

Abstract
To extend our knowledge of the site and mechanism of action of L-type Ca2+ channel antagonists on 5-HT3 receptors, whole-cell voltage clamp electrophysiology was used to investigate the action of one of these compounds, diltiazem, on the recombinant receptor expressed in human embryonic kidney (HEK) 293 cells.
Application of diltiazem with 5-HT (30 μm) caused an increase in the rate of receptor current decay, but did not significantly affect peak current (Ip), the EC50 or the Hill coefficient, indicating a non-competitive mechanism of action. Pre-application of the antagonist had no effect indicating that diltiazem mediates its effects by binding preferentially to the open state of the 5-HT3 receptor.
To examine the effects of diltiazem on the open state of the receptor in more detail we used 10 mM 5-hydroxyindole (5-OHi) to reduce receptor desensitisation. These experiments showed that diltiazem causes a rapid, reversible, block in the presence of agonist but can become trapped in the unliganded state of the receptor by prior washout of agonist. Dose-inhibition data yielded an IC50 of 5.5 μm and a Hill coefficient of 0.96; inhibition was slightly voltage dependent as the degree of blockade at +60 mV was reduced.
The Hill coefficient of near unity suggests a single molecule of diltiazem mediates inhibition and, indeed, kinetic analysis verified that the interaction of diltiazem with the 5-HT3 receptor was well described by a bimolecular reaction scheme. The results suggest that diltiazem acts by causing open-channel block of the 5-HT3 receptor.
5-Hydroxytryptamine3 (5-HT3) receptors are members of the nicotinic-acetylcholine (nACh) receptor-like family of ligand-gated ion channels, and share many functional and structural characteristics with these proteins (Maricq et al. 1991). Neuronally located 5-HT3 receptors mediate fast-activating depolarising responses; agonists cause rapid activation of the receptor and a transient opening of the integral ion channel, before the receptor closes or becomes desensitised (Derkach et al. 1989; Lambert et al. 1989; Yang, 1990). The ion channel is cation selective, although it discriminates poorly between monovalent ions such as Na+, K+ and Cs+ (Yang, 1990), and it also allows Ca2+ to permeate (Yang, 1990; Yang et al. 1992; Hargreaves et al. 1994). During experiments to assess the Ca2+ permeability of the 5-HT3 receptor using fura-2 Ca2+ imaging, we established that although the receptor native to N1E-115 mouse neuroblastoma cells was Ca2+ permeable (Hargreaves et al. 1994), several L-type Ca2+ channel antagonists almost completely abolished Ca2+ signals in these cells (Hargreaves et al. 1996). Subsequent experiments established a direct inhibitory action of these compounds on recombinant 5-HT3 receptors expressed in human embryonic kidney (HEK) 293 cells (Hargreaves et al. 1996).
Our experiments demonstrated that examples from each of the classes of L-type Ca2+ channel antagonists had similar effects: (+)verapamil, (-)verapamil, diltiazem and nimodipine all inhibited 5-HT3 receptor-mediated increases in intracellular Ca2+ in HEK 293 cells in a concentration-dependent manner (IC50 values, 2.5-6.5 μm; Hargreaves et al. 1996). Similarly, our whole-cell patch clamp experiments showed that these compounds blocked 5-HT3 receptor-evoked responses with similar IC50 values (3.0-6.8 μm) and apparently similar mechanisms; all of the compounds accelerated the rate of decay of 5-HT-evoked currents when co-applied with the agonist. Radioligand binding studies showed that (+)verapamil, (-)verapamil and diltiazem (but not nimodipine) reduced [3H]1-(m-chlorophenyl)-biguanide (mCPBG) and [3H]granisetron binding to the 5-HT3 receptor, and that this was likely to be due to an allosteric (non-competitive) mechanism, although it is not yet clear whether the compounds have one or more sites of action (see Hargreaves et al. 1996). These results suggested that the L-type Ca2+ channel antagonists exert their effects via a site on the 5-HT3 receptor that is distinct from the agonist binding site.
To gain insight into the mechanism of action of these compounds on the 5-HT3 receptor, in the current study we examined the action of one of these compounds, diltiazem, on the 5-HT3 receptor expressed in HEK 293 cells, using the whole-cell recording configuration of the patch clamp technique. The results support a model in which the binding of a single molecule of diltiazem per receptor causes inhibition of the 5-HT3 receptor by open-channel block; diltiazem is therefore the first open-channel blocker of the 5-HT3 receptor to be identified, opening the way for the development of drugs to selectively target active 5-HT3 receptors in the central and peripheral nervous systems.
METHODS
Reagents
HEK 293 cells were obtained from the European Collection of Animal Cell Cultures (Porton Down, UK). Cell culture reagents were obtained from Gibco BRL, except fetal calf serum which was from Sigma. 5-HT hydrochloride was from Research Biochemicals International. All other reagents were obtained from Sigma.
Cell culture
HEK 293 cells stably expressing 5-HT3 receptors were developed using the eukaryotic expression vector pRc/CMV (InVitrogen, Abingdon, UK) containing the complete coding sequence for the 5-HT3-A(b) subunit from N1E-115 cells as previously described (Hargreaves et al. 1996). Cells were routinely grown until confluent (3-5 days) in a 1:1 mix of Dulbecco's modified Eagle's medium and F12 medium containing 10 % fetal calf serum and 500 mg ml−1 geneticin in 7 % CO2 and then passaged. For experiments cells were plated in 35 mm dishes and used 1-3 days after passage.
Electrophysiological procedures
Whole-cell currents were recorded at 20-22°C using an EPC-9 amplifier (HEKA Elektronic, Darmstadt, Germany) controlled by Pulse software (HEKA, version 7.85). Briefly, culture dishes were continuously superfused (3-5 ml min−1) with extracellular solution (130 mM NaCl, 5.4 mM KCl, 2 mM MgCl2, 1.8 mM CaCl2, 30 mM glucose and 10 mM Hepes, pH 7.2 with NaOH). Patch pipettes (3-5 MΩ) were made from thin-walled borosilicate glass capillary tubing (GC120F-10, Clark Electromedical) and filled with intracellular solution (140 mM CsCl, 1 mM MgCl2, 0.1 mM CaCl2, 1.1 mM EGTA (∼10 nM free Ca2+) and 10 mM Hepes, pH 7.2 with CsOH). Cells were held at -60 mV unless otherwise stated. Compounds were applied using a DAD-12 perfusion device (ALA Scientific Instruments Inc.) controlled via a computer interface with DADPORT software.
