

Abstract
The role of actin polymerization in the regulation of smooth muscle contractility was investigated in canine trachealis muscle strips. The effect of contractile activation on the content of monomeric globular (G)-actin was estimated by the method of DNase I inhibition. The G-actin content was 30 % lower in extracts of muscle strips activated with 10−4 M acetylcholine (ACh) than in extracts from unstimulated muscle strips. The decrease in G-actin in response to contractile stimulation was prevented by latrunculin-A, an agent that prevents actin polymerization by binding to G-actin monomers.
The inhibition of actin polymerization by latrunculin-A markedly depressed force development in response to ACh but had no effect on ACh-induced myosin light chain (MLC) phosphorylation. Latrunculin also suppressed the length sensitivity of force during ACh-induced isometric contractions. The actin-capping agent cytochalasin-D also markedly inhibited force and caused only a slight decrease in MLC phosphorylation. Cytochalasin-D also inhibited force in α-toxin-permeabilized muscle strips that were activated either by Ca2+ or by ACh at constant pCa. No disorganization of smooth muscle cell ultrastructure was detected by electron microscopy or by immunofluorescence microscopy of muscles treated with either agent.
The results suggest that the polymerization of actin is stimulated by the contractile activation of tracheal smooth muscle and that this actin polymerization contributes directly to force development. In addition, actin filament remodelling contributes to the length sensitivity of tracheal smooth muscle contractility.
The objective of this study was to evaluate the role of actin filament polymerization in the contraction of smooth muscle. The activation of a number of non-muscle cells including neutrophils, platelets and fibroblasts has been shown to stimulate the rapid polymerization of actin from monomeric globular (G) actin to filamentous (F) actin (Cano et al. 1991; Symons & Mitchison, 1991; Hartwig, 1992; Theriot, 1994; Iwig et al. 1995). In unstimulated smooth muscle tissues, actin exists predominantly in filamentous form (Lehman et al. 1996); however, there is little evidence as to whether additional actin polymerization is stimulated in smooth muscle in response to the contractile activation.
Previous work from our laboratory and others has demonstrated that the physical length of smooth muscle at the time of contractile activation has long-lasting effects on its mechanical properties for the duration of the period of contractile activation (Gunst, 1986; Harris & Warshaw, 1991; Gunst et al. 1993; Meiss, 1993). Differences in mechanical properties caused by activation of the muscle at different lengths can be observed by subsequently measuring its contractile properties at the same muscle length during the same contraction. These persistent effects of length history are not the result of mechanically induced alterations in myosin light chain phosphorylation (Mehta et al. 1996). We have hypothesized that differences in smooth muscle contractility that are established by contracting muscles at different muscle lengths may result from changes in the organization of the actin filament network that enable the cell to optimize the organization of its contractile apparatus to its mechanical environment at the time of contractile activation (Gunst et al. 1993, 1995; Pavalko et al. 1995; Wang et al. 1996; Tang et al. 1999). Changes in the organization of the actin cytoskeleton might occur via the remodelling of actin filaments and/or through changes in the sites of actin filament attachment to the smooth muscle membrane (Pavalko et al. 1995; Wang et al. 1996; Tang et al. 1999). Thus the polymerization of even a small pool of G-actin might hypothetically have significant physiological effects on smooth muscle contractility.
In non-muscle cells, mechanical strain is sensed by transmembrane integrins and transduced by integrin-associated cytoskeletal proteins into signalling pathways that regulate strain-sensitive cellular processes (Burridge & Chrzanowska-Wodnicka, 1996; Shyy & Chien, 1997; Ingber, 1997). In a number of cell types, cytoskeletal remodelling can be stimulated by an integrin-associated mechano-transduction pathway (Wang et al. 1993; Banes et al. 1995; Shyy & Chien, 1997; Glogauer et al. 1998). Alterations in cell shape or stiffness in response to mechanical stimuli may be mediated by remodelling the length or attachment sites of actin filaments (Hartwig, 1992; Theriot, 1994). Analogous signalling pathways may mediate cytoskeletal remodelling in response to mechanical stimuli in smooth muscle cells, and this may contribute to length-sensitive changes in contractility. A mechanosensitive signal transduction pathway involving integrin-associated cytoskeletal proteins is present in tracheal smooth muscle. The tyrosine phosphorylation of the integrin-associated focal adhesion proteins paxillin and focal adhesion kinase (FAK) is regulated in activated tracheal smooth muscle in response to changes in mechanical strain (Tang et al. 1999).
The objective of the present study was to evaluate the effects of contractile activation on actin polymerization in tracheal smooth muscle, and to determine the role of actin polymerization in force development. We investigated whether actin filament dynamics contribute to the changes in smooth muscle contractility that occur in response to changes in muscle length. Two approaches were used to assess the role of actin polymerization in smooth muscle contraction. First, the effects of inhibiting actin filament polymerization on myosin light chain phosphorylation and force development were evaluated using cytochalasin-D and latrunculin-A. Cytochalasin-D and latrunculin-A act by different mechanisms to inhibit actin filament polymerization (Cooper, 1987; Coue et al. 1987). Second, changes in the concentration of G-actin in response to smooth muscle contraction were assessed using the DNase I inhibition assay (Blikstad et al. 1978).
METHODS
Tissue preparation and measurement of contractile force
Mongrel dogs (20-25 kg) were anaesthetized with pentobarbital sodium (150 mg kg−1i.v.) and killed by rapid exsanguination, as approved by the Indiana University Animal Care and Use Committee. A 10-15 cm segment of extrathoracic trachea was immediately removed and immersed in physiological saline solution (PSS) at 22°C of the following composition (mM): 110 NaCl, 3.4 KCl, 2.4 CaCl2, 0.8 MgSO4, 25.8 NaHCO3, 1.2 KH2PO4 and 5.6 glucose. The solution was aerated with 95 % O2-5 % CO2 to maintain a pH of 7.4. Rectangular strips of trachealis muscle 12-15 mm long and 2-3 mm wide were dissected from the trachea after removal of the epithelium and connective tissue layer. Muscle strips were equilibrated for approximately 90 min after being mounted in a tissue bath and attached to a force transducer (Grass) at a resting tension of 2 g. The optimal length for maximal active force (Lo) was determined by increasing muscle length progressively during successive stimulations with 10−5 M acetylcholine (ACh) until the force of active contraction reached a maximum.
After the determination of Lo, muscle strips were maintained in PSS containing the vehicle (0.1 % DMSO), incubated for 1 h with cytochalasin-D (0.5, 1.0 or 10 μm), or incubated for 45 min with latrunculin-A (0.1, 0.5 or 1 μm) dissolved in 0.1 % DMSO. The strips were then stimulated with 10−4 M ACh for 5 min after which they were quickly frozen with liquid N2-cooled tongs for the measurement of MLC phosphorylation. Up to 14 muscle strips from a single trachea were studied concurrently. Duplicate muscle strips were used for each measurement.
The effects of cytochalasin-D and latrunculin-A on the length dependence of force and MLC phosphorylation were assessed at muscle lengths between Lo and 0.6Lo as follows. Muscle strips were contracted isometrically at the predetermined lengths repeatedly using 10−5 M ACh until they developed constant force at that length. They were then incubated with 1 μm cytochalasin-D or 0.5 μm latrunculin-A for 45 min. Strips were then stimulated with 10−5 M ACh for 5 min and contractile force was determined. In some experiments, strips were then frozen for the measurement of MLC phosphorylation.
