

Abstract
More than 30 years ago, Douglas (Douglas & Rubin, 1961; Douglas, 1968) proposed that intracellular Ca2+ controls stimulus-secretion coupling in endocrine cells, and Katz & Miledi (1967; Katz, 1969) proposed that intracellular Ca2+ ions control the rapid release of neurotransmitters from synapses. These related hypotheses have been amply confirmed in subsequent years and for students of excitable cells, they dominate our teaching and research. Calcium controls regulated exocytosis. On the other hand, many studies of epithelial and blood cell biology emphasize Ca2+-independent regulation of secretion of mucin, exocytotic delivery of transporters and degranulation. The evidence seems good. Are these contrasting conclusions somehow mistaken, or are the dominant factors controlling exocytosis actually different in different cell types? In this essay, we try to reconcile these ideas and consider classes of questions to ask and hypotheses to test in seeking a more integrated understanding of excitation-secretion coupling. Our review is conceptual and narrowly selective of a few examples rather than referring to a broader range of useful studies in the extensive literature. The examples are taken from mammals and are documented principally by citing other reviews and two of our own studies. The evidence shows that protein phosphorylation by kinases potentiates Ca2+-dependent exocytosis and often suffices to induce exocytosis by itself. Apparently, protein phosphorylation is the physiological trigger in a significant number of examples of regulated exocytosis. We conclude that although sharing many common properties, secretory processes in different cells are specialized and distinct from each other.
The mainstream analysis in endocrine cells and synapses
The regulation of exocytosis has been most precisely studied by single-cell biophysical methods, which provide several strong tools. Membrane capacitance, amperometry, lipophilic dyes and postsynaptic currents are used as measures of exocytosis and transmitter release. Intracellular calcium-sensitive dyes, calcium buffers and caged calcium measure and control the intracellular Ca2+ concentration, [Ca2+]i. Whole-cell patch clamping controls membrane potential and monitors Ca2+ fluxes electrically.
Typically, exocytosis from excitable cells is triggered by Ca2+ entry through voltage-gated Ca2+ channels. The most complete studies are with adrenal chromaffin cells and several types of synapses. They lead to the following well-known conclusions (Heinemann et al. 1994; Roberts, 1994; Parsons et al. 1995; Zucker, 1996; von Gersdorff & Matthews, 1999). Exocytosis occurs whenever the local [Ca2+]i rises into the 10-100 μm range. Its rate is proportional to [Ca2+]i raised to a higher power, e.g. 3 or 4, implying that the cytoplasmic calcium receptors for exocytosis require several bound Ca2+ ions to initiate the final event of fusion (Dodge & Rahamimoff, 1967). When [Ca2+]i is raised suddenly and then held high by an experimental manoeuvre, the rate of exocytosis rises immediately but then begins to fall again within 5–100 ms, presumably reflecting consumption of a pool of readily releasable vesicles. The multi-exponential time course of this slowing is interpreted to mean that the readily releasable pool is slowly being replenished from a series of preceding pools of less ready vesicles, which in turn may also become partially depleted (Fig. 1). The readily releasable pool of vesicles, as gauged by the initial burst of exocytosis during steps of [Ca2+]i or hyperosmotic shocks, is typically estimated to comprise only about 1 % of all vesicles, even in a rested cell. This small pool size may be limited by the scarcity of some protein components essential for forming a docked and primed vesicle. In this case, refilling of the pool might have to wait until a departing vesicle liberates some of these scarce components.
Figure 1. Kinetic pools of vesicles in the regulated secretory pathway of an excitable cell.
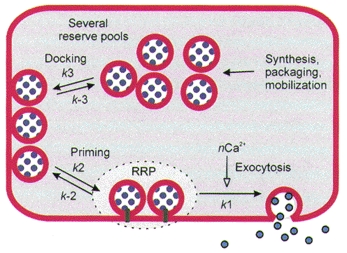
Steps of vesicle traffic towards exocytosis are defined by kinetic analysis of responses to Ca2+ steps and evoked Ca2+ rises in endocrine cells and synapses. Exocytosis (step k1) is triggered by binding of n Ca2+ ions. The small readily releasable pool (RRP) of vesicles is formed by priming (step k2) of docked vesicles. Docked vesicles are formed (step k3) by mobilization and transport from deeper intracellular pools.
The emphasis of these biophysical experiments has been on steps that are completed in a few seconds. For the release of small synaptic vesicles, the experiments place Ca2+ in a final controlling step that is so fast (< 100 μs) it is commonly supposed that there is insufficient time to break covalent bonds in releasing the brake on fast chemical transmission. In the secretion of large, dense core vesicles, the time scale is somewhat longer (Kasai, 1999). In addition, Ca2+ influences several steps that precede (or follow) the critical fusion event.
In parallel with biophysical kinetic analysis, there has been much success in enumerating molecular components of the secretory machinery (Rothman & Wieland, 1996; Hanson et al. 1997; Avery et al. 1999). Soluble proteins include N-ethylmaleimide-sensitive factor (NSF), soluble NSF attachment proteins (SNAPs) and various ras-like GTPases (rabs and others). Membrane components include SNAP receptors (SNAREs), synaptotagmin and many others. The importance and universality of all of these proteins was revealed by reconstitution assays in mammalian cells and mutant analysis in yeast, Caenorhabditis elegans, Drosophila and mouse. The importance of SNAREs was reinforced by their identification as the sole substrates for cleavage by the clostridial metalloproteases botulinum toxin and tetanus toxin, an action that abolishes secretion in many cell types. Homologues of the synaptic SNAREs are used in vesicle fusion steps at many earlier stages in intracellular membrane traffic, such as transport between endoplasmic reticulum and Golgi complex or between Golgi complex and lysosomes. The SNAREs and synaptotagmins, which participate in specific membrane recognition, fusion and regulation of fusion, are actually large gene families with cell type-specific expression of selected members. Thus, although usually composed of the same types of proteins, the secretory machinery does differ in specific molecular composition in different cells. There is not yet agreement on which molecules participate in the brakes that prevent regulated exocytosis in cells at rest and permit it during appropriate stimulation. It is also not known in molecular terms what the special state of the release machinery is in the readily releasable pool of vesicles.