The rate of solution exchange at the cell membrane, using the DAD-12 system, was calibrated according to the following protocol. The current of a cell voltage clamped at -60 mV was monitored during application of diluted (1:10 with water) extracellular solution via the DAD-12 micromanifold (Fig. 1). The relaxation, due to the reduction in the electrochemical driving force across the cell membrane, could be fitted with a single exponential (time constant (τ) = 21 ± 2 ms, n = 5) and the time taken from the onset of the decrease in current until attainment of a new stable value was estimated to be 61 ± 4 ms (n = 5). The latter value was taken as an estimate of the time for complete solution exchange at the cell membrane. The rate of solution exchange at the tip of an open patch pipette, 6.0 ± 0.2 ms (n = 5; Fig. 1), was much faster than at the cell membrane and probably reflects the perfusion of a large surface area in comparison to the smaller opening of the tip of a patch pipette.
Figure 1. Fast perfusion using the DAD-12 micromanifold system.
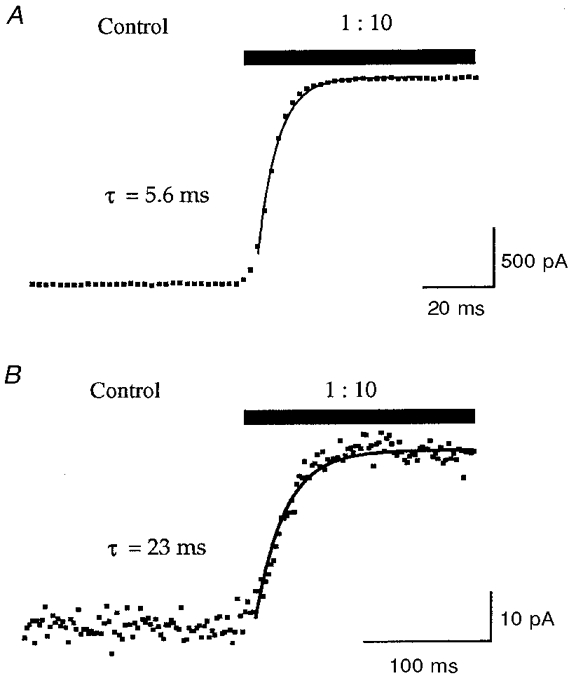
A, change in current recorded using an open patch pipette voltage clamped at 0 mV during a switch from normal extracellular solution to a diluted (1:10 with water) extracellular solution using the DAD-12 perfusion system. The relaxation due to a change in junction potential was well fitted by a single exponential function; in this example the time constant (τ) was 5.6 ms. B, whole-cell recording of current passing across the membrane of a HEK 293 cell voltage clamped at -60 mV and subjected to a concentration jump via the DAD-12 perfusion system. Application of a diluted (1:10 with water) extracellular solution to the cell caused a reduction in the electrochemical driving force across the cell membrane, the relaxation of which was fitted with a single exponential function. In this example τ was 23 ms. These data were used to estimate the time for complete solution exchange at the cell membrane (see Methods).
Data analysis
The decay phase of whole-cell current traces, relaxations due to solution exchanges, and the kinetics of diltiazem blockade of the 5-HT3 receptor were fitted with an exponential function using Igor (WaveMetrics) by least-squares regression analysis according to the equation:
| (1) |
where It is the current at time t, I0 is the current amplitude at t = 0, I0 - I∞ is the amplitude of the time dependent component of the response at the start of relaxation and τ is the time constant of decay.
Dose-response data were fitted to the Hill equation using Kaleidagraph (Abelbeck Software):
| (2) |
where Rmax is the maximal response, EC50 is the concentration of agonist required for half-maximal effect, [A] is the concentration of agonist and nH is the Hill coefficient.
Inhibition data were fitted using Kaleidagraph to the Hill equation:
| (3) |
where Rmax is the maximal response, Rmin is the minimal response, IC50 is the concentration of antagonist required to decrease the response by 50 %, [A] is the concentration of antagonist and nH is the Hill coefficient.
All data are quoted as the mean ±s.e.m. for n independent experiments. Where appropriate, Student's t test for unpaired data was used and values of P < 0.05 were regarded as significant.
RESULTS
Diltiazem inhibition of the 5-HT3 receptor
Whole-cell currents, recorded in response to the application of 30 μm 5-HT to HEK 293 cells stably expressing the 5-HT3 receptor, activated rapidly (10-90 % rise time = 41 ± 5 ms, n = 11) and desensitised monoexponentially in the continued presence of agonist (τ= 3.4 ± 0.5 s, n = 5; Fig. 2A-C). Simultaneous application of diltiazem with 5-HT (30 μm) caused an increase in the rate of 5-HT3 receptor current decay, which could also be fitted by a monoexponential function (Fig. 2C). The inhibition observed was reversible and caused little or no effect upon the peak current (Ip; Fig. 2D). 5-HT dose-response curves constructed in the presence or absence of 30 μm diltiazem were similar (Fig. 2E): the EC50 for 5-HT was 3.7 ± 0.1 and 4.0 ± 0.1 μm and the Hill coefficient was 2.3 ± 0.1 and 2.6 ± 0.2 for experiments conducted in the absence and presence of 30 μm diltiazem, respectively (n = 3-5), indicating that diltiazem acts in a non-competitive manner. 5-HT-evoked responses in which 10 μm diltiazem was pre-applied (10 s) to (closed) receptors prior to simultaneous application with agonist were indistinguishable from co-application experiments (Fig. 2A-D). In both cases, the rate of current activation (10-90 % rise time = 50 ± 17 and 56 ± 3 ms for pre-application and co-application experiments, respectively, n = 3) and the peak currents obtained were not significantly different from control. These results suggest that diltiazem is unlikely to bind to the closed state of the receptor but binds readily to the open or active state.
Figure 2. Diltiazem inhibition of the 5-HT3 receptor.