Measurement of myosin light chain phosphorylation
Frozen muscle strips were immersed in acetone containing 10 % (w/v) trichloroacetic acid and 10 mM dithiothreitol (DTT) (acetone- TCA-DTT) cooled to -80°C with crushed dry ice. Strips were thawed in acetone-TCA-DTT at room temperature and then washed with acetone-DTT. Myosin light chains were extracted for 60 min in 8 M urea, 20 mM Tris, 22 mM glycine and 10 mM DTT. Proteins were separated by glycerol-urea polyacrylamide gel electrophoresis and blotted onto nitrocellulose. MLCs were specifically labelled with polyclonal rabbit anti-myosin light chain 20 antibody. The primary antibody was detected with 125I-labelled recombinant Protein A (New England Nuclear). Unphosphorylated and phosphorylated bands of myosin light chains were localized on nitrocellulose membranes by autoradiography. Bands were cut out and counted in a gamma counter. Background counts were subtracted and fractional phosphorylation was calculated as the ratio of phosphorylated myosin light chains to total myosin light chains.
Permeabilization of muscle strips
A modification of the method of Kitazawa et al (1989) was used to permeabilize the muscle strips. Muscle strips (0.1-0.2 mm wide and 7-10 mm long) were incubated at 22°C in a relaxing solution composed of (mM): 8.5 Na2ATP, 4 K-EGTA, 1 DTT, 10 sodium creatinine phosphate, 20 imidazole, 8.9 magnesium acetate, 100.5 potassium acetate and 1 mg ml−1 creatine phosphokinase (Sigma cat. no. C 3755; 310 U mg−1). After 20 min, the strips were then incubated in the same solution with the addition of α-toxin (350 u ml−1) (Calbiochem), plus 0.1 mg ml−1 phosphocreatine kinase and 1 μm leupeptin (a protease inhibitor) for another 20-25 min. Intracellular Ca2+ stores were depleted by incubating the strips in 10 μm calcium ionophore A23187 in relaxing solution. An algorithm of Fabiato & Fabiato (1979) was used to calculate the composition of relaxing or contracting solutions containing free Ca2+ from pCa 9 to pCa 5. For the measurement of isometric force, permeabilized muscle strips were mounted in tissue baths and attached to Gould GM-2 force transducers. In each experiment, permeabilization of the strips was verified by contracting the muscles with contracting solution at pCa 5.
Measurement of monomeric actin (G-actin) in tracheal muscle strips
The content of G-actin in smooth muscle strips was estimated by measuring the inhibition of DNase I activity by G-actin (Blikstad et al. 1978). Muscle strips were subjected to treatment with ACh, latrunculin, or cytochalasin-D and then frozen and pulverized under liquid N2. The pulverized powder was transferred to dry ice-cooled centrifuge tubes for the extraction of protein. After the addition of 800 μl of extraction buffer the sample was quickly vortexed. The extraction buffer (pH 6.9) contained 60 mM Pipes, 25 mM Hepes, 10 mM EGTA, 2 MgCl2, 0.5 % Triton X-100, 0.1 mM DTT, 0.5 mM phenylmethylsulfonylfluoride (PMSF) and protease inhibitors (0.01 mg ml−1 each of chymotrypsin, leupeptin, aprotinin and pepstatin). The sample was then kept at 4°C for 5 min after which it was centrifuged at 14000 g for 8 min. The supernatant was then transferred to another tube for the measurement of G-actin content.
The DNase I inhibition assay was performed at 25°C. The same time course for protein extraction and G-actin determination was maintained for each sample. One millilitre of DNA solution (100 μg of calf thymus DNA dissolved in 0.1 M Tris-HCl (pH 7.4), 4 mM MgSO4 and 1.8 mM CaCl2) was added to 10 μl of DNase I solution (1 μg of enzyme in 0.05 M Tris-HCl (pH 7.4), 0.01 mM PMSF, and 0.1 mM CaCl2). The production of DNA oligonucleotides due to hydrolysis of DNA was then monitored by recording the hyperchromicity at 260 nm as a function of time using a Beckman UV spectrophotometer. The concentration of G-actin in different volumes (5-30 μl) of muscle extract was determined using multiple aliquots from each muscle extract sample. Extract samples were mixed with DNase I solution for 10 s before the addition of DNA solution and the reaction rate was followed for up to 3 min. Muscle extract volumes were chosen to allow 30-70 % inhibition of DNase activity. DNase activity was also measured in the presence of the same volume of sample buffer without the addition of muscle extract. The concentration of G-actin in the muscle extract that caused 50 % inhibition of DNase activity was estimated from a standard curve that was determined using known amounts of purified actin. The concentration of protein in each sample of muscle extract was estimated by using a standard micro protein assay (Pierce). G-actin content of the muscle extract was then normalized as a fraction of soluble protein. The accuracy of the assay in smooth muscle extracts was confirmed by adding known amounts of purified G-actin to samples of muscle extracts and verifying that this resulted in predicted increases in the inhibition of DNase I activity.
In separate experiments, the proportion of G-actin to total actin in tracheal muscle strips was estimated by quantifying the actin content contained in the supernatant of muscle extracts relative to the actin contained in total muscle homogenates, assuming that most of the actin in the supernatant was in the form of G-actin. Proteins in the supernatant and in total muscle homogenates were separated by 15 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis, transferred to nitrocellulose, and blocked with a 5 % solution of non-fat dry milk. The membranes were probed with mouse monoclonal G-actin antibody (Sigma) and then with horseradish peroxidase and anti-mouse immunoglobulin (Amersham) for visualization by chemiluminescence. Actin in the supernatant and total muscle homogenate was quantified by densitometry and calculated as a ratio to determine the proportion of G-actin to total actin.
Electron microscopy of tracheal smooth muscle tissues
Muscle strips were fixed at room temperature in 2 % glutaraldehyde in 0.075 M cacodylate containing 1.2 mM calcium and 4.5 % sucrose at pH 7.4. The tissues were postfixed in 2 % osmium in 0.1 M cacodylate buffer for 2 h, rinsed with buffer and in block stained with saturated aqueous uranyl acetate for 90 min. They were then dehydrated in graded alcohols and embedded in Spurr's resin. Longitudinal sections 60-70 nm thick were then cut with a Sorwall MT5000 ultramicrotome with the muscle tissue oriented longitudinally parallel to the edge of the Dupont diamond microtome knife. The tissue sections were picked up on uncoated grids and viewed at a magnification of ×10000 under the electron microscope.
Fluorescence imaging of smooth muscle cells
Tracheal muscle strips (1 mm wide, 10-20 mm long) were transferred to 5 ml of dissociation solution (composition, mM: 125 NaCl, 4.7 KCl, 0.25 CaCl2, 1.0 MgCl2, 10 Hepes, 0.25 EDTA, 10 D-glucose, 10 taurine, pH 7), with the addition of collagenase (400 U ml−1, type I), papain (30 U ml−1, type IV) bovine serum albumin (1 mg ml−1) and DTT (1 mM). All enzymes were from Sigma. The strips were then maintained in a 37°C shaking water bath at 80 oscillations min. After 40 min, the strips were washed several times with Hepes-buffered saline solution (composition, mM: 130 NaCl, 5 KCl, 1.0 CaCl2, 1.0 MgCl2, 20 Hepes, 10 D-glucose, pH 7.4) and gently triturated to liberate individual smooth muscle cells (Halayko et al. 1996). Dissociated cells were allowed to settle onto coverslips for 40 min.
