

Abstract
The objective of this study was to clarify the relationships between loss of mitochondrial potential and the perturbation of neuronal Ca2+ homeostasis induced by a toxic glutamate challenge. Digital fluorescence imaging techniques were employed to monitor simultaneously changes in cytoplasmic Ca2+ concentration ([Ca2+]i) and mitochondrial potential (ΔΨm) in individual hippocampal neurones in culture coloaded with fura-2 AM or fura-2FF AM and rhodamine 123 (Rh 123).
In most cells (96 %) at 6-7 days in vitro (DIV) and in a small proportion of cells (29 %) at 11-17 DIV the [Ca2+]i increase induced by exposure to 100 μm glutamate for 10 min was associated with a small mitochondrial depolarisation, followed by mitochondrial repolarisation, and a degree of recovery of [Ca2+]i following glutamate washout. In the majority of neurones at 11-17 DIV (71 %), exposure to glutamate for 10 min induced a profound mono- or biphasic mitochondrial depolarisation, which was clearly correlated with a sustained [Ca2+]i plateau despite the removal of glutamate.
Addition of glutamate receptor antagonists (15 μm MK-801 plus 75 μm 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX)) to the washout solution did not affect the post-glutamate [Ca2+]i plateau in neurones exhibiting a profound mitochondrial depolarisation but greatly improved [Ca2+]i recovery in those neurones undergoing only a small mitochondrial depolarisation, suggesting that the release of endogenous glutamate delays [Ca2+]i recovery in the postglutamate period.
Cyclosporin A (500 nM) or N-methyl Val-4-cyclosporin A (200 nM) delayed or even prevented the development of the second phase of mitochondrial depolarisation in cells at 11-17 DIV and increased the proportion of neurones exhibiting a small monophasic mitochondrial depolarisation and [Ca2+]i recovery upon glutamate removal.
We have thus described a striking correlation between mitochondrial depolarisation and the failure of cells to restore [Ca2+]i following a toxic glutamate challenge. These data suggest that mitochondrial dysfunction plays a major role in the deregulation of [Ca2+]i associated with glutamate toxicity.
A large part of the delayed neuronal death that follows episodes of anoxia and ischaemia in the mammalian CNS is due to the accumulation of glutamate in the extracellular space. The delayed neuronal death induced by the overstimulation of glutamate receptors depends critically on a sustained increase in cytoplasmic Ca2+ concentration ([Ca2+]i), which may in turn persist far beyond the termination of the exposure to glutamate (Ogura et al. 1988; Manev et al. 1989; Glaum et al. 1990; De Erausquin et al. 1990; Khodorov et al. 1993). Elevation of [Ca2+]i during and following a prolonged (10-15 min) glutamate exposure is accompanied by complex changes in cellular homeostasis: a sustained increase in cytoplasmic Na+ concentration ([Na+]i) (Kiedrowski et al. 1994; Pinelis et al. 1994), an intracellular acidification (Pinelis et al. 1992; Hartley & Dubinsky, 1993; Wang et al. 1994) and a decrease in the cellular ATP content ([ATP]) (Bogachev et al. 1992; Budd & Nicholls, 1996; Pinelis et al. 1997). Recent studies have shown that during and following a prolonged glutamate challenge, [Ca2+]i may remain at a high plateau level despite a progressive decrease in Ca2+ permeability of the neuronal membrane (Khodorov et al. 1996a), suggesting that the neuronal Ca2+ overload results primarily from impaired Ca2+ extrusion from the cell. Indeed, both systems responsible for Ca2+ extrusion - the Na+-Ca2+ exchanger and the Ca2+-H+ pump of the neuronal membrane appear to be impaired during and following a toxic glutamate challenge (Khodorov et al. 1993, 1996a; Kiedrowski et al. 1994; Pinelis et al. 1994).
A number of recent reports have also described changes in mitochondrial function associated with glutamate toxicity. Thus, in a variety of models, neuronal mitochondria depolarise in response to toxic glutamate exposures (White & Reynolds, 1996; Schinder et al. 1996; Nieminen et al. 1996). In these studies, however, glutamate-induced changes in [Ca2+]i and mitochondrial potential (ΔΨm) were not measured in the same cells. Therefore neither the specific relationships between changes in [Ca2+]i and mitochondrial depolarisation during and following a toxic glutamate exposure, nor the consequence of mitochondrial depolarisation for [Ca2+]i homeostasis have been explored. In a recent study (Khodorov et al. 1996b), cerebellar granule cells in culture were coloaded with fura-2 and rhodamine 123 (Rh 123), and the optics were switched rather infrequently between the two signals. This study revealed a close correlation between the degree of mitochondrial depolarisation during glutamate exposure and the failure of recovery of the [Ca2+]i signal. Thus, those cells in which glutamate induced only a small mitochondrial depolarisation showed substantial recovery from a Ca2+ load, while those cells in which glutamate treatment induced a profound mitochondrial depolarisation exhibited a high [Ca2+]i plateau in the post-glutamate period, leading to the conclusion that mitochondrial de-energisation may underlie the neuronal Ca2+ overload following a prolonged glutamate challenge.
The purpose of the present work was to carry out a detailed analysis of the relationships between changes in ΔΨm and [Ca2+]i during and after a toxic glutamate challenge in cultured rat hippocampal neurones by using digital imaging techniques to measure both signals simultaneously in single cells.
METHODS
Tissue culture
Hippocampal neurones were grown in mixed neuronal and glial primary cultures from rat pups aged 2-4 days post-partum. Animals were killed by decapitation, hippocampi were dissected and placed in ice-cold Gey's salt solution (Life Technologies, UK) with 20 μg ml−1 gentamycin. Gey's salt solution contained (g l−1): 0.22 CaCl.2H2O, 0.37 KCl, 0.03 KH2PO4, 0.21 MgCl2.6H2O, 0.07 MgSO4.7H2O, 8 NaCl, 0.227 NaHCO3, 0.12 Na2HPO4 and 1 D-glucose. After mincing the tissue with fine scissors, the tissue was placed in Ca2+/Mg2+-free Hanks’ buffered saline (HBSS) with 0.1 % trypsin for 15 min at 36°C. HBSS (Life Technologies) contained (g l−1): 0.4 KCl, 0.06 KH2PO4, 8 NaCl, 0.35 NaHCO3, 0.048 Na2HPO4, 1 D-glucose and 0.01 Phenol Red. Trypsin was inactivated by washing with normal HBSS. Cells were dissociated by trituration and pelleted in HBSS. They were then resuspended in Minimal Essential Medium with Earle's salts (MEM; Life Technologies) containing 10 % horse serum (Life Technologies) and plated onto 24 mm sterile circular glass coverslips which had been previously coated with poly-D-lysine. The cultures were maintained at 36°C in a humidified atmosphere of 5 % CO2 and 95 % air and fed bi-weekly with MEM containing 10 % horse serum. Cultures were treated on day 3 with 10 μm cytosine arabinoside (Life Technologies) for 24 h to prevent glial proliferation. Neurones were grown for a minimum of 6 days before experimental use to ensure the expression of glutamate receptors and sufficient sensitivity to excitotoxic treatment and were discarded after 17 days in vitro (DIV). Neurones were easily distinguishable from glial cells in these cultures: they appeared phase-bright, had smooth rounded somata and distinct processes, and usually lay just above the focal plane of the glial layer.
Imaging of [Ca2+]i and ΔΨm
Hippocampal cultures were loaded for 30 min at room temperature with 5 μm fura-2 AM (Molecular Probes) or fura-2FF AM (Teflabs, Austin, TX, USA) and 0.005 % Pluronic in a standard physiological recording saline containing (mM): 156 NaCl, 3 KCl, 2 MgSO4, 1.25 KH2PO4, 2 CaCl2, 10 glucose and 10 Hepes, pH adjusted to 7.35 with NaOH. For simultaneous measurements of [Ca2+]i and ΔΨm, Rh 123 (26 μm, equivalent to 10 μg ml−1; Molecular Probes) was added into the culture during the last 15 min of the fura-2 or fura-2FF loading period.
Fluorescence measurements were obtained on an epifluorescence inverted microscope equipped with a × 20 fluorite objective. [Ca2+]i and ΔΨm were monitored in single cells using excitation light provided by a Xenon arc lamp, the beam passing sequentially through 10 nm bandpass filters centred at 340, 380 and 490 nm housed in a computer-controlled filter wheel (Cairn Research, Kent, UK). Emitted fluorescence light was reflected through a 515 nm long-pass filter to a frame transfer cooled CCD camera with an active sensor of 800 × 600 pixels and digitised to 12 bit resolution (Digital Pixel Ltd, UK). All imaging data were collected and analysed using Kinetic Imaging software (Liverpool, UK). A computer-controlled shutter kept photodamage of the cells to a minimum by allowing exposure to excitation light only when required for imaging. The exposure time to excitation varied between experiments and was 200-400 ms and 400-800 ms for 340 nm and 380 nm light, respectively, and 100-200 ms for 490 nm light. The fluorescence data were acquired at intervals of 15 to 20 s.