Regulation in epithelia
Unlike nerve and endocrine cells, epithelial cells lack action potentials and Ca2+ influx mechanisms triggered by depolarization. Instead, hormones acting through G-protein-coupled receptors and intracellular second messengers and protein kinases typically initiate regulated membrane fusion and secretion. The time scale of responses may be as long as minutes rather than milliseconds, and the secretion itself is slow and tonic rather than being a brief intense burst typical of excitable cells. Much of this literature lacks rapid assays of secretion, measurements with rapid steps of second messengers or kinases, and the ability to measure several variables simultaneously from single cells. Because of the differences in vantage points, detailed differences or similarities in the control of exocytosis in comparison with excitable cells are often not clearly established. Since the secretory response seems slow, the rate-limiting step(s) may well be much earlier in the vesicle trafficking cycle than in a synapse. It is even possible that exocytosis per se is constitutive in some epithelia and that all regulation is on mobilization of vesicles to regions of exocytosis. The concept of a readily releasable pool of docked and primed vesicles would not be so useful if the rate-limiting steps precede these late events. Also pool sizes may be difficult to define if it remains impossible to phosphorylate the targets of kinases on a millisecond time scale.
We consider first regulated membrane protein traffic. A fascinating example of regulated membrane fusion occurs in kidney collecting ducts, where water channels are inserted into the apical membrane of principal cells in response to vasopressin (reviewed by Brown et al. 1998). As originally proposed in the shuttle hypothesis (Wade et al. 1981), the water channels (aquaporin 2) of the collecting ducts reside in membranes of intracellular vesicles at rest. The vesicle membranes fuse reversibly with the apical plasma membrane surface under the action of vasopressin, a process that uses the machinery of vesicular exocytosis including SNAREs and rab GTPases. The number of water channels at the cell surface is viewed as a dynamic balance of continual exocytosis and endocytosis. This is an example of membrane trafficking induced by cyclic AMP (cAMP)-dependent protein kinase A (PKA). Vasopressin stimulates adenylyl cyclase and the hormone action can be mimicked by activating the cyclase directly with forskolin or by adding membrane-permeant analogues of cAMP. The response requires protein phosphorylation by PKA, one essential target being the aquaporin channel itself. In rat kidney duct cells, the proteins associated with vesicular membranes include SNAP-23, VAMP/synaptobrevin2 and syntaxin 4 (Inoue et al. 1998) and in the canine kidney epithelial cell line (MDCK cells), proteins associated with apical membrane trafficking include SNAP-23, VAMP/synaptobrevin7 and syntaxin 3 (Lafont et al. 1999).
Apparently quite similar is the stimulation of HCl production in parietal cells of the stomach, perhaps the first recognized example of regulated insertion of transporters (Forte et al. 1977; reviewed by Urushidani & Forte, 1997). Again the primary regulator seems to be cAMP. At rest, the acid-secreting H+,K+-ATPase resides on intracellular vesicular (tubulovesicular) membranes. Activation of acid secretion requires fusion of these vesicles with the plasma membrane and possibly with each other to bring the transporters topologically to the cell surface. These events are activated by histamine via coupling of H2 receptors to adenylyl cyclase. In rabbit but not dog, there is also an accompanying rise of [Ca2+]i. However, this rise is considered only a potentiator of the actions of cAMP rather than the initiator of secretion. Activation of muscarinic or cholecystokinin (CCK) receptors, which couple to phospholipase C and make a [Ca2+]i rise without cAMP, does not activate secretion by itself.
Now let us turn to regulated exocytosis itself. A well-studied epithelial example is the secretion of the glycoprotein mucin from large, dense core granules in airway, gastrointestinal tract and related cell lines (reviewed by Forstner, 1995; Davis, 1997). It is often found that mucin secretion may be augmented by two different signalling systems: cAMP and phopholipase C. For example, secretion is stimulated by raising cAMP via β-adrenergic agonists, vasoactive intestinal peptide (VIP), secretin, forskolin or cholera toxin or by incubation with cAMP analogues in submandibular gland, gastric, small intestine and pancreatic duct cells (but not airway goblet cells). Mucin secretion from the same cells can also usually be stimulated by agents that activate phospholipase C, such as muscarinic agonists, ATP and α1-adrenergic agonists (e.g. Hong et al. 1997; Nguyen et al. 1998). Such stimulation activates PKC and may cause a rise of [Ca2+]i by release from intracellular stores. In airway goblet cells and a related cell line SPOC1, both of which can respond to ATP, mucin secretion can be evoked independently by raising [Ca2+]i in permeabilized cells or by adding phorbol ester under buffered, low-Ca2+ conditions (Davis, 1997; Scott et al. 1998). In much of the literature, tests for the action of PKC involve addition of a phorbol ester and perhaps blockade of the phorbol action by a PKC inhibitor. These criteria of mimicry show that a powerful, simultaneous activation of many subtypes of PKC can evoke exocytosis, but by themselves do not show that PKC is the signal used by any physiological pathway.