Dissociated smooth muscle cells adhering to coverslips were incubated in Hepes-buffered saline containing 0.1 % DMSO, 10 μm cytochalasin-D, or 1 μm latrunculin for 45 min after which they were fixed with 3.7 % (v/v) paraformaldehyde in phosphate-buffered saline (composition, mM: 137 NaCl, 4.3 Na2HPO4, 1.4 KH2PO4, 2.7 KCl, pH 7.4) for 10 min. The cells were washed three times in Tris-buffered saline (TBS) containing 50 mM Tris, 100 mM NaCl, 0.1 % NaN3 and then permeabilized for 2 min with 0.2 % Triton X-100 dissolved in TBS. Cells were rinsed again three times in TBS and labelled with rhodamine-phalloidin (1:400, Molecular Probes) at room temperature. After 25 min the cells were washed again to remove excess label and coverslips were mounted onto slides using 10-15 μl mounting medium containing antifade agents (gelvatol with glycerol, n-propyl gallate and sodium azide). Cells were then viewed using a BioRad 1024 MRC laser confocal microscope and viewed with a × 100 oil immersion objective (numerical aperture 1.4).
Data analysis
Comparisons among different groups were performed by one-way analysis of variance or Kruskal-Wallis one-way analysis of variance. Differences between the two groups were analysed by Student's t test or Dunn's method. Statistical analysis was performed using SigmaStat software. Values of n represent the number of experiments used to obtain each value. P < 0.05 was considered to be significant.
RESULTS
Effect of latrunculin-A and cytochalasin-D on active force and MLC phosphorylation during isometric contraction of tracheal muscle strips with acetylcholine
Force and MLC phosphorylation in response to 10−4 M ACh were assessed in muscle strips treated for 45 min with 0, 0.1, 0.5 or 1 μm of latrunculin-A, an agent that binds to monomeric actin and prevents its assembly into actin filaments (Fig. 1A and B). Force was significantly reduced in all latrunculin-A treated muscle strips as compared to that in untreated muscle strips. The reduction in force increased when the concentration of latrunculin was increased from 0.1 to 1.0 μm. Latrunculin-A had no effect on MLC phosphorylation in ACh-stimulated or unstimulated muscles.
Figure 1. Effects of latrunculin (A and B) and cytochalasin-D (C and D) on force and myosin light chain (MLC) phosphorylation in tracheal muscle strips stimulated with 100 μm ACh under isometric conditions.
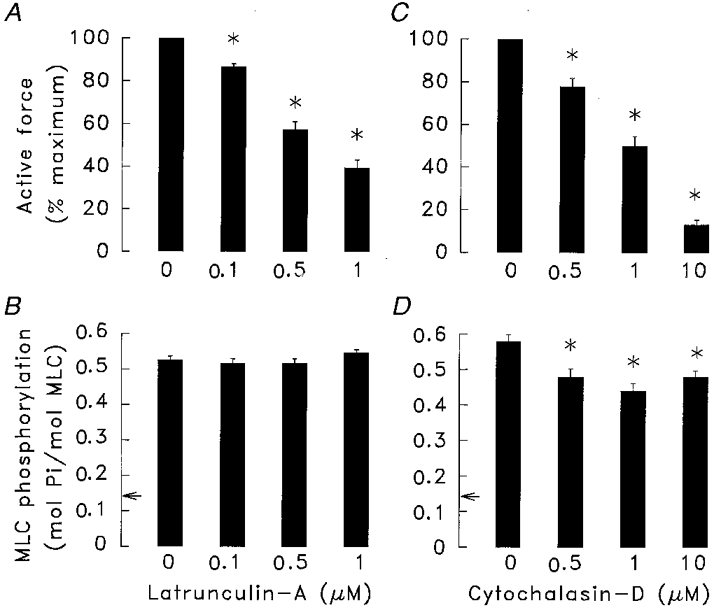
Latrunculin (0.1, 0.5, or 1 μm) significantly reduced force development; however it had no significant effect on MLC phosphorylation (n = 5). Cytochalasin (0.5, 1 or 10 μm) significantly reduced both force development and MLC phosphorylation (n = 5). Neither cytochalasin-D nor latrunculin affected MLC phosphorylation in unstimulated muscles. Arrows represent the mean value of MLC phosphorylation in unstimulated muscle strips. Force is normalized to the maximal force obtained in response to 100 μm ACh in the absence of cytochalasin or latrunculin (untreated strips). Data are means ±s.e.m.* Values significantly different from untreated muscle strips (P < 0.05).
Force and MLC phosphorylation in response to 10−4 M ACh were also assessed in five muscle strips incubated for 1 h in 0, 0.5, 1.0 or 10 μm cytochalasin-D (Fig. 1C and D). Force development in response to ACh was significantly depressed in all muscle strips treated with cytochalasin-D as compared to that in untreated muscle strips. The magnitude of the effect of cytochalasin-D on force in response to ACh increased with increases in the concentration of cytochalasin. Cytochalasin-D significantly reduced ACh-induced MLC phosphorylation below that obtained in untreated muscle strips; however, the effect of cytochalasin-D on MLC phosphorylation was small and was not dose dependent. Cytochalasin-D had no effect on MLC phosphorylation in unstimulated muscle strips.
Effect of cytochalasin-D on isometric contraction in permeabilized tracheal smooth muscle strips
Permeabilized muscle strips were used to evaluate whether the effects of cytochalasin-D on force were due to effects on Ca2+ signalling. Permeabilized muscle strips were incubated in 0, 1, or 10 μm cytochalasin-D in Ca2+-free buffer (pCa 9) for 15 min. Strips were then isometrically contracted with contracting solution at pCa 5 or with 10−4 M ACh at pCa 7. Force development in response to an increase in Ca2+ was significantly depressed in muscle strips treated with 1 or 10 μm cytochalasin-D as compared to that in untreated muscle strips (Fig. 2). Contractions induced by ACh at constant Ca2+ (pCa 7) were also significantly depressed in muscle strips treated with either 1 or 10 μm cytochalasin-D.
Figure 2. Effect of cytochalasin-D on contractions induced by increasing [Ca2+] to pCa 5 (A) or by adding 10−4 M ACh at pCa 7 (B) in α-toxin-permeabilized muscle strips.
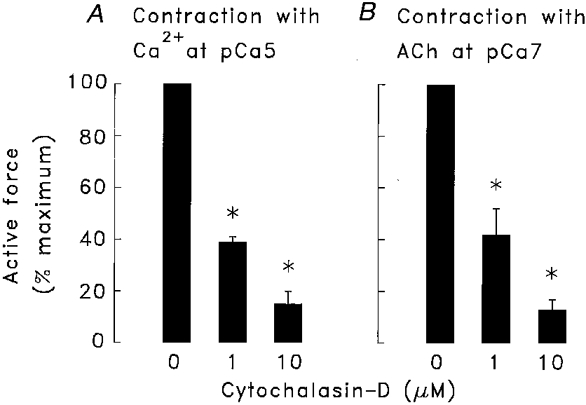
Cytochalasin-D (1 or 10 μm) caused significant inhibition of force in muscle strips stimulated with 10 μm Ca2+or with ACh (n = 3). Force was normalized to the maximal force obtained in response to Ca2+ or ACh in muscle strips not treated with cytochalasin. Data are means ±s.e.m.* Values significantly different from untreated muscle strips (P < 0.05).
Effect of latrunculin and cytochalasin-D on the length-dependent modulation of active force and MLC phosphorylation in tracheal smooth muscle
Muscle strips were set to lengths of 0.6Lo, 0.8Lo or Lo and then incubated for 45 min in PSS with or without 1 μm cytochalasin-D or 0.5 μm latrunculin. The strips were then stimulated with 10−5 M ACh for 5 min after which they were frozen for the measurement of MLC phosphorylation. Figure 3 illustrates the effect of treatment with cytochalasin and latrunculin on force and MLC phosphorylation for contractions induced at Lo, 0.8Lo or 0.6Lo. Values shown for force are normalized to active force at Lo in the untreated muscle strips.