The fura-2 and fura-2FF data have not been calibrated in terms of [Ca2+]i because of the uncertainty arising from the use of different calibration techniques. These data are presented as the ratio of light excited at 340 nm/380 nm. Rh 123 is a lipophilic cationic dye which is concentrated in mitochondria, where its fluorescence is quenched. Upon mitochondrial depolarisation the dye is released, its fluorescence dequenched and a rapid increase in fluorescence is observed (Duchen & Biscoe, 1992). Leakage of Rh 123 across the plasma membrane was sufficiently slow for the probe to be retained within the cells for the duration of the experiment, even when the plasma membrane was depolarised by glutamate. The dye appears not to respond to changes in plasma membrane potential per se, as changes in Rh 123 fluorescence following depolarisation of the plasma membrane in voltage-clamped neurons (Duchen, 1992) are due solely to changes in ΔΨm, secondary to changes in [Ca2+]i. Because Rh 123 is a single-wavelength dye and its initial fluorescence intensity may therefore vary between cells, all Rh 123 data have been normalised to the resting intensity in each cell and the resting fluorescence has been expressed as 100 %. To show the extent of glutamate-induced mitochondrial depolarisation, the uncoupler carbonyl cyanide p-trifluoro-methoxyphenyl hydrazone (FCCP, 1 μm), which is expected to collapse the mitochondrial potential completely, was applied at the end of each experiment, after which the cells were discarded.
Experimental procedure
The coverslips with cell culture were placed into the 700 μl experimental chamber. Cells were continuously perfused with normal recording saline at room temperature (22°C). This could be changed to specified solutions from one of four reservoirs via a solenoid switching mechanism, allowing bath application of glutamate, glutamate antagonists or FCCP as required. The rate of perfusion was 2.5 ml min−1. In experiments with cyclosporin A (CsA), cells were preincubated with CsA for 30 min.
Mathematical analysis
Statistical analysis and exponential curve fitting were performed with the aid of Origin 4.1 (Microcal Software Inc., Northampton, MA, USA) software. Means are expressed ± the standard error of the mean (s.e.m.) and comparisons were made using Student's paired t test.
RESULTS
Patterns of [Ca2+]i recovery following a glutamate challenge
In agreement with previous reports (Manev et al. 1990; Limbrick et al. 1995; Adamec et al. 1998) we observed a considerable difference in the time course of [Ca2+]i recovery following short-term (1 min) and prolonged (10 min) glutamate exposures. After a pulse of glutamate for 1 min, [Ca2+]i usually decayed back to baseline with a time course that could be well fitted by a double-exponential function. The time constant (τ) and relative amplitude (A) of the fast component were τfast= 0.39 ± 0.04 min and Afast= 0.90 ± 0.02 (n = 24 neurones); and of the slow component, τslow= 12.40 ± 2.54 min and Aslow= 0.10 ± 0.001 (n = 24 neurones). Figure 1A shows an example of such a response in a single cell.
Figure 1. The kinetics of [Ca2+]i recovery following a glutamate challenge varies between cells and depends on the duration of glutamate exposure.
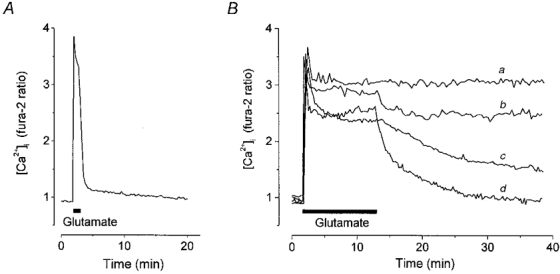
Cultured hippocampal neurones were stimulated with 100 μm glutamate (in a solution containing 0 Mg2+ and 10 μm glycine). A, representative trace showing the response of a neurone at 7 DIV to stimulation for 1 min. The decay phase showed biphasic kinetics of [Ca2+]i recovery and was well fitted by a double exponential process with time constants τfast= 0.48 min and τslow= 14.47 min and relative amplitudes (A) of the fast and slow components Afast= 0.92 and Aslow= 0.08. B, representative traces showing the responses of 4 individual neurones at 11 DIV (a and b) and 7 DIV (c and d) with different patterns of [Ca2+]i recovery following glutamate stimulation for 10 min: a, ‘plateau response’; b and c, delayed and d, complete [Ca2+]i recovery.
The pattern of [Ca2+]i recovery following a prolonged (10 min) glutamate challenge varied systematically with the age of the culture. If maintained in culture for 11-17 days (i.e. days in vitro, DIV) most neurones failed to restore [Ca2+]i to resting levels after a 10 min exposure to glutamate, and [Ca2+]i remained at a high plateau level over the time course of the experiment (20-50 min; Fig. 1Ba). In contrast, at 6-8 DIV, [Ca2+]i recovered either completely or partially in most cells following a 10 min glutamate exposure (Fig. 1Bb-d). The proportion of cells exhibiting these different post-glutamate [Ca2+]i dynamics varied among the culture series.
Relationships between changes in [Ca2+]i and ΔΨm during and following a glutamate challenge
Cell by cell comparisons between changes in [Ca2+]i and ΔΨm appeared to reveal a clear relationship between the two. Thus, in the majority (96 %) of neurones at 6-8 DIV (n = 130/135) and in only 29 % of neurones at 11-17 DIV (n = 79/275), glutamate application produced only a relatively small mitochondrial depolarisation (the Rh 123 fluorescence increased no more than 2-fold) associated with a degree (either partial or complete) of [Ca2+]i recovery. Some examples illustrating these relationships are presented in Fig. 2A and B.
Figure 2. Relationships between [Ca2+]i and mitochondrial potential (ΔΨm) during and after exposure to glutamate for 10 min.
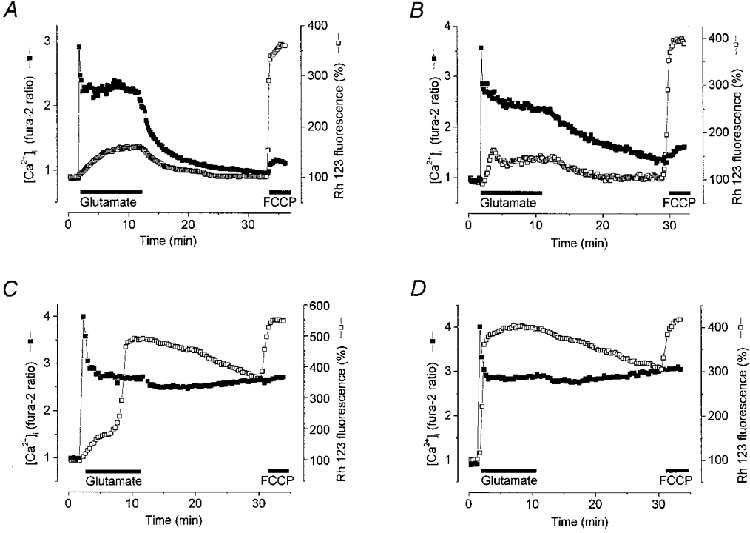
Simultaneous measurements of changes in the fura-2 ratio (▪) and relative Rh 123 fluorescence (□) were made from single neurones. Glutamate (100 μm) and glycine (10 μm) were applied in a Mg2+-free solution. An increase in Rh 123 fluorescence reflects mitochondrial depolarisation. A and B show different dynamics of [Ca2+]i recovery in single neurones at 7 DIV (A) and 8 DIV (B) exhibiting small mitochondrial depolarisations. C and D present exemplar neurones at 16 DIV in which large glutamate-induced mitochondrial depolarisations were seen. A large mitochondrial depolarisation was associated with a post-glutamate [Ca2+]i plateau. The mitochondrial uncoupler FCCP (1 μm) was applied at the end of this and subsequent experiments to indicate the level of fluorescence of Rh 123 that reflected complete dissipation of ΔΨm.
In the majority (71 %) of neurones at 11-17 DIV (n = 196/275), the glutamate-induced mitochondrial depolarisation was profound and showed a time course which could be either mono- or biphasic. The first phase of a biphasic mitochondrial depolarisation was usually small (the Rh 123 fluorescence increased no more than 2-fold). After a variable delay, this initial depolarisation was superseded by a second phase of depolarisation - an up to 6-fold increase in Rh 123 fluorescence, equivalent to that seen with the mitochondrial uncoupler FCCP, which is expected to dissipate the mitochondrial potential completely. Indeed, in a number of cells the glutamate-induced mitochondrial depolarisation could not be further substantially increased by application of FCCP. Thus, glutamate seemed, in these cells, to cause an almost complete collapse of the mitochondrial potential (Fig. 2C and D). Note that in these and all subsequent experiments, FCCP was applied at the end of the period of observation. This provides a demonstration of the available dynamic range of the Rh 123 signal from ‘rest’ to complete depolarisation, and is also important in establishing whether a fall in signal is due to a true recovery of mitochondrial potential or whether it simply reflects dye leakage from the cell (in which case addition of uncoupler will have no further effect). In some experiments (not shown) we applied FCCP before glutamate exposure. The fluorescence of Rh 123 in response to FCCP increased to 353 ± 32 % (n = 15) of basal level when FCCP was applied to resting cells and to 335 ± 32 % (n = 15) at the end of the experiments. Since there was no significant difference (P > 0.05, n = 15) between the amplitude of FCCP responses before and after glutamate application, in most of our experiments we used FCCP application only at the end of each experiment.
The duration of the delay between the first and second phases of mitochondrial depolarisation varied among the cells from seconds to minutes. Figure 2D presents an example of a large and apparently monophasic mitochondrial depolarisation. It is reasonable to consider such an apparently monophasic mitochondrial response only as a particular case of the biphasic depolarisation with a very short (practically indistinguishable given the relatively low rates of image acquisition) delay between its first and second phases.
A systematic comparison of the mitochondrial and [Ca2+]i responses showed that the majority of neurones at 11-17 DIV showing a large mitochondrial depolarisation also exhibited a high [Ca2+]i plateau (n = 186/196) in the post-glutamate period (see Fig. 3). Such a plateau response was infrequently observed in association with a small mitochondrial depolarisation. Thus, of 79 cells at 11-17 DIV that showed a small mitochondrial response (< 2-fold increase in Rh 123 signal), only 25 showed a sustained [Ca2+]i plateau phase. Among the cells studied at 6-8 DIV, both a high [Ca2+]i plateau and a large mitochondrial depolarisation were observed only very rarely. These correlations are summarised in Fig. 3.