Our recent unpublished work on dog pancreatic duct cells has used a biophysical approach on single cells. Secretory vesicles were loaded artificially with an exogenous detectable molecule, dopamine, by soaking the epithelial cells in solutions containing millimolar concentrations of dopamine (Kim et al. 1998). Then quantal secretion of individual dopamine-filled vesicles could be detected by amperometry as spikes of oxidation current at a polarized carbon fibre electrode placed just outside a cell. At the same time, [Ca2+]i was monitored optically using the fluorescent Ca2+ indicator indo-1. Figure 2 shows an example of the results. Initially we see a continuous background of spontaneous exocytotic events (raw data in Fig. 2A and summary in Fig. 2B). The endoplasmic reticulum Ca2+-ATPase inhibitor thapsigargin is applied to promote emptying of intracellular Ca2+ stores. This slightly augments the rate of exocytosis. When forskolin is subsequently added to stimulate synthesis of cAMP, exocytosis is strongly stimulated. The forskolin effect develops in 1–2 min and persists for > 5 min after the forskolin is removed. In this experiment, cAMP stimulates exocytosis fourfold without a detectable rise in [Ca2+]i (Fig. 2C). Related experiments confirm that this stimulation requires activation of PKA and that phorbol ester, acting via PKC, can also stimulate exocytosis within 1–2 min, but not as strongly as PKA. Finally, elevating [Ca2+]i by exposure to ionomycin and Ca2+-containing solutions also leads to brisk exocytosis. Such single-cell biophysical experiments confirm and strengthen the conclusion reached by studies of mucin secretion in multicellular preparations that exocytotic secretion from duct cells can be stimulated by any of three messenger systems: cAMP acting through PKA, diacylglycerols acting through PKC, and Ca2+.
Figure 2. Forskolin-stimulated exocytosis in an epithelial cell.
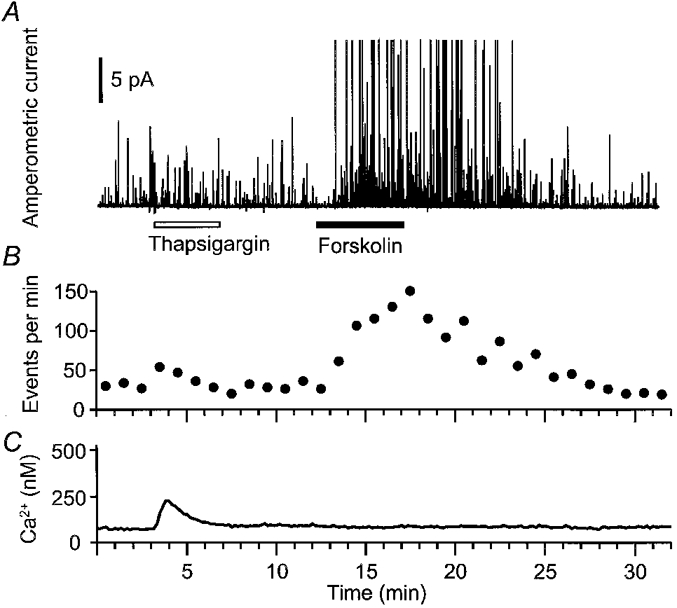
Subconfluent dog pancreatic duct epithelial cells were loaded with 1 μm indo-1 AM for 30 min, then 70 mm dopamine-containing saline solution for 40 min at room temperature. A carbon fibre electrode polarized to +600 mV lightly touched a cell to detect oxidation of quantally released exogenous dopamine upon exocytosis. A, amperometric currents during application of 5 μm thapsigargin (open bar) and 20 μm forskolin (filled bar). Currents > 24 pA have been truncated. B, rate of exocytosis plotted as number of events per minute. C, [Ca2+]i measured with indo-1 ratio photometry. Data are from a single cell.
Haematopoietic cells
Several cells of the haematopoietic lineage store large intracellular secretory granules and degranulate in response to antigenic stimuli (reviewed by Lindau & Gomperts, 1991). Despite much study, the final signal controlling such exocytotic degranulation in neutrophils, mast cells and eosinophils, for example, is not well understood. It seems not to be Ca2+, PKA or PKC (Neher & Almers, 1986; Almers & Neher, 1987; Scepek et al. 1998). Gomperts (1983) discovered that permeabilized mast cells will degranulate in response to non-hydrolysable GTP analogues and proposed that exocytosis is controlled by an as yet unidentified G-protein, which he dubbed GE. Both GTP analogues and the secretagogue compound 4880, stimulate Ca2+ transients in mast cells, but the Ca2+ transients are not temporally related or necessary for the exocytotic events (Neher & Almers, 1986). These cells lack several of the common isoforms of the proteins of the exocytotic machinery including rab-3 (Lacy et al. 1995), and should present an interesting extension of our understanding of secretory regulation when more is known.
