

Abstract
Substance P and bradykinin, endothelium-dependent vasodilators of pig coronary artery, trigger in endothelial cells a rise in cytosolic Ca2+ concentration ([Ca2+]i) and membrane hyperpolarization. The aim of the present study was to determine the type of Ca2+-dependent K+ (KCa) currents underlying the endothelial cell hyperpolarization.
The substance P-induced increase in [Ca2+]i was 30 % smaller than that induced by bradykinin, although the two peptides triggered a membrane hyperpolarization of the same amplitude. The two agonists evoked a large outward K+ current of the same conductance at maximal stimulation. Agonists applied together produced the same maximal current amplitude as either one applied alone.
Iberiotoxin (50 nM) reduced by about 40 % the K+ current activated by bradykinin without modifying the substance P response. Conversely, apamin (1 μm) inhibited the substance P-induced K+ current by about 65 %, without affecting the bradykinin response. Similar results were obtained on peptide-induced membrane hyperpolarization.
Bradykinin-induced, but not substance P-induced, endothelium-dependent relaxation resistant to NG-nitro-L-arginine and indomethacin was partly inhibited by 3 μm 17-octadecynoic acid (17-ODYA), an inhibitor of cytochrome P450 epoxygenase. Similarly, the bradykinin-induced K+ current was reduced by 17-ODYA.
Our results show that responses to substance P and bradykinin result in a hyperpolarization due to activation of different KCa currents. A current consistent with the activation of large conductance (BKCa) channels was activated only by bradykinin, whereas a current consistent with the activation of small conductance (SKCa) channels was stimulated only by substance P. The observation that a similar electrical response is produced by different pools of channels implies distinct intracellular pathways leading to KCa current activation.
Endothelial cells play a major role in the local control of the vascular tone by producing powerful vasoactive agents, like nitric oxide (NO), prostacyclin, and the still unidentified endothelium-derived hyperpolarizing factor (EDHF). The activation of endothelial cells by circulating mediators leads to an intracellular Ca2+ concentration ([Ca2+]i) rise, due to Ca2+ release from intracellular Ca2+ stores and Ca2+ influx from the extracellular space (Himmel et al. 1993). In many cases, this effect is associated with a membrane hyperpolarization (for review see Nilius et al. 1997), which plays an important role in the production of endothelial vasoactive substances by increasing the driving force for Ca2+ entry (Lückhoff & Busse, 1990).
The [Ca2+]i rise seems to be directly responsible for the membrane hyperpolarization, as shown by the hyperpolarizing effect of Ca2+ ionophores (Colden-Stanfield et al. 1990; von der Weid & Bény, 1992), which strongly suggests the involvement of Ca2+-dependent K+ (KCa) channels. Furthermore, agonist-induced membrane hyperpolarization also results from the activation of KCa channels (for review see Nilius et al. 1997). Single channel analysis indicated that many types of KCa channels are stimulated by various agonists in endothelial cells (Nilius et al. 1997); however, the relative contribution of the different K+ channels in endothelial cell hyperpolarization has been poorly studied.
The two peptides substance P and bradykinin are both endothelium-dependent vasodilators of the pig coronary artery, producing NO and EDHF as relaxing agents (Pacicca et al. 1992). In coronary artery endothelial cells, both peptides produced a transient hyperpolarization associated with an increase of [Ca2+]i (Brunet & Bény, 1989; Sharma & Davis, 1994; Baron et al. 1996). The intracellular second messenger cascade triggered by substance P- and bradykinin-receptor binding involves the activation of phospholipase C and the production of inositol 1,4,5-trisphosphate (IP3), which in turn release Ca2+ from IP3-sensitive Ca2+ stores (Farmer & Burch, 1992; Regoli et al. 1994). In this context, it is relevant to learn whether such a common pathway leads to the stimulation of different KCa channels. d-Tubocurarine, a non-specific inhibitor of KCa channels, differentially reduced the hyperpolarization provoked by substance P and bradykinin in pig coronary artery (von der Weid & Bény, 1992; Baron et al. 1996). This suggested that the hyperpolarization produced by both agonists possibly resulted from activation of different KCa channels. To test this hypothesis, we performed whole-cell patch clamp experiments, intracellular microelectrode membrane potential recordings, and [Ca2+]i measurements in endothelial cells in primary culture. We took advantage of specific KCa channel inhibitors in order to discriminate between the different K+ conductances stimulated by substance P and bradykinin.
METHODS
Endothelial cell primary culture
Left anterior descending branches of freshly killed domestic pig Sus scrofa coronary arteries were obtained at the slaughterhouse. The endothelial cells were collected by gentle rubbing of the internal face of the vessel with a scalpel, and centrifuged at 800 g for 8 min in culture medium consisting of: M199 medium (Gibco) supplemented with 20 % fetal calf serum, 2 mM glutamine, non-essential amino acids (13 ml added to 1 l of M199; Gibco), MEM vitamin solution (13 ml added to 1 l of M199; Gibco) and gentamicin (50 mg l−1). The cell pellet was resuspended in culture medium M199 and plated on collagen-coated culture Petri dishes or glass coverslips. Cells were cultured at 37°C under 5 % CO2. Culture medium was changed 3 times a week. Cells were used after 2-5 days of primary culture. Endothelial cells were identified by their morphology, fusiform growing cells forming islets during 4 to 5 days, and a monolayer of polygonal cells (cobblestone-like) after 5-6 days of culture.
Whole-cell patch clamp recordings
We used the whole-cell configuration of the patch clamp technique (Hamill et al. 1981). Endothelial cells were observed with an inverted microscope (Nikon Diaphot 200, Tokyo, Japan). Borosilicate glass patch pipettes were pulled with a BB-CH-PC puller (Mecanex SA, Nyon, Switzerland) and had a resistance of 3-5 MΩ. Patch clamp recordings were made using a List EPC-7 amplifier (EPC7; List Medical, Darmstadt, Germany). Current was filtered with a low-pass filter at 1 kHz, digitized by an IT16 interface (Instrutech Corporation, Great Neck, NY, USA) and stored by a Macintosh II vx computer (Pulse; HEKA Electronik, Lambrecht, Germany). Recordings were performed on single cells, or small islets never exceeding four cells, to avoid space clamp problems.