Figure 3. Relationship between the post-glutamate [Ca2+]i plateau, the degree of mitochondrial depolarisation and the age of the culture.
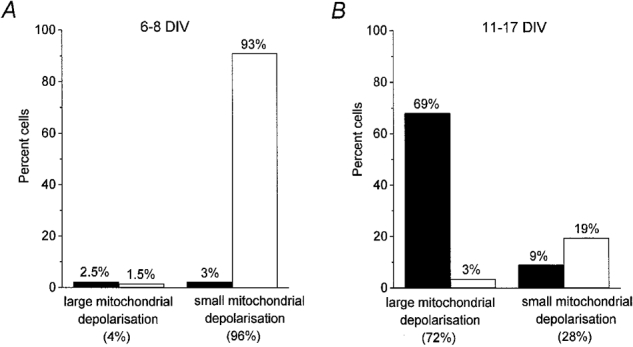
Neurones at each developmental stage (6-8 and 11-17 DIV) were subdivided into two groups. The total number of neurones was taken as 100 %. Neurones showing a less than 2-fold increase in Rh 123 fluorescence during glutamate exposure were included in the groups with small mitochondrial depolarisation; neurones in which glutamate produced an up to 6-fold increase of basal Rh 123 fluorescence were included in the groups with large mitochondrial depolarisation. The columns show the proportion of neurones exhibiting a [Ca2+]i plateau (▪) or [Ca2+]i recovery (□). The groups of neurones with a [Ca2+]i recovery include cells exhibiting either a partial or complete recovery (see Fig. 1B, traces b-d). A and B represent data from 135 neurones at 6-8 DIV and 275 neurones at 11-17 DIV. Clearly a [Ca2+]i plateau (‘no recovery’) was observed in the majority of cells with a large mitochondrial depolarisation, while a degree of [Ca2+]i recovery occurred in the majority of cells with a small mitochondrial depolarisation.
Figure 4A shows the relationship between the degree of [Ca2+]i recovery and mitochondrial depolarisation. The Rh 123 data have been normalised against the response to FCCP, such that a ratio of 1 indicates a complete mitochondrial depolarisation.
Figure 4. Relationship between the degree of mitochondrial depolarisation during glutamate exposure and the postglutamate [Ca2+]i dynamics in the presence and absence of glutamate receptor antagonists.
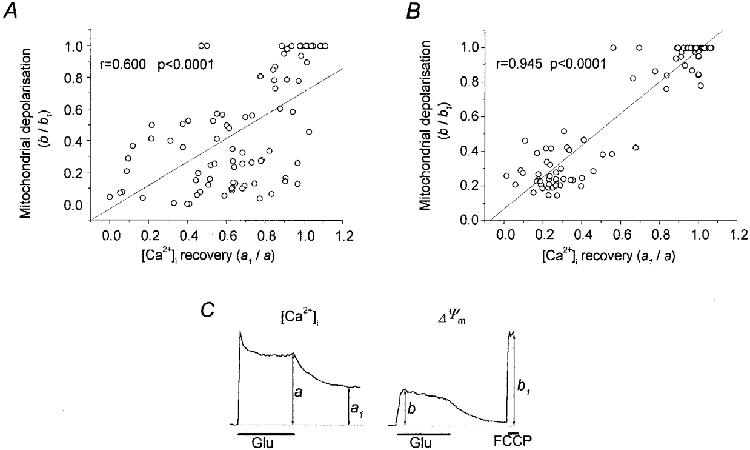
Each symbol in A and B corresponds to an individual hippocampal neurone. A, data from 7 experiments (86 neurones) in which the washout solution did not contain glutamate receptor antagonists. B, data from 6 experiments (103 neurones) in which the washout solution contained 15 μm MK-801 and 75 μm CNQX. In C, a schematic representation is shown to illustrate how the measurements of maximum glutamate-induced mitochondrial depolarisation and the extent of [Ca2+]i recovery were made; a and a1, fura-2 fluorescence ratio (340 nm/380 nm) at the end of the glutamate challenge and 10 min after glutamate washout, respectively; b and b1, maximal Rh 123 fluorescence during glutamate exposure and in response to a pulse of FCCP (1 μm).
Note that while there is a clear tendency for the points to lie close to the line of correlation, the clustering of cells away from the relationship - particularly cells with a ‘poor’[Ca2+]i recovery but with a small mitochondrial response - degrades the quality of fit. Thus, in this series of experiments, in some cells the correlation between [Ca2+]i recovery and mitochondrial response seemed not to apply. Hence, a significant number of cells showed a delayed [Ca2+]i recovery even though the mitochondrial depolarisation was small and reversible. We will return to Fig. 4B below.
Contribution of endogenous excitatory amino acids to the rate of [Ca2+]i recovery
It has been shown previously that glutamate application to neuronal cultures induces a delayed release of endogenous excitatory amino acids (enEAA), which in turn may cause the secondary activation of both NMDA and non-NMDA receptors (Hartley & Choi, 1989; Khodorov et al. 1996c). To clarify a possible contribution of enEAA to the deterioration of [Ca2+]i recovery in the post-glutamate period, we examined changes in the dynamics of [Ca2+]i recovery following the addition of glutamate antagonists to the washout solution, using (+)-MK-801 maleate (MK-801, 15 μm) and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, 75 μm) to inhibit NMDA and non-NMDA glutamate receptors, respectively.
In the experiments illustrated in Fig. 5, we compared the impact of glutamate antagonists on the dynamics of [Ca2+]i recovery in cells which exhibited a small (Fig. 5A and B) and a large (Fig. 5C and D) mitochondrial depolarisation. In a control group of cells exhibiting small mitochondrial depolarisation, the [Ca2+]i signal showed only a very slight and slow recovery after glutamate washout (Fig. 5Ab). Addition of the glutamate receptor antagonists to a sister culture dramatically enhanced the rate of recovery (Fig. 5Bb) in a group of cells that also showed small mitochondrial responses. The recovery phases in individual records could in this case be well fitted by two components: τfast= 0.80 ± 0.02 min, Afast= 0.56 ± 0.03; τslow= 18.88 ± 1.09 min, Aslow= 0.43 ± 0.05 (n = 11). In the control culture (Fig. 5Ab), the fast component of [Ca2+]i recovery was either very small or absent. The fitting of the slow component of recovery showed that [Ca2+]i decreased to a plateau level (with some variability between the neuronal population) with τ= 11.90 ± 0.46 min.
Figure 5. Contribution of endogenous excitatory amino acids (enEAA) to delayed [Ca2+]i recovery on glutamate washout.
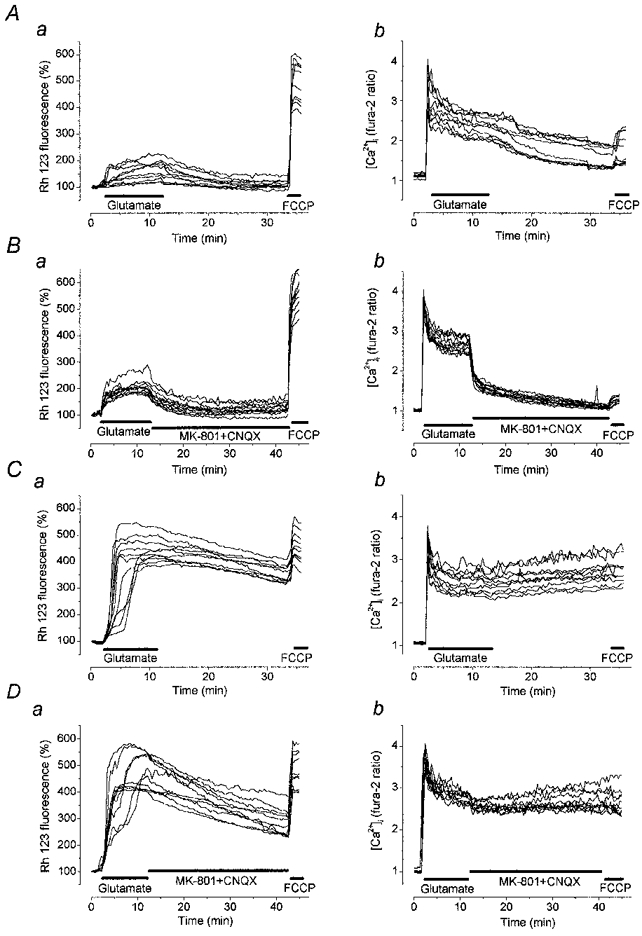
These traces show simultaneous recordings of Rh 123 fluorescence (left-hand panels, a) and [Ca2+]i (right-hand panels, b) in cultures at 13 DIV. The records from neurones exhibiting small and large mitochondrial depolarisation in response to glutamate were plotted separately in A and B, and in C and D, respectively. Aa and b show the responses to glutamate typical of neurones with a small mitochondrial depolarisation followed by washout with control saline. In B, the glutamate receptor antagonists MK-801 (15 μm) and CNQX (75 μm) were added to the washout, and greatly enhanced recovery of the [Ca2+]i signal. C, in neurones with a large mitochondrial depolarisation, [Ca2+]i remained at a high plateau in the postglutamate period and washout of glutamate with the glutamate receptor antagonists (D) had no effect on the postglutamate [Ca2+]i plateau.