Regulation in excitable cells again
Let us reconsider the situation in excitable cells. Is Ca2+ always the message? The five endocrine cell types of the anterior pituitary are excitable secretory cells capable of making action potentials with voltage-gated Na+ and Ca2+ channels. Pituitary gonadotropes secrete gonadotropins from large, dense core vesicles in response to gonadotropin-releasing hormone (GnRH). The GnRH receptor couples to the G-protein Gq, activating the PLC cascade with production of inositol trisphosphate (IP3) and oscillatory release of Ca2+ from intracellular stores (reviewed by Stojilkovic & Catt, 1995; Hille et al. 1995). PKC is also activated. As studied with biophysical techniques in single cells, secretion from gonadotropes exhibits a tight temporal correlation with each Ca2+ elevation and a steep, cubic power dependence on the [Ca2+]i (Tse et al. 1993, 1997). Dialysing a cell with Ca2+ chelators blocks hormone-induced secretion, and uncaging IP3 or Ca2+ within the cell can elicit secretion. Caged Ca2+ experiments reveal a readily releasable pool of about 200 secretory granules that can be secreted within 100 ms when [Ca2+]i is stepped up to high levels. This exocytotic burst is followed by several progressively slower phases of secretion. Thus gonadotropes satisfy all the criteria of classical Ca2+-dependent stimulus-secretion coupling in Fig. 1 - with the twist that virtually all of the trigger Ca2+ comes from intracellular stores rather than from the extracellular medium.
Despite the clear physiological necessity and sufficiency of the hormone-induced Ca2+ signal in gonadotropes, there is also extensive literature showing that treatments with phorbol esters can release several pituitary hormones from pituitary cells (Vale et al. 1977; van der Merwe et al. 1990). We have studied this treatment as well. In Fig. 3, cells secreting luteinizing hormone (LH) in response to various treatments are identified by a reverse haemolytic plaque assay using antibodies to LH. The natural secretagogue, GnRH, raises the number of secreting cells well above the basal level in a 1 h assay. A phorbol ester, phorbol myristate acetate (PMA) without or with GnRH stimulates the same number of cells as GnRH alone, and an inactive phorbol analogue does not stimulate secretion (Fig. 3A). Further experiments showed that the phorbol ester induces secretion by signalling mechanisms different from those used by GnRH. Thus, the phorbol ester-induced secretion of luteinizing hormone is much more sensitive to the PKC inhibitor bisindolylmaleimide I (BIS) than the GnRH-induced secretion is (Fig. 3B), and it is much less sensitive to block by Cd2+ or Ca2+ chelators (Fig. 3C). In a 10-20 s exposure, GnRH makes large oscillations of [Ca2+]i, whereas during a 1 h exposure phorbol ester makes no detectable [Ca2+]i elevation (Billiard et al. 1997). The full secretory response, measured by the amperometric technique on dopamine-loaded cells, also develops within 10-20 s with GnRH but takes over 100 s to develop during incubation with phorbol ester. Thus PKC mediates significant exocytosis without a [Ca2+]i rise in this excitable cell, but the physiological, more rapid secretion in response to GnRH seems almost entirely due to the rise in [Ca2+]i.
Figure 3. Releasing hormone and kinase-stimulated exocytosis in pituitary cells.
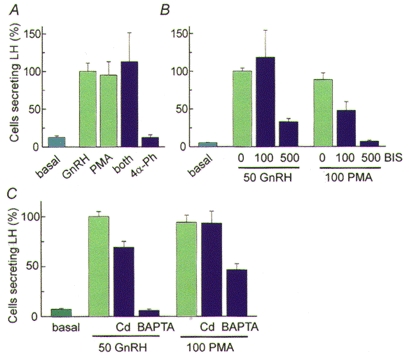
Cells were cultured following enzymatic dispersal from the anterior pituitary of a castrated male rat. The number of cells secreting luteinizing hormone (LH) in 1 h was counted using a reverse haemolytic plaque assay with antibodies against LH and normalized to the mean number of cells secreting in response to the natural releasing hormone GnRH. Note that this assay does not report the amount of LH secreted, rather the number of cells secreting above a certain threshold amount. A, similar stimulation of LH secretion by 50 nM GnRH, 100 nM phorbol myristate acetate (PMA), and both together (both); and no stimulation by 1 μm 4α-phorbol-12,13-didecanoate (4α-Ph). B, greater sensitivity of PMA-induced secretion vs. GnRH-induced secretion to block by the PKC inhibitor bisindolylmaleimide I (BIS, 100 and 500 nM). C, greater sensitivity of GnRH-induced secretion vs. PMA-induced secretion to block by 100 μm extracellular Cd2+ or intracellular BAPTA (loading in 10 μm BAPTA-AM solution for 15 min). (After Billiard et al. 1997.)
Pancreatic β-cells present a similar picture. Secretion of insulin is synchronous with elevations of [Ca2+]i, but activating protein kinases can lead to secretion as well. A large amount of literature documents how high bathing glucose concentrations lead to plateau and burst membrane depolarizations that open voltage-gated Ca2+ channels of β-cells, allowing entry of the Ca2+ that triggers insulin release (reviewed by Wollheim et al. 1996). Secretion of insulin tracks the changes of [Ca2+]i very closely (Gilon & Henquin, 1995; reviewed by Henquin et al. 1998). This leads to a classical view of electrical control of secretion via the effect of membrane potential on [Ca2+]i. However, more recent literature has emphasized modulation or stimulation of secretion by PKA and PKC. For example, the depolarization-induced increases of membrane area (membrane capacitance) in isolated β-cells are augmented by pretreatment with forskolin, cAMP analogues, a phosphodiesterase inhibitor, phorbol esters or phosphatase inhibitors (Gillis & Misler, 1993; Ämmäla et al. 1994). This potentiation is not due to enhanced [Ca2+]i transients. High glucose can still induce insulin secretion in calcium-depleted conditions if both PKA and PKC have been stimulated (Komatsu et al. 1995). It seems plausible that control via Ca2+ is the principal coupling for energy-regulated insulin release and that receptor-mediated control via kinases superimposes a modulatory bias from the autonomic nervous system.