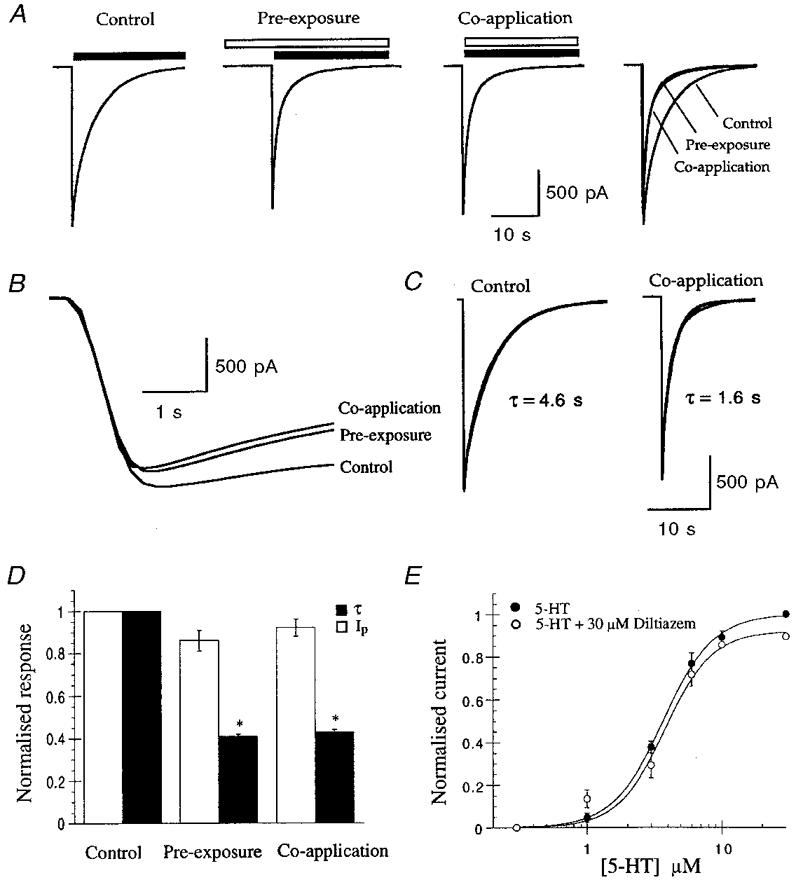
A, whole-cell currents recorded in response to application of 5-HT (30 μm, filled bar) with or without diltiazem (10 μm, open bar). The extent and time course of inhibition were not affected by pre-exposure to 10 μm diltiazem, as indicated by overlay of the traces. Results are from one experiment and are typical of three others. B, data from A on an expanded time scale showing that there was no difference in the rise time of the response in the presence or absence of diltiazem. These data also demonstrate that the rise time was slower than the solution exchange time shown in Fig. 1. C, 5-HT-evoked control responses were well fitted by monoexponential functions. In this example τ was 4.6 s for the control and 1.6 s in the presence of diltiazem. D, normalised data for the experiments illustrated in A.* Significantly different from control, n = 3. E, diltiazem did not affect the dose-response curve for 5-HT activation of the receptor. The curve fits show the best fit of the Hill equation, which yielded an EC50 of 3.7 and 4 μm and a Hill coefficient of 2.3 and 2.6 for experiments conducted in the presence and absence of 30 μm diltiazem, respectively.
In all experiments described above, drugs were applied by perfusion via the extracellular solution. To investigate whether or not diltiazem could produce similar effects on the 5-HT3 receptor when applied intracellularly, experiments were conducted with 100 μm diltiazem included in the pipette solution (Fig. 3). In these experiments diltiazem was without effect, as demonstrated by a subsequent application of the same drug concentration to the extracellular surface via the conventional perfusion system. Therefore, diltiazem appears unable to block the receptor channel when applied intracellularly.
Figure 3. Diltiazem binds to the extracellular side of the 5-HT3 receptor.
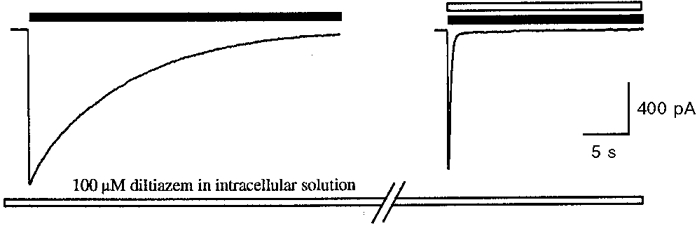
Sample traces showing responses to extracellular application of 5-HT (30 μm, filled bar) with or without 100 μm diltiazem (open bar) co-applied externally with 100 μm diltiazem included in the patch pipette solution. A delay of 5 min was allowed after the whole-cell configuration had been established to allow complete dialysis of cell contents. Results are from a single experiment from one cell, and are typical of three experiments on different cells. The example shown was chosen to emphasise the difference in the time course of the response between control and diltiazem-modified responses, and the kinetics of the decay of the control response are within the range observed for other control responses.
Action of 5-hydroxyindole on the recombinant 5-HT3 receptor
To perform more detailed experiments to investigate the interaction of diltiazem with the open state of the 5-HT3 receptor, we used 5-hydroxyindole (5-OHi), a compound shown to reduce 5-HT3 receptor desensitisation (Kooyman et al. 1993a, 1994; van Hooft et al. 1997). Application of 5-OHi (≤ 30 mM) to HEK 293 cells expressing 5-HT3 receptors was without effect (data not shown). Co-application of 1 mM 5-OHi with 5-HT (30 μm) reduced the rate of receptor desensitisation by 397 ± 45 % (n = 8), compared with control experiments (agonist alone), and this was further reduced to 636 ± 109 % (n = 6) by 10 mM 5-OHi (Fig. 4). These concentrations of 5-OHi caused no significant effect on the Ip compared with control experiments; Ip= 106 ± 4 % for 1 mM 5-OHi (n = 8), Ip= 100 ± 5 % for 10 mM 5-OHi (n = 6). Increasing the 5-OHi concentration to 30 mM resulted in an inhibition of Ip (33 ± 6 %, n = 3; Fig. 4). Concentrations of ≥ 10 mM 5-OHi also led to the appearance of ‘tail’ currents (Fig. 4A) which were probably due to the unbinding of 5-OHi from the receptor (Fig. 4C). Based on the above characterisation of the actions of 5-OHi on the recombinant 5-HT3 receptor, 10 mM 5-OHi was used in further experiments to investigate the mechanism of action of diltiazem.
