

Abstract
Coronary flow elevation from enhanced perfusion pulsatility is synergistically amplified by adenosine. This study determined the specificity of this interaction and its potential mechanisms.
Mean and phasic coronary flow responses to increasing pulsatile perfusion were assessed in anaesthetized dogs, with the anterior descending coronary artery servoperfused to regulate real-time physiological flow pulsatility at constant mean pressure. Pulsatility was varied between 40 and 100 mmHg. Hearts ejected into the native aorta whilst maintaining stable loading.
Increasing pulsatility elevated mean coronary flow +11.5 ± 1.7% under basal conditions. Co-infusion of adenosine sufficient to raise baseline flow 66% markedly amplified this pulsatile perfusion response (+82.6 ± 14.3% increase in mean flow above adenosine baseline), due to a leftward shift of the adenosine-coronary flow response curve at higher pulsatility. Flow augmentation with pulsatility was not linked to higher regional oxygen consumption, supporting direct rather than metabolically driven mechanisms.
Neither bradykinin, acetylcholine nor verapamil reproduced the synergistic amplification of mean flow by adenosine and higher pulsatility, despite being administered at doses matching basal flow change with adenosine.
ATP-sensitive potassium (KATP) activation (pinacidil) amplified the pulse-flow response 3-fold, although this remained significantly less than with adenosine. Co-administration of the phospholipase A2 inhibitor quinacrine virtually eliminated adenosine-induced vasodilatation, yet synergistic interaction between adenosine and pulse perfusion persisted, albeit at a reduced level.
Thus, adenosine and perfusion pulsatility specifically interact to enhance coronary flow. This synergy is partially explained by KATP agonist action and additional non-flow-dependent mechanisms, and may be important for modulating flow reserve during exercise or other high output states where increased flow demand and higher perfusion pulsatility typically co-exist.
In vivo coronary artery flow is modulated by biochemical/ion channel mediators such as nitric oxide (NO), adenosine and ATP-sensitive potassium channels (KATP), and by mechanical forces stemming from pulsatile perfusion. In intact hearts, increasing coronary flow pulsatility at the same mean pressure, cardiac workload and energy consumption raises mean flow by nearly 15% (Saeki et al. 1995). Studies have further shown that this mechanical effect can be biochemically amplified by adenosine, and partially inhibited by Nω-monomethyl-L-arginine (L-NMMA), a competitive inhibitor of nitric oxide synthase (Recchia et al. 1996). These findings suggest important interactions of perfusion pulsatility with adenosine which could be due to changes in basal flow, nitric oxide signalling and/or specific mechano/humoral synergies. Understanding the mechanisms for this interaction is important as both signals rise during exertional demands.
One mechanism for an adenosine-flow pulsatility synergy is that by enhancing basal shear and lowering vascular tone, adenosine augments cyclic shear and distension-related signals (Goto et al. 1996). If true, then similar interactions would be predicted with alternative dilators such as verapamil, or submaximal bradykinin and acetylcholine. Alternatively, the adenosine response might depend upon specific agonist activity on KATP channels (Akatsuka et al. 1994; Kleppisch & Nelson, 1995). Support for this hypothesis stems from in vitro data showing that flow-induced NO-dependent dilatation is enhanced by adenosine via KATP channel activation (Kuo & Chancellor, 1995). Other mechanisms of adenosine dilatation, such as protein kinase A-linked activation of large-conductance calcium-activated potassium (KCa) channels (Hartley & Kozlowski, 1996; Shinoda et al. 1997), or A1 receptor-coupled effects (Merkel et al. 1992), might contribute. Lastly, adenosine dilates predominantly distal resistance vessels (< 150 μm) (Kanatsuka et al. 1989) and this site selectivity may be important for enhancing proximal arteriole responses to greater pulsatility.
The present study was designed to test the specificity of the synergistic interaction of perfusion pulsatility and adenosine, and clarify the likely mechanisms for this interaction in vivo. Studies were conducted by employing a novel model in open-chest anaesthetized dogs, in which the left anterior descending coronary artery is cannulated and perfused with blood by a computer servosystem that independently controls mean and pulsatile perfusion pressure. The effect of steady-state increases in pulse pressure on phasic and mean epicardial coronary flow were determined under basal conditions and after administration of equi-dilating concentrations of agents that acted via differing mechanisms and sites of action within the coronary vascular bed.