Figure 3. Effect of latrunculin (A and B) or cytochalasin (C and D) on the length sensitivity of force and MLC phosphorylation in muscle strips contracted isometrically at 0.6Lo, 0.8Lo or Lo with 10 μm ACh.
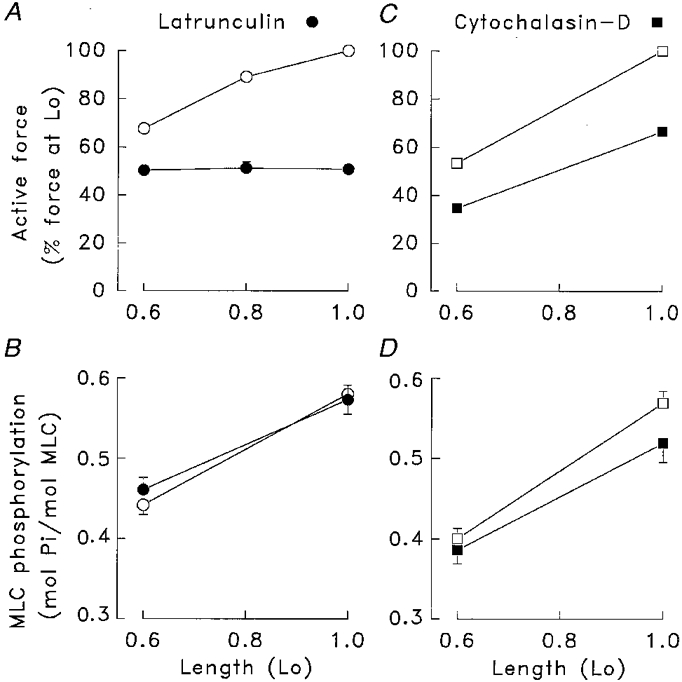
○, □, muscle strips not treated with cytochalasin or latrunculin; •, ▪, strips treated with cytochalasin or latrunculin. In untreated muscle strips, force was significantly different at lengths of 0.6Lo, 0.8Lo and Lo (A and C) (n = 8-18). In the presence of latrunculin, no significant differences in muscle force were observed at these muscle lengths (n = 8-18), whereas in the presence of cytochalasin force was significantly different at Lo and at 0.6Lo (n = 8). Differences in MLC phosphorylation at 0.6Lo and Lo were statistically significant in untreated muscle strips as well as in strips treated with cytochalasin or latrunculin (n = 8-10). Forces at 0.6Lo and at 0.8Lo were normalized to the force obtained at Lo in untreated muscle strips.
In latrunculin-treated muscle strips, active force in response to ACh was not significantly different in muscle strips contracted at 0.6Lo, 0.8Lo or Lo (Fig. 3A), indicating that latrunculin inhibited the length sensitivity of muscle force (P < 0.05). Thus, the inhibition of force by latrunculin was greater at longer muscle lengths: at Lo, latrunculin inhibited force by 49.2 ± 2.0 % (n = 18); at 0.8Lo, latrunculin inhibited force by 34.7 ± 4.8 % (n = 8); whereas at 0.6Lo, force was inhibited by only 25.2 ± 3.0 % (n = 18). Differences in the effect of latrunculin on force at different muscle lengths were statistically significant. In contrast, cytochalasin had no effect on the length sensitivity of muscle force. Cytochalasin inhibited force by 33.4 ± 2.7 % at Lo and by 35.2 ± 8.3 % at 0.6Lo (n = 8-10) (Fig. 3C).
MLC phosphorylation was significantly lower in muscle strips isometrically contracted at 0.6Lo as compared to Lo in all muscle strips regardless of treatment (Fig. 3B and D), demonstrating that the length sensitivity of MLC phosphorylation was not affected by either cytochalasin or latrunculin. Treatment with cytochalasin did result in a small but significant depression of MLC phosphorylation at both Lo and 0.6Lo.
The tension and strain experienced by muscle cells both increase when the muscle is contracted at longer muscle lengths. We therefore evaluated whether the greater inhibition of force by latrunculin at longer muscle lengths results from the effect of the increased active tension or the longer muscle length per se (Fig. 4). In each experiment, one pair of muscle strips was contracted at different lengths (Lo and 0.6Lo) using concentrations of ACh that resulted in similar levels of tension (Fig. 4C and D). Another pair of strips was contracted at the same muscle length (Lo) using concentrations of ACh that resulted in different levels of tension (Fig. 4A and B). In strips contracted to different levels of tension at the same length, the percentage inhibition of muscle force by latrunculin was not significantly different (Fig. 4B). In contrast, when strips were contracted at different lengths to comparable levels of tension, the inhibition of muscle force by latrunculin was much greater at the longer length. This indicates that increased muscle length rather than greater tension is the primary stimulus for the increased sensitivity of muscle force to latrunculin during isometric contraction at longer lengths.
Figure 4. Comparison of the effects of differences in active tension (A and B) or differences in muscle length (C and D) on the sensitivity of contraction to latrunculin.
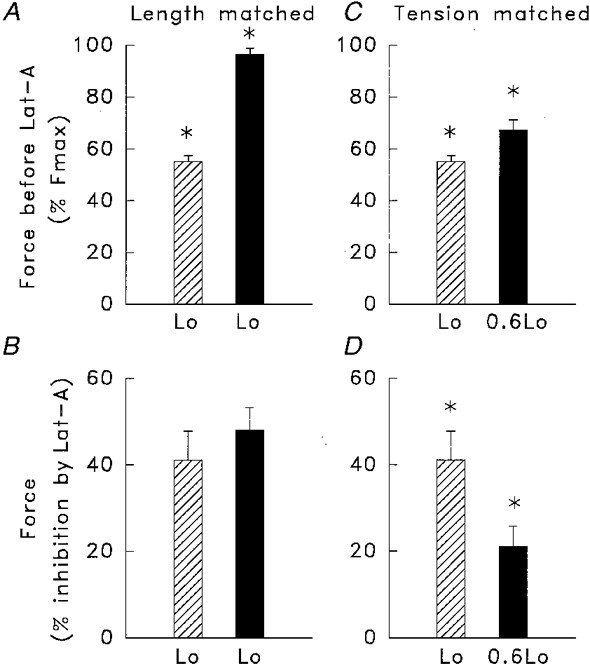
In strips contracted to different tensions at the same muscle length (A), the inhibitory effect of latrunculin (Lat-A) on force was similar (B) (n = 6). In strips contracted to similar tensions at different lengths, Lo and 0.6Lo (C), latrunculin caused significantly more force inhibition at Lo (D) (n = 6). ▪, contraction with 10 μm ACh;  , contraction with 0.1 μm ACh. Force was normalized to the force obtained in response to 10 μm ACh at Lo in the absence of latrunculin. Percentage inhibition of force by latrunculin was calculated as the percentage reduction in force after treatment. * Values significantly different from each other (P < 0.05).
, contraction with 0.1 μm ACh. Force was normalized to the force obtained in response to 10 μm ACh at Lo in the absence of latrunculin. Percentage inhibition of force by latrunculin was calculated as the percentage reduction in force after treatment. * Values significantly different from each other (P < 0.05).
Effect of contractile stimulation, cytochalasin and latrunculin on the G-actin content of tracheal smooth muscle strips
We used the DNase I inhibition assay to assess the effects of contractile activation and muscle length on G-actin content (Fig. 5A). Strips were assayed for G-actin after being maintained at either 0.6Lo or Lo without contractile stimulation (unstimulated), or after 5 min isometric contraction with ACh at either Lo or 0.6Lo (stimulated). At both muscle lengths, stimulation with ACh caused the G-actin content to decrease significantly by about 30 %. The mean G-actin content of muscles stimulated at 0.6Lo was slightly higher than that at Lo, but the difference was not statistically significant. The level of G-actin in unstimulated muscle strips was similar at Lo and at 0.6Lo.