To determine the current-potential relationship, repetitive 300 ms voltage pulses were applied throughout the recording, usually reaching 30, 50 and 80 mV above the holding potential (varying between -60 and -20 mV). To normalize the results, we expressed the current conductance as density (pS pF−1), and thus membrane capacitance was measured before each experiment by applying a 10 mV voltage step. The capacitive current transients were fitted with a single exponential (Pulsefit; HEKA Electronik), and the cell membrane capacitance (Cm) was calculated according to the following equations (de Roos et al. 1996):
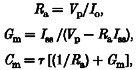 |
where Ra is the access resistance, Vp the applied voltage step (10 mV), Io the peak current, Iss the steady-state current, Gm the membrane conductance and τ the decay constant of the transient.
Experiments were performed at room temperature (20-22°C). Cells were superfused with a solution containing (mM): 130 NaCl, 5.6 KCl, 1 MgCl2, 2 CaCl2, 11 glucose, 8 Hepes (pH 7.5 with NaOH). In ionic selectivity experiments, the concentration of NaCl was changed to 95.6 mM and that of KCl was changed to 40 mM. In Ca2+-free solution, CaCl2 and MgCl2 were omitted, and 2 mM EGTA added. The standard pipette solution contained (mM): 130 KCl, 1 MgCl2, 5 MgATP, 10 Hepes (pH 7.3 with NaOH). MgCl2 was omitted and 5 mM BAPTA added to assess the Ca2+ dependence of the activated currents.
Cytosolic Ca2+ measurements
Fura-2 loading
Cultured endothelial cells were loaded with fura-2 acetoxymethylester (fura-2 AM, 10 μm) for 1 h in a Hepes-buffered solution containing (mM): 145 NaCl, 5 KCl, 1 CaCl2, 0.5 MgSO4, 1 NaH2PO4, 20 Hepes and 10.1 glucose at pH 7.4 (37°C), in the presence of pluronic F-127 (25 %) and probenecid (1 mM) in order to improve fura-2 loading. Cells were then rinsed for half an hour with the same Krebs-Hepes solution used for experiments.
Fura-2 fluorescence measurements
Endothelial cells were observed on an inverted microscope (Diaphot TMD, Nikon). Cells were continuously perfused with an oxygenated Krebs-buffered solution containing (mM): 118.7 NaCl, 4.7 KCl, 2.5 CaCl2, 1.2 MgSO4, 1.2 KH2PO4, 24.8 NaHCO3, 10.1 glucose, pH 7.4, gassed with a mixture of 75 % N2, 20 % O2 and 5 % CO2, since high O2 concentrations greatly reduce the intensity of fura-2 fluorescence. Experiments were done at 32-33°C. Images were recorded through an Extended Isis Intensified CCD camera (Photonic Science Ltd, East Sussex, UK) and interfaced with a Macintosh II fx computer. Fura-2 was excited with a high pressure mercury lamp at dual excitation wavelengths of 340 and 380 nm, via a rotating filter wheel. The emitted fluorescence was measured at 510 nm. 256-Greyscale images were stored, processed and pseudocoloured with the image analysis software IPLab Spectrum (Signal Analytics Corporation, Vienna, VA, USA). A pair of images was acquired every 3 s.
The process of ratioing was done on each pixel by dividing fluorescence obtained for 340 nm excitation by fluorescence obtained for 380 nm excitation.
Intracellular free calcium concentration was calculated from the fluorescence ratio according to the following equation (Grynkiewicz et al. 1985):
where Rmin and Rmax are the minimal and maximal values of fluorescence ratio R when all the fura-2 is in the Ca2+-free form or saturated with Ca2+, respectively. The factor Sf,2/Sb,2 is the ratio of fluorescence intensity at 380 nm when all the fura-2 is in the Ca2+-free form divided by the fluorescence intensity when the fura-2 is bound to Ca2+. Like Rmin and Rmax, this factor was determined from calibration experiments. To perform a calibration, endothelial cells were exposed to 4-bromo-A23187 (1.7 μm), in the presence of 2.5 mM extracellular Ca2+ (Rmax), and in a Krebs solution containing no Ca2+ and 2 mM EGTA (Rmin). The Kd of fura-2 for Ca2+ was taken as 225 nM.
Membrane potential measurements by intracellular microelectrode
Endothelial cells were observed on an inverted microscope (Diaphot TMD, Nikon). Intracellular membrane potential measurements were made at room temperature in Krebs solution, pH 7.4, and gassed with a mixture of 95 % O2 and 5 % CO2. The cultured cell membrane potential was measured with a conventional glass microelectrode containing 3 M KCl (90-150 MΩ) and connected to an amplifier (WPI Intra707) with an Ag-AgCl half-cell. The membrane potential was monitored on a digital oscilloscope (Gould, type 1425) and recorded on a potentiometric recorder (W + W Electronics, USA 312). Criteria for accepting a record were a stable membrane potential below -20 mV, and a sharp penetration and withdrawal.
Tension measurements
The coronary artery lumina were rinsed by injection of cold, oxygenated (95 % O2-5 % CO2) Krebs solution. Segments of the coronary artery were cleaned of adherent tissue, and cut into rings of about 2 mm width. The rings were then cut to give strips of about 5 mm long. Ligatures were attached to both ends of the strips, which were mounted with a resting isometric tension of about 10 mN in an 85 μl tissue bath as previously described (Pacicca et al. 1992). Strips were precontracted with 10 μm prostaglandin F2α (PGF2α) in the presence of 1 μmNG-nitro-L-arginine (L-NA) and 10 μm indomethacin in order to inhibit nitric oxide synthase and cyclo-oxygenase, respectively. Concentration- response curves were obtained by adding agonists in a non-cumulative manner. In order to block the cytochrome P450 epoxygenase activity, 17-octadecynoic acid (17-ODYA) was added for at least 25 min before the first application of substance P or bradykinin.
Chemicals and drugs
Bradykinin and substance P were purchased from Bachem Feinchemikalien AG (Bubendorf, Switzerland). Pluronic F-127 was obtained from Calbiochem. Fura-2 AM, probenecid, PGF2α, indomethacin and 17-ODYA were purchased from Sigma. Apamin and iberiotoxin were purchased from Alomone Labs (Jerusalem, Israel).
Statistics
Experimental data were expressed as means ± 1 standard error of the mean (s.e.m.), n referring to the number of measurements. Student's t test was used to compare results, with P < 0.05 taken as the level of significance. The EC50 was calculated for each concentration-response curve by interpolating between two points on either side of the half-maximal response, and reading the corresponding concentration on the logarithmic scale.
