-NMDA glutamate receptor antagonist CNQX (10 μm; Fig. 1A) by 94.0 ± 1.4 % (n = 13), whereas IACs were inhibited by the GABAA receptor antagonist BMI (30 μm; Fig. 1B) by 90.7 ± 2.4 % (n = 5).
Figure 1. Noradrenaline inhibits excitatory, but not inhibitory, autaptic currents.

Two different microisland neurons were depolarized from a holding potential of -70 mV to 0 mV for 1 ms. A, excitatory autaptic current (EAC) traces obtained before, during and after application of noradrenaline, and in the presence of CNQX. B, inhibitory autaptic current (IAC) traces obtained before, during and after application of noradrenaline, and in the presence of bicuculline methiodide (BMI). C, the effects of 1 μm noradrenaline on EACs (n = 23), on IACs (n = 11), and on EACs in neurons treated with 100 ng ml−1 pertussis toxin for 24 h (EAC/PTX; n = 7); horizontal lines indicate arithmetic means. D, glutamate was applied to a microisland neuron which displayed EACs before, during, and after the application of 1 μm noradrenaline; the membrane potential was clamped at -70 mV.
Noradrenaline (1 μm) reduced EACs (Fig. 1A) in an entirely reversible manner. This effect varied considerably between single neurons, and the inhibition ranged from as little as 12 % to complete abolition of EACs. On average, the inhibition of EACs by 1 μm noradrenaline amounted to 61.4 ± 6.8 % (Fig. 1C). IACs, on the contrary, were not affected by noradrenaline (Fig. 1B), and current amplitude in the presence of the amine amounted to 100.4 ± 1.5 % of the amplitude in its absence (Fig. 1C).
The reduction of autaptic currents described above may arise at either a presynaptic or a postsynaptic site of action. To determine whether noradrenaline interferes with the function of non-NMDA glutamate receptors, which mediate EACs, glutamate (100 μm) was applied directly to excitatory microisland neurons in either the absence or the presence of 1 μm noradrenaline (Fig. 1D). Glutamate-evoked currents were not altered in the presence of the amine, and peak current amplitudes amounted to 102.6 ± 8.1 % (n = 5) of those of glutamate-induced currents recorded in its absence. Hence, noradrenaline did not affect glutamate receptors and thus reduced autaptic currents via a presynaptic site of action.
Receptors for noradrenaline belong to the superfamily of G protein-coupled receptors (Summers & McMartin, 1993), and presynaptic α2-adrenergic inhibition is most frequently mediated by proteins of the Gi/Go class (Boehm et al. 1996). To investigate whether this type of G protein mediated the inhibitory action of noradrenaline in excitatory microisland neurons, cultures were treated with pertussis toxin (PTX; 100 ng ml−1 for 24 h). This treatment either attenuated or, more commonly, abolished the inhibitory action of noradrenaline on EACs (Fig. 1C). Thus, noradrenaline inhibited EACs via Gi or Go type G proteins.
Noradrenaline inhibits EACs via α2-adrenoceptors
The inhibitory action of noradrenaline on EACs was concentration dependent with half-maximal effects at 0.11 ± 0.06 μm and a maximum inhibition of 65.0 ± 6.3 %. The selective α2-adrenergic agonist UK 14 304 reduced EACs by up to 59.0 ± 7.0 %, the effect being half-maximal at 0.08 ± 0.04 μm. Clonidine blocked EACs in the same range of concentration (half-maximal inhibition at 0.08 ± 0.23 μm), but the maximum of inhibition amounted to only 36.5 ± 4.8 % (Fig. 2A). To verify that clonidine acted as a partial agonist, its action at 1 μm was compared with the actions of noradrenaline and UK 14 304 at the same concentration in one set of neurons, and clonidine turned out to be significantly less effective than the other two agonists (Fig. 2B). The effect of noradrenaline on EACs was further compared with that of methoxamine, an α1-adrenoceptor-preferring agonist, and isoprenaline, a β-adrenoceptor agonist: these two agents failed to reduce EACs in neurons which displayed a pronounced inhibition in the presence of noradrenaline (Fig. 2C).
Figure 2. Pharmacological characterization of the receptor mediating the inhibition of excitatory autaptic currents.
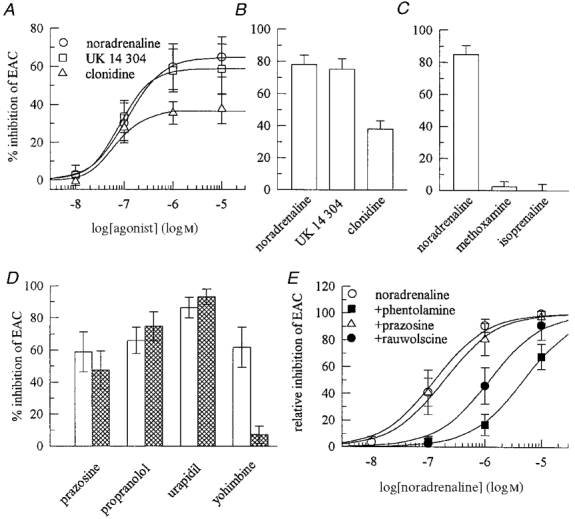
Excitatory autaptic currents (EACs) were recorded as in Fig. 1A. A, concentration-response curves for the inhibition of EACs by noradrenaline, UK 14 304 and clonidine (n = 5-8). B, the effects of noradrenaline, UK 14 304 and clonidine (all at 1 μm) on EACs in 14 microisland neurons; the effect of clonidine is significantly different (P < 0.001) from those of the other agonists. C, the effects of noradrenaline, methoxamine and isoprenaline (all at 1 μm) on EACs in 8 neurons. D, the inhibition of EACs by 1 μm noradrenaline determined in the absence (□) or presence  ) of prazosine (n = 5), propranolol (n = 9), urapidil (n = 4) and yohimbine (n = 5) (all antagonists at 1 μm); with yohimbine, there is a significant difference ( P < 0.05) between the two columns. E, concentration-response curves for the inhibition of EACs by noradrenaline applied either alone or together with 1 μm prazosine, 0.3 μm phentolamine or 0.3 μm rauwolscine (n = 6-8). Since inhibitory actions of noradrenaline may vary between single neurons (as in D), inhibitory effects are shown as a percentage of the inhibition exerted by 10 μm noradrenaline in the same neuron.