Even fast chemical synaptic transmission is significantly stimulated by protein kinases. Both excitatory and inhibitory synapses are usually potentiated by activation of PKA or PKC. The quantal content of evoked transmitter release is increased, and the frequency of spontaneous quantal transmitter release is raised as well (Malenka et al. 1986; Shapira et al. 1987; Capogna et al. 1995).
Hypotheses to consider
Where do the kinases act?Figure 4 presents several testable variations of the standard kinetic description of exocytosis. Each of these plausible ideas has been suggested before and probably applies in some specific cases. Figure 4A reiterates the standard description of Fig. 1 with the small addition that the action of kinases simply causes a Ca2+ transient. This would be a trivial explanation that says kinases have no effect on the secretory mechanism - they are just another trigger of Ca2+ transients. This possibility must always be considered by measuring [Ca2+]i directly and by eliminating any such transients. Indeed, in our unpublished work on dog pancreatic duct epithelial cells, we quite often found that phorbol esters or stimuli that raise cAMP did elevate [Ca2+]i transiently. However, the elevation could be averted by preincubation with thapsigargin (Fig. 2) or with membrane-permeant precursors of Ca2+ chelators without eliminating the stimulation of secretion. Permeabilized cells and low-Ca2+ buffers would offer an alternative experimental approach to the same question. A related hypothesis for excitable cells is that kinases enhance the action potential-evoked [Ca2+]i transient by potentiating evoked Ca2+ entry. Phorbol esters do in fact relieve a tonic inhibition of Ca2+ channels (Swartz, 1993; Yang & Tsien, 1993), and they could also prolong presynaptic action potentials. Nevertheless, in several cases the potentiation of synaptic transmission is known not to be due to a larger presynaptic [Ca2+]i transient (e.g. Yawo, 1999).
Figure 4. Possible sites of action of protein kinases on the secretory pathway.
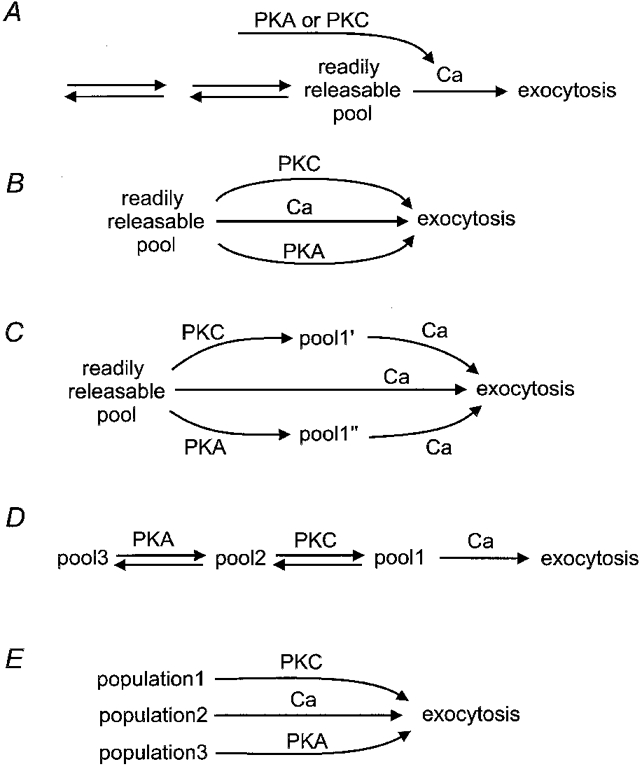
Kinases may act indirectly by augmenting the Ca2+ signal in the cytoplasm (A), directly on the conventional Ca2+-sensitive pathway (B-D) or directly on populations of vesicles that are not Ca2+ sensitive (E).
Figure 4B hypothesizes that the brake for exocytosis of the docked and primed, readily releasable pool can be relieved directly by protein phosphorylation instead of by Ca2+. As virtually all of the proteins known to participate in the last stages of exocytosis have consensus sites for protein phosphorylation, this idea is quite plausible. For the same reason and because we do not know which proteins form the brake, it could be quite difficult to prove. Figure 4C is a slight modification suggesting that kinases may modify vesicles in the readily releasable pool, not by removing the brake fully but by changing the sensitivity to Ca2+ so that some exocytosis occurs even at physiological [Ca2+]i levels. An alternative and conservative approach is to consider that the pathway towards exocytosis remains linear, with a Ca2+-sensitive final step and kinases affecting the rates and equilibria of the preceding steps (Fig. 4D). Then the size of the readily releasable pool might become larger, and its refilling during continuous exocytosis faster, even if the final step remains the same. This mechanism might make changes observable in a morphometric approach that uses quantitative electron microscopy to look for changes in the number of vesicles in different cellular domains. At early steps of mobilization and recruitment of vesicles, many investigators have considered regulatory actions on cytoskeleton and cytoskeletal links, which might be a barrier to mobilization or which might assist in the directed movement of vesicles towards sites of release. The first and classical example of such regulation at synapses is the actin-associated, vesicle-binding family of synapsin proteins (Llinás et al. 1985; Hilfiker et al. 1999). Phosphorylation of synapsin 1 allows mobilization of synaptic vesicles from a tethered condition in reserve pools. One recent study of capacitance changes in melanotropes concludes that cAMP treatments actually enlarge the secretory granule size, thus increasing exocytosis (Sikdar et al. 1998). Biophysical approaches to distinguish the hypotheses of Fig. 4B-D are to measure Ca2+ sensitivity, kinetic pool size and refilling rates, using controlled [Ca2+]i increases, hyperosmotic shocks and amphipathic membrane dyes. In hippocampal cultures, Stevens & Sullivan (1998) showed that PKC augments the size of the vesicle pool releasable by hyperosmotic shock and increases the rate of refilling of this pool. Similarly in chromaffin cells, Gillis et al. (1996) found that PKC augments the readily releasable pool defined by fast capacitance increases following step elevations of [Ca2+]i.