RESULTS
Intracellular Ca2+ rise induced by substance P and bradykinin
Primary cultured endothelial cells showed a basal [Ca2+]i level of 96.4 ± 8.3 nM (n = 90; 24 experiments). Substance P triggered an increase of [Ca2+]i reaching a maximal amplitude of 702.3 ± 50.7 nM (n = 19; 5 experiments). Bradykinin-evoked increase of [Ca2+]i reached a significantly higher peak value of 961.4 ± 32.0 nM (n = 71; 19 experiments; P < 0.05vs. substance P). The response duration, estimated by the half-time of return to the basal Ca2+ level, was 211.1 ± 20.0 s (n = 39; 16 experiments) for bradykinin, and about 50 % shorter, 112.7 ± 18.4 s (n = 15; 5 experiments; P < 0.05), for substance P. Figure 1A shows representative responses induced by substance P (100 nM, filled squares) and bradykinin (100 nM, open circles) obtained on two different cells. In the absence of extracellular Ca2+, both responses became more transient with half-times of 35.7 ± 3.2 s and 60.0 ± 7.7 s, and reduced amplitudes of 314.4 ± 33.1 nM (n = 16; 4 experiments) and 474.4 ± 54.6 nM (n = 18; 7 experiments), for substance P and bradykinin, respectively (Fig. 1B), showing the importance of Ca2+ influx in the agonist-induced cellular Ca2+ rise.
Figure 1. Substance P- and bradykinin-induced cytoplasmic Ca2+ rise in presence (A) and absence of extracellular Ca2+ (B).
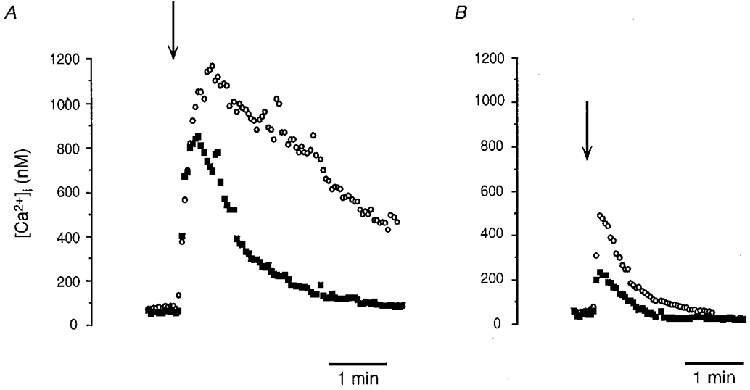
Effect of substance P (100 nM, ▪) and bradykinin (100 nM, ○) on cytosolic Ca2+ concentration ([Ca2+]i) measured with fura-2. Experiments were performed in the presence (A) and absence (B) of extracellular Ca2+. Recordings were obtained from four different cells. Peptides were applied at the time indicated by the arrows and maintained until the end of the recordings.
It should be noted that the substance P-evoked cell stimulation declined with the age of the culture. The largest response to substance P was obtained after 2 days of culture with 85.6 % of responding cells. The amplitude of the response decreased on day 3, and reached 267 ± 47 nM (n = 4) on day 4, with only 11 % of responding cells. On the contrary, the response to bradykinin remained unchanged even after 4-5 days of culture (data not shown). Consequently, all subsequent experiments using substance P were performed on 2-day-old cultures.
K+ current activated by substance P and bradykinin
Figure 2A and B shows typical currents induced by successive application of substance P (100 nM) and bradykinin (100 nM) on the same cell. Following the cell stimulation, an outward current developed rapidly and then gradually decreased with time. The maximal current evoked by the two peptides was of similar amplitude. Estimated by the half-time of return to the basal current level, the substance P response duration was 32.4 ± 3.9 s (n = 28) compared with 70.0 ± 10.7 s (n = 32) for bradykinin (P < 0.05), corresponding to a 54 % reduction.
Figure 2. Whole-cell currents induced by substance P and bradykinin in pig coronary artery endothelial cells.
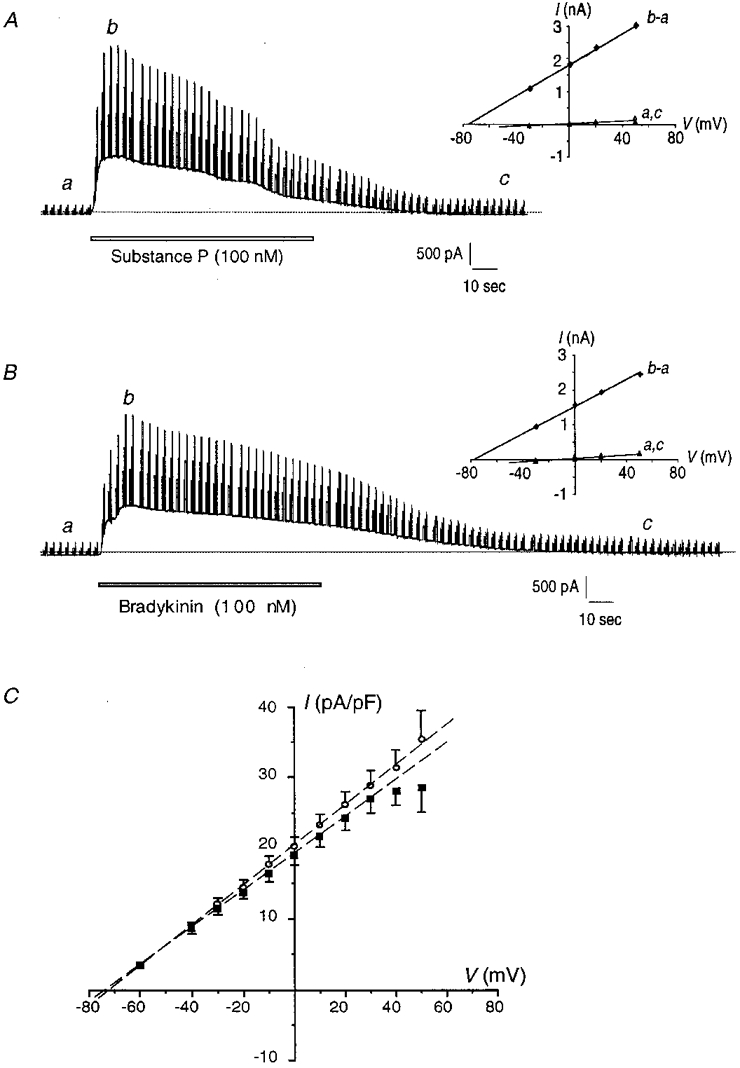
A and B, outward current elicited by 100 nM substance P (A) followed by 100 nM bradykinin (B) on the same recorded cell, separated by a 5 min washout period. The holding potential was -30 mV, and voltage steps were of 30, 50 and 80 mV above the holding potential. Peptides were applied in the bathing solution for the period indicated by the open bar. Corresponding current-potential curves are shown on the right insets, with a being obtained in control condition, c after peptide washout and b - a corresponding to the stimulated current. Dotted lines indicate the zero current. C, mean current-potential curves of K+ current stimulated by substance P (▪) and bradykinin (○). Maximal currents are represented, normalized by membrane capacitance. Data were obtained with a standard bathing solution containing 130 mM NaCl and 2 mM CaCl2. Data are means ±s.e.m.