The effect of glutamate antagonists on the postglutamate [Ca2+]i dynamics in neurones with profound mitochondrial depolarisation is shown in Fig. 5C and D. All of these neurones exhibited a high [Ca2+]i plateau in the post-glutamate period. During the recovery phase, when glutamate was no longer present, addition of the glutamate receptor antagonists had no impact on either the mitochondrial or [Ca2+]i dynamics. Qualitatively similar results were obtained in eight other analogous experiments. Thus, release of enEAA following glutamate exposure makes a significant contribution to the delay in [Ca2+]i recovery in neurones exhibiting a small mitochondrial depolarisation, but makes no noticeable contribution to the [Ca2+]i signal in cells exhibiting a large mitochondrial depolarisation in response to glutamate. This is illustrated further in Fig. 4B, in which we have plotted an equivalent data set to that seen in Fig. 4A, but this time with the addition of glutamate antagonists to the washout solution. Clearly, the correlation between [Ca2+]i recovery and mitochondrial depolarisation was much closer, suggesting that one important complicating variable had been removed. Note, however, that in a number of neurones with small mitochondrial depolarisations, the post-glutamate [Ca2+]i recovery was incomplete even in the presence of glutamate antagonists in the washout solution. This evidently indicates that there are some other factors which contribute to the deregulation of [Ca2+]i homeostasis caused by a toxic glutamate challenge.
Recently it has been shown that [Ca2+]i can reach 50-100 μm in microdomains of stimulated cells (Llinas, 1992), well above the sensitivity range of fura-2, which has a Kd∼0.264 μm (Augustine & Neher, 1992; Petrozzino et al. 1995). Fura-2 fails to measure [Ca2+]i accurately when high Ca2+ microdomains are present because a large portion of the indicator pool is saturated. In glutamate excitotoxicity specifically, high-affinity calcium indicators tend to underestimate increases in [Ca2+]i (Stout & Reynolds, 1999). It thus seemed necessary to use a low-affinity Ca2+ indicator - e.g. fura-2FF (Hyrc et al. 1998) with a Kd of 35 μm - for measurements in which [Ca2+]i may rise to very high levels. We have therefore performed some similar experiments with neurones coloaded with fura-2FF and Rh 123. In terms of the recovery kinetics, at least, the results proved to be in essence similar to those obtained in analogous experiments with fura-2. Thus, in the majority of cells (n = 42/43) with a large mitochondrial depolarisation, [Ca2+]i failed to recover following removal of glutamate from the external solution despite the presence of glutamate receptor antagonists (Fig. 6A). In contrast, in cells showing small mitochondrial depolarisations, glutamate removal prompted mitochondrial repolarisation accompanied by complete [Ca2+]i recovery (n = 35/41) in the presence of glutamate receptor antagonists (Fig. 6B).
Figure 6. Relationships between [Ca2+]i and ΔΨm in hippocampal neurones coloaded with fura-2FF and Rh 123.
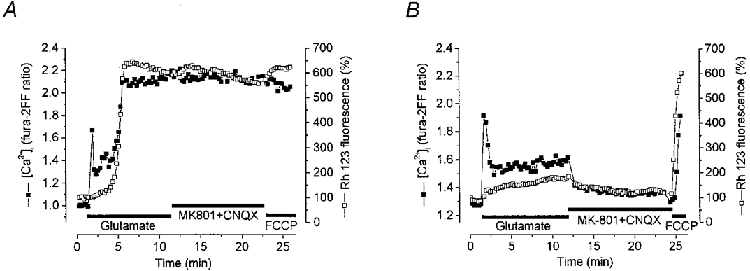
Simultaneous measurements of [Ca2+]i (▪) using fura-2FF and of ΔΨm (□) in neurones at 16 DIV (A) and 8 DIV (B). Glutamate application caused a brisk transient rise in [Ca2+]i followed by a secondary rise to a high plateau level which was sustained on glutamate washout despite the presence of the glutamate receptor antagonists. The secondary rise in [Ca2+]i was accompanied by a profound mitochondrial depolarisation. In the record shown in B, glutamate caused a transient increase in [Ca2+]i very similar in amplitude to that seen in A but associated with a very small, slow mitochondrial depolarisation and a sustained low [Ca2+]i signal which recovered completely on glutamate washout with the addition of receptor antagonists.
It was interesting to note that in most neurons exhibiting a small mitochondrial depolarisation and [Ca2+]i recovery in the postglutamate period, application of FCCP raised [Ca2+]i. The amplitude of this response varied considerably among the cells. The FCCP-induced increase in [Ca2+]i was independent of the age of the cell culture or of the Ca2+ indicator used (fura-2 or fura-2FF). Thus, in experiments with cells at 7 DIV loaded with fura-2, application of FCCP in the postglutamate period produced a large increase in [Ca2+]i in 37 of 87 neurons. The amplitude of this response was comparable to the glutamate-induced increase in [Ca2+]i (see for example Fig. 9Ad, trace 3). A similar result was obtained in an experiment with a sister culture loaded with fura-2FF: a large increase in [Ca2+]i in response to FCCP was seen in 16 of 71 cells (see Fig. 6B), while the remaining 55 cells showed either a very small or no change in [Ca2+]i. This FCCP-induced increase in [Ca2+]i may reflect release of Ca2+ from mitochondria to the cytosol and, if so, should reflect mitochondrial Ca2+ loading and the rate of mitochondrial Ca2+ efflux during the postglutamate recovery period.
Figure 9. Effects of cyclosporin A (CsA) on glutamate-induced mitochondrial depolarisation and [Ca2+]i dynamics.
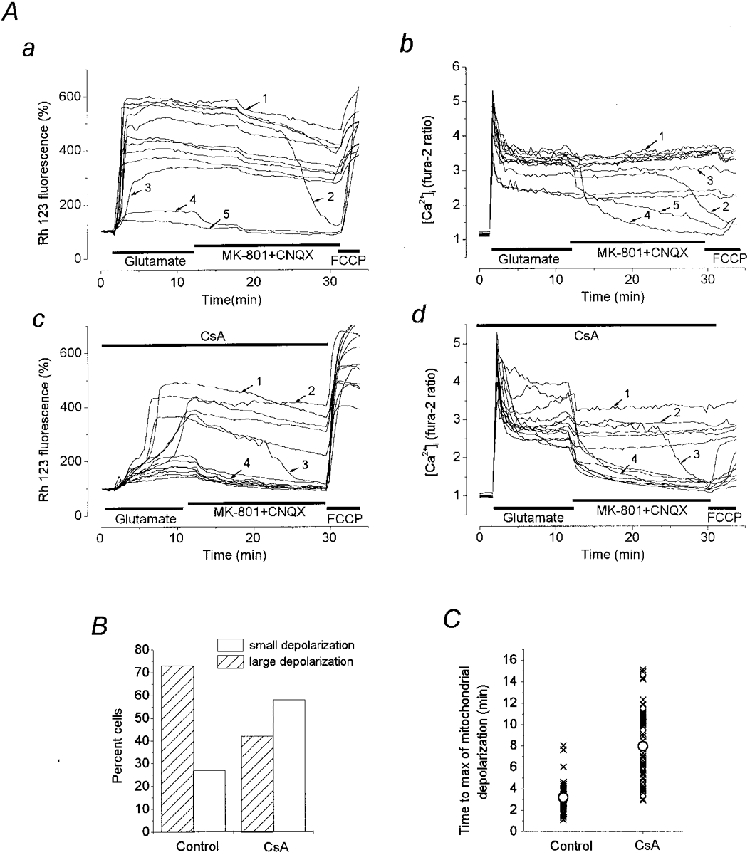
Simultaneous measurements of [Ca2+]i and ΔΨm in neurones at 16 DIV are shown. The glutamate antagonists MK-801 (15 μm) and CNQX (75 μm) were added to the washout solution to exclude the effect of enEAA on the post-glutamate [Ca2+]i recovery. Aa and b show [Ca2+]i and ΔΨm dynamics during and after glutamate exposure in a matched control experiment while Ac and d show [Ca2+]i and ΔΨm dynamics in neurones of a sister culture preincubated with 0.5 μm CsA for 50 min at room temperature. CsA was present in all the solutions used during the experiment. B, histogram showing that CsA decreased the number of neurones exhibiting a large glutamate-induced mitochondrial depolarisation (from 73 % in control culture to 43 % in CsA-treated culture) and correspondingly increased the percentage of neurones with a small mitochondrial depolarisation. C shows the scatter of time-to-peak measurements of the glutamate-induced mitochondrial depolarisation (measured from the beginning of the glutamate application) in the control cells and in cells treated with CsA. Each symbol (×) corresponds to the individual neurone with ○ being the mean value in each group. The mean value was 3.16 ± 0.20 min (n = 46) in the control, and 7.96 ± 0.44 min (n = 57) in the CsA treated group.
Mitochondrial repolarisation and [Ca2+]i recovery
Changes in the mitochondrial potential in the post-glutamate period deserve attention. In most cells washout of glutamate prevented further mitochondrial depolarisation and usually initiated a repolarisation. In neurones exhibiting small mitochondrial depolarisation, the post-glutamate repolarisation was fast and complete (see Fig. 2A and B). In contrast, in most cells with a large mono- or biphasic depolarisation (79 %) the mitochondria remained strongly depolarised during the time course of the experiment (20-40 min) (Fig. 2C and D).
In other cells (21 % of neurones exibiting a strong mitochondrial depolarisation) the initial slow repolarisation after a certain delay gave place to a fast, often complete, repolarisation, which in turn was followed by [Ca2+]i recovery. In the measurements presented in Fig. 7A and B the time interval (d) between the termination of a glutamate exposure and the onset of a fast mitochondrial repolarisation was about 2.5 and 12.5 min, respectively. In other neurones (n = 28) d varied between 0 and 29 min.