Finally, the three regulatory signals, Ca2+, PKA and PKC, may be acting on quite different populations of vesicles to induce exocytosis by entirely distinct mechanisms (Fig. 4E). In two of the examples above, secretion of LH from gonadotropes and secretion of mucin from epithelia, the same secretory product is released by different stimuli, so the hypothesis of different vesicle populations seems unlikely. However, this hypothesis would make sense for cells that package multiple products that are secreted independently in response to different signals. Possibly all cells have multiple regulated secretory pathways. For example, when a cell has both small, clear and large, dense core secretory vesicles, independent control seems quite probable. In mast cells, Almers & Neher (1987) and Kirillova et al. (1993) distinguish a degranulation of large granules, which is stimulated by GTP analogues, from another significant membrane surface area increase, which is due entirely to a fusion of small vesicles and can be stimulated by step increases of [Ca2+]i. Even when both vesicle types are thought to have a Ca2+-dependent trigger, the regulation and nature of the Ca2+ signal may be distinguishable (Kasai, 1999).
Conclusions
The physiological regulation of exocytosis has been most extensively studied in electrically excitable cells and in epithelia. One body of literature emphasizes Ca2+ and the other, serine-threonine protein kinases. The standards of the work are sufficiently high that one must accept that these two cell types typically use different strategies. Neurons exploit the only message that could act on a submillisecond time scale, the Ca2+ ion, and epithelia use a system that is slow and longer lasting. In either case, we do not actually know the critical molecular target(s) of the relevant signal. Quite probably additional signals will be found as more becomes known about the secretion that follows activation of tyrosine kinase pathways in haematopoietic and other cells. In almost every case studied, protein kinases can potentiate exocytosis in excitable cells and Ca2+ can evoke secretion in epithelia. Thus the secretory machinery, which comprises different protein subtypes from homologous families of molecules, retains some sensitivity to all of the possible stimuli, but is specially tuned to respond well to signals relevant to that cell type and to that cargo. It may even be that intracellular fusion events and pathways, usually thought of as constitutive, could also be accelerated by some of these signals.
Acknowledgments
We thank Dr Wolfhard Almers for helpful suggestions on the manuscript, and L. Miller and D. Stienmier for technical help. Our work is supported by grants from the National Institutes of Health (HD 12629, AR 17803).
References
- Almers W, Neher E. Gradual and stepwise changes in the membrane capacitance of rat peritoneal mast cells. The Journal of Physiology. 1987;386:205–217. doi: 10.1113/jphysiol.1987.sp016530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ammala C, Eliasson L, Bokvist K, Berggren PO, Honkanen RE, Sjøholm A, Rorsman P. Activation of protein kinases and inhibition of protein phosphatases play a central role in the regulation of exocytosis in mouse pancreatic beta cells. Proceedings of the National Academy of Sciences of the USA. 1994;91:4343–4347. doi: 10.1073/pnas.91.10.4343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery J, Jahn R, Edwardson JM. Reconstitution of regulated exocytosis in cell-free systems: a critical appraisal. Annual Review of Physiology. 1999;61:777–807. doi: 10.1146/annurev.physiol.61.1.777. [DOI] [PubMed] [Google Scholar]
- Billiard J, Koh D-S, Babcock DF, Hille B. Protein kinase C as a signal for exocytosis. Proceedings of the National Academy of Sciences of the USA. 1997;94:12192–12197. doi: 10.1073/pnas.94.22.12192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D, Katsura T, Gustafson CE. Cellular mechanisms of aquaporin trafficking. American Journal of Physiology. 1998;275:F328–331. doi: 10.1152/ajprenal.1998.275.3.F328. [DOI] [PubMed] [Google Scholar]
- Capogna M, Gähwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. Journal of Neuroscience. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CW. Goblet cells: Physiology and pharmacology. In: Rogers D, Letham MI, editors. Airway Mucus: Basic Mechanisms and Clinical Perspective. Basel: Birkhäuser Verlag; 1997. pp. 149–177. [Google Scholar]
- Dodge FA, Jr, Rahamimoff R. Co-operative action of calcium ions in transmitter release at the neuromuscular junction. The Journal of Physiology. 1967;193:419–432. doi: 10.1113/jphysiol.1967.sp008367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas WW. Stimulus-secretion coupling: the concept and clues from chromaffin and other cells. British Journal of Pharmacology. 1968;34:453–474. doi: 10.1111/j.1476-5381.1968.tb08474.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Douglas WW, Rubin RP. The role of calcium in the secondary response of the adrenal medulla to acetylcholine. The Journal of Physiology. 1961;159:40–47. doi: 10.1113/jphysiol.1961.sp006791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forstner G. Signal transduction, packaging and secretion of mucins. Annual Review of Physiology. 1995;57:585–605. doi: 10.1146/annurev.ph.57.030195.003101. [DOI] [PubMed] [Google Scholar]