In order to compare the results obtained on different cells, the slope conductance of the activated currents was expressed as a function of the cell capacitance. The membrane capacitance was 25.8 ± 1.6 pF (n = 30) for a single cell, and 43.9 ± 2.3 pF (n = 19) and 84.0 ± 3.4 pF (n = 22) for two and three electrically coupled cells, respectively.
The current-potential curves of the maximal currents activated by substance P and bradykinin are represented in Fig. 2C. The slope conductances were 258.7 ± 17.7 pS pF−1 (n = 40) for substance P, and 268.5 ± 18.7 pS pF−1 (n = 46) for bradykinin, showing that the amplitude of the activated currents was not significantly different between responses (P = 0.7). With 5.6 mM extracellular K+, the current reversal potentials were -72.9 ± 0.8 mV (n = 40) for substance P, and -73.7 ± 1.0 mV (n = 46) for bradykinin (P = 0.4), close to the K+ equilibrium potential of -80 mV. Changing the ionic composition of the bathing solution confirmed that these currents were selective for K+ ions. With 40 mM K+ in the bath, the current reversal potentials were then -25.8 ± 0.7 mV (n = 5) for substance P, and -27.2 ± 1 mV (n = 5) for bradykinin (P = 0.3; bradykinin vs. substance P), for a calculated K+ equilibrium potential of -30 mV.
In the absence of intra- and extracellular Ca2+ (2 mM EGTA in the bath and 5 mM BAPTA in the patch pipette), no currents were elicited by the two peptides (data not shown), thus indicating that KCa currents are implicated in endothelial cell stimulation.
Inhibitory effect of apamin and iberiotoxin on the K+ current
In order to discriminate between the different KCa currents underlying the cellular activation, we tested different specific KCa channel inhibitors on the substance P and bradykinin responses. The application of apamin (1 μm), a well-known blocker of small conductance KCa (SKCa) channels (Latorre et al. 1989), strongly reduced the slope conductance of the substance P-evoked current to 92.8 ± 37.5 pS pF−1 (n = 5; P < 0.05 vs. control 258.7 ± 17.7 pS pF−1) corresponding to an inhibition of about 65 % (Fig. 3A and B). Iberiotoxin (50 nM), a selective blocker of large conductance KCa (BKCa) channels (Galvez et al. 1990), did not significantly modify the substance P response (249.4 ± 34.4 pS pF−1; n = 11; P =0.8 vs. control; Fig. 3A and B). The combination of apamin and iberiotoxin produced the same inhibition as apamin alone on the substance P-evoked current (100.1 ± 42.3 pS pF−1; n = 5; P = 0.9 vs. apamin 92.8 ± 37.5 pS pF−1; Fig. 3B).
Figure 3. Effect of KCa channel blockers on the K+ current activated by substance P (A and B) and bradykinin (C and D).
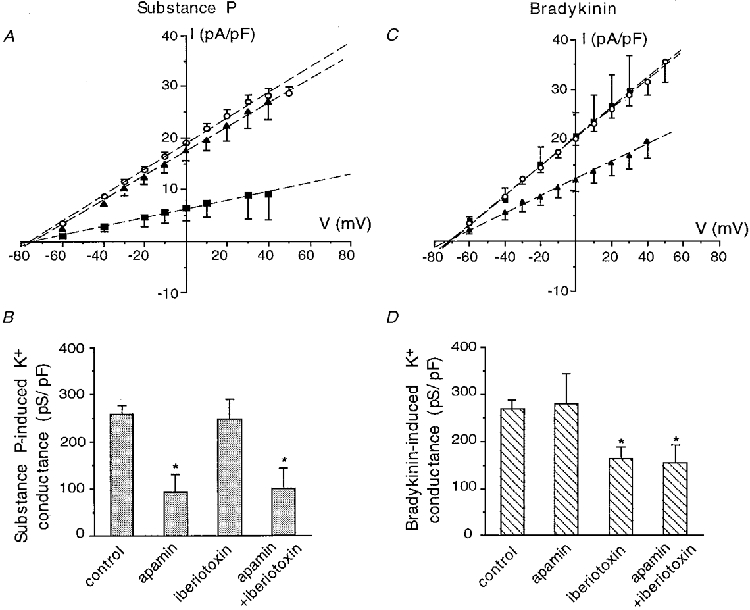
A and C, current-potential curves for 100 nM substance P (A) and 100 nM bradykinin (C) obtained in control conditions (○), and in the presence of 1 μm apamin (▪) or 50 nM iberiotoxin (▴). Data were linearly fitted, and are means ±s.e.m.B and D, maximal K+ current conductances activated by 100 nM subtance P (B) and 100 nM bradykinin (D) are represented in control conditions, and in the presence of apamin (1 μm), iberiotoxin (50 nM) or a combination of both inhibitors. Both points and columns represent the mean of 5 experiments in the presence of inhibitors, and a mean of 40-46 experiments in control conditions. Data are means ±s.e.m. * P < 0.05.
Conversely, the bradykinin response was not significantly affected by apamin; the current slope conductance was then 280.0 ± 63.6 pS pF−1 (n = 5; P = 0.8 vs. control 268.5 ± 18.7 pS pF−1; Fig. 3C and D), whereas iberiotoxin decreased the current conductance to 164.1 ± 24.2 pS pF−1 (n = 6; P < 0.05 vs. control; Fig. 3C and D), thus reducing by about 40 % the K+ current activated by bradykinin. Here again, apamin plus iberiotoxin did not produce a higher inhibition than iberiotoxin alone: 154.2 ± 35.7 pS pF−1 (n = 7; P = 0.8 vs. iberiotoxin 164.1 ± 24.2 pS pF−1; Fig. 3D). Neither apamin nor iberiotoxin shifted the current reversal potential of the currents activated by substance P or bradykinin; when both inhibitors were applied, the current reversal potential of the substance P-induced outward current was slightly shifted from -73 mV to -66 ± 3.02 mV (n = 5; P < 0.05).