METHODS
Surgical procedure
The protocol was reviewed and approved by the Animal Care and Use Committee of the Johns Hopkins University. The servosystem preparation for in situ coronary pulsatile perfusion control has been reported previously (Recchia et al. 1996). Briefly, adult mongrel dogs (n = 45) were anaesthetized with pentobarbitone (30 mg kg−1i.v.) and fentanyl (50 μg kg−1i.v.) and ventilated with enhanced inspired oxygen. Arterial pH, PCO2 and PO2 were maintained in the physiological range. Supplemental anaesthesia was provided continuously during the procedure (pentobarbitone, 3 mg kg−1 h−1, fentanyl, 8–10 μg kg−1 h−1). Adequacy of anaesthesia was confirmed by absence of eye-blink response and loss of jaw tension. Femoral, left carotid and left subclavian arteries were cannulated to withdraw oxygenated blood to the servoperfusion system, to monitor pressure in the aortic root and left ventricular cavity, and for blood sampling. After administration of pancuronium bromide (0.3 mg kg−1i.v.), the chest was opened via a left lateral thoracotomy, the heart placed in a pericardial cradle and the animal heparinized (8000 i.u. bolus, 1000 i.u. h−1). Heart rate and blood pressure monitored during the surgical procedure were unchanged, confirming the adequacy of anaesthesia during this period of transient paralysis. The proximal left anterior descending artery (LAD) was dissected free and a short metal cannula fitted with an in-line volume flowmeter (Transonics, 2N) was inserted into the vessel. Left carotid artery blood was diverted through the cannula, maintaining perfusion to the distal bed until flow could be provided by the servopump. In subsets of animals, a distal coronary vein draining the servoperfused region was also cannulated. Adequacy of venous blood sampling was verified by transiently lowering mean coronary pressure and confirming a decline in venous O2 saturation. The product of the arterial-venous O2 difference and coronary flow provided a measure of regional myocardial oxygen consumption (MV̇O2). At the conclusion of the study, animals were killed by a barbiturate overdose followed by KCl cardioplegia.
Servopump system
The servoperfusion system was modified from that previously reported (Recchia et al. 1996). Arterial blood from a femoral artery was pressurized to a constant 100 mmHg by being pumped into and out of a small balloon reservoir submerged under pressurized water. The blood then passed through a filter, heat exchanger and one-way valve, and into a chamber with a movable floor coupled to a linear motor (Ling Electronics, 411). The motor was controlled by real-time digital feedback to generate the desired pulse pressure. Blood exited the chamber into a non-distensible conduit perfusing the LAD, with luminal pressure measured by micromanometer (SPC350, Millar Instr). Central aortic pressure was digitized and stored to computer memory, and then modified to adjust mean and pulse pressure amplitude to a desired level. This signal, which remained synchronized with left ventricle (LV) contraction, served as the servocommand for digital feedback control of the perfusion pressure. Thus, coronary pulse pressure (PP) could be independently varied from mean pressure or cardiac chamber function. Perfusate blood was maintained at 37°C, with arterial gases matching those of the systemic circulation.
Experimental protocol
After establishing servoperfusion of the isolated LAD bed, coronary flow was stabilized at a constant mean pressure of 100 mmHg and PP amplitude of 40 mmHg. PP was then varied between 40 and 100 mmHg (2.5-fold rise), holding mean pressure and ventricular ejection constant. Phasic coronary flow and pressure, aortic flow and pressure, and left ventricular pressure were measured at steady state, 4–5 min after a PP change, with the primary output variable being phasic and mean coronary flow to the isolated LAD bed. In 15 studies, the mass of perfused myocardium was quantified by tissue staining (Monastral Blue) to convert flow to millilitres per gram per minute.
The effect of raising PP under baseline conditions was assessed in all animals. Subsets of animals provided data with various concomitant pharmacological interventions. Adenosine (1–150 μg min−1, 0.03- 0.8 μg ml−1)-coronary flow dose-response curves were measured at both PPs (n = 8). From these data, we determined the level of adenosine-induced flow elevation (μ60%) which maximally amplified subsequent flow changes from increasing PP. This magnitude of basal flow elevation was matched by alternative agents: verapamil (30-60 ng ml−1; n = 10) acting diffusely on smooth muscle tone (Kung et al. 1995); acetylcholine (80- 140 ng ml−1; n = 10), acting principally on proximal resistance arterioles by NO-dependent and -independent pathways (Kuo et al. 1995); and pinacidil (5–12 ng ml−1; n = 7), a KATP channel agonist that dilates principally distal resistance arterioles (Sato et al. 1994). One or at most two agents were tested in each animal. To further define dose responses for each interaction, additional studies were performed at higher doses of each agent (in μg ml−1: adenosine, 0.25–0.45; verapamil, 0.05–0.16; acetylcholine, 0.12–0.17; and bradykinin, 0.02–0.06; n = 7), each raising the mean flow by 100-150% at basal (40 mmHg) pulsatility. To test whether the adenosine-PP interaction was specific to intravascular administration, perhaps belying facilitated mass transfer of the nucleoside to target vessels, studies were also performed in which intracellular adenosine catabolism was inhibited by iodotubercidin (ITC; 5–15 μg min−1, 0.04–0.1 μg ml−1) and erythro-9-(2-hydroxy-3-nonyl) adenosine (EHNA; 15-60 μg min−1, 0.125–0.4 μg ml−1) (Ely et al. 1992; Stepp et al. 1996). These data compared with equi-dilating doses of exogenous adenosine. Lastly, the phospholipase A2 (PLA2) inhibitor quinacrine was first infused (6.7 μg kg−1 min−1× 20 min intracoronary (i.c.)) to inhibit adenosine-mediated flow elevation (Kimura & Satoh, 1985). Adenosine was then combined with quinacrine before and after augmenting pulsatile perfusion to further test the role of basal flow in the adenosine-PP synergy.