Figure 5. Effect of muscle length (A) and cytochalasin-D or latrunculin (B) on the G-actin content of muscle strips after 5 min stimulation with 100 μm ACh.
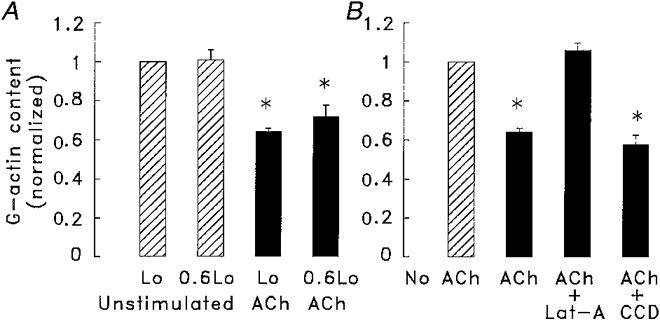
A, at muscle lengths of either Lo or 0.6Lo, stimulation with ACh caused a significant decrease in G-actin content. Differences in G-actin content in muscles at 0.6Lo or Lo were not significant in stimulated or in unstimulated muscles (n = 8). B, the G-actin content in muscles stimulated with ACh in the presence of 1 μm latrunculin (Lat-A) was not significantly different from that in unstimulated muscles, whereas the G-actin content of muscles stimulated with ACh in the presence of 10 μm cytochalasin (CCD) was significantly lower than that in unstimulated muscles. Values of G-actin content are normalized to values at Lo in unstimulated strips (n = 8). * Values significantly different from values at Lo in unstimulated tissues.
We also evaluated the effects of cytochalasin (10 μm) and latrunculin (1 μm) on the G-actin content of muscle strips stimulated with ACh. Latrunculin prevented the decrease in G-actin during stimulation with ACh; G-actin levels in latrunculin-treated muscle strips stimulated with ACh were similar to those in unstimulated strips. Latrunculin had no effect on G-actin in unstimulated strips. In contrast, cytochalasin did not prevent the decrease in G-actin that occurs with contractile activation (Fig. 5B). In separate experiments, we estimated that G-actin represented 31 ± 1.1 % (n = 4) of the total actin content of the muscle. Thus a 30 % decrease in the G-actin content represents polymerization of approximately 10 % of the total actin in the cell.
Effect of cytochalasin and latrunculin on the ultrastructure of tracheal smooth muscle
Sections of tracheal smooth muscle strips were viewed under the electron microscope to determine the effects of treatment with 10 μm cytochalasin-D or 1 μm latrunculin-A on the ultrastructure of the muscle cells and the organization of contractile filaments (Fig. 6). No differences in contractile filament organization or ultrastructure could be detected in electron micrographs taken from strips treated with cytochalasin or latrunculin as compared with untreated muscle strips.
Figure 6.
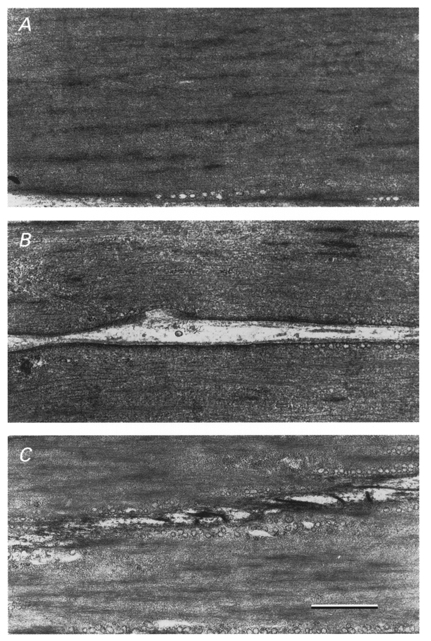
Representative electron micrographs of 60 nm thick longitudinal sections of unstimulated tracheal muscle strips. A, untreated muscle strips; B, strips treated with 10 μm cytochalasin-D; C, strips treated with 1 μm latrunculin. Scale bar, 0.5 μm.
We also evaluated the effects of cytochalasin and latrunculin on cell morphology and F-actin fluorescence in isolated dissociated tracheal smooth muscle cells. Unstimulated cells were incubated in 10 μm cytochalasin-D or 1 μm latrunculin for 45 min, and then fixed and stained with rhodamine-phalloidin. No significant differences in cell morphology or F-actin fluorescence were detected among untreated, cytochalasin-treated and latrunculin-treated smooth muscle cells (Fig. 7).
Figure 7.
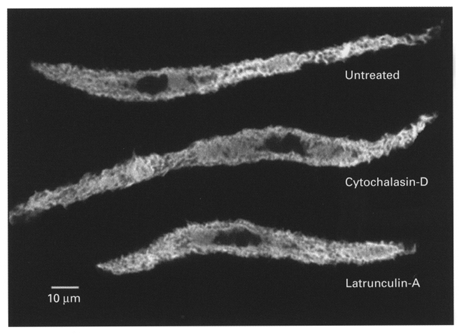
Representative confocal images of single dissociated unstimulated tracheal smooth muscle cells stained with rhodamine-phalloidin. Top, untreated; middle, treated with 10 μm cytochalasin-D; bottom, treated with 1 μm latrunculin-A.
DISCUSSION
Summary
The results of this study demonstrate that the contractile activation of tracheal smooth muscle causes a decrease in the content of G-actin, and that the inhibition of actin filament polymerization inhibits force development. These results suggest that actin polymerization is stimulated by the contractile activation of tracheal smooth muscle, and that this polymerization is necessary for force development. Our results also indicate that actin filament dynamics are sensitive to muscle length; latrunculin has a greater inhibitory effect on force development at a long muscle length than at a short muscle length. Thus, the regulation of actin dynamics may contribute to the length dependence of smooth muscle contractility. Our data clearly demonstrate that the inhibition of contractile force by agents that inhibit actin filament polymerization does not result from the disruption of signalling pathways that regulate myosin light chain phosphorylation, or from the disorganization of smooth muscle cell ultrastructure. The results of this study are consistent with our hypothesis that contractile stimulation initiates strain-sensitive signalling pathways that regulate the attachment and polymerization of actin filaments in smooth muscle cells, and that this contributes to the length-dependent regulation of smooth muscle contractility.
Effect of contractile stimulation on actin polymerization, force development and contractile protein activation
These studies provide several lines of evidence that contractile activation stimulates actin polymerization in tracheal smooth muscle. The contraction of tracheal muscle with acetylcholine decreases the content of G-actin by approximately 30 %. This represents the polymerization of about 10 % of the total cellular actin (Fig. 5). In addition, both latrunculin and cytochalasin inhibit force development. Cytochalasin-D and latrunculin-A are both inhibitors of actin polymerization, although they act by distinct mechanisms. Cytochalasin caps existing actin filaments, preventing their growth at the barbed end, whereas latrunculin binds to actin monomers and prevents their assembly into filamentous actin (Cooper, 1987; Coue et al. 1987). At the optimal muscle length, Lo, we found that both cytochalasin-D and latrunculin-A depressed ACh-induced force development in a dose-dependent manner. Latrunculin also prevented the decrease in G-actin content in response to contractile stimulation. This is consistent with its inhibitory effect on actin polymerization through the inactivation of G-actin monomers. In the presence of cytochalasin, ACh induced a decrease in the G-actin content. Actin polymerization in the presence of cytochalasin may have resulted from the accelerated nucleation of new actin filaments caused by the capping of existing filaments (Saito & Karaki, 1996; Sugidachi et al. 1998).