These results showed that substance P and bradykinin stimulated in a large proportion different types of KCa currents. However the stimulation of endothelial cells with a combination of substance P and bradykinin did not result in a larger outward current than either peptide alone, the slope conductance being 230.5 ± 38.2 pS pF−1 (n = 20; P = 0.4 vs. substance P 258.7 ± 17.7 pS pF−1 or bradykinin 268.5 ± 18.7 pS pF−1 alone). We thus speculate that one agonist produced an inhibition on the channels stimulated by the other. To test this hypothesis we applied simultaneously substance P and bradykinin in the presence of iberiotoxin, in order to block BKCa channels stimulated by bradykinin and putatively revealed an inhibition of the total current. In this condition the K+ current was 144.3 ± 37.3 pS pF−1 (n = 8) corresponding to a reduction of about 40 % compared with the K+ current stimulated by substance P in the presence of iberiotoxin (249.4 ± 34.4 pS pF−1; P = 0.057). Although not reaching statistical significance, this result strongly suggested that bradykinin somehow inhibited a part of the substance P-induced KCa current.
Membrane hyperpolarization induced by substance P and bradykinin
The putative role played by SKCa and the BKCa channels in the response due to substance P and bradykinin, respectively, was further investigated by measuring the change in membrane potential elicited by both peptides, using intracellular microelectrode recording. Endothelial cells had a resting membrane potential of -31.1 ± 1.6 mV (n = 68). Figure 4A shows the electrical response of an endothelial cell following application of substance P and then bradykinin. Typically, the two agonists induced hyperpolarizations of similar amplitude; the membrane potential change was 27.0 ± 1.7 mV (n = 31) for substance P and 28.1 ± 1.6 mV (n = 46) for bradykinin (P = 0.6, substance P vs. bradykinin, Fig. 4B). Similar to data obtained on K+ current and [Ca2+]i rise, the response duration evoked by substance P was about 50 % shorter, with a half-time of 109.7 ± 11.0 s (n = 25), compared with 224.8 ± 34.2 s (n = 27; P < 0.05) with bradykinin. The application of 1 μm apamin strongly reduced the substance P response without affecting the hyperpolarization due to bradykinin, as shown on the original recording in Fig. 4A. On average, the substance P-induced hyperpolarization was reduced by 60 %, to a value of 10.0 ± 3.0 mV (n = 7; P < 0.05 vs. control 27.0 ± 1.7 mV; Fig. 4B). Conversely, iberiotoxin (50 nM) significantly reduced the bradykinin-induced hyperpolarization (19.8 ± 3.3 mV; n = 9; P < 0.05 vs. control 28.1 ± 1.6 mV), without affecting the response to substance P. Neither apamin nor iberiotoxin affected the resting membrane potential.
Figure 4. Effect of apamin on the endothelial cell hyperpolarization due to substance P and bradykinin.
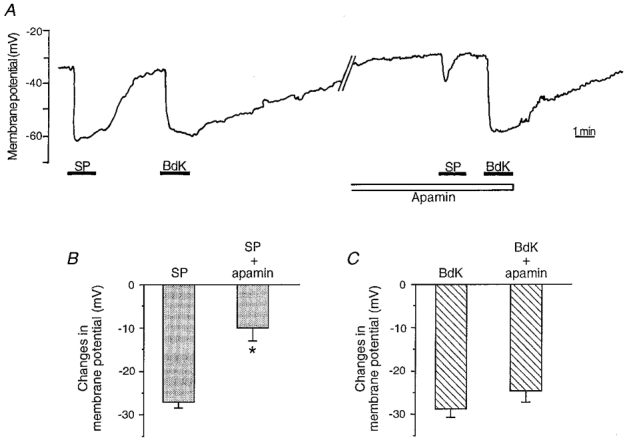
A, change in membrane potential measured by an intracellular microelectrode, following stimulation by 100 nM substance P (SP) and 100 nM bradykinin (BdK) in control conditions and after application of apamin (1 μm). The entire recording was performed on the same cell; the time between the first bradykinin application and the second substance P application was 26 min. B and C, mean values of the subtance P- (B) and bradykinin-induced (C) cell hyperpolarization in control or in the presence of apamin. n ranges from 4 to 7 for each value in the presence of apamin, and from 31 to 37 in controls. Data are means ±s.e.m. * P < 0.05.
Involvement of cytochrome P450 epoxygenase in the endothelium-dependent relaxation and K+ current due to substance P and bradykinin
Epoxyeicosatrienoic acids (EETs), produced by cytochrome P450 epoxygenase following endothelial cell stimulation (Harder et al. 1995), have been shown to modulate smooth muscle cell KCa channel activity (Hu & Kim, 1993; Li & Campell, 1997). Furthermore, EETs relax and hyperpolarize smooth muscles, thus leading some authors to propose that they are the putative EDHF (Hecker et al. 1994; Campbell et al. 1996). We have previously demonstrated that EETs enhance the open state probability of a 285 pS BKCa channel activated by bradykinin in pig coronary artery endothelial cells (Baron et al. 1997). In addition, EETs are produced by porcine coronary artery endothelial cells following stimulation by bradykinin (Hecker et al. 1994; Popp et al. 1996). In this context, we wondered if bradykinin or substance P could stimulate cytochrome P450 and, consequently, if EETs could explain the difference of KCa channel activation. To address this question, we looked at the involvement of cytochrome P450-derived compounds as relaxing factors. Concentration-response curves were performed on intact tissues precontracted with 10 μm PGF2α, in the presence of 10 μm indomethacin and 100 μm L-NA, to reveal the L-NA/indomethacin-resistant component of the endothelium-dependent relaxation. The application of 17-ODYA (3 μm), a suicide substrate of cytochrome P450, did not influence the substance P response: EC50= 0.27 ± 0.04 nM (n = 11) in control conditions and 0.32 ± 0.04 nM (n = 8; P = 0.4; Fig. 5A) in the presence of inhibitor. Conversely, 17-ODYA caused a rightward shift in the concentration-response curve to bradykinin: EC50= 3.1 ± 0.6 nM (n = 9) in control conditions, and 15.1 ± 6.1 nM (n = 7; P < 0.05) in the presence of 17-ODYA; and it reduced the maximal relaxation by about 20 % (Fig. 5B), thus suggesting that cytochrome P450 was stimulated by bradykinin but not by substance P.
Figure 5. Effect of 17-ODYA on the substance P- (A) and bradykinin-induced (B) endothelium-dependent relaxation.