Figure 4. Effects of 5-OHi on the 5-HT3 receptor.
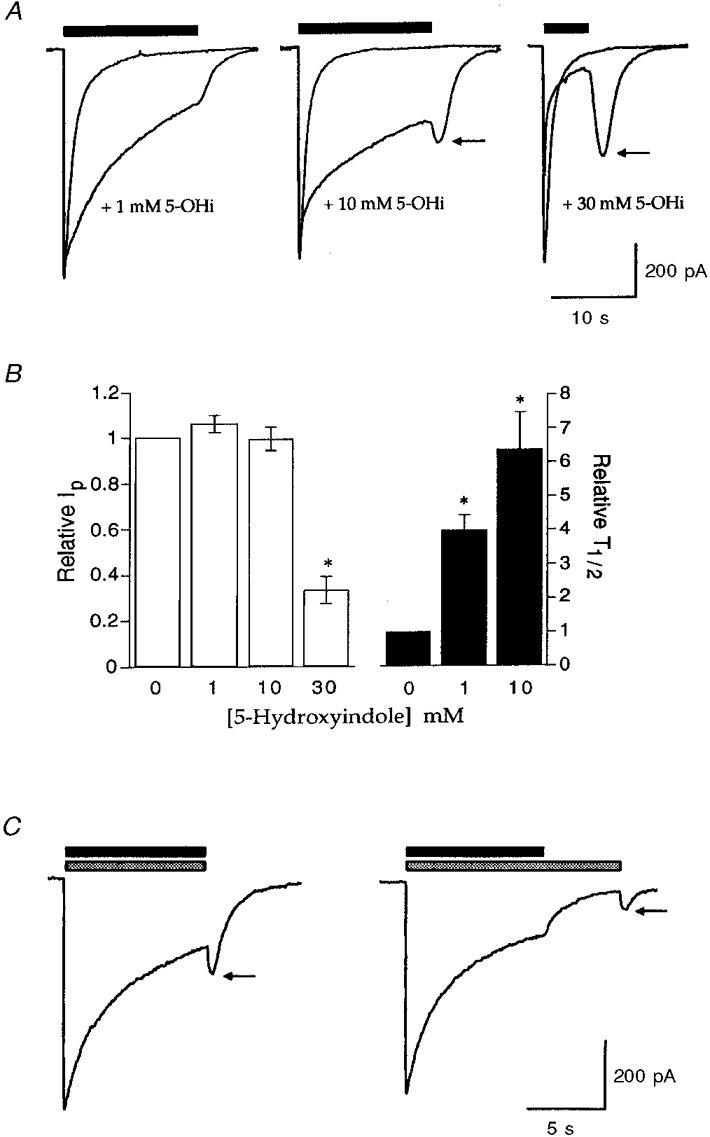
A, sample traces illustrating the effect of 1, 10 or 30 mM 5-OHi upon 5-HT-evoked whole-cell currents. 5-OHi was co-applied with 5-HT (30 μm, filled bar) for the duration indicated. The effects of 5-OHi on the 5-HT3 receptor were completely reversible. Tail currents obtained upon termination of 5-HT and 5-OHi application are indicated by arrows. Traces are from single experiments and are representative of at least three others. B, graphs summarising the data obtained from the experiments illustrated in A showing the effects of 5-OHi upon peak current amplitude and receptor desensitisation (T½, half-time of recovery of whole-cell current). * Significantly different from control (0 mM 5-OHi), n = 3-9. C, investigation of the tail current reveals that it was observed following termination of application and subsequent wash-off of 5-HT (30 μm, filled bar) and 5-OHi (10 mM, stippled bar) but prevented by continuous application of 5-OHi; subsequent removal resulted in its re-appearance. This suggests that the tail current is due to the unbinding of 5-OHi from the receptor.
Diltiazem inhibition of the open state of the 5-HT3 receptor
Subsequent application of diltiazem (1-100 μm) to 5-HT-activated (30 μm) receptors, gated in the presence of 10 mM 5-OHi, caused a rapid inhibition of whole-cell current (Fig. 5A). Inhibition was readily reversible as indicated by the re-appearance of the inward current when diltiazem application was terminated (Fig. 5A). The dose-inhibition relationship constructed for diltiazem inhibition of whole-cell currents elicited by 30 μm 5-HT co-applied with 10 mM 5-OHi is shown in Fig. 5B. The curve fit to this data yielded an IC50 of 5.5 μm and an empirical Hill coefficient of approximately 1 (nH= 0.96). The latter finding is consistent with a single binding site for diltiazem action and suggests a simple bimolecular reaction for diltiazem block, according to Scheme 1:
Figure 5. Diltiazem reversibly inhibits open 5-HT3 receptors.
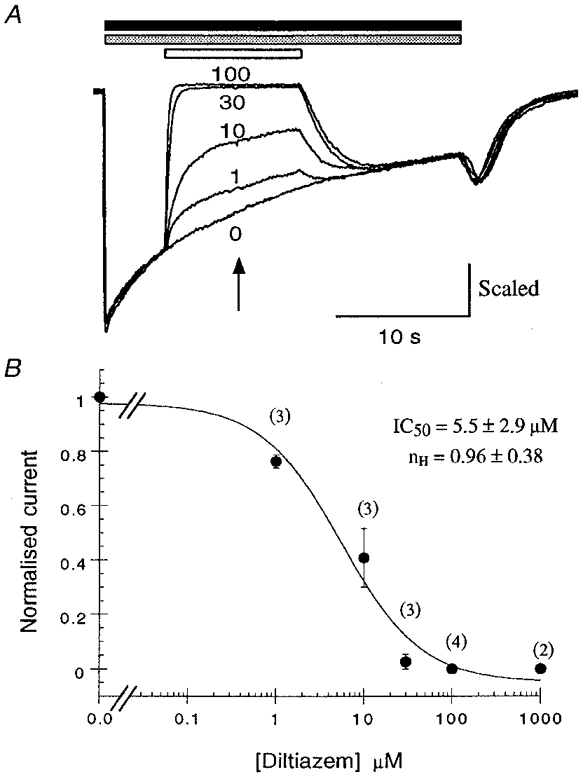
Inhibition of 5-HT-evoked (30 μm, filled bar) currents by diltiazem (1-100 μm, open bar) in the presence of 10 mM 5-OHi (stippled bar). A, sample traces from experiments to investigate the concentration-effect relationship for diltiazem inhibition of the open 5-HT3 receptor. Diltiazem application (in the presence of 10 mM 5-OHi) led to a fast, reversible, block of the 5-HT-evoked whole-cell current (see Fig. 6 for kinetic analysis); 100 μm diltiazem was sufficient to cause complete blockade of the ion current. Traces are from one experiment and are representative of three similar experiments. B, graph of the data obtained from the experiments described in A; each data point was calculated from the number of cells shown in parentheses. The normalised current (Idiltiazem/Icontrol) at the mid-point of drug application (arrow) was used to construct a dose-response relationship for diltiazem inhibition. The data were fitted with an IC50 curve and the parameters obtained are shown.