Figure 7. Relationships between the dynamics of mitochondrial repolarisation and postglutamate [Ca2+]i recovery.
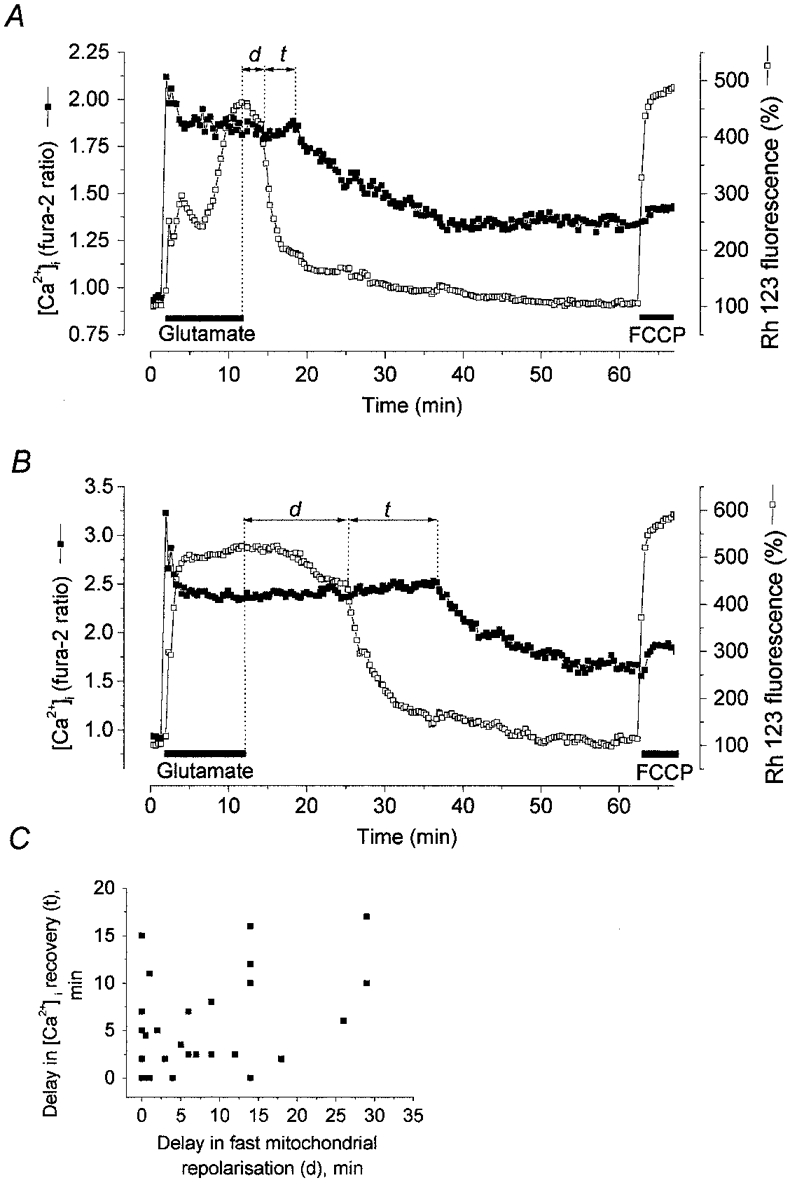
[Ca2+]i (▪) and ΔΨm (□) were monitored simultaneously in cells at 14 DIV coloaded with fura-2 and Rh 123. Representative records are shown to illustrate mitochondrial repolarisation with accompanying [Ca2+]i recovery. A and B show examples of a partial [Ca2+]i recovery following fast mitochondrial repolarisation after a short (A) and long (B) delay. C, relationship between the delay of a fast mitochondrial repolarisation (d) and the delay of [Ca2+]i recovery (t) following a 10 min glutamate exposure; each symbol corresponds to the d and t in the individual cells. The large mitochondrial response to 1 μm FCCP at the end of the experiment testifies that the decrease in Rh 123 fluorescence reflected mitochondrial repolarisation and not dye leakage from the cell.
It is noteworthy, though, that [Ca2+]i never recovered before the recovery of ΔΨm. The delay between the onset of recovery of ΔΨm and the onset of recovery of [Ca2+]i (t in Fig. 7) was variable. Thus in Fig. 7A[Ca2+]i recovery started ∼4 min after the beginning of the fast mitochondrial repolarisation, while in the trace illustrated in Fig. 7B this delay was about 12 min. In other cells t varied between 1 and 22 min. Figure 7C demonstrates the relationships between d and t in 28 neurones exhibiting both mitochondrial repolarisation and [Ca2+]i recovery (partial or complete). It can be seen that in most cells a delay in [Ca2+]i recovery following the mitochondrial repolarisation did not depend on the time delay to the onset of the mitochondrial repolarisation. Only in five cells did [Ca2+]i recovery begin almost simultaneously with the fast mitochondrial repolarisation. The fact that [Ca2+]i recovery never preceded the mitochondrial repolarisation strongly favours the hypothesis that mitochondrial repolarisation is required for [Ca2+]i recovery. However, considerable variations in the delay in [Ca2+]i recovery following the fast mitochondrial repolarisation indicate that mitochondrial repolarisaton is not the only determinant of [Ca2+]i recovery. Indeed in 21 neurones, we failed to observe [Ca2+]i recovery during the 10-13 min after glutamate washout despite a complete post-glutamate mitochondrial repolarisation. Possibly in these cells, the delay in [Ca2+]i recovery was longer than the period of observation. Alternatively, one could suppose that in these cells the Ca2+ overload was irreversible.
Effects of repeated short-term glutamate application on mitochondrial potential and [Ca2+]i dynamics
Glaum et al. (1990) showed that in cultured hippocampal neurones a high [Ca2+]i plateau appeared not only after a prolonged glutamate exposure, but also sometimes following a short glutamate pulse. Moreover in about 2 % of neurones a sustained increase in [Ca2+]i was already apparent after a single 1 min glutamate pulse. In order to clarify the origin of this deterioration of [Ca2+]i homeostasis we have examined the effects of repeated short-term applications of glutamate on both [Ca2+]i recovery dynamics and the mitochondrial response in cells at different stages of development in culture. In all neurones at 6-8 DIV (n = 20) the first 1 min pulse of glutamate induced a transient [Ca2+]i elevation associated with small mitochondrial depolarisation (Fig. 8A). An increase in the duration of subsequent pulses to 3 and 4 min allowed mitochondrial depolarisation to rise to larger values which, however, did not prevent a complete [Ca2+]i recovery during the washout period (Fig. 8A and E). In contrast, in cells at 16 DIV a transient [Ca2+]i and a small mitochondrial depolarisation in response to the first glutamate pulse were observed in 10 of 21 neurones (Fig. 8B). In nine remaining neurones of this group, even the first 1 min glutamate pulse evoked a large transient mitochondrial depolarisation (more than 2-fold increase in Rh 123 fluorescence) associated with a delay in the [Ca2+]i recovery (Fig. 8C). In this example, the subsequent mitochondrial repolarisation was followed by a partial [Ca2+]i recovery. Finally, in two cells a large and sustained mitochondrial depolarisation and a high [Ca2+]i plateau were apparent after only the first 1 min pulse (Fig. 8D). The sustained mitochondrial depolarisation and high [Ca2+]i plateau following the second (Fig. 8C) or third (Fig. 8B) glutamate pulse were observed in four and 11 cells, respectively (Fig. 8F).
Figure 8. Relationships between changes in [Ca2+]i and ΔΨm induced by repeated glutamate pulses of different duration.
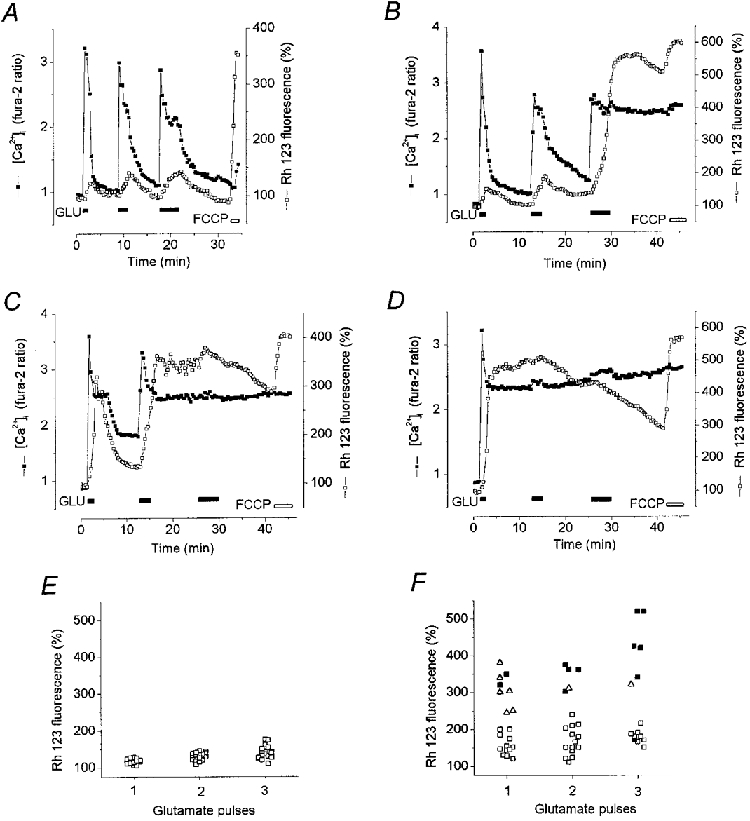
Neurones at 6 DIV (A) and 16 DIV (B-D) were stimulated with sequential 1, 2 and 4 min pulses of 100 μm glutamate separated by 6 min (A) and 10 min (B-D) intervals. [Ca2+]i (▪) and ΔΨm (□) were monitored simultaneously. The record in A shows responses representative of those seen in cells at 6 DIV (n = 20). An increase in the glutamate pulse duration from 1 to 2 and then to 4 min slowed but did not prevent [Ca2+]i recovery. B-D show representative records from a similar experiment with neurones at 16 DIV(n = 21). The plots shown in E and F summarise the data of 2 experiments on neurones at 6 (E) and 16 DIV (F) presented in A-D in terms of the [Ca2+]i dynamics. Each symbol shows the response of an individual neurone to the first, second and third glutamate pulses: □, transient [Ca2+]i response; ▵, delayed [Ca2+]i recovery (as shown in C);▪, [Ca2+]i plateau response. Ordinate, degree of mitochondrial depolarisation (Rh 123 fluorescence in %).