) of prazosine (n = 5), propranolol (n = 9), urapidil (n = 4) and yohimbine (n = 5) (all antagonists at 1 μm); with yohimbine, there is a significant difference ( P < 0.05) between the two columns. E, concentration-response curves for the inhibition of EACs by noradrenaline applied either alone or together with 1 μm prazosine, 0.3 μm phentolamine or 0.3 μm rauwolscine (n = 6-8). Since inhibitory actions of noradrenaline may vary between single neurons (as in D), inhibitory effects are shown as a percentage of the inhibition exerted by 10 μm noradrenaline in the same neuron.
The above results suggest that noradrenaline inhibited EACs via an α2-adrenoceptor subtype. To verify this conclusion, noradrenaline (1 μm) was applied to excitatory microisland neurons either alone or together with prazosine or urapidil, α1-adrenoceptor antagonists, with propranolol, a β-adrenoceptor antagonist, and with yohimbine, an α2-adrenoceptor antagonist, all at 1 μm. Amongst these antagonists, only yohimbine caused a significant attenuation of the noradrenaline-induced inhibition of EACs (Fig. 2D).
The subfamily of α2-adrenoceptors comprises at least four different members (α2A, α2B, α2C, α2D), and one can discriminate between these subtypes by using several antagonists (Bylund et al. 1994; MacKinnon et al. 1994), in particular phentolamine, prazosine and rauwolscine (e.g. Nörenberg, 1997). Prazosine (1 μm) failed to cause a significant alteration in the concentration-response curve for the noradrenaline-induced inhibition of EACs (the calculated rightward shift was 1.4-fold), whereas both, rauwolscine and phentolamine, both at 0.3 μm, induced an apparent 9- and 36-fold rightward shifts, respectively (Fig. 2E). Assuming competitive antagonism, one may calculate apparent affinities for these antagonists by the formula log (CR - 1) = logB - logKB, where CR is the ratio of equieffective agonist concentrations in the presence and absence of the antagonist concentration (B) (Arunlakshana & Schild, 1959). Calculation of the apparent antagonist affinities (KB) by this method gives the following values: 2.6 μm for prazosine, 33 nM for rauwolscine and 8.5 nM for phentolamine.
α2-Adrenoceptors reduce voltage-activated Ca2+ currents at the somata of excitatory neurons
Presynaptic receptors of central neurons are believed to reduce transmitter release most commonly via an inhibition of voltage-gated Ca2+ channels (Miller, 1998). Furthermore, noradrenaline is known to block Ca2+ channels in several types of neurons (Hille, 1994). Therefore it was investigated whether noradrenaline might regulate the function of Ca2+ channels in excitatory as well as inhibitory microisland neurons. Noradrenaline reduced EACs and inhibited Ca2+ currents recorded from the somata of the very same neurons (Fig. 3A). In contrast, IACs were not inhibited by noradrenaline, and in these cells the amine failed to reduce Ca2+ currents (Fig. 3B). To exclude that the failure of noradrenaline to inhibit Ca2+ currents in inhibitory neurons was due to some technical problems, its action was compared with that of the GABAB receptor agonist baclofen which is known to inhibit excitatory as well as inhibitory transmission in the hippocampus (Scanziani et al. 1992). In a set of five excitatory and four inhibitory microisland neurons, baclofen reduced both types of autaptic currents and blocked Ca2+ currents in both types of neurons (Fig. 3C). Noradrenaline, in contrast, reduced EACs, but not IACs, and blocked Ca2+ currents in excitatory, but not in inhibitory, microisland neurons (Fig. 3C).
Figure 3. Effects of noradrenaline on voltage-activated Ca2+ currents in excitatory and inhibitory microisland neurons.