- Forte TM, Machen TE, Forte JG. Ultrastructural changes in oxyntic cells associated with secretory function: a membrane-recycling hypothesis. Gastroenterology. 1977;73:941–955. [PubMed] [Google Scholar]
- Gillis KD, Misler S. Enhancers of cytosolic cAMP augment depolarization-induced exocytosis from pancreatic B-cells: evidence for effects distal to Ca2+ entry. Pflügers Archiv. 1993;424:195–197. doi: 10.1007/BF00374612. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Gilon P, Henquin JC. Distinct effects of glucose on the synchronous oscillations of insulin release and cytoplasmic Ca2+ concentration measured simultaneously in single mouse islets. Endocrinology. 1995;136:5725–5730. doi: 10.1210/endo.136.12.7588329. [DOI] [PubMed] [Google Scholar]
- Gomperts BD. Involvement of guanine nucleotide-binding protein in the gating of Ca2+ by receptors. Nature. 1983;306:64–66. doi: 10.1038/306064a0. [DOI] [PubMed] [Google Scholar]
- Hanson PI, Heuser JE, Jahn R. Neurotransmitter release - four years of SNARE complexes. Current Opinion in Neurobiology. 1997;7:310–315. doi: 10.1016/s0959-4388(97)80057-8. [DOI] [PubMed] [Google Scholar]
- Heinemann C, Chow RH, Neher E, Zucker RS. Kinetics of the secretory response in bovine chromaffin cells following flash photolysis of caged Ca2+ Biophysical Journal. 1994;67:2546–2557. doi: 10.1016/S0006-3495(94)80744-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Jonas JC, Gilon P. Functional significance of Ca2+ oscillations in pancreatic beta cells. Diabetes and Metabolism. 1998;24:30–36. [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philosophical Transactions of the Royal SocietyB. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B, Tse A, Tse FW, Bosma MM. Signaling mechanisms during the response of pituitary gonadotropes to GnRH. Recent Progress in Hormone Research. 1995;50:75–95. doi: 10.1016/b978-0-12-571150-0.50008-1. [DOI] [PubMed] [Google Scholar]
- Hong DH, Forstner JF, Forstner GG. Protein kinase C-ε eis the likely mediator of mucin exocytosis in human colonic cell lines. American Journal of Physiology. 1997;272:G31–37. doi: 10.1152/ajpgi.1997.272.1.G31. [DOI] [PubMed] [Google Scholar]
- Inoue T, Nielsen S, Mandon B, Terris J, Kishore BK, Knepper MA. SNAP-23 in rat kidney: colocalization with aquaporin-2 in collecting duct vesicles. American Journal of Physiology. 1998;275:F752–760. doi: 10.1152/ajprenal.1998.275.5.F752. [DOI] [PubMed] [Google Scholar]
- Kasai H. Comparative biology of Ca2+-dependent exocytosis: implications of kinetic diversity for secretory function. Trends in Neurosciences. 1999;22:88–93. doi: 10.1016/s0166-2236(98)01293-4. [DOI] [PubMed] [Google Scholar]
- Katz B. The Release of Neural Transmitter Substances. Springfield, IL, USA: Charles Thomas; 1969. [Google Scholar]
- Katz B, Miledi R. The timing of calcium action during neuromuscular transmission. The Journal of Physiology. 1967;189:535–544. doi: 10.1113/jphysiol.1967.sp008183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim K-T, Koh D-S, Hille B. Loading of false transmitters into secretory vesicles for detection by carbon-fiber amperometry. Society for Neuroscience Abstracts. 1998;24:1316. doi: 10.1523/JNEUROSCI.20-20-j0001.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirillova J, Thomas P, Almers W. Two independently regulated secretory pathways in mast cells. Journal de Physiologie. 1993;87:203–208. doi: 10.1016/0928-4257(93)90031-n. [DOI] [PubMed] [Google Scholar]
- Komatsu M, Schermerhorn T, Aizawa T, Sharp GW. Glucose stimulation of insulin release in the absence of extracellular Ca2+ and in the absence of any increase in intracellular Ca2+ in rat pancreatic islets. Proceedings of the National Academy of Sciences of the USA. 1995;92:10728–10732. doi: 10.1073/pnas.92.23.10728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lacy P, Thompson N, Tian M, Solari R, Hide I, Newman TM, Gomperts BD. A survey of GTP-binding proteins and other potential key regulators of exocytotic secretion in eosinophils. Apparent absence of rab3 and vesicle fusion protein homologues. Journal of Cell Science. 1995;108:3547–3556. doi: 10.1242/jcs.108.11.3547. [DOI] [PubMed] [Google Scholar]
- Lafont F, Verkade P, Galli T, Wimmer C, Louvard D, Simons K. Raft association of SNAP receptors acting in apical trafficking in Madin-Darby canine kidney cells. Proceedings of the National Academy of Sciences of the USA. 1999;96:3734–3738. doi: 10.1073/pnas.96.7.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindau M, Gomperts BD. Techniques and concepts in exocytosis: focus on mast cells. Biochimica et Biophysica Acta. 1991;1071:429–471. doi: 10.1016/0304-4157(91)90006-i. [DOI] [PubMed] [Google Scholar]