Two important conclusions follow from the analysis of these data. (1) The dynamics of [Ca2+]i recovery following a short-term glutamate challenge depends strongly on the mitochondrial depolarisation induced by the glutamate pulse. (2) A small and reversible mitochondrial depolarisation is associated with [Ca2+]i recovery, while a strong and prolonged mitochondrial depolarisation is followed by a [Ca2+]i plateau irrespective of the timing of glutamate application.
Effect of CsA on mitochondrial responses and [Ca2+]i recovery
CsA may delay the development of NMDA-induced mitochondrial depolarisation (Nieminen et al. 1996) and may decrease the fraction of neurones undergoing a glutamate-induced mitochondrial depolarisation (White & Reynolds, 1996; Schinder et al. 1996). The authors interpreted this effect of CsA as evidence in favour of the notion that the mitochondrial permeability transition pore (PTP) opening plays a central role in the mechanism of the glutamate-induced collapse of the mitochondrial potential. However, the effects of CsA on glutamate-induced Ca2+ overload have not yet been described. The objective of the experiments illustrated in Fig. 9 was to elucidate the effects of CsA on the relationship between ΔΨm and [Ca2+]i dynamics during and after glutamate exposure. Neurones cultured for 11-17 DIV were preincubated for 30-60 min with 500 nM CsA, which was then present in all solutions used in the experiments. In all experiments of this series, CsA pretreatment decreased the population of neurones undergoing a profound mitochondrial depolarisation from 73 % in control (n = 120 neurones, 7 dishes) to 42 % (n = 153 neurones, 9 dishes) (Fig. 9B). In those neurones in which a profound mitochondrial depolarisation still occured, its development was significantly delayed when compared to control responses. The time to maximum mitochondrial depolarisation was 3.16 ± 0.20 min (n = 46) in control experiments and 7.96 ± 0.44 min (n = 52) in CsA-treated cells (Fig. 9C).
In the example shown in Fig. 9A, in the absence of CsA (a and b) only two of 12 neurones exhibited small mitochondrial depolarisations followed by substantial [Ca2+]i recovery upon glutamate washout. The remainder of the neuronal population exhibited a large mitochondrial depolarisation and a prolonged high [Ca2+]i plateau. In the CsA-treated sister culture (c and d), the majority of neurones now underwent only a small mitochondrial depolarisation and showed pronounced [Ca2+]i recovery (7 out of 13 neurones). In the remaining cells in the CsA-treated population, a significant delay developed in the onset of a large mitochondrial depolarisation. Similar effects of CsA treatment were seen in five other equivalent experiments. In three experiments CsA prevented development of the second phase of mitochondrial depolarisation in all of the neurones studied. If the washout solution also contained MK-801 (15 μm) and CNQX (75 μm) (n = 4 experiments), then cells showing a small mitochondrial depolarisation in response to glutamate also showed complete recovery of [Ca2+]i on glutamate washout (as above). In contrast, neurones showing a glutamate-induced profound mono- or biphasic mitochondrial depolarisation exhibited a high [Ca2+]i plateau in the post-glutamate period regardless of whether glutamate antagonists were present or absent in the washout medium. Thus CsA prevented the appearance of a [Ca2+]i plateau only in those neurones in which it prevented the profound mitochondrial depolarisation occurring during glutamate exposure. CsA-induced delay in the mitochondrial depolarisation by itself did not prevent the appearance of a [Ca2+]i plateau. The interpretation of these results is complicated by the lack of specificity of CsA which, in addition to its blockade of the PTP in the mitochondrial membrane, also interacts with calcineurin and in this way inhibits NO synthesis (Dawson et al. 1993; Ankarcrona et al. 1996). We have found, however, that the CsA analogue N-methyl Val-4-cyclosporin A (200 nM), which lacks the effect of CsA on calcineurin (Schreier et al. 1993), also delayed a development of the second phase of mitochondrial depolarisation and increased the fraction of neurones exhibiting small monophasic mitochondrial depolarisation (n = 121 neurones). The relationships between changes in mitochondrial potential and [Ca2+]i proved to be qualitatively similar to those obtained in experiments with CsA.
DISCUSSION
The relationship between glutamate-induced changes in mitochondrial potential and the restoration of [Ca2+]i homeostasis
The objective of the present work was to explore the relationship between mitochondrial dysfunction and the deregulation of neuronal [Ca2+]i homeostasis caused by toxic stimulation of glutamate receptors. Our main and most consistent finding has been the clear correlation between a glutamate-induced profound mitochondrial depolarisation and a failure of the neurone to recover from the Ca2+ load. Hence, in the majority of nerve cells exhibiting complete loss of mitochondrial potential, [Ca2+]i remained at a high plateau level despite the removal of glutamate. In contrast, in the majority of cells in which glutamate exposure produced only a small, reversible mitochondrial depolarisation, the [Ca2+]i recovered after glutamate washout. Qualitatively similar relationships have recently been described in cerebellar granule cells (Khodorov et al. 1996b;Kiedrowski, 1998).
In our experiments we routinely saw a striking correlation between the time for which the cells were kept in culture and the pattern of change in mitochondrial potential and [Ca2+]i recovery following a toxic glutamate challenge. Thus, small, reversible mitochondrial depolarisation in conjunction with post-glutamate [Ca2+]i recovery proved to be characteristic for ‘young’ neurones - between 6 and 8 days in culture. In contrast, profound mitochondrial depolarisation in conjunction with a lack of [Ca2+]i recovery was typical for ‘older’ neurones - between 11 and 17 days in culture. The basis for these differences may be very instructive and will be the subject of further study. In the intact CNS, neurones show a maturation-dependent increase in the vulnerability to injury. Recently, Adamec et al. (1998) showed that the age in culture of both cultured hippocampal neurones and cerebellar granular cells had a profound influence on the ability of cells to recover from a glutamate-induced [Ca2+]i load. Our data suggest that the difference in the postglutamate [Ca2+]i dynamics in neurons of different ages may reflect a difference in the glutamate-induced mitochondrial response. Alternatively, both mitochondrial depolarisation and a failure of [Ca2+]i recovery may be determined by the calcium load, which in turn depends on the expression of glutamate (predominantly NMDA) receptors over time in culture.
Our experiments also showed that the correlation between mitochondrial depolarisation and failure of [Ca2+]i recovery in cells at 11-17 DIV was not dependent on the duration of glutamate stimulation. Thus the responses to repeated short-duration glutamate stimulation showed that pulses that caused only a small transient mitochondrial depolarisation were associated with [Ca2+]i recovery, pulses that caused a collapse of ΔΨm were associated with the failure of [Ca2+]i recovery and in most cells, repeated pulses caused progressively larger mitochondrial responses and progressively delayed recovery of [Ca2+]i.
Perhaps most strikingly, CsA pretreatment to delay or suppress the second phase of glutamate-induced mitochondrial depolarisation in 11-17 DIV cells was clearly associated with an increase in the proportion of cells which recovered from the Ca2+ load in the post-glutamate period.
Contribution of endogenous excitatory amino acids
A direct relationship between the glutamate-induced mitochondrial depolarisation and the post-glutamate deregulation of [Ca2+]i homeostasis was seen most clearly when the glutamate receptor antagonists MK-801 and CNQX were added to the washout solution. These agents greatly improved [Ca2+]i recovery only in those cells in which glutamate induced only a relatively small mitochondrial depolarisation, and had no significant effect in cells exhibiting a high [Ca2+]i plateau associated with a profound mitochondrial depolarisation. These data suggest that the release of enEAA is one of the factors which delays the recovery of [Ca2+]i, but only in those neurones that exhibit a small glutamate-induced mitochondrial depolarisation. Nerve and glial cells express two mechanisms of enEAA release: (1) Ca2+-dependent vesicular release; and (2) Na+- and voltage-dependent non-vesicular glutamate release, operating through reversal of the glutamate uptake carrier that occurs under certain pathological conditions (Nicholls & Attwell, 1990; Takahashi et al. 1997). As glutamate application causes a considerable increase in both [Ca2+]i and [Na+]i, it seems plausible that a delay in [Ca2+]i recovery in cells exhibiting small mitochondrial depolarisation may well result from the activation of both vesicular and non-vesicular mechanisms of enEAA release, and also from the activation of reverberating neuronal circuits in the culture.