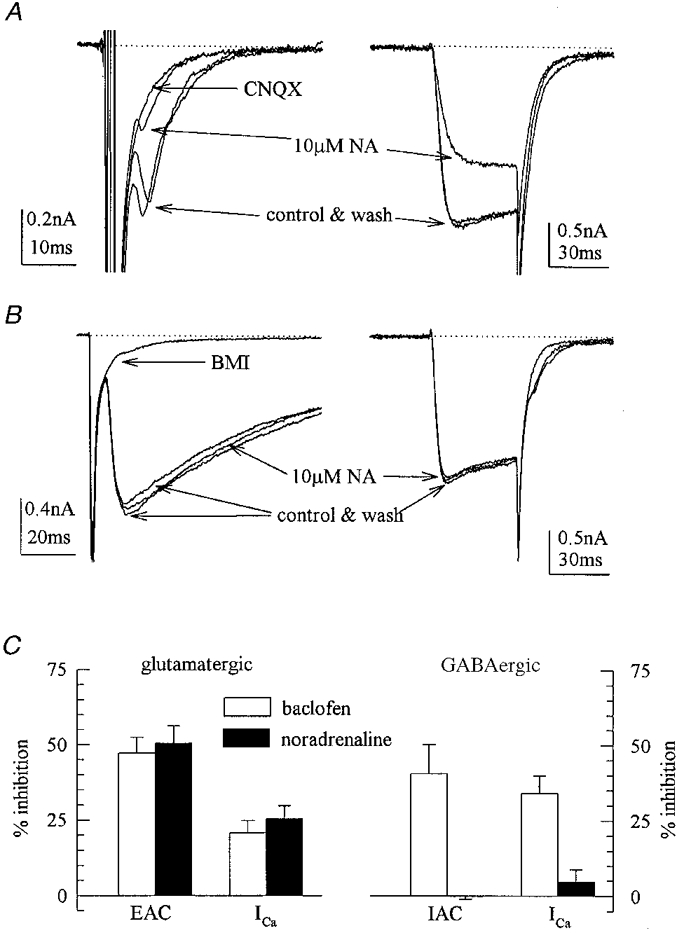
A and B, microisland neurons were first depolarized for 1 ms to evoke EACs and IACs, respectively, and then for 30 ms in the presence of 1 μm tetrodotoxin to evoke voltage-activated Ca2+ currents. The current traces shown were obtained before, during and after the application of noradrenaline (NA). C, the effects of noradrenaline and baclofen (each at 10 μm) on EACs and IACs, and on voltage-activated Ca2+ currents (ICa) in four GABAergic and five glutamatergic microisland neurons.
To determine the subtype of adrenoceptor mediating the inhibition of Ca2+ currents by noradrenaline, Ca2+ current recordings were performed in mass culture neurons (Fig. 4A). Noradrenaline caused an unequivocal reduction of Ca2+ current amplitudes in 14 of 22 neurons tested (mean inhibition 32.5 %). In the remaining eight neurons, an eventual inhibition by noradrenaline amounted to less than 5 % (mean inhibition, -0.8 %). In 11 neurons treated with pertussis toxin (100 ng ml−1 for 24 h), noradrenaline failed to inhibit Ca2+ currents and the mean inhibition amounted to -1.2 % (Fig. 4B). The inhibition of Ca2+ currents by noradrenaline was concentration dependent, the effect being half-maximal at 0.17 ± 0.05 μm. UK 14 304 and clonidine were as potent as noradrenaline in reducing Ca2+ currents with half-maximal effects at 0.12 ± 0.03 and 0.12 ± 0.08 μm, respectively. Clonidine acted again as a partial agonist, since its maximal inhibitory effect was only one-third of the inhibition exerted by UK 14 304 or noradrenaline (Fig. 4C). The inhibitory action of noradrenaline (1 μm) on Ca2+ currents was not altered in the presence of propranolol or urapidil, but largely attenuated by yohimbine (1 μm; Fig. 4D). Furthermore, the inhibitory effect of 1 μm noradrenaline was not altered in the presence of prazosine, reduced by about 50 % by rauwolscine, and almost abolished by phentolamine, all at 0.3 μm (Fig. 4E).
Figure 4. Pharmacological characterization of the receptor mediating the inhibition of voltage-activated Ca2+ currents in mass culture neurons.
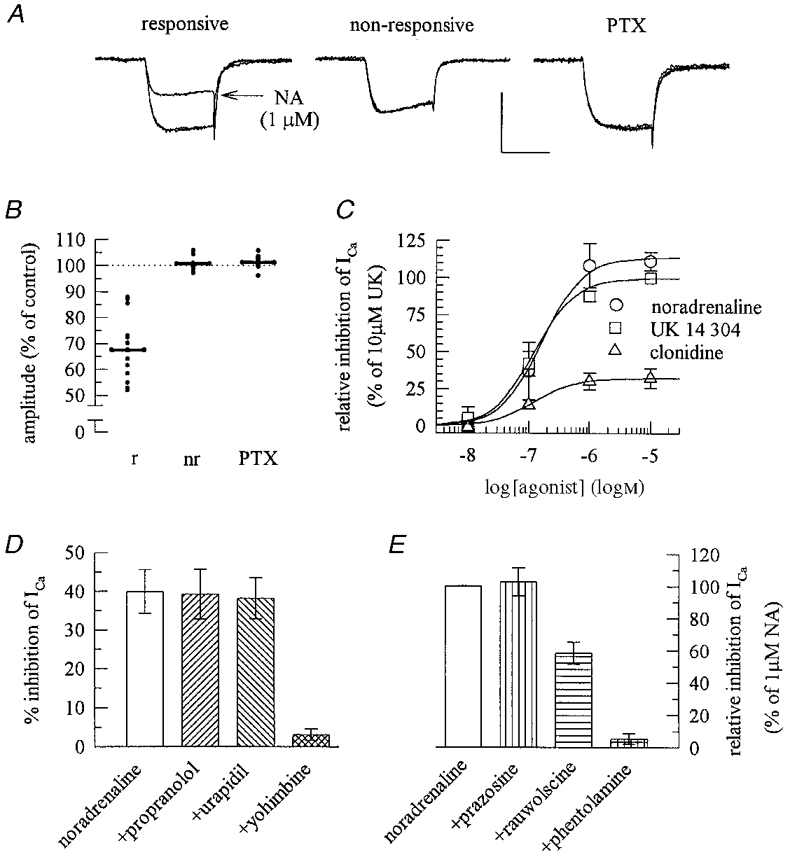
A, representative voltage-dependent Ca2+ currents elicited by 30 ms depolarizations from -80 mV to 0 mV. Each of the three panels shows three traces obtained before, during and after the application of 1 μm noradrenaline (NA) in a responsive (> 10 % inhibition) and a non-responsive (< 5 % inhibition) neuron (calibration: 1 nA, 20 ms) and in a neuron treated with 100 ng ml−1 pertussis toxin (PTX) for 24 h (calibration: 0.5 nA, 20 ms). B, relative amplitudes of currents (elicited in a similar way to those in A) in the presence of NA in responsive (r; n = 14), non-responsive (nr; n = 8) and PTX-treated (n = 11) mass culture neurons. C, concentration-response curves for the inhibition of Ca2+ currents by noradrenaline, UK 14 304 and clonidine; since the inhibitory actions of these agonists may vary between single neurons (as in B) inhibitory effects are shown as a percentage of the inhibition exerted by 10 μm UK 14 304 in the same neuron (n = 4-6). D, noradrenaline (1 μm) was applied to five mass culture neurons either alone or together with propranolol, urapidil or yohimbine (all at 1 μm); the effect obtained in the presence of yohimbine is significantly different (P < 0.01) from that of noradrenaline alone. E, noradrenaline (1 μm) was applied again to five mass culture neurons either alone or together with prazosine, rauwolscine or phentolamine (all at 0.3 μm); to account for differences in the action of noradrenaline in different neurons (as in B), inhibitory effects are shown as a percentage of the inhibition exerted by 1 μm noradrenaline in the same neuron; the effect obtained in the presence of rauwolscine is significantly different (P < 0.05) from those of either noradrenaline alone or of noradrenaline applied together with phentolamine.