- Llinás R, McGuinness TL, Leonard CS, Sugimori M, Greengard P. Intraterminal injection of synapsin I or calcium/calmodulin-dependent protein kinase II alters neurotransmitter release at the squid giant synapse. Proceedings of the National Academy of Sciences of the USA. 1985;82:3035–3039. doi: 10.1073/pnas.82.9.3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malenka RC, Madison DV, Nicoll RA. Potentiation of synaptic transmission in the hippocampus by phorbol esters. Nature. 1986;321:175–177. doi: 10.1038/321175a0. [DOI] [PubMed] [Google Scholar]
- Neher E, Almers W. Fast calcium transients in rat peritoneal mast cells are not sufficient to trigger exocytosis. EMBO Journal. 1986;5:51–53. doi: 10.1002/j.1460-2075.1986.tb04176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TD, Moody MW, Savard CE, Lee SP. Secretory effects of ATP on nontransformed dog pancreatic duct epithelial cells. American Journal of Physiology. 1998;275:G104–113. doi: 10.1152/ajpgi.1998.275.1.G104. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst, and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Roberts WM. Localization of calcium signals by a mobile calcium buffer in frog saccular hair cells. Journal of Neuroscience. 1994;14:3246–3262. doi: 10.1523/JNEUROSCI.14-05-03246.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieland FT. Protein sorting by transport vesicles. Science. 1996;272:227–234. doi: 10.1126/science.272.5259.227. [DOI] [PubMed] [Google Scholar]
- Scepek S, Coorssen JR, Lindau M. Fusion pore expansion in horse eosinophils is modulated by Ca2+ and protein kinase C via distinct mechanisms. EMBO Journal. 1998;17:4340–4345. doi: 10.1093/emboj/17.15.4340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott CE, Abdullah LH, Davis CW. Ca2+ and protein kinase C activation of mucin granule exocytosis in permeabilized SPOC1 cells. American Journal of Physiology. 1998;275:C285–292. doi: 10.1152/ajpcell.1998.275.1.C285. [DOI] [PubMed] [Google Scholar]
- Shapira R, Silberberg SD, Ginsburg S, Rahamimoff R. Activation of protein kinase C augments evoked transmitter release. Nature. 1987;325:58–60. doi: 10.1038/325058a0. [DOI] [PubMed] [Google Scholar]
- Sikdar SK, Kreft M, Zorec R. Modulation of the unitary exocytic event amplitude by cAMP in rat melanotrophs. The Journal of Physiology. 1998;511:851–859. doi: 10.1111/j.1469-7793.1998.851bg.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Sullivan JM. Regulation of the readily releasable vesicle pool by protein kinase C. Neuron. 1998;21:885–893. doi: 10.1016/s0896-6273(00)80603-0. [DOI] [PubMed] [Google Scholar]
- Stojilkovic SS, Catt KJ. Novel aspects of GnRH-induced intracellular signaling and secretion in pituitary gonadotrophs. Journal of Neuroendocrinology. 1995;7:739–757. doi: 10.1111/j.1365-2826.1995.tb00711.x. [DOI] [PubMed] [Google Scholar]
- Swartz KJ, Merritt A, Bean BP, Lovinger DM. Protein kinase C modulates glutamate receptor inhibition of Ca2+ channels and synaptic transmission. Nature. 1993;361:165–168. doi: 10.1038/361165a0. [DOI] [PubMed] [Google Scholar]
- Tse A, Tse FW, Almers W, Hille B. Rhythmic exocytosis stimulated by GnRH-induced calcium oscillations in rat gonadotropes. Science. 1993;260:82–84. doi: 10.1126/science.8385366. [DOI] [PubMed] [Google Scholar]
- Tse FW, Tse A, Hille B, Horstmann H, Almers W. Local Ca2+ release from internal stores controls exocytosis in pituitary gonadotrophs. Neuron. 1997;18:121–132. doi: 10.1016/s0896-6273(01)80051-9. [DOI] [PubMed] [Google Scholar]
- Urushidani T, Forte JG. Signal transduction and activation of acid secretion in the parietal cell. Journal of Membrane Biology. 1997;159:99–111. doi: 10.1007/s002329900274. [DOI] [PubMed] [Google Scholar]
- Vale W, Rivier C, Rivier J, Brown M. Diverse roles of hypothalamic regulatory peptides. In: Mathieu J, editor. Medicinal Chemistry V: Proceedings of the 5th International Symposium on Medicinal Chemistry. Amsterdam: Elsevier; 1977. pp. 25–62. Paris, July 19-22, 1976. [Google Scholar]
- van der Merwe PA, Millar RP, Davidson JS. Calcium stimulates luteinizing-hormone (lutropin) exocytosis by a mechanism independent of protein kinase C. Biochemical Journal. 1990;268:493–498. doi: 10.1042/bj2680493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Electrophysiology of synaptic vesicle cycling. Annual Review of Physiology. 1999;61:725–752. doi: 10.1146/annurev.physiol.61.1.725. [DOI] [PubMed] [Google Scholar]
- Wade JB, Stetson DL, Lewis SA. ADH action: evidence for a membrane shuttle mechanism. Annals of the New York Academy of Sciences. 1981;372:106–117. doi: 10.1111/j.1749-6632.1981.tb15464.x. [DOI] [PubMed] [Google Scholar]
- Wollheim CB, Lang J, Regazzi R. The exocytotic process of insulin secretion and its regulation by Ca2+ and G-proteins. Diabetes Reviews. 1996;4:276–297. [Google Scholar]
- Yang J, Tsien RW. Enhancement of N- and L-type calcium channel currents by protein kinase C in frog sympathetic neurons. Neuron. 1993;10:127–136. doi: 10.1016/0896-6273(93)90305-b. [DOI] [PubMed] [Google Scholar]
- Yawo H. Two components of transmitter release from the chick ciliary presynaptic terminal and their regulation by protein kinase C. The Journal of Physiology. 1999;516:461–470. doi: 10.1111/j.1469-7793.1999.0461v.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. [DOI] [PubMed] [Google Scholar]