Causal link between the glutamate-induced mitochondrial depolarisation and impaired [Ca2+]i recovery following the glutamate challenge
Glutamate-induced deterioration of [Ca2+]i homeostasis may reflect one or both of the following: (i) impaired neuronal Ca2+ extrusion mechanisms (Na+-Ca2+ exchange and Ca2+ pump), and (ii) inhibition of mitochondrial electrophoretic Ca2+ uptake. Collapse of ΔΨm will abolish and reverse two major mitochondrial functions, ATP synthesis and uptake of cytoplasmic Ca2+ (for a review see Nicholls, 1986; Duchen, 1999). Therefore it seems logical to assume that a correlation between the glutamate-induced mitochondrial depolarisation and the cell's inability to restore [Ca2+]i homeostasis reflects the causal link between these two processes. However, a more detailed analysis of the existing data suggested that these relationships may be more complex.
Inhibition of mitochondrial Ca2+ uptake
Collapse of ΔΨm inhibits or reverses the uniporter-mediated electrophoretic Ca2+ uptake. What is the contribution of this aspect of mitochondrial dysfunction to changes in [Ca2+]i following a glutamate challenge? In experiments with repeated short-term glutamate applications, [Ca2+]i recovery following each of these pulses was strongly dependent on the magnitude and duration of the mitochondrial depolarisation: a small transient mitochondrial depolarisation in response to a 1 min glutamate pulse did not prevent fast [Ca2+]i recovery. In contrast, a large transient mitochondrial depolarisation was associated with a delay in [Ca2+]i recovery, which was only renewed following a subsequent mitochondrial repolarisation. It is tempting to interpret all these changes in [Ca2+]i following a glutamate pulse as a direct result of corresponding variations in mitochondrial Ca2+ uptake and release in response to changes in ΔΨm.
Qualitatively similar relationships were observed in experiments with prolonged glutamate challenges: fast mitochondrial repolarisation observed in some cells in the postglutamate period was usually followed by [Ca2+]i recovery. This occurred, however, after a delay which varied widely between cells, from seconds to several minutes. This could be explained if mitochondrial depolarisation not only prevents Ca2+ uptake but also induces inactivation of the uniporter. During a subsequent mitochondrial repolarisation the uniporter could then reactivate slowly, renewing Ca2+ uptake and enhancing [Ca2+]i recovery. If so, the duration of the delay between the onset of mitochondrial repolarisation and the beginning of [Ca2+]i recovery would be defined mainly by the rate of reactivation of the uniporter. The most obvious alternative would be to suppose that mitochondrial repolarisation is followed by re-establishment of ATP synthesis, and that the variable delay reflects variability in the degree of ATP depletion during mitochondrial depolarisation.
ATP depletion
Profound mitochondrial depolarisation reverses the mitochondrial ATP synthase which both suppresses mitochondrial ATP production and promotes ATP hydrolysis (for reviews, see Nicholls, 1986; Duchen, 1999; and see Leyssens et al. 1996). Since both the major Ca2+ extrusion systems, the Na+-Ca2+ exchanger and the Ca2+-H+ pump, effectively require ATP (see for reviews DiPolo & Beauge, 1988; Sheu & Blaustein, 1992), it would be logical to speculate that ATP depletion (or a decrease in the ATP/ADP ratio) is the major cause of impaired post-glutamate [Ca2+]i recovery. However, the available data about the effects of prolonged exposure of cultured cerebellar granule cells to glutamate on ATP levels (Bogachev et al. 1992; Pinelis et al. 1997) or on the ATP/ADP ratio (Budd & Nicholls, 1996) do not support this conclusion, although specific data defining the rates of ATP depletion in our own preparations of hippocampal neurones are not yet available. Nevertheless, in cultured cerebellar granule cells, exposure to glutamate for 15 min decreased [ATP] only by 40-50 % of its control value. This [ATP] decrease was maintained in the post-glutamate period (Bogachev et al. 1992; Pinelis et al. 1997). Even at 37°C, a 60 min glutamate treatment of cerebellar granule cells decreased the ATP/ADP ratio only to 35 % of parallel controls (Budd & Nicholls, 1996). It is highly implausible that such a reduction of [ATP] or the ATP/ADP ratio would suffice to suppress Ca2+ extrusion from the cell, as the Kd of the plasmalemmal Ca2+-ATPase for ATP lies in the micromolar range.
Moreover, it does seem unlikely that the experiments with short-term glutamate applications could be explained in terms of neuronal ATP depletion. Indeed, in some cells, even a single 1 min glutamate pulse could produce a profound mitochondrial depolarisation followed by a high postglutamate [Ca2+]i plateau. Given the data cited above, it seems unlikely that a 1 min glutamate pulse could decrease the neuronal [ATP] sufficiently to impair Ca2+extrusion, as even application of an uncoupler (FCCP) for 2 min failed to produce ATP depletion in forebrain neurones (White & Reynolds, 1995), indicating the presence of well-developed glycolytic pathways for ATP production in these cultured cells (see McConnell et al. 1992), and limited mitochondrial ATP consumption.
Glutamate-induced deterioration of neuronal Ca2+ extrusion systems may be produced not only by [ATP] depletion but also by other factors, including free radicals, which have been implicated in this pathology. Indeed, there is growing evidence to implicate the generation of free radicals, including nitric oxide (NO), peroxynitrite and superoxide anion in glutamate toxicity (Coyle & Puttfarcken, 1993; Bindokas, 1996; Kashii et al. 1996). There are many potential sites of free radical induced cell damage that could contribute to the picture that emerges from our experimental data: (i) reactive oxygen species (ROS) are known to impair plasma membrane ionic transport systems: ion channels, ion pumps and ion exchangers (for review, see Kourie, 1998); (ii) together with high intramitochondrial [Ca2+], ROS may trigger the mitochondrial permeability transition pore (Zoratti & Szabo, 1995; Ankarcrona et al. 1996), which is blocked by CsA and N-methyl Val-4-cyclosporin A; (iii) NO and peroxynitrite also impair mitochondrial respiration (for review, see Peuchen et al. 1997), which could lead to mitochondrial depolarisation. Clearly, free radicals may produce damage to both of the Ca2+ homeostatic regulation systems that we have considered - to the mitochondria and to the Ca2+ extrusion mechanisms.
Given all these considerations, it is difficult to conclude, from the currently available data, that any one process dominates in defining the correlation between the glutamate-induced mitochondrial depolarisation and deregulation of neuronal [Ca2+]i homeostasis. The inhibitory effect of mitochondrial depolarisation on ATP synthesis and mitochondrial Ca2+ uptake may contribute, but both depolarisation of mitochondria and the inhibition of neuronal membrane Ca2+ extrusion systems may have a common origin, perhaps being induced by high [Ca2+]i and ROS generated in mitochondria and cytosol. A further clarification of the relative importance of each of these contributory mechanisms awaits more direct examination.
Acknowledgments
This study was supported by The Wellcome Trust and The Royal Society. We are grateful to Miss D. Lilian Patterson for her help with preparation and maintenance of cell cultures.
References
- Adamec E, Didier M, Nixon R. Developmental regulation of the recovery process following glutamate-induced calcium rise in rodent primary neuronal cultures. Developmental Brain Research. 1998;108:101–110. doi: 10.1016/s0165-3806(98)00034-0. [DOI] [PubMed] [Google Scholar]
- Ankarcrona M, Dypbukt JM, Orrenius S, Nicotera P. Calcineurin and mitochondrial function in glutamate-induced neuronal cell death. FEBS Letters. 1996;394:321–324. doi: 10.1016/0014-5793(96)00959-3. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Neuronal Ca2+ signalling takes the local route. Current Opinion in Neurobiology. 1992;2:302–307. doi: 10.1016/0959-4388(92)90119-6. [DOI] [PubMed] [Google Scholar]
- Bindokas VP, Jordan J, Lee CC, Miller RJ. Superoxide production in rat hippocampal neurons: selective imaging with hydroethidine. Journal of Neuroscience. 1996;16:1324–1336. doi: 10.1523/JNEUROSCI.16-04-01324.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogachev A, Bykova L, Khodorov B, Andreeva N, Khaspekov L, Pinelis V, Victorov I. Stable decrease in ATP contents in cultured cerebellar and hippocampal neurons during and after toxic glutamate treatment. Biological Membranes. 1992;9:1057–1059. in Russian. [Google Scholar]
- Budd SL, Nicholls DG. Mitochondria, calcium regulation, and acute glutamate excitotoxicity in cultured cerebellar granule cells. Journal of Neurochemistry. 1996;67:2282–2291. doi: 10.1046/j.1471-4159.1996.67062282.x. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders. Science. 1993;262:689–695. doi: 10.1126/science.7901908. [DOI] [PubMed] [Google Scholar]
- Dawson TM, Steiner JP, Dawson VL, Dinerman JL, Uhl GR, Snyder SH. Immunosuppressant FK506 enhances phosphorylation of nitric oxide synthase and protects against glutamate neurotoxicity. Proceedings of the National Academy of Sciences of the USA. 1993;90:9808–9812. doi: 10.1073/pnas.90.21.9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Erausquin GA, Manev H, Guidotti A, Costa E, Brooker G. Gangliosides normalize distorted single-cell intracellular free Ca2+ dynamics after toxic doses of glutamate in cerebellar granule cells. Proceedings of the National Academy of Sciences of the USA. 1990;87:8017–8021. doi: 10.1073/pnas.87.20.8017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiPolo R, Beauge L. Ca2+ transport in nerve fibers. Biochimica et Biophysica Acta. 1988;947:549–569. doi: 10.1016/0304-4157(88)90007-x. [DOI] [PubMed] [Google Scholar]