α2-Adrenoceptors do not modulate voltage-activated K+ or miniature excitatory postsynaptic currents
Presynaptic receptors may reduce transmitter release not only by an inhibition of Ca2+ channels, but also by a facilitation of voltage-activated K+ channels (Miller, 1998). Accordingly, EACs and outward K+ currents in excitatory microislands neurons were recorded. As described above, noradrenaline (10 μm) markedly reduced EACs, but failed to affect outward currents evoked by ramp depolarizations from -70 to 50 mV (Fig. 5). Previously, noradrenaline had been reported to inhibit afterhyperpolarizations in hippocampal slices (Madison & Nicoll, 1986), an effect that counteracts rather than contributes to presynaptic inhibition. In mass cultures of hippocampal neurons, however, noradrenaline (10 μm) did not alter afterhyperpolarization currents (IAHP), the amplitude in its presence being 99.3 ± 2.1 % of that in its absence (n = 4).
Figure 5. Noradrenaline does not affect voltage-activated K+ currents in excitatory microisland neurons.

In microisland neurons showing EACs in response to 1 ms depolarizations (A), outward currents were elicited by 200 ms ramp depolarizations from -70 to + 50 mV (B) in the presence of 1 μm TTX; the current traces shown were obtained before, during, and after the application of noradrenaline (NA). Results equivalent to those shown in this figure were obtained in five different microisland neurons.
Agonists at various types of presynaptic receptor in hippocampal neurons may reduce glutamate release independently of voltage-gated ion channels by a direct effect on vesicle exocytosis (e.g. Scholz & Miller, 1992; Scanziani et al. 1995; Boehm & Betz, 1997). Therefore, miniature excitatory postsynaptic currents (mEPSC) were recorded in mass cultures, and noradrenaline (10 μm) was applied (Fig. 6). Under control conditions, mEPSCs were detected at a mean frequency of 3.2 ± 0.7 Hz and displayed an average amplitude of 38.6 ± 4.4 pA (n = 6). Noradrenaline failed to alter the mEPSC frequency significantly (91 ± 5.0 % of control) and did not affect mEPSC amplitudes (99.3 ± 1.8 % of control; n = 6). For comparison, the A1 adenosine receptor agonist cyclopentyladenosine (1 μm) was tested in two cells which did not respond to the application of noradrenaline, and this agent reduced the mEPSC frequency by 42 % without causing an inhibition of mEPSC amplitudes (see also Scholz & Miller, 1992).
Figure 6. Noradrenaline does not affect miniature excitatory postsynaptic currents in mass culture neurons.

Miniature excitatory postsynaptic currents were recorded at a potential of -70 mV in a mass culture neuron superfused with 1 μm TTX and 30 μm bicuculline methiodide. The traces shown were obtained before, during, and after the application of noradrenaline, and in the presence of the non-NMDA glutamate receptor antagonist CNQX.
DISCUSSION
Mechanisms of excitatory and inhibitory neurotransmission can be studied in isolation by recording autaptic currents from single glutamatergic or GABAergic neurons growing on microislands of glial cells (e.g. Bekkers & Stevens, 1991; Boehm & Betz, 1997). Using this method, the present study demonstrates that noradrenaline affects exclusively glutamatergic, but not GABAergic, neurotransmission in hippocampal neurons.