Scheme 1.

This reaction scheme predicts that the macroscopic blocking and unblocking actions of diltiazem will proceed with monoexponential kinetics, the macroscopic pseudo-first order rate constant of blocking (kon) will depend linearly on diltiazem concentration (as well as a constant, C) and the macroscopic unblocking rate (koff) will be independent of diltiazem concentration, i.e.:
| (4) |
| (5) |
Experiments to examine the kinetics of the diltiazem inhibition support these predictions (Fig. 6). The macroscopic blocking (τon) and unblocking (τoff) rates of diltiazem inhibition of the 5-HT3 receptor were both well fitted by monoexponential functions, and as these rates were more than one order of magnitude greater than the underlying decaying 5-HT current response, the latter was not considered to affect the data. The rate of onset of channel block by diltiazem increased linearly with diltiazem concentration: τon was 820 ± 93, 382 ± 89 and 132 ± 16 ms for experiments conducted with 10, 30 and 100 μm diltiazem, respectively (n = 3). The data for 1 μm diltiazem were not included in these calculations as the rate here may have been affected by the decay of the 5-HT current response. In contrast τoff did not change significantly with diltiazem concentration; the mean τoff obtained from the three diltiazem concentrations shown in Fig. 6 was 1.45 s. The rate of onset of diltiazem blockade using 10 or 30 μm diltiazem is therefore slower than the rate of 5-HT3 receptor activation (time to peak = 88 ± 11 ms, n = 11) consistent with the lack of effect on the Ip of whole-cell currents in co-application experiments. The rate of block using 100 μm diltiazem begins to approach the rate of 5-HT3 receptor activation and therefore may be expected to cause a small reduction in Ip.
Figure 6. Kinetics of diltiazem interaction with the 5-HT3 receptor.
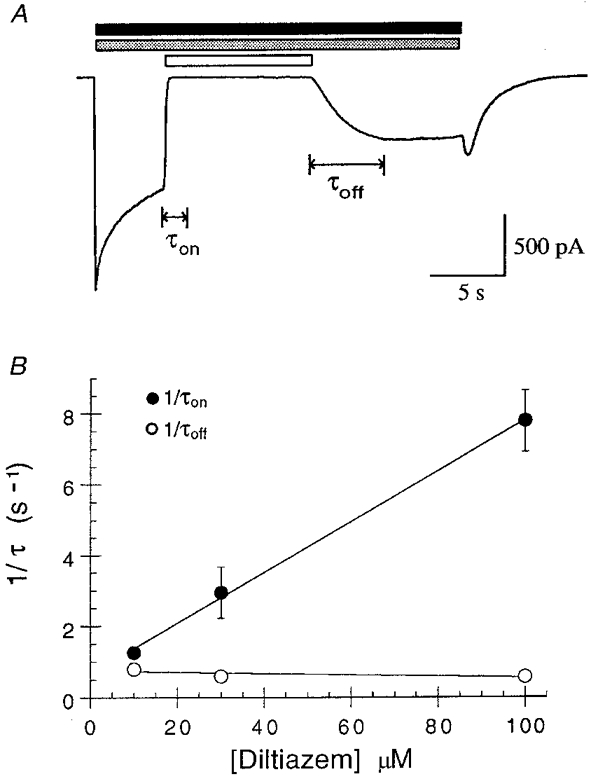
A, sample trace showing the fast onset (τon) of diltiazem inhibition (100 μm, open bar) and slower recovery from inhibition (τoff) of 5-HT-evoked (30 μm, filled bar) currents in the presence of 10 mM 5-OHi (stippled bar). B, diltiazem association with and dissociation from the 5-HT3 receptor were well fitted by monoexponential functions to obtain the on-time constant (τon) and unblocking time constant (τoff) for diltiazem inhibition. The values of τon were: 10 μm, 820 ± 93 ms; 30 μm, 382 ± 89 ms; and 100 μm, 132 ± 16 ms (n = 3). The concentration dependence of 1/τon and 1/τoff are shown; 1/τon changed linearly with the concentration of diltiazem and 1/τoff did not change significantly with diltiazem concentration.
According to a one binding site model,
| (6) |
and
| (7) |
The estimate of kon obtained from the gradient of the 1/τon curve was 7.17 × 104 M−1 s−1. The mean koff (i.e.1/τoff) was 0.69 s−1, a value consistent with the estimate of 0.65 s−1 for koff which can be obtained from the Y-axis intercept of the 1/τon curve (Fig. 6). The validity of the bimolecular reaction scheme proposed can be determined by estimating the equilibrium dissociation constant (Ki) for diltiazem action from the following equation:
| (8) |
This equation yields a Ki value of 9.6 μm which is similar to the apparent Ki (IC50) determined by direct experimentation (5.5 μm). The results are therefore consistent with a single molecule of diltiazem binding rapidly to a single site in the 5-HT-gated channel.