- Duchen MR. Ca2+-dependent changes in the mitochondrial energetics in single dissociated mouse sensory neurons. Biochemical Journal. 1992;283:41–50. doi: 10.1042/bj2830041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Contributions of mitochondria to animal physiology: from homeostatic sensor to calcium signalling and cell death. The Journal of Physiology. 1999;516:1–17. doi: 10.1111/j.1469-7793.1999.001aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. The Journal of Physiology. 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glaum SR, Scholz WK, Miller RJ. Acute- and long-term glutamate-mediated regulation of [Ca2+]i in rat hippocampal pyramidal neurons in vitro. Journal of Pharmacology and Experimental Therapeutics. 1990;253:1293–1302. [PubMed] [Google Scholar]
- Hartley DM, Choi DW. Delayed rescue of N-methyl-D-aspartate receptor-mediated neuronal injury in cortical culture. Journal of Pharmacology and Experimental Therapeutics. 1989;250:752–758. [PubMed] [Google Scholar]
- Hartley Z, Dubinsky JM. Changes in intracellular pH associated with glutamate excitotoxicity. Journal of Neuroscience. 1993;13:4690–4699. doi: 10.1523/JNEUROSCI.13-11-04690.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyrc KL, Bownik JM, Goldberg MP. BTC, fura-2FF and mag-fura-2 - in search of an ideal low affinity calcium indicator. Society for Neuroscience Abstracts. 1998;24:1059. [Google Scholar]
- Kashii S, Mandai M, Kikuchi M, Honda Y, Tamura Y, Kaneda K, Akaike A. Dual actions of nitric oxide in N-methyl-D-aspartate receptor-mediated neurotoxicity in cultured retinal neurons. Brain Research. 1996;711:93–101. doi: 10.1016/0006-8993(95)01330-x. [DOI] [PubMed] [Google Scholar]
- Khodorov BI, Fayuk DA, Koshelev SG, Vergun OV, Pinelis VG, Vinskaya NP, Storozhevikh TP, Arsenyeva EN, Khaspekov LG, Lyzhin AP, Isaev N, Victorov IV, Dubinsky JM. Effect of a prolonged glutamate challenge on plasmalemmal calcium permeability in mammalian central neurones. Mn2+ as a tool to study calcium influx pathways. International Journal of Neuroscience. 1996a;88:215–241. doi: 10.3109/00207459609000616. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Golovina V, Fajuk D, Andreeva N, Uvarova T, Khaspekov L, Victorov I. On the origin of a sustained increase in the cytosolic Ca2+ concentration after a toxic glutamate treatment of the nerve cell culture. FEBS Letters. 1993;324:271–273. doi: 10.1016/0014-5793(93)80132-e. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Storozhevokh T, Vergun O, Vinskaya N. Dominant role of mitochondria in protection against a delayed neuronal Ca2+ overload induced by endogenous excitatory amino acids following a glutamate pulse. FEBS Letters. 1996c;393:135–138. doi: 10.1016/0014-5793(96)00873-3. [DOI] [PubMed] [Google Scholar]
- Khodorov B, Pinelis V, Vergun O, Storozhevikh T, Vinskaya N. Mitochondrial deenergization underlies neuronal calcium overload following a prolonged glutamate challenge. FEBS Letters. 1996b;397:230–234. doi: 10.1016/s0014-5793(96)01139-8. [DOI] [PubMed] [Google Scholar]
- Kiedrowski L. The difference between mechanisms of kainate and glutamate excitotoxicity in vitro: osmotic lesion versus mitochondrial depolarization. Restorative Neurology and Neuroscience. 1998;12:71–79. [PubMed] [Google Scholar]
- Kiedrowski L, Wroblewski JT, Costa E. Intracellular sodium concentration in cultured cerebellar granule cells challenged with glutamate. Molecular Pharmacology. 1994;45:1050–1054. [PubMed] [Google Scholar]
- Kourie JI. Interaction of reactive oxygen species with ion transport mechanisms. American Journal of Physiology. 1998;275:C1–24. doi: 10.1152/ajpcell.1998.275.1.C1. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. The Journal of Physiology. 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limbrick DD, Churn SB, Sombati S, DeLorenzo RJ. Inability to restore resting intracellular calcium levels as an early indicator of delayed neuronal cell death. Brain Research. 1995;690:145–156. doi: 10.1016/0006-8993(95)00552-2. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M, Silver RB. Presynaptic calcium concentration microdomains and transmitter release. Journal de Physiologie. 1992;86:135–138. doi: 10.1016/s0928-4257(05)80018-x. [DOI] [PubMed] [Google Scholar]
- McConnell HM, Owicki JC, Parce JW, Miller DL, Baxter GT, Wada HG, Pitchford S. The cytosensor microphysiometer: biological applications of silicon technology. Science. 1992;257:1906–1912. doi: 10.1126/science.1329199. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, DeFrausquin GA, Guidotti A, Brooker G, Costa E. Destabilization of ionized Ca2+ homeostasis in excitatory amino acids neurotoxicity: antagonism by glycosphyngolipids. Cell Biology International Reports. 1990;14:3–14. doi: 10.1016/0309-1651(90)90066-8. [DOI] [PubMed] [Google Scholar]
- Manev H, Favaron M, Guidotti A, Costa E. Delayed increase of Ca2+ influx elicited by glutamate: role in neuronal death. Molecular Pharmacology. 1989;36:106–112. [PubMed] [Google Scholar]
- Nicholls DG. Intracellular calcium homeostasis. British Medical Bulletin. 1986;42:353–358. doi: 10.1093/oxfordjournals.bmb.a072152. [DOI] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends in Pharmacological Science. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. [DOI] [PubMed] [Google Scholar]
- Nieminen AL, Petrie TG, Lemasters JJ, Selman WR. Cyclosporin A delays mitochondrial depolarization induced by N-methyl-D-aspartate in cortical neurons: evidence of the mitochondrial permeability transition. Neuroscience. 1996;75:993–997. doi: 10.1016/0306-4522(96)00378-8. [DOI] [PubMed] [Google Scholar]
- Ogura A, Miyamoto M, Kudo Y. Neuronal death in vitro: parallelism between survivability of hippocampal neurones and sustained elevation of cytosolic Ca2+ after exposure to glutamate receptor agonist. Experimental Brain Research. 1988;73:447–458. doi: 10.1007/BF00406601. [DOI] [PubMed] [Google Scholar]
- Petrozzino JJ, Pozzo-Miller LD, Connor JA. Micromolar Ca2+ transients in dendritic spines of hippocampal pyramidal neurons in brain slice. Neuron. 1995;14:1223–1231. doi: 10.1016/0896-6273(95)90269-4. [DOI] [PubMed] [Google Scholar]
- Peuchen S, Bolanos JP, Heales SJ, Almeida A, Duchen MR, Clark JB. Interrelationships between astrocyte function, oxidative stress and antioxidant status within the central nervous system. Progress in Neurobiology. 1997;52:261–281. doi: 10.1016/s0301-0082(97)00010-5. [DOI] [PubMed] [Google Scholar]
- Pinelis V, Khodorov B, Fajuk D, Zagulova D, Andreeva N, Uvarova T, Khaspekov L, Golovina V, Victorov I. A persistent calcium-dependent decrease in cytoplasmic pH in cultured nerve cells induced by toxic glutamate treatment. Biological Membranes. 1992;9:1050–1051. in Russian. [Google Scholar]
- Pinelis V, Segal M, Greenberger V, Khodorov B. Changes in cytosolic sodium caused by a toxic glutamate treatment of cultured hippocampal neurons. Biochemistry and Molecular Biology International. 1994;32:1475–1482. [PubMed] [Google Scholar]
- Pinelis VG, Bykova LP, Bogachev AP, Isaev NK, Victorov IV, Khodorov BI. Toxic glutamate treatment of cerebellar granule cells decreases the intracellular ATP content. Role of Ca2+ ions. Bulluten Experimentalnoy Biologii i Medicini. 1997;123:162–164. in Russian. [PubMed] [Google Scholar]
- Schinder AF, Olson EC, Spitzer NC, Montal M. Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. Journal of Neuroscience. 1996;16:6125–6133. doi: 10.1523/JNEUROSCI.16-19-06125.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreier MH, Baumann G, Zenke G. Inhibition of T-cell signaling pathways by immunophilin drug complexes: are side effects inherent to immunosuppressive properties? Transplantation Proceedings. 1993;25:502–507. [PubMed] [Google Scholar]
- Sheu S-S, Blaustein MP. Sodium/calcium exchange and control of cell calcium and contractility in cardiac and vascular smooth muscles. In: Fozzard HA, Haber E, Jennings RB, Katz AM, Morgan HE, editors. The Heart and Cardiovascular System. Raven Press; 1992. pp. 509–537. [Google Scholar]
- Stout AK, Reynolds IJ. High-affinity calcium indicators underestimate increases in intracellular calcium concentrations associated with excitotoxic glutamate stimulations. Neuroscience. 1999;89:91–100. doi: 10.1016/s0306-4522(98)00441-2. [DOI] [PubMed] [Google Scholar]
- Takahashi M, Billups B, Rossi D, Sarantis M, Hamann M, Attwell D. The role of glutamate transporters in glutamate homeostasis in the brain. Journal of Experimental Biology. 1997;200:401–409. doi: 10.1242/jeb.200.2.401. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+ loads. Journal of Neurophysiology. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- White RJ, Reynolds I. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. Journal of Neuroscience. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White RJ, Reynolds IJ. Mitochondrial depolarization in glutamate-stimulated neurons: an early signal specific to excitotoxin exposure. Journal of Neuroscience. 1996;16:5688–5697. doi: 10.1523/JNEUROSCI.16-18-05688.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoratti M, Szabo I. The mitochondrial permeability transition. Biochimica et Biophysica Acta. 1995;1241:139–176. doi: 10.1016/0304-4157(95)00003-a. [DOI] [PubMed] [Google Scholar]