Several investigators have reported the inhibition of force development by cytochalasin in other smooth muscle tissues (Adler et al. 1983; Obara & Yabu, 1994; Saito et al. 1996; Tseng et al. 1997), although the effects of latrunculin on smooth muscle have not previously been reported. In endothelial cells, fibroblasts and platelets, cytochalasin inhibits Ca2+ signalling (Bourguignon et al. 1993; Ribeiro et al. 1997; Holda & Blatter, 1997), which suggests that actin polymerization is necessary for Ca2+ signalling in these cells. If this were also true in smooth muscle tissues, the effects of cytochalasin or latrunculin on force development might be accounted for by an inhibition of Ca2+-calmodulin-dependent myosin light chain phosphorylation. However, we found that while latrunculin-A caused a marked inhibition of force development, it had no effect on ACh-induced myosin light chain phosphorylation (Fig. 1). Although cytochalasin-D caused a slight depression of ACh-induced MLC phosphorylation, the reduction was insufficient to account for the marked inhibition of force that occurred at higher concentrations of this agent (Fig. 1). Furthermore, cytochalasin-D inhibited force in α-toxin-permeabilized muscle strips that were activated either by an increase in pCa or by the addition of ACh at constant pCa (Fig. 2). This indicates that cytochalasin was still effective at inhibiting force development even when the intracellular Ca2+ concentration was experimentally maintained. Thus, our results demonstrate that the inhibition of force development caused by cytochalasin-D and latrunculin-A in canine tracheal muscle does not result from effects on signalling pathways leading to Ca2+-dependent MLC phosphorylation. These results therefore suggest that actin polymerization plays a direct role in force development in smooth muscle that is independent of signalling events regulating the phosphorylation of myosin light chains.
The results of previous studies investigating the mechanism for the effects of cytochalasin on smooth muscle contraction are conflicting. Tseng et al. (1997) reported that in bovine tracheal smooth muscle, the inhibition of force caused by cytochalasin-B, a less specific agent than cytochalasin-D, was associated with a decrease in myosin light chain phosphorylation. They concluded that the inhibitory effect of cytochalasin on force was due to the inhibition of Ca2+ signalling and MLC phosphorylation caused by disruption of the actin cytoskeleton. In contrast, Saito et al. (1996) found that cytochalasin-D inhibited contractions induced by norepinephrine and K+ in rat aorta smooth muscle without affecting intracellular Ca2+ or MLC phosphorylation. Consistent with this, Obara & Yabu (1994) found no effect of cytochalasin-B on voltage-dependent Ca2+ currents and membrane potentials in taenia coli smooth muscle. Our present results demonstrating that smooth muscle contraction is inhibited by latrunculin, which has no effect on myosin light chain phosphorylation, provide unequivocal evidence that the inhibition of actin polymerization per se inhibits smooth muscle contraction.
Role of actin polymerization in smooth muscle contraction
The activation of cells such as platelets, neutrophils and fibroblasts results in the rapid polymerization of monomeric globular (G) actin into filamentous (F) actin (Cano et al. 1991; Symons & Mitchison, 1991; Hartwig, 1992; Iwig et al. 1995). This polymerization occurs primarily at the ‘barbed end’ of the actin filament, where the filament links to transmembrane integrins (Cooper, 1987, 1991; Schafer & Cooper, 1995). In these cells, actin polymerization is regulated by the availability of free barbed ends of actin filaments as well as by the pool of available actin monomers. Most actin filament barbed ends are not freely accessible in resting cells (Hartwig, 1992; Hug et al. 1995) as they are blocked from elongation by capping proteins such as gelsolin and CapZ (Barkalow et al. 1996). These proteins are also present in differentiated smooth muscle (Pollard & Cooper, 1986; Schafer & Cooper, 1995). The activation of platelets and other non-muscle cells causes the uncapping of actin filaments, thereby increasing the number of free barbed ends and enabling actin polymerization to proceed (Symons & Mitchison, 1991; Hartwig, 1992, 1995; Nachmias et al. 1996). In platelets, the interaction of the endogenous capping protein gelsolin with actin can be inhibited by PIP2, a by-product of phosphatidyl inositol breakdown that is also produced in smooth muscle in response to stimulation (Janmey, 1994). The capping of the barbed ends of actin filaments may be coupled to detachment of the filament from the membrane (Carlier, 1998).
Cytochalasin-D binds to the barbed (fast-growing) ends of actin filaments and acts as a capping protein (Cooper, 1987). As the binding of cytochalasin is not regulated by PIP2, actin filaments capped by cytochalasin do not become uncapped during activation of the cell. Thus they do not undergo polymerization at the barbed end, and they may not attach to the integrin complex and participate in force transmission during cell activation. Thus the effects of cytochalasin on force in smooth muscle may be related to its ability to block the attachment of actin filaments to the integrin complex, as well as to its ability to prevent filament polymerization at the barbed end. In contrast, the latrunculin does not interfere with the uncapping or attachment of filamentous actin to the integrin complex, but prevents actin polymerization by complexing with free actin monomers and thereby inactivating the pool of monomeric actin (Coue et al. 1987).
Role of actin filament dynamics in the length sensitivity of smooth muscle force
A striking finding of our study was that the inhibitory effect of latrunculin on contractile force was length sensitive, suggesting that mechanical strain may stimulate the rate of actin polymerization. The inhibition of force caused by latrunculin was greatest when the strips were activated at the longest length, Lo, and smallest when the muscles were activated at 0.6Lo. As a result, in the presence of latrunculin, there were no significant differences in force development at muscle lengths between 0.6Lo and Lo in response to acetylcholine (Fig. 3). Myosin light chain phosphorylation is length sensitive in smooth muscle, and the length sensitivity of contractile activation has been proposed as a mechanism to account for the length-dependent modulation of contractility (Rembold & Murphy, 1990; Mehta et al. 1996). However, latrunculin did not affect the length sensitivity of myosin light chain phosphorylation; thus the differences in its effect on contraction at different muscle lengths could not have resulted from a suppression of the mechanosensitivity of myosin activation. We observed a slight difference in the mean levels of G-actin in muscles activated with ACh at Lo and 0.6Lo using the DNase inhibition assay, but this difference was not statistically significant (Fig. 5A). This suggests that differences in the amount of actin polymerized at different lengths may be small and difficult to discriminate using this assay. Alternatively, mechanical strain may affect the dynamics of actin polymerization rather than the total amount of G-actin polymerized into filaments during contractile activation.
We evaluated whether differences in the sensitivity of contractions at Lo and 0.6Lo to inhibition by latrunculin were related directly to the difference in muscle length or to the differences in active tension at these muscle lengths. Our results indicated that muscle length rather than tension is the primary determinant of latrunculin sensitivity (Fig. 4). These results are consistent with our recent report that the muscle length rather than the tension is the stimulus for the length-sensitive increases in the tyrosine phosphorylation of paxillin and focal adhesion kinase during the contractile activation of tracheal smooth muscle (Tang et al. 1999). These proteins have been implicated in the signalling pathway for cytoskeletal remodelling in non-muscle cells (Burridge & Chrzanowska-Wodnicka, 1996; Shyy & Chien, 1997; Schmidt et al. 1998; Glogauer et al. 1998). Taken in sum, our observations suggest a mechanosensitive process in smooth muscle in which externally imposed strain activates an integrin-mediated signalling pathway leading to actin filament polymerization and cytoskeletal reorganization. Actin polymerization in smooth muscle cells might involve the lengthening of existing actin filaments and/or the nucleation of new actin filaments. This could enable the smooth muscle cell to adapt the arrangement of its contractile apparatus to externally imposed changes in the shape of the cell. In tracheal smooth muscle, contractile activation at different lengths results in persistent differences in stiffness that suggest differences in cell structure (Gunst et al. 1995; Gunst & Wu, 1996; Gunst, 1999).