Diltiazem inhibition of the receptor was also shown to be voltage dependent. Application of 30 μm diltiazem was sufficient for almost complete block (97 ± 1 %, n = 6) of 5-HT-activated currents at -60 mV but only 71 ± 4 % (n = 4) block when the same experiments were performed at +60 mV (Fig. 7). Determination of the rate constants for the onset and offset of block for paired data sets revealed that there was a significant difference in τoff at the two holding potentials, but not in τon: at +60 mV τoff was 0.81 ± 0.12 s, whilst at -60 mV τoff was 2.48 ± 0.29 s (n = 3). Values of τon for the same data sets were 527 ± 85 and 485 ± 91 ms for +60 and -60 mV, respectively.
Figure 7. Diltiazem inhibition of the 5-HT3 receptor is voltage dependent.
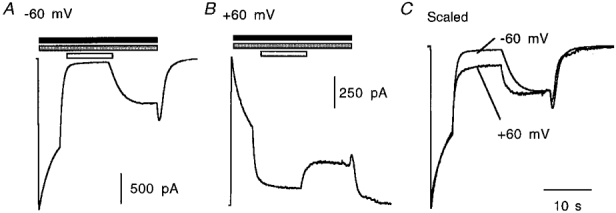
A and B, inhibition of 5-HT-evoked (30 μm, filled bar) currents by diltiazem (30 μm, open bar) in the presence of 10 mM 5-OHi (stippled bar) at -60 and +60 mV, respectively. C, the degree of inhibition at -60 mV was 97 ± 1 % (n = 6) but at +60 mV the same protocol caused only 71 ± 4 % (n = 4) inhibition (significantly different from -60 mV data). The sample traces are from one experiment and are representative of at least three others.
Diltiazem block was reversible in the continued presence of agonist (Figs 5–7). Removal (and washout) of agonist prior to removal of diltiazem did not result in the appearance of a current, which might have indicated unbinding of diltiazem from open channels (Fig. 8A). Experiments were also conducted to test whether or not diltiazem could be trapped within the closed, unliganded, 5-HT3 receptor. In these experiments (Fig. 8B), re-application of agonist (with 5-OHi) caused reversal of the current block, and current re-appeared with similar kinetics to that previously estimated for the unbinding of diltiazem rather than the rapid current activation which would be expected for the re-activation of closed (unblocked) 5-HT3 receptors. Surprisingly, 5-OHi alone was also sufficient for partial reversal of diltiazem blockade (Fig. 8C). This effect of 5-OHi (since this compound has no agonist activity) could be reconciled as an allosteric effect upon the diltiazem binding site: it may simply hold the receptor in a conformation from which diltiazem can dissociate. Overlay of the recovery portion of the traces indicated that the extent of recovery was less with 5-OHi alone compared with 5-HT and 5-OHi (Fig. 8D). However, the rate of reversal of block in the two cases was similar (Fig. 8E) and is therefore likely to correspond to the same molecular event: unbinding of diltiazem from its binding site in the open form of the 5-HT3 receptor. The results therefore suggest that diltiazem can be trapped within the closed receptor and are consistent with diltiazem acting by a mechanism of open-channel block.
Figure 8. Reversal of diltiazem inhibition of the 5-HT3 receptor.
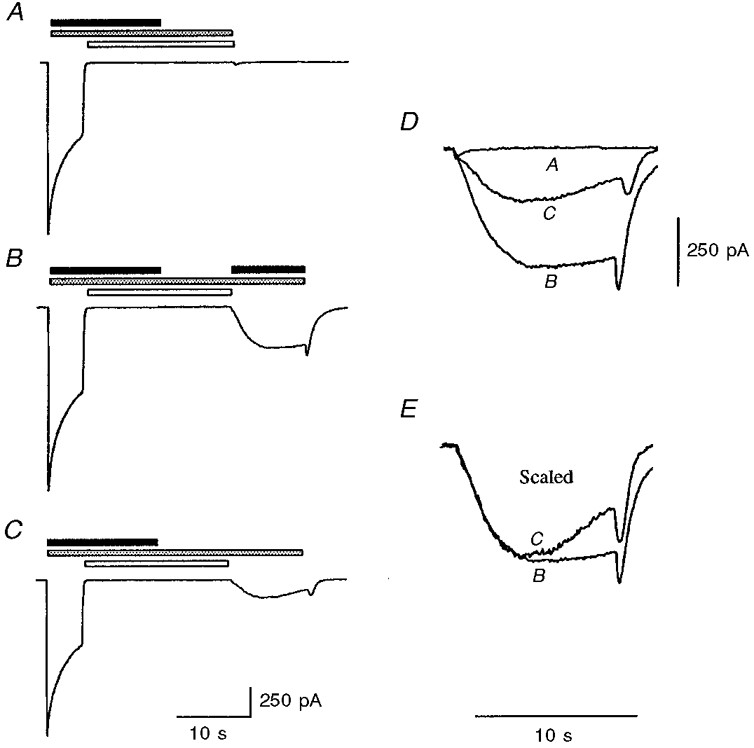
Sample traces illustrating the effect of 5-HT (30 μm, filled bar), 5-OHi (10 mM, stippled bar) and diltiazem (100 μm, open bar) as indicated. Traces in A-C are to the same scale. Traces are from a single experiment but are representative of three others. A, diltiazem application (with 5-OHi) was maintained for 10 s following agonist application. Using this protocol diltiazem inhibition was not reversed when diltiazem was removed. B, re-application of agonist (with 5-OHi) caused reversal of diltiazem inhibition. C, application of 5-OHi alone could also partially reverse diltiazem inhibition. D, overlay of enlarged portions of the recovery phases of the experiments shown in A-C. E, overlay of the recovery phases of B and C scaled to allow comparison of the kinetics of reversal of diltiazem inhibition.