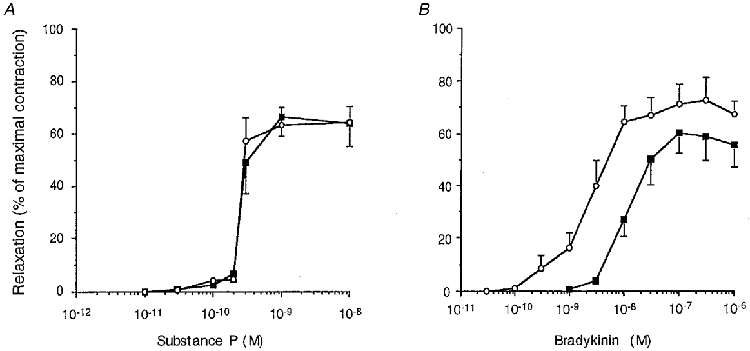
Concentration-response curves to substance P (A) and bradykinin (B) on coronary artery strips precontracted with 10 μm PGF2α, in the presence of 10 μm indomethacin and 100 μm L-NA. Experiments were done either in the absence (○) or in the presence (▪) of 3 μm 17-ODYA. Each point is the mean of 7-11 experiments. Data are means ±s.e.m.
We then tested the effect of 17-ODYA (3 μm) on the bradykinin-induced K+ current. In the presence of the inhibitor, the response due to bradykinin was reduced to 178.9 ± 36.8 pS pF−1 (n = 11; P < 0.05 vs. control 268.5 ± 18.7 pS pF−1), corresponding to an inhibition of about 35 %. On 12 experiments only one response was more than 2 times higher than mean control value and consequently was not considered. The substance P response was not affected by the application of 17-ODYA (248.7 ± 63.7 pS pF−1; n = 7; P = 0.3 vs. control 258.7 ± 17.7 pS pF−1), which gives support to the previous finding that bradykinin but not substance P stimulated the production of EETs.
DISCUSSION
Our present results, obtained on cultured endothelial cells, showed that in the same experimental conditions, cellular responses elicited by substance P and bradykinin were very similar. Substance P and bradykinin evoked large outward currents reaching a conductance at maximal stimulation of around 260 pS pF−1. In the nominal absence of intra- and extracellular Ca2+ no currents were observed. This is in agreement with a previous report (Vaca et al. 1996) and confirms the central role of [Ca2+]i in agonist-induced endothelial cell activations. The reversal potential of the kinin-induced outward currents closely corresponds to the K+ equilibrium potential, which supports the assertion that substance P (Sharma & Davis, 1994) and bradykinin (Colden-Stanfield et al. 1990) mainly activate KCa currents, responsible for the change of membrane potential. Membrane hyperpolarization activated by substance P or bradykinin reached similar amplitudes, although substance P triggered a 30 % smaller increase of [Ca2+]i. At this point, the most striking difference was the response duration: K+ current, membrane hyperpolarization and [Ca2+]i were about 50 % shorter when elicited by substance P. The shorter substance P-induced membrane hyperpolarization has already been observed (Brunet & Bény, 1989). Although it highlighted a significant difference between substance P- and bradykinin-induced endothelial cell activations, it was not further investigated at that time.
Bradykinin stimulated iberiotoxin-sensitive BKCa current
Large conductance iberiotoxin-sensitive BKCa channels were activated by bradykinin, whereas they did not seem to be stimulated by substance P. Single channel analysis has shown that bradykinin activates BKCa channels of rabbit aorta and porcine coronary artery endothelial cells (Rusko et al. 1992; Baron et al. 1996). BKCa channel involvement only during bradykinin-induced cellular activation may be explained by considering two parameters. Firstly, bradykinin produced a higher [Ca2+]i increase compared with substance P (Fig. 1A), and BKCa channels typically required a high Ca2+ concentration to be activated (Latorre et al. 1989), as was noticed in endothelial cells, with an EC50 of about 1 μm Ca2+ at +20 mV (Rusko et al. 1992; Baron et al. 1996). Secondly, EET production by endothelial cells could play a major role in BKCa channel activation. The present study suggests that bradykinin, as opposed to substance P, activates cytochrome P450 epoxygenase. The application of 17-ODYA, a suicide substrate for cytochrome P450 epoxygenase (Zou et al. 1994), caused a rightward shift and reduced the maximal amplitude of the concentration-response curve to bradykinin, but it did not modify the substance P response. As no major non-specific inhibitory effects were observed with 17-ODYA, even at concentrations above 3 μm (Edwards et al. 1996), and no inhibition was observed for the substance P response (Fig. 5A), we conclude that the shift of the bradykinin concentration-response curve is due to the inhibition of cytochrome P450. Much evidence supports bradykinin as a stimulator of cytochrome P450 epoxygenase in pig coronary artery, and bovine aorta and coronary artery (Hecker et al. 1994; Graier et al. 1995; Popp et al. 1996). However, few data concern the possible activation of cytochrome P450 epoxygenase by substance P, but Wallerstadt & Bodelsson (1997) suggested that this enzyme was not activated by substance P on human omental artery. Interestingly, we have previously shown that at constant Ca2+ concentration, the P450-derived EETs enhanced the open state probability of a 285 pS BKCa channel activated by bradykinin in pig coronary artery endothelial cells (Baron et al. 1997). Therefore, the higher [Ca2+]i increase and the EET production caused by bradykinin would contribute to BKCa channel activation. As substance P probably did not induce EET synthesis, and triggered a lower Ca2+ increase, the probability of activating BKCa channels is very low. In addition our results obtained on K+ current activated in the presence of 17-ODYA confirmed this statement. 17-ODYA reduced the K+ current evoked by bradykinin to a similar extent to those produced by iberiotoxin (about 35 %), suggesting that the activation of the cytochrome P450 is necessary for BKCa channel stimulation. By contrast, the substance P-induced K+ current was not affected by the preincubation with 17-ODYA. Thus, in physiological conditions, both an increase in [Ca2+]i and EET production seem to be required for the stimulation of BKCa in endothelial cells.