In our study, the effects of cytochalasin-D contrasted with those of latrunculin in that cytochalasin inhibited force development proportionally during contraction at all muscle lengths whereas latrunculin inhibited proportionally more at long muscle lengths (Fig. 3). The absence of an effect of cytochalasin on the length sensitivity of active force has been previously reported for bovine tracheal muscle (Youn et al. 1998). The difference in the inhibitory effect of latrunculin and cytochalasin on the length sensitivity of active force can be interpreted in terms of the mechanisms of action of these agents. Latrunculin prevents the polymerization of new actin filaments or the lengthening of existing filaments. This should prevent the strain-sensitive remodelling of actin filaments, although existing filaments could continue to participate in force transmission. In contrast, cytochalasin caps existing F-actin filaments and may therefore inhibit their attachment and participation in force transmission (Carlier, 1998). Only the actin filaments left uncapped by cytochalasin-D could continue to participate in force transmission. Therefore, in the presence of cytochalasin, force development at all muscle lengths should be proportionally inhibited.
Effect of cytochalasin and latrunculin on smooth muscle cell structure
We used electron microscopy and immunofluorescence microscopy to evaluate the effects of cytochalasin and latrunculin on actin filament integrity and the ultrastructure of tracheal smooth muscle. Electron micrographs of tracheal muscle strips treated with cytochalasin or latrunculin revealed no differences in contractile filament organization or ultrastructure as compared with untreated muscle strips (Fig. 6). We also detected no significant differences in the morphology of untreated, cytochalasin-treated or latrunculin-treated smooth muscle cells stained for F-actin using rhodamine-phalloidin (Fig. 7). Thus the effects of these agents on force are unlikely to result from the disruption of actin filament integrity or disorganization of the contractile apparatus.
Conclusion
In conclusion, our results suggest that the contractile activation of tracheal smooth muscle stimulates actin polymerization, and that this actin polymerization contributes directly to force development. The polymerization of actin in response to contractile stimuli may modulate the organization or length of actin filaments as well as the attachment of actin filaments to the cell membrane for the transmission of force. The inhibition of force by inhibitors of actin polymerization does not result from the disruption of signalling pathways leading to myosin light chain phosphorylation, or from the disorganization of cell structure. Actin polymerization may also play an important role in regulating the length sensitivity of force development.
Acknowledgments
We are grateful to Dr Simon Atkinson for his advice and assistance with the DNase inhibition assay. We also thank Ming-Fang Wu for his assistance with experiments and for his help in preparing the figures. This work was supported by a US PHS grant HL29289, the Midwest Affiliate of the American Heart Association, and the Showalter Foundation.
References
- Adler KB, Krill J, Alberghini TV, Evans JN. Effect of cytochalasin D on smooth muscle contraction. Cell Motility. 1983;3:545–551. doi: 10.1002/cm.970030521. [DOI] [PubMed] [Google Scholar]
- Banes AJ, Tsuzaki M, Yamamoto J, Fischer T, Brigman B, Brown T, Miller L. Mechanoreception at the cellular level: the detection, interpretation, and diversity of responses to mechanical signals. Biochemistry and Cell Biology. 1995;73:349–365. doi: 10.1139/o95-043. [DOI] [PubMed] [Google Scholar]
- Barkalow K, Witke W, Kwiatkowski DJ, Hartwig JH. Coordinated regulation of platelet actin filament barbed ends by gelsolin and capping protein. Journal of Cell Biology. 1996;134:389–399. doi: 10.1083/jcb.134.2.389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blikstad I, Markey F, Carlsson L, Persson T, Lindberg U. Selective assay of monomeric and filamentous actin in cell extracts, using inhibition of deoxyribonuclease I. Cell. 1978;15:935–943. doi: 10.1016/0092-8674(78)90277-5. [DOI] [PubMed] [Google Scholar]
- Bourguignon LY, Iida N, Jin H. The involvement of the cytoskeleton in regulating IP3 receptor-mediated internal Ca2+ release in human blood platelets. Cell Biology International. 1993;17:751–758. doi: 10.1006/cbir.1993.1136. [DOI] [PubMed] [Google Scholar]
- Burridge K, Chrzanowska-Wodnicka M. Focal adhesions, contractility, and signaling. Annual Review of Cell and Developmental Biology. 1996;12:463–518. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- Cano ML, Lauffenburger DA, Zigmond SH. Kinetic analysis of F-actin depolymerization in polymorphonuclear leukocyte lysates indicates that chemoattractant stimulation increases actin filament number without altering the filament length distribution. Journal of Cell Biology. 1991;115:677–687. doi: 10.1083/jcb.115.3.677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlier MF. Control of actin dynamics. Current Opinion in Cell Biology. 1998;10:45–51. doi: 10.1016/s0955-0674(98)80085-9. [DOI] [PubMed] [Google Scholar]
- Cooper JA. Effects of cytochalasin and phalloidin on actin. Journal of Cell Biology. 1987;105:1473–1478. doi: 10.1083/jcb.105.4.1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper JA. The role of actin polymerization in cell motility. Annual Review of Physiology. 1991;53:585–605. doi: 10.1146/annurev.ph.53.030191.003101. [DOI] [PubMed] [Google Scholar]
- Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Letters. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. Journal de Physiologie. 1979;75:463–505. [PubMed] [Google Scholar]
- Glogauer M, Arora P, Chou D, Janmey PA, Downey GP, McCulloch CA. The role of actin-binding protein 280 in integrin-dependent mechanoprotection. Journal of Biological Chemistry. 1998;273:1689–1698. doi: 10.1074/jbc.273.3.1689. [DOI] [PubMed] [Google Scholar]
- Gunst SJ. Effect of length history on contractile behavior of canine tracheal smooth muscle. American Journal of Physiology. 1986;250:C146–154. doi: 10.1152/ajpcell.1986.250.1.C146. [DOI] [PubMed] [Google Scholar]
- Gunst SJ. Applicability of sliding filament/crossbridge paradigm to smooth muscle. Reviews of Physiology Biochemistry and Pharmacology. 1999;134:7–61. doi: 10.1007/3-540-64753-8_2. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Meiss RA, Wu MF, Rowe M. Mechanisms for the mechanical plasticity of tracheal smooth muscle. American Journal of Physiology. 1995;268:C1267–1276. doi: 10.1152/ajpcell.1995.268.5.C1267. [DOI] [PubMed] [Google Scholar]
- Gunst SJ, Wu MF. Canine tracheal smooth muscle stiffness during stretch depends on muscle length at contraction onset. Biophysical Journal. 1996;70:A45. [Google Scholar]
- Gunst SJ, Wu MF, Smith DD. Contraction history modulates isotonic shortening velocity in smooth muscle. American Journal of Physiology. 1993;265:C467–476. doi: 10.1152/ajpcell.1993.265.2.C467. [DOI] [PubMed] [Google Scholar]