Presynaptic α2-adrenoceptors inhibit glutamate release
The reduction of excitatory autaptic currents by noradrenaline did not involve a postsynaptic mechanism, as the amine failed to modulate glutamate-evoked currents. Furthermore, noradrenaline did not alter mEPSC amplitudes, a result that also argues against a postsynaptic site of action. Hence, noradrenaline must have inhibited excitatory autaptic currents by reducing glutamate release via a presynaptic receptor. Adrenoceptors are G protein-coupled receptors, and in the present experiments the actions of noradrenaline were abolished by pretreatment of neurons with pertussis toxin, which prevents the receptor-dependent activation of Gi or Go type G proteins. Amongst adrenoceptors, it is the α2-subtype which is most commonly linked to Gi/o proteins (Summers & McMartin, 1993; Ruffolo et al. 1994). Hence, the pertussis toxin sensitivity of the noradrenaline-induced inhibition of glutamatergic autaptic currents suggests that this effect was mediated by α2-adrenoceptors.
This conclusion was further supported by pharmacological results. (i) The selective α2-adrenergic agonists UK 14 304 and clonidine mimicked the actions of noradrenaline. (ii) Neither the β-adrenergic agonist isoprenaline nor the α1-adrenergic agonist methoxamine exerted inhibitory effects on excitatory autaptic currents. (iii) The inhibitory effect of noradrenaline on these synaptic currents was attenuated by the α2-adrenoceptor antagonists phentolamine, rauwolscine and yohimbine, but not by urapidil or propranolol, antagonists at α1- and β-adrenoceptors, respectively.
Currently, four different subtypes of α2-adrenoceptor are known, and these are designated α2A to α2D (Bylund et al. 1994; MacKinnon et al. 1994). However, in any given species only three different subtypes are expressed, and the α2A and α2D subtypes which show > 89 % sequence identity are believed to represent species orthologues with α2A being expressed, for instance, in man and pig, and α2D being expressed, for example, in rats and cattle (O'Rourke et al. 1994). Presynaptic α2-adrenoceptors, in particular the presynaptic α2-autoreceptors, generally belong to the genetic α2A/D subtype (Trendelenburg et al. 1993). In the rat hippocampus, transcripts for α2A/D- and α2C-, but not for α2B-receptors have been detected (Nicholas et al. 1996). The α-adrenoceptor antagonist prazosine differentiates between α2-adrenoceptor subtypes in the following manner: α2B- and α2C-receptors show affinities for prazosine between 3 and 135 nM, whereas the affinities of α2A- and α2D-receptors for this antagonist are in the micromolar range (MacKinnon et al. 1994). Thus, the α2-adrenoceptor located at glutamatergic nerve terminals, as described above, is either an α2A- or an α2D-subtype, since the calculated affinity of prazosine for this receptor was > 1 μm. α2A- and α2D-subtypes can be discriminated from each other by using rauwolscine and phentolamine, since the order of affinities for these two antagonists is rauwolscine > phentolamine at α2A and phentolamine > rauwolscine at α2D (O'Rourke et al. 1994; Nörenberg et al. 1997). Here, phentolamine (8.5 nM) had about 4-fold higher apparent affinity for the presynaptic α2-adrenoceptors than rauwolscine (33 nM), which indicates that this receptor displays pharmacological characteristics of the α2D-subtype. This is in accordance with the notion that rats express the α2D- rather than the α2A-subtype (O'Rourke et al. 1994).
Previously, noradrenaline has been reported to reduce excitatory synaptic transmission in slice cultures of the rat hippocampus via α1-adrenoceptors (Scanziani et al. 1993). In that report, the action of noradrenaline was mimicked by the α1-adrenoceptor agonist phenylephrine and antagonized by phentolamine and prazosine (see above), but not by the α2-antagonist idazoxan. Similar results have also been obtained in rat prefrontal cortex slices, where the noradrenergic inhibition of excitatory postsynaptic potentials was abolished by prazosine, but not by yohimbine (Law-Tho et al. 1993). In contrast, in synaptosomes derived from rat anterior cortex and hippocampus, glutamate release was reduced by the activation of α2-adrenoceptors (Kamisaki et al. 1992). In situ hybridization studies have revealed that besides α2C and α2A/D, α1D subtypes of adrenoceptors can be found in the hippocampus. Amongst these adrenoceptor subtypes, the α1D and α2C receptors are expressed at the highest levels, whereas α2A/D receptors are expressed at moderate levels only (Nicholas et al. 1996). In light of the lack of effect of α1-adrenergic ligands in the present study, it remains to be established by alternative experiments, such as immunocytochemistry with subtype-specific antisera (e.g. Stone et al. 1998), whether glutamatergic hippocampal neurons, either in situ or in cell culture, do express different (and possibly more than one) subtypes of presynaptic α-adrenoceptors.
Signalling mechanisms of presynaptic α2-adrenoceptors
At least three different mechanisms may contribute to presynaptic inhibition, namely a blockade of Ca2+ currents, a facilitation of K+ currents, and a direct modulation of vesicle exocytosis (Miller, 1998). Of these effects, only the first, the inhibition of voltage-gated Ca2+ currents, was detectable in the present study: noradrenaline reduced Ca2+ currents at the somata of glutamatergic, but not of GABAergic neurons. Baclofen, for comparison, reduced excitatory as well as inhibitory autaptic currents and blocked Ca2+ currents in glutamatergic as well as GABAergic neurons. Thus, the inhibition of Ca2+ currents at neuronal somata correlates with the presynaptic inhibition of transmitter release.