Substance P-stimulated apamin-sensitive SKCa
Our results demonstrate that SKCa channels are not involved in the bradykinin-evoked K+ current, but appear to be involved in the substance P-induced response. Similarly to the effects produced on K+ current, apamin strongly reduced the substance P-induced hyperpolarization, whereas no significant inhibition was produced on the bradykinin-induced response. In pig coronary artery, Sharma & Davis (1994) showed that substance P stimulated a 23 pS KCa channel, that corresponds to a SKCa channel, even though its apamin sensitivity was not determined. Regarding changes in [Ca2+]i induced by substance P and the high Ca2+ sensitivity of the SKCa channels (Latorre et al. 1989), their activation by this peptide is not surprising. However, their absence of involvement during cell stimulation by bradykinin is unexpected, and is not in agreement with previous reports. In pig aorta, bradykinin activated a 9 pS KCa channel, responsible for most of the K+ current elicited by bradykinin in these cells (Groschner et al. 1992). In bovine aorta, Vaca et al. (1996) have shown that noxiustoxin, an inhibitor of SKCa channels in this tissue (Vaca et al. 1993), completely inhibited the hyperpolarization due to bradykinin. In the same tissue, bradykinin stimulated KCa channels with conductances of 30-40 pS (Sauvéet al. 1990; Vaca et al. 1992), but their apamin sensitivity was not investigated. Conversely, apamin only slightly reduced, by 15 %, the bradykinin-evoked membrane hyperpolarization in guinea-pig coronary artery (Mehrke & Daut, 1990). Thus, depending on the tissue, SKCa channels are stimulated differently by bradykinin. In pig coronary artery endothelial cells, we cannot exclude the possibility that bradykinin stimulated a particular type of SKCa channel insensitive to apamin, which accounts for a part of the K+ current insensitive to iberiotoxin. For instance Marchenko & Sage (1996) have described in rat aorta an 18 pS KCa channel, a conductance that corresponds to the usual classification for SKCa channels (Latorre et al. 1989), that is insensitive to apamin.
One important question was to determine if the SKCa channels were not activated by bradykinin, or if they were secondarily inhibited following cell stimulation by bradykinin. If distinct KCa channels were activated by substance P and bradykinin, and if no inhibition of SKCa channels occurred, then endothelial cell stimulation by a combination of substance P and bradykinin would have produced a larger activated K+ current, compared with the application of one peptide alone. Our results demonstrated that this was not the case, as substance P and bradykinin together produced a K+ current of the same amplitude as that with either peptide applied alone. Furthermore, the K+ current stimulated by substance P and bradykinin in the presence of iberiotoxin was reduced compared with that stimulated by substance P and iberiotoxin alone. Even if this inhibition did not reach statistical significance, probably reflecting the high heterogeneity of K+ channels present in cultured endothelial cells, this revealed that bradykinin secondarily inhibited some of the channels usually activated by substance P, likely to be the SKCa channels. Thus it could explain why when both peptides were applied, the K+ current was not enhanced. In this condition we suggested that bradykinin stimulated BKCa channels at the same time as it inhibited SKCa channels, leading to the total K+ current being unaltered. The difference in KCa channel stimulation implies an intracellular signalling pathway different for substance P and bradykinin, which requires further investigation to be elucidated.
Other studies have shown that a combination of KCa channel blockers potentiates the effect of either inhibitor applied alone. This was mainly studied for the endothelium-dependent hyperpolarization of smooth muscle cells, with the combination of apamin and charybdotoxin (Chen & Cheung, 1997; Petersson et al. 1997). Such potentiating effects were not observed in the present study. The combination of iberiotoxin and apamin resulted in a similar inhibition compared with that in the presence of the effective inhibitor alone (iberiotoxin for bradykinin and apamin for substance P). These discrepancies could be due to different preparations used, i.e. cultured endothelial cells (present work) and intact strips with two cell types, endothelial and smooth muscle cells, in the above-mentioned studies.
Regarding the strong inhibition of endothelial cell K+ current produced by apamin or iberiotoxin, we can probably suppose that vasoactive factor production will be altered (Lückhoff & Busse, 1990). As already stressed by Mombouli & Vanhoutte (1997), particular care has to be taken when addressing the question of K+ channels underlying the endothelium-dependent smooth muscle cell hyperpolarization in intact tissue, by applying K+ channel blockers that can act either at the level of endothelial or smooth muscle cells, or most likely on both cell types.
In conclusion, we have shown that substance P and bradykinin, both endothelium-dependent vasodilators, stimulated endothelial cells in apparently the same way. Looking at the membrane potential and outward K+ current induced by the two peptides, no major differences were observed, except for the response durations. Nevertheless, different populations of KCa currents were stimulated by substance P and bradykinin. Substance P mainly evoked a current consistent with the activation of small conductance, apamin-sensitive SKCa channels, which accounted for about 65 % of the total K+ current. A current consistent with the activation of large conductance iberiotoxin-sensitive BKCa channels did not appear to be stimulated by this agonist, but contributed about 40 % of the total bradykinin-evoked K+ current. The production of EETs by bradykinin only is likely to explain the differential BKCa channel stimulations. Apamin-sensitive SKCa channels were not activated, and possibly were inhibited, by bradykinin, by an as yet undetermined intracellular second messenger. Our observation that a similar electrical response is produced by different pools of channels implies distinct intracellular pathways leading to KCa current activations.
Acknowledgments
This work was supported by the Swiss National Science Foundation, grant 31-49163.96. We thank D. Solomos for preparing the cell cultures, F. Gribi for her excellent technical assistance, R. Henauer for his technical competency, Dr A. Baron for helpful discussion and critical comments on the manuscript and Dr R. Peck for helping to improve the manuscript.