- Halayko AJ, Salari H, Ma X, Stephens NL. Markers of airway smooth muscle cell phenotype. American Journal of Physiology. 1996;270:L1040–1051. doi: 10.1152/ajplung.1996.270.6.L1040. [DOI] [PubMed] [Google Scholar]
- Harris DE, Warshaw DM. Length vs. active force relationship in single isolated smooth muscle cells. American Journal of Physiology. 1991;260:C1104–1112. doi: 10.1152/ajpcell.1991.260.5.C1104. [DOI] [PubMed] [Google Scholar]
- Hartwig JH. Mechanisms of actin rearrangements mediating platelet activation. Journal of Cell Biology. 1992;118:1421–1442. doi: 10.1083/jcb.118.6.1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartwig JH, Bokoch GM, Carpenter CL, Janmey PA, Taylor LA, Toker A, Stossel TP. Thrombin receptor ligation and activated Rac uncap actin filament barbed ends through phosphoinositide synthesis in permeabilized human platelets. Cell. 1995;82:643–653. doi: 10.1016/0092-8674(95)90036-5. [DOI] [PubMed] [Google Scholar]
- Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Letters. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Hug C, Jay PY, Reddy I, McNally JG, Bridgman PC, Elson EL, Cooper JA. Capping protein levels influence actin assembly and cell motility in Dictyostelium. Cell. 1995;81:591–600. doi: 10.1016/0092-8674(95)90080-2. [DOI] [PubMed] [Google Scholar]
- Ingber DE. Tensegrity: the architectural basis of cellular mechanotransduction. Annual Review of Physiology. 1997;59:575–599. doi: 10.1146/annurev.physiol.59.1.575. [DOI] [PubMed] [Google Scholar]
- Iwig M, Czeslick E, Muller A, Gruner M, Spindler M, Glaesser D. Growth regulation by cell shape alteration and organization of the cytoskeleton. European Journal of Cell Biology. 1995;67:145–157. [PubMed] [Google Scholar]
- Janmey PA. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annual Review of Physiology. 1994;56:169–191. doi: 10.1146/annurev.ph.56.030194.001125. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ Journal of Biological Chemistry. 1989;264:5339–5342. [PubMed] [Google Scholar]
- Lehman W, Vibert P, Craig R, Barany M. Actin and the structure of smooth muscle thin filaments. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. San Diego, CA, USA.: Academic Press Inc.; 1996. pp. 47–60. [Google Scholar]
- Mehta D, Wu MF, Gunst SJ. Role of contractile protein activation in the length-dependent modulation of tracheal smooth muscle force. American Journal of Physiology. 1996;270:C243–252. doi: 10.1152/ajpcell.1996.270.1.C243. [DOI] [PubMed] [Google Scholar]
- Meiss RA. Persistent mechanical effects of decreasing length during isometric contraction of ovarian ligament smooth muscle. Journal of Muscle Research and Cell Motility. 1993;14:205–218. doi: 10.1007/BF00115455. [DOI] [PubMed] [Google Scholar]
- Nachmias VT, Golla R, Casella JF, Barron-Casella E. Cap Z, a calcium insensitive capping protein in resting and activated platelets. FEBS Letters. 1996;378:258–262. doi: 10.1016/0014-5793(95)01474-8. [DOI] [PubMed] [Google Scholar]
- Obara K, Yabu H. Effect of cytochalasin B on intestinal smooth muscle cells. European Journal of Pharmacology. 1994;255:139–147. doi: 10.1016/0014-2999(94)90092-2. [DOI] [PubMed] [Google Scholar]
- Pavalko FM, Adam LP, Wu MF, Walker TL, Gunst SJ. Phosphorylation of dense-plaque proteins talin and paxillin during tracheal smooth muscle contraction. American Journal of Physiology. 1995;268:C563–571. doi: 10.1152/ajpcell.1995.268.3.C563. [DOI] [PubMed] [Google Scholar]
- Pollard TD, Cooper JA. Actin and actin-binding proteins. A critical evaluation of mechanisms and functions. Annual Review of Biochemistry. 1986;55:987–1035. doi: 10.1146/annurev.bi.55.070186.005011. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Murphy RA. Muscle length, shortening, myoplasmic [Ca2+], and activation of arterial smooth muscle. Circulation Research. 1990;66:1354–1361. doi: 10.1161/01.res.66.5.1354. [DOI] [PubMed] [Google Scholar]
- Ribeiro CM, Reece J, Putney JW., Jr Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. Journal of Biological Chemistry. 1997;272:26555–26561. doi: 10.1074/jbc.272.42.26555. [DOI] [PubMed] [Google Scholar]
- Saito SY, Hori M, Ozaki H, Karaki H. Cytochalasin D inhibits smooth muscle contraction by directly inhibiting contractile apparatus. Journal of Smooth Muscle Research. 1996;32:51–60. doi: 10.1540/jsmr.32.51. [DOI] [PubMed] [Google Scholar]
- Saito S, Karaki H. A family of novel actin-inhibiting marine toxins. Clinical and Experimental Pharmacology and Physiology. 1996;23:743–746. doi: 10.1111/j.1440-1681.1996.tb01770.x. [DOI] [PubMed] [Google Scholar]
- Schafer DA, Cooper JA. Control of actin assembly at filament ends. Annual Review of Cell and Developmental Biology. 1995;11:497–518. doi: 10.1146/annurev.cb.11.110195.002433. [DOI] [PubMed] [Google Scholar]
- Schmidt C, Pommerenke H, Durr F, Nebe B, Rychly J. Mechanical stressing of integrin receptors induces enhanced tyrosine phosphorylation of cytoskeletally anchored proteins. Journal of Biological Chemistry. 1998;273:5081–5085. doi: 10.1074/jbc.273.9.5081. [DOI] [PubMed] [Google Scholar]
- Shyy JY, Chien S. Role of integrins in cellular responses to mechanical stress and adhesion. Current Opinion in Cell Biology. 1997;9:707–713. doi: 10.1016/s0955-0674(97)80125-1. [DOI] [PubMed] [Google Scholar]
- Sugidachi A, Ogawa T, Asai F, Saito S, Ozaki H, Fusetani N, Karaki H, Koike H. Inhibition of rat platelet aggregation by mycalolide-B, a novel inhibitor of actin polymerization with a different mechanism of action from cytochalasin-D. Thrombosis and Haemostasis. 1998;79:614–619. [PubMed] [Google Scholar]
- Symons MH, Mitchison TJ. Control of actin polymerization in live and permeabilized fibroblasts. Journal of Cell Biology. 1991;114:503–513. doi: 10.1083/jcb.114.3.503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang DC, Mehta D, Gunst SJ. Mechanosensitive tyrosine phosphorylation of paxillin and focal adhesion kinase in tracheal smooth muscle. American Journal of Physiology. 1999;276:C250–258. doi: 10.1152/ajpcell.1999.276.1.C250. [DOI] [PubMed] [Google Scholar]
- Theriot JA. Regulation of the actin cytoskeleton in living cells. Seminars in Cell Biology. 1994;5:193–199. doi: 10.1006/scel.1994.1024. [DOI] [PubMed] [Google Scholar]
- Tseng S, Kim R, Kim T, Morgan KG, Hai CM. F-actin disruption attenuates agonist-induced [Ca2+], myosin phosphorylation, and force in smooth muscle. American Journal of Physiology. 1997;272:C1960–1967. doi: 10.1152/ajpcell.1997.272.6.C1960. [DOI] [PubMed] [Google Scholar]
- Wang N, Butler JP, Ingber DE. Mechanotransduction across the cell surface and through the cytoskeleton. Science. 1993;260:1124–1127. doi: 10.1126/science.7684161. [DOI] [PubMed] [Google Scholar]
- Wang Z, Pavalko FM, Gunst SJ. Tyrosine phosphorylation of the dense plaque protein paxillin is regulated during smooth muscle contraction. American Journal of Physiology. 1996;271:C1594–1602. doi: 10.1152/ajpcell.1996.271.5.C1594. [DOI] [PubMed] [Google Scholar]
- Youn T, Kim SA, Hai CM. Length-dependent modulation of smooth muscle activation: effects of agonist, cytochalasin, and temperature. American Journal of Physiology. 1998;274:C1601–1607. doi: 10.1152/ajpcell.1998.274.6.C1601. [DOI] [PubMed] [Google Scholar]