DISCUSSION
In this study we investigated the inhibitory action of the L-type Ca2+ channel antagonist diltiazem on the recombinant 5-HT3 receptor expressed in HEK 293 cells. Our results are consistent with a mechanism of open-channel block of the 5-HT3 receptor mediated by the binding of a single diltiazem molecule to the extracellular domain of the receptor. Many compounds have been identified that act as open-channel blockers of other ligand-gated ion channels. For example, for the closely related nACh receptor, hexamethonium causes open-channel block of ganglionic nACh receptors (Gurney & Rang, 1984); amantadine, memantine and hexamethonium block the α4β2 neuronal nACh receptor (Buisson & Bertrand, 1998) and QX222 has been shown to block the Torpedo nACh receptor (Charnet et al. 1990). To date, no open-channel blockers of the 5-HT3 receptor have been identified, even though a number of compounds have been reported to modulate its function. The mode of action of some of these compounds is known; for example, tetraethylammonium inhibits 5-HT3 receptor currents and prevents desensitisation via the agonist recognition site (Kooyman et al. 1993b), and alcohols such as trichloroethanol have recently been shown to potentiate 5-HT3 receptor currents by altering channel gating (Zhou et al. 1998). However, for most other 5-HT3 receptor modulators, including ketamine (Peters et al. 1991), 5-OHi (Kooyman et al. 1993a, 1994; van Hooft et al. 1997) and ifenprodil (McCool & Lovinger, 1995), neither the site nor the mechanism of action is known.
Our data show that diltiazem binds rapidly to the open state, but not the closed (unliganded) state of the 5-HT3 receptor (Fig. 2), and that it has a non-competitive mechanism of action since it had little effect on the concentration-effect relationship for 5-HT action (Fig. 2). To further investigate the interaction of diltiazem with the open state of the 5-HT3 receptor, experiments were conducted in the presence of 5-OHi. This compound has previously been shown to cause a concentration-dependent reduction in the desensitisation of 5-HT3 receptors native to N1E-115 cells (Kooyman et al. 1993a, 1994; van Hooft et al. 1997). Our data show that 5-OHi acts similarly on recombinant 5-HT3 receptors; 1 and 10 mM 5-OHi enhance receptor function whereas 30 mM 5-OHi causes inhibition. Co-application of 10 mM 5-OHi with 5-HT (30 μm) slowed receptor desensitisation sufficiently to demonstrate that diltiazem causes a rapid block of whole-cell current which readily reverses in the presence of agonist. The IC50 for this inhibition was 5.5 μm and the Hill coefficient was close to unity, suggesting a single binding site for diltiazem action. Kinetic analysis of the binding and unbinding of diltiazem to the 5-HT3 receptor gave results consistent with a bimolecular reaction scheme; the pseudo-first order blocking rate constant (kon) and unblocking constant (koff) of diltiazem inhibition were well fitted by monoexponential functions: 1/τon was linearly dependent on the concentration of diltiazem and 1/τoff was not significantly affected by diltiazem concentration. From the values of 1/τon and 1/τoff obtained, kon and koff were determined and yielded an estimate of 9.6 μm for the theoretical equilibrium dissociation constant Ki, similar to the IC50 obtained from direct experimentation. These results are therefore consistent with a single molecule of diltiazem binding rapidly to, and inhibiting, the 5-HT-gated channel.
The phenomenon of use dependence is often cited as evidence for open-channel blockade (Buisson & Bertrand, 1998). It arises due to an accumulation of receptors in a blocked state due to trapping of the antagonist within the receptor (closed state). The experiments described in Fig. 8 demonstrate that diltiazem can be trapped within the closed state of the receptor. However, observation of use dependence requires the dissociation rate (koff) for the antagonist to be much slower than the dissociation rate of the agonist from the receptor; alternatively, a high frequency of agonist application (with a similar order of magnitude to koff) should result in an accumulation of block. Unfortunately, the relatively fast dissociation of diltiazem from the receptor (koff= 1.45 s−1) compared with the dissociation rate of 5-HT (6.9 s; Neijt et al. 1989) combined with the slow recovery of the 5-HT3 receptor from desensitisation (approximately 60 s for complete recovery from desensitisation; Neijt et al. 1989; van Hooft & Vijverberg, 1996) preclude such experiments from being conducted.
Voltage dependency of inhibition is also often cited as evidence for a mechanism of open-channel block. It suggests that the drug in question binds to a binding site which experiences the electric field across the membrane bilayer, i.e. a site within the transmembrane region of the channel. Diltiazem is a tertiary amine and therefore its charge is dependent on the external pH; at pH > 8.5 it is uncharged whilst at pH < 6.0 it has a charge of +1. At physiological pH diltiazem is therefore only partially charged, although within the confines of the pore of the receptor, the proportion of charged molecules is difficult to predict as there are likely to be local pH variations. The fact, however, that diltiazem was less potent at more depolarised potentials (+60 mV), presumably due largely to the increase in τoff, is consistent with it binding to a site which is at least partially exposed to the transmembrane electric field.
Insight into the location of the diltiazem binding site was also provided by experiments to investigate the action of diltiazem when applied to the intracellular side of the 5-HT3 receptor. The lack of effect of diltiazem when it was applied intracellularly indicates that the site from which it mediates its inhibitory action is not accessible from the intracellular side of the receptor. Thus diltiazem probably binds either on the extracellular side of the channel, or in its extracellular vestibule.
Experiments to investigate the reversal of diltiazem inhibition showed that although diltiazem can freely dissociate from the active receptor, prior removal (and washout) of agonist can trap diltiazem within the closed state of the receptor (Fig. 8). Subsequent re-application of agonist or 5-OHi was required for at least partial reversal of diltiazem blockade. The latter finding was initially surprising as 5-OHi has no agonist activity (this study; Kooyman et al. 1993a); however, since 5-OHi has been suggested to stabilise the open state of the 5-HT3 receptor (van Hooft et al. 1997), it is conceivable that 5-OHi may hold the receptor in a conformation from which diltiazem can dissociate. The observation that the rate of re-appearance of the whole-cell current was similar to the τoff of diltiazem blockade, rather than the fast activation that would be expected for the re-activation of unblocked channels, suggests that recovery due to application of 5-HT (with 5-OHi) or 5-OHi is likely to correspond to the same molecular event: unbinding of diltiazem from its binding site in the open state of the 5-HT3 receptor.
Diltiazem is therefore the first open-channel blocker of the 5-HT3 receptor to be identified. Although 5-HT3 receptors are unlikely to be inhibited by clinically effective concentrations of L-type Ca2+ channel antagonists (typically < 0.3 μm; Spedding & Paoletti, 1992), the blockade of 5-HT3 receptors by modest (2.5-6.8 μm) concentrations of antagonist raises the possibility that drugs, based on the structures of diltiazem and the other L-type Ca2+ channel antagonists, could be devised to selectively target active 5-HT3 receptors. Such drugs could be useful in the selective attenuation of pathways in which 5-HT3 receptors are overactive, as might be the case for the limbic or mesolimbic systems in certain psychiatric disorders (Tricklebank, 1989).