Similarly to the situation in microisland neurons, noradrenaline inhibited Ca2+ currents only in a fraction (14/22) of mass culture neurons. Nevertheless, the receptor involved in the inhibition of Ca2+ currents at neuronal somata showed pharmacological characteristics identical to those of the presynaptic receptor which mediated the inhibition of EACs: (i) by means of antagonist pharmacology, both receptors unequivocally belong to the α2-subfamily; (ii) the α2-adrenoceptor agonists exerted their half-maximal effects at the two receptors at similar concentrations; (iii) clonidine acted as partial agonist at the presynaptic as well as at the somatic receptor; (iv) the receptors in both types of location display the same order of apparent antagonist affinities: phentolamine > rauwolscine >> prazosine. Taken together, the above results strongly suggest that a modulation of Ca2+ entry at presynaptic nerve terminals is involved in the presynaptic inhibition elicited by noradrenaline, even though such a mechanism could not be determined directly by the methods used in this study.
A number of presynaptic receptors in the hippocampus, including A1 adenosine (Scholz & Miller, 1992), muscarinic (Scanziani et al. 1995) and somatostatin (Boehm & Betz, 1997) receptors, reduce spontaneous vesicle exocytosis by a mechanism independent of transmembrane Ca2+ entry. However, in accordance with previous results obtained in organotypic slice cultures of rat hippocampus (Scanziani et al. 1993), noradrenaline failed to alter the frequency of mEPSCs. Furthermore, the catecholamine did not modulate K+ currents. Thus, the noradrenaline-dependent inhibition of glutamate release appears to rely exclusively on the reduction of voltage-dependent Ca2+ currents.
Mechanisms of α2-adrenoceptor-mediated inhibition of transmitter release have been studied in detail in peripheral, sympathetic, neurons: the presynaptic inhibition by α2-adrenergic agonists is lost when transmitter release is associated with transmembrane Ca2+ entry occuring independently of voltage-gated Ca2+ channels (Boehm & Huck, 1995, 1996). In sympathetic neurons of chick embryos, the α2-autoreceptor-dependent reduction of noradrenaline is based exclusively on the inhibition of one specific type of voltage-gated Ca2+ channel, of the ω-conotoxin GVIA-sensitive N-type channel (Boehm & Huck, 1996). N-Type Ca2+ channels also contribute to synaptic transmission at hippocampal synapses (Wheeler et al. 1994), but the subtypes of Ca2+ channels modulated by noradrenaline were not identified in the present study. Nevertheless, one may conclude that presynaptic α2-adrenoceptors, whether they are auto- or heteroreceptors, whether they are located in central or peripheral neurons, inhibit transmitter release through a modulation of voltage-gated Ca2+ channels.
Functional significance of presynaptic α2-adrenoceptors on glutamatergic nerve terminals
The present results show that presynaptic α2-adrenoceptors mediate an inhibition of glutamate release in hippocampal neurons in cell culture. Provided that this mechanism also occurs in vivo, these α2-adrenoceptors may be important in several pathological conditions. The noradrenergic ascending sytem of the CNS has been suggested to inhibit the spread of epileptic activity via α2-adrenoceptors (Gellmann et al. 1987), and the authors concluded that this antiepileptogenic effect was mediated by postsynaptic receptors. Considering that glutamate release is drastically enhanced due to enhanced firing during paroxysmal depolarizations within epileptic foci (Dichter & Ayala, 1987), the inhibition of glutamate release via presynaptic α2-adrenoceptors may form the basis for the reported antiepileptogenic activity of noradrenaline. Furthermore, a reduction of glutamate release may also underlie the neuroprotective activity of α2-adrenoceptor agonists in cerebral ischaemia (Hoffmann et al. 1991), since enhanced glutamate release is a key pathological event in brain ischaemia (Choi, 1990). Interestingly, endogenous noradrenaline release is significantly increased during epileptic as well as ischaemic episodes (Kokaia et al. 1989; Globus et al. 1989). Thus, presynaptic α2-adrenoceptors as described here might subserve an important neuroprotective function.
Acknowledgments
This study was supported by a grant from the Austrian Science Fund (FWF; P-12997-MED). I wish to thank A. Motejlek for excellent technical assistance and Dr M. Freissmuth for critical reading of this manuscript.