References
- Baron A, Frieden M, Bény J-L. Epoxyeicosatrienoic acids activate a high-conductance, Ca2+-dependent K+ channel on pig coronary artery endothelial cells. The Journal of Physiology. 1997;504:537–543. doi: 10.1111/j.1469-7793.1997.537bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baron A, Frieden M, Chabaud F, Bény J-L. Ca2+-dependent non-selective cation and potassium channels activated by bradykinin in pig coronary artery endothelial cells. The Journal of Physiology. 1996;493:691–706. doi: 10.1113/jphysiol.1996.sp021415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunet PC, Bény J-L. Substance P and bradykinin hyperpolarize pig coronary artery endothelial cells in primary culture. Blood Vessels. 1989;26:228–234. doi: 10.1159/000158770. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Chen G, Cheung DW. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. American Journal of Physiology. 1997;272:H2306–2312. doi: 10.1152/ajpheart.1997.272.5.H2306. [DOI] [PubMed] [Google Scholar]
- Colden-Stanfield M, Schilling WP, Possani LD, Kunze DL. Bradykinin-induced potassium current in cultured bovine aortic endothelial cells. Journal of Membrane Biology. 1990;116:227–238. doi: 10.1007/BF01868462. [DOI] [PubMed] [Google Scholar]
- De Roos ADG, Van Zoelen EJJ, Theuvenet APR. Determination of gap junctional intercellular communication by capacitance measurements. Pflügers Archiv. 1996;431:556–563. doi: 10.1007/BF02191903. [DOI] [PubMed] [Google Scholar]
- Edwards G, Zygmunt PM, Högestätt ED, Weston AH. Effects of cytochrome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. British Journal of Pharmacology. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer SG, Burch RM. Biochemical and molecular pharmacology of kinin receptors. Annual Review of Pharmacology and Toxicology. 1992;32:511–536. doi: 10.1146/annurev.pa.32.040192.002455. [DOI] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben JP, Roy-Contancin L, Feigenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium-activated potassium channel from venom of the scorpion Buthus tamulus. Journal of Biological Chemistry. 1990;265:11083–11090. [PubMed] [Google Scholar]
- Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. The Journal of Physiology. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groschner K, Graier WF, Kukovetz WR. Activation of small-conductance Ca2+-dependent K+ channel contributes to bradykinin-induced stimulation of nitric oxide synthesis in pig aortic endothelial cells. Biochimica et Biophysica Acta. 1992;1137:162–170. doi: 10.1016/0167-4889(92)90198-k. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sackmann B, Sigworth FJ. Improved patch-clamp techniques for the high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Harder DR, Campbell WB, Roman RJ. Role of cytochrome P-450 enzymes and metabolites of arachidonic acid in the control of vascular tone. Journal of Vascular Research. 1995;32:79–92. doi: 10.1159/000159080. [DOI] [PubMed] [Google Scholar]
- Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. The Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himmel HM, Whorton AR, Strauss HC. Intracellular calcium, currents, and stimulus-response coupling in endothelial cells. Hypertension. 1993;21:112–127. doi: 10.1161/01.hyp.21.1.112. [DOI] [PubMed] [Google Scholar]
- Hu S, Kim HS. Activation of K+ channel in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. European Journal of Pharmacology. 1993;230:215–221. doi: 10.1016/0014-2999(93)90805-r. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annual Review of Physiology. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- Li P-L, Campbell WB. Epoxyeicosatrienoic acids activate K+ channels in coronary smooth muscle through a guanine nucleotide binding protein. Circulation Research. 1997;80:877–884. doi: 10.1161/01.res.80.6.877. [DOI] [PubMed] [Google Scholar]
- Lückhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Archiv. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- Marchenko SM, Sage SO. Calcium-activated potassium channels in the endothelium of intact rat aorta. The Journal of Physiology. 1996;492:53–60. doi: 10.1113/jphysiol.1996.sp021288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehrke G, Daut J. The electrical response of cultured guinea-pig coronary endothelial cells to endothelium-dependent vasodilators. The Journal of Physiology. 1990;430:251–272. doi: 10.1113/jphysiol.1990.sp018290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mombouli J-V, Vanhoutte PM. Endothelium-derived hyperpolarizing factor(s): updating the unknown. Trends in Pharmacological Sciences. 1997;18:252–256. [PubMed] [Google Scholar]
- Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annual Review of Physiology. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- Pacicca C, Von Der Weid P-Y, Bény J-L. Effect of nitro-L-arginine on endothelium-dependent hyperpolarizations and relaxations of pig coronary arteries. The Journal of Physiology. 1992;457:247–256. doi: 10.1113/jphysiol.1992.sp019376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson J, Zygmunt PM, Högestätt ED. Characterization of potassium channels involved in EDHF-mediated relaxation in cerebral arteries. British Journal of Pharmacology. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. The Journal of Physiology. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoli D, Boudon A, Fauchère J-L. Receptors and antagonists for substance P and related peptides. Pharmacological Reviews. 1994;46:551–599. [PubMed] [Google Scholar]
- Rusko J, Tanzi F, Van Breemen C, Adams DJ. Calcium-activated potassium channels in native endothelial cells from rabbit aorta: conductance, Ca2+ sensitivity and block. The Journal of Physiology. 1992;455:601–621. doi: 10.1113/jphysiol.1992.sp019318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sauvé R, Chahine M, Tremblay J, Hamet P. Single-channel analysis of the electrical response of bovine aortic endothelial cells to bradykinin stimulation: contribution of a Ca2+-dependent K+ channel. Journal of Hypertension. 1990;8:S193–201. [PubMed] [Google Scholar]
- Sharma NR, Davis MJ. Mechanism of substance P-induced hyperpolarization of porcine coronary artery endothelial cells. American Journal of Physiology. 1994;266:H156–164. doi: 10.1152/ajpheart.1994.266.1.H156. [DOI] [PubMed] [Google Scholar]
- Vaca L, Gurrola GB, Possani LD, Kunze DL. Blockade of a KCa channel with synthetic peptides from noxiustoxin: a K+ channel blocker. Journal of Membrane Biology. 1993;134:123–129. doi: 10.1007/BF00232748. [DOI] [PubMed] [Google Scholar]
- Vaca L, Licea A, Possani LD. Modulation of cell membrane potential in cultured vascular endothelium. American Journal of Physiology. 1996;270:C819–824. doi: 10.1152/ajpcell.1996.270.3.C819. [DOI] [PubMed] [Google Scholar]
- Vaca L, Schilling WP, Kunze DL. G-protein-mediated regulation of a Ca2+-dependent K+ channel in cultured vascular endothelial cells. Pflügers Archiv. 1992;422:66–74. doi: 10.1007/BF00381515. [DOI] [PubMed] [Google Scholar]
- Von Der Weid P-Y, Bény J-L. Effect of Ca2+ ionophores on membrane potential of pig coronary artery endothelial cells. American Journal of Physiology. 1992;262:H1823–1831. doi: 10.1152/ajpheart.1992.262.6.H1823. [DOI] [PubMed] [Google Scholar]
- Wallerstedt SM, Bodelsson M. Endothelium-dependent relaxation by substance P in human isolated omental arteries and veins: relative contribution of prostanoids, nitric oxide and hyperpolarization. British Journal of Pharmacology. 1997;120:25–30. doi: 10.1038/sj.bjp.0700879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A-P, Ma Y-H, Sui Z-H, Ortiz De Montellano PR, Clark JE, Masters BS, Roman RJ. Effects of 17-octadecynoic acid, a suicide-substrate inhibitor of cytochrome P450 fatty acid ω-hydroxylase, on renal function in rats. Journal of Pharmacology and Experimental Therapeutics. 1994;268:474–481. [PubMed] [Google Scholar]