Acknowledgments
This work was supported by a grant from The Wellcome Trust. M.J.G. was a MRC student and S.C.R.L. is a Wellcome Senior Research Fellow in Basic Biomedical Science.
References
- Buisson B, Bertrand D. Open-channel blockers at the human α4β2 neuronal nicotinic acetylcholine receptor. Molecular Pharmacology. 1998;53:555–563. doi: 10.1124/mol.53.3.555. [DOI] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Leonard RJ, Vogelaar NJ, Czyzyk L, Gouin A, Davidson N, Lester HA. An open-channel blocker interacts with adjacent turns of α-helices in the nicotinic acetylcholine receptor. Neuron. 1990;4:87–95. doi: 10.1016/0896-6273(90)90445-l. [DOI] [PubMed] [Google Scholar]
- Derkach V, Surprenant A, North RA. 5-HT3 receptors are membrane ion channels. Nature. 1989;339:706–709. doi: 10.1038/339706a0. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Rang HP. The channel-blocking action of methonium compounds on rat submandibular ganglion cells. British Journal of Pharmacology. 1984;82:623–642. doi: 10.1111/j.1476-5381.1984.tb10801.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hargreaves AC, Gunthorpe MJ, Taylor CW, Lummis SCR. Direct inhibition of 5-hydroxytryptamine3 receptors by antagonists of L-type Ca2+ channels. Molecular Pharmacology. 1996;50:1284–1294. [PubMed] [Google Scholar]
- Hargreaves AC, Lummis SCR, Taylor CW. Ca2+ permeability of cloned and native 5-hydroxytryptamine type 3 receptors. Molecular Pharmacology. 1994;46:1120–1128. [PubMed] [Google Scholar]
- Kooyman AR, van Hooft JA, Vanderheijden PM, Vijverberg HPM. Competitive and non-competitive effects of 5-hydroxyindole on 5-HT3 receptors in N1E-115 neuroblastoma cells. British Journal of Pharmacology. 1994;112:541–546. doi: 10.1111/j.1476-5381.1994.tb13107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooyman AR, van Hooft JA, Vijverberg HPM. 5-Hydroxyindole slows desensitization of the 5-HT3 receptor-mediated ion current in N1E-115 neuroblastoma cells. British Journal of Pharmacology. 1993a;108:287–289. doi: 10.1111/j.1476-5381.1993.tb12795.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kooyman AR, Zwart R, Vijverberg HP. Tetraethylammonium ions block 5-HT3 receptor-mediated ion current at the agonist recognition site and prevent desensitization in cultured mouse neuroblastoma cells. European Journal of Pharmacology. 1993b;246:247–254. doi: 10.1016/0922-4106(93)90038-b. [DOI] [PubMed] [Google Scholar]
- Lambert JJ, Peters JA, Hales TG, Dempster J. The properties of 5-HT3 receptors in clonal cell lines studied by patch-clamp techniques. British Journal of Pharmacology. 1989;97:27–40. doi: 10.1111/j.1476-5381.1989.tb11920.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCool BA, Lovinger DM. Ifenprodil inhibition of the 5-hydroxytryptamine3 receptor. Neuropharmacology. 1995;34:621–629. doi: 10.1016/0028-3908(95)00030-a. [DOI] [PubMed] [Google Scholar]
- Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D. Primary structure and functional expression of the 5-HT3 receptor, a serotonin-gated ion channel. Science. 1991;254:432–437. doi: 10.1126/science.1718042. [DOI] [PubMed] [Google Scholar]
- Neijt HC, Plomp JJ, Vijverberg HPM. Kinetics of the membrane current mediated by serotonin 5-HT3 receptors in cultured mouse neuroblastoma cells. The Journal of Physiology. 1989;411:257–269. doi: 10.1113/jphysiol.1989.sp017572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JA, Malone HM, Lambert JJ. Ketamine potentiates 5-HT3 receptor-mediated currents in rabbit nodose ganglion neurones. British Journal of Pharmacology. 1991;103:1623–1625. doi: 10.1111/j.1476-5381.1991.tb09837.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spedding M, Paoletti R. Classification of calcium channels and the sites of action of drugs modifying channel function. Pharmacological Reviews. 1992;44:363–376. [PubMed] [Google Scholar]
- Tricklebank MD. Interactions between dopamine and 5-HT3 receptors suggest new treatments for psychosis and drug addiction. Trends in Pharmacological Sciences. 1989;10:127–129. doi: 10.1016/0165-6147(89)90157-0. [DOI] [PubMed] [Google Scholar]
- van Hooft JA, van der Haar E, Vijverberg HPM. Allosteric potentiation of the 5-HT3 receptor-mediated ion current in N1E-115 neuroblastoma cells by 5-hydroxyindole and analogues. Neuropharmacology. 1997;36:649–653. doi: 10.1016/s0028-3908(97)00045-2. [DOI] [PubMed] [Google Scholar]
- van Hooft JA, Vijverberg HPM. Selection of distinct conformational states of the 5-HT3 receptor by full and partial agonists. British Journal of Pharmacology. 1996;117:839–846. doi: 10.1111/j.1476-5381.1996.tb15269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. Ion permeation through 5-hydroxytryptamine-gated channels in neuroblastoma N18 cells. Journal of General Physiology. 1990;96:1177–1198. doi: 10.1085/jgp.96.6.1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mathie A, Hille B. 5-HT3 receptor channels in dissociated rat superior cervical ganglion neurons. The Journal of Physiology. 1992;448:237–256. doi: 10.1113/jphysiol.1992.sp019039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Verdoorn TA, Lovinger DM. Alcohols potentiate the function of 5-HT3 receptor-channels on NCB-20 neuroblastoma cells by favouring and stabilizing the open channel state. The Journal of Physiology. 1998;507:335–352. doi: 10.1111/j.1469-7793.1998.335bt.x. [DOI] [PMC free article] [PubMed] [Google Scholar]