References
- Andreasen M, Lambert JDC. Noradrenaline receptors participate in the regulation of GABAergic inhibition in area CA1 of the rat hippocampus. The Journal of Physiology. 1991;439:649–669. doi: 10.1113/jphysiol.1991.sp018686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arunlakshana O, Schild HO. Some quantitative uses of drug antagonists. British Journal of Pharmacology. 1959;14:48–58. doi: 10.1111/j.1476-5381.1959.tb00928.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proceedings of the National Academy of Sciences of the USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S. Selective inhibition of M-type K+ channels in rat sympathetic neurons by uridine nucleotide preferring receptors. British Journal of Pharmacology. 1998;124:1261–1269. doi: 10.1038/sj.bjp.0701956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Betz H. Somatostatin inhibits excitatory transmission at rat hippocampal synapses via presynaptic receptors. Journal of Neuroscience. 1997;17:4066–4075. doi: 10.1523/JNEUROSCI.17-11-04066.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Harvey RJ, Von Holst A, Rohrer H, Betz H. Glycine receptors in cultured chick sympathetic neurons are excitatory and trigger neurotransmitter release. The Journal of Physiology. 1997;504:683–694. doi: 10.1111/j.1469-7793.1997.683bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm S, Huck S. α2-Adrenoceptor-mediated inhibition of acetylcholine-induced noradrenaline release from rat sympathetic neurons: an action at voltage-gated Ca2+ channels. Neuroscience. 1995;69:221–231. doi: 10.1016/0306-4522(95)00235-b. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S. Inhibition of N-type calcium channels: the only mechanism by which presynaptic α2-autoreceptors control sympathetic transmitter release. European Journal of Neuroscience. 1996;8:1924–1931. doi: 10.1111/j.1460-9568.1996.tb01336.x. [DOI] [PubMed] [Google Scholar]
- Boehm S, Huck S, Motejlek A, Drobny H, Singer EA, Freissmuth M. Cholera toxin induces cyclic AMP-independent downregulation of Gsα and sensitization of α2-autoreceptors in chick sympathetic neurons. Journal of Neurochemistry. 1996;66:1019–1026. doi: 10.1046/j.1471-4159.1996.66031019.x. [DOI] [PubMed] [Google Scholar]
- Bylund DB, Eikenberg DC, Hieble JP, Langer SZ, Lefkowitz RJ, Minneman KP, Molinoff PB, Ruffolo RR, Jr, Trendelenburg U. IV International Union of Pharmacology. Nomenclature of Adrenoceptors. Pharmacological Reviews. 1994;46:121–136. [PubMed] [Google Scholar]
- Chauvel P, Trottier S. Role of noradrenergic ascending system in extinction of epileptic phenomena. Advances in Neurology. 1986;44:475–487. [PubMed] [Google Scholar]
- Choi DW. Cerebral hypoxia: Some new approaches and unanswered questions. Journal of Neuroscience. 1990;10:2493–2501. doi: 10.1523/JNEUROSCI.10-08-02493.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delean A, Munson PJ, Rodbard D. Simultaneous analysis of families of sigmoidal curves: Application to bioassay, radioligand assay, and physiological dose-response curves. American Journal of Physiology. 1978;235:E97–102. doi: 10.1152/ajpendo.1978.235.2.E97. [DOI] [PubMed] [Google Scholar]
- Dichter MA, Ayala GF. Cellular mechanisms of epilepsy: a status report. Science. 1987;237:157–164. doi: 10.1126/science.3037700. [DOI] [PubMed] [Google Scholar]
- Dolphin AC. Noradrenergic modulation of glutamate release in the cerebellum. Brain Research. 1982;252:111–116. doi: 10.1016/0006-8993(82)90983-0. [DOI] [PubMed] [Google Scholar]
- Doze VA, Cohen GA, Madison DV. Synaptic localization of adrenergic disinhibition in the rat hippocampus. Neuron. 1991;6:889–900. doi: 10.1016/0896-6273(91)90229-s. [DOI] [PubMed] [Google Scholar]
- Gellman RL, Kallianos JA, Mcnamara JO. Alpha-2 receptors mediate an endogenous noradrenergic suppression of kindling development. Journal of Pharmacology and Experimental Therapeutics. 1987;241:891–898. [PubMed] [Google Scholar]
- Globus M Y-T, Busto R, Dietrich WD, Martinez E, Valdes I, Ginsberg MD. Direct evidence for acute and massive norepinephrine release in the hippocampus during transient ischemia. Journal of Cerebral Blood Flow and Metabolism. 1989;9:892–896. doi: 10.1038/jcbfm.1989.123. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflüger's Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hille B. Modulation of ion channel function by G protein-coupled receptors. Trends in Neurosciences. 1994;17:531–536. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- Hoffman WE, Kochs E, Werner C, Thomas C, Albrecht RF. Dexmedetomidine improves neurologic outcome from incomplex ischemia in the rat. Anesthesiology. 1991;75:328–332. doi: 10.1097/00000542-199108000-00022. [DOI] [PubMed] [Google Scholar]
- Kamisaki Y, Hamahashi T, Hamada T, Maeda K, Itoh T. Presynaptic inhibition by clonidine of neurotransmitter amino acid release in various brain regions. European Journal of Pharmacology. 1992;217:57–63. doi: 10.1016/0014-2999(92)90511-2. [DOI] [PubMed] [Google Scholar]
- Kokaia M, Kalen P, Bengzon J, Lindvall O. Noradrenaline and 5-hydroxytryptamine release in the hippocampus during seizures induced by hippocampal kindling stimulation: an in vivo microdialysis study. Neuroscience. 1989;32:647–656. doi: 10.1016/0306-4522(89)90286-8. [DOI] [PubMed] [Google Scholar]
- Kondo S, Marty A. Differential effects of noradrenaline on evoked, spontaneous and miniature IPSCs in rat cerebellar stellate cells. The Journal of Physiology. 1998;509:233–243. doi: 10.1111/j.1469-7793.1998.233bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law-Tho D, Crepel F, Hirsch JC. Noradrenaline decreases transmission of NMDA- and non-NMDA-receptor mediated monosynaptic epsps in rat prefrontal neurons in vitro. European Journal of Neuroscience. 1993;5:1494–1500. doi: 10.1111/j.1460-9568.1993.tb00217.x. [DOI] [PubMed] [Google Scholar]
- Loy R, Koziell DA, Lindsey JD, Moore RY. Noradrenergic innervation of the adult rat hippocampal formation. Journal of Comparative Neurology. 1980;189:699–710. doi: 10.1002/cne.901890406. [DOI] [PubMed] [Google Scholar]
- MacKinnon AC, Spedding M, Brown CM. α2-Adrenoceptors: more subtypes but fewer functional differences. Trends in Pharmacological Sciences. 1994;15:119–123. doi: 10.1016/0165-6147(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Actions of noradrenaline recorded intracellularly in rat hippocampal CA1 parymidal neurones in vitro. The Journal of Physiology. 1986;372:221–244. doi: 10.1113/jphysiol.1986.sp016006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Norepinephrine decreases synpatic inhibition in the rat hippocampus. Brain Research. 1988;442:131–138. doi: 10.1016/0006-8993(88)91440-0. [DOI] [PubMed] [Google Scholar]
- Miller RJ. Presynaptic receptors. Annual Review of Pharmacology and Toxicology. 1998;38:201–227. doi: 10.1146/annurev.pharmtox.38.1.201. [DOI] [PubMed] [Google Scholar]
- Nicholas AP, Hökfelt T, Pieribone VA. The distribution and significance of CNS adrenoceptors examined with in situ hybridization. Trends in Pharmacological Sciences. 1996;17:245–255. doi: 10.1016/0165-6147(96)10022-5. [DOI] [PubMed] [Google Scholar]
- Nörenberg W, Schöffel E, Szabo B, Starke K. Sybtype determination of soma-dendritic α2-autoreceptors in slices of rat locus coeruleus. Naunyn-Schmiedeberg's Archives of Pharmacology. 1997;356:159–165. doi: 10.1007/pl00005036. [DOI] [PubMed] [Google Scholar]
- O'Rourke MF, Iversen LJ, Lomasney JW, Bylund DB. Species orthologs of the alpha-2A adrenergic receptor: the pharmacological properties of the bovine and rat receptors differ from the human and porcine receptors. Journal of Pharmacology and Experimental Therapeutics. 1994;271:735–740. [PubMed] [Google Scholar]
- Pittaluga A, Raiteri M. GABAergic nerve terminals in rat hippocampus possess α2-adrenoceptors regulating GABA release. Neuroscience Letters. 1987;76:363–367. doi: 10.1016/0304-3940(87)90430-7. [DOI] [PubMed] [Google Scholar]
- Ruffolo RR, Stadel JM, Hieble JP. α-Adrenoceptors: recent developments. Medicinal Research Reviews. 1994;14:229–270. doi: 10.1002/med.2610140204. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Capogna M, Gähwiler BH, Thompson SM. Presynaptic inhibition of miniature excitatory synaptic currents by baclofen and adenosine in the hippocampus. Neuron. 1992;9:919–927. doi: 10.1016/0896-6273(92)90244-8. [DOI] [PubMed] [Google Scholar]
- Scanziani M, Gähwiler BH, Thompson SM. Presynaptic inhibition of excitatory synaptic transmission mediated by adrenergic receptors in area CA3 of the rat hippocampus in vitro. Journal of Neuroscience. 1993;13:5393–5401. doi: 10.1523/JNEUROSCI.13-12-05393.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanziani M, Gähwiler BH, Thompson SM. Presynaptic inhibition of excitatory transmission by muscarinic and metabotropic glutamate receptor activation in the hippocampus: are Ca2+ channels involved? Neuropharmacology. 1995;34:1549–1557. doi: 10.1016/0028-3908(95)00119-q. [DOI] [PubMed] [Google Scholar]
- Scholz KP, Miller RJ. Inhibition of quantal trannsmitter release in the absence of Ca2+ influx by a G protein linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- Segal M. The action of norepinephrine in the rat hippocampus: intracellular studies in the slice preparation. Brain Research. 1981;206:107–128. doi: 10.1016/0006-8993(81)90104-9. [DOI] [PubMed] [Google Scholar]
- Stone LS, Broberger C, Vulchanova L, Wilcox GL, Hökfelt T, Riedl MS, Elde R. Differential distribution of a2A and a2C adrenergic receptor immunoreactivity in the rat spinal cord. Journal of Neuroscience. 1998;18:5928–5937. doi: 10.1523/JNEUROSCI.18-15-05928.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers RJ, McMartin LR. Adrenoceptors and their second messenger systems. Journal of Neurochemistry. 1993;60:10–23. doi: 10.1111/j.1471-4159.1993.tb05817.x. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Limberger N, Starke K. Presynaptic alpha 2-autoreceptors in brain cortex: alpha 2D in the rat and alpha 2A in the rabbit. Naunyn-Schmiedeberg's Archives of Pharmacology. 1993;348:35–45. doi: 10.1007/BF00168534. [DOI] [PubMed] [Google Scholar]
- Verhage M, Ghijsen WEJM, Boomsma F, Lopes Da Silva FH. Endogenous noradrenaline and dopamine in nerve terminals of the hippocampus: differences in levels and release kinetics. Journal of Neurochemistry. 1992;59:881–887. doi: 10.1111/j.1471-4159.1992.tb08326.x. [DOI] [PubMed] [Google Scholar]
- Wheeler DB, Randall A, Tsien RW. Roles of N-type and Q-type Ca2+ channels in supporting hippocampal synaptic transmission. Science. 1994;264:107–111. doi: 10.1126/science.7832825. [DOI] [PubMed] [Google Scholar]