Pharmaceuticals
Adenosine (Adenocard, Fujisawa), acetylcholine, bradykinin and quinacrine (Sigma), EHNA and ITC (RBI), and verapamil (ARL, Inc.) were each freshly dissolved in isotonic saline at physiological temperature. Pinacidil (RBI) was dissolved in dimethylsulphoxide (DMSO) and isotonic saline, with a final DMSO concentration of less than 0.01%. All drugs were administered by an intracoronary route.
Data analysis and statistics
Haemodynamic data were digitized at 200 Hz and analysed off-line using custom software. Tests of the effects of raising PP were performed by Student's paired t test. Between-intervention effects were first tested by one-way ANOVA, and then individual comparisons were made by Student's non-paired t test, using a Bonferroni correction for multiple comparisons. Data are presented as means ±s.e.m.
RESULTS
Haemodynamics and basal coronary flow
Mean heart rate was 117 ± 5 beats min−1, and resting ventricular systolic pressure, cardiac output and maximal rate of LV pressure rise (dP/dtmax) were 109 ± 4 mmHg, 32 ± 3 ml s−1 and 1520 ± 53 mmHg s−1, respectively. There were no significant changes in these baseline parameters with the various intracoronary infusions, except for high-dose verapamil, which induced a slight negative inotropic effect. Under no conditions were the parameters affected by varying coronary perfusion pulsatility. Basal coronary flow in the LAD territory at 100 mmHg mean and 40 mmHg pulse pressure was 44 ± 3 ml min−1 (1.3 ± 0.1 ml min−1 g−1).
Maximal PP potentiation by low-dose adenosine
Figure 1 displays example coronary flow responses to enhanced perfusion pulsatility under basal conditions (Fig. 1A) and during co-administration of low-dose adenosine (Fig. 1B). At baseline, the higher PP (arrow) resulted in a modest increase in mean flow. For the group data, this averaged 11.5 ± 1.7%, ranging from 41.0 ± 4.1 to 45.5 ± 4.5 ml min−1 (P < 0.0001). However, when adenosine was co-infused to raise basal flow by +21%, the flow augmentation observed after initiating higher pulsatility was markedly amplified (Fig. 1B). In this example, mean flow rose to 136 ml min−1 after 15 s, and reached a plateau at a steady-state value of 92 ml min−1 (124% above baseline).
Figure 1.
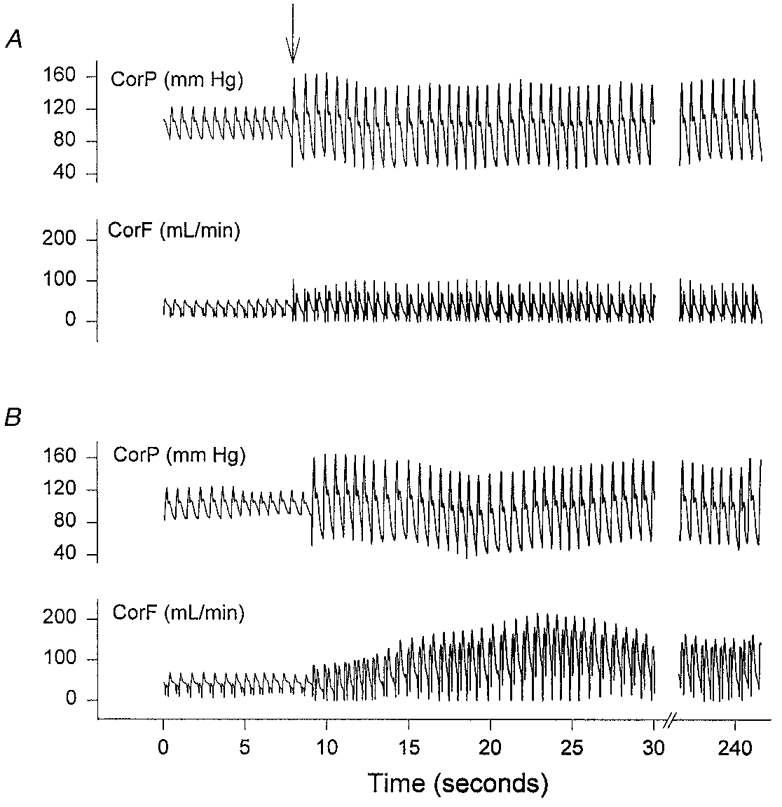
A, effect of elevating coronary perfusion pulse pressure (PP) on coronary flow under basal conditions. CorP, coronary pressure; CorF, coronary flow in the left anterior descending arterial bed. The sudden change to a higher PP (arrow) was accompanied by a modest but stable rise in mean flow, averaging 11%. B, synergy of adenosine and PP on coronary flow augmentation. Data are from the same heart as shown in A. Adenosine was first administered i.c. to raise baseline flow by 21% and then PP was increased similarly. In this case, there was a marked rise in coronary flow, peaking at 3.3 times baseline after 25 s, and reaching a plateau at a steady-state value 2.2 times that of the control.
The magnitude of adenosine amplification of flow pulsatility responses depended upon the dose. Figure 2A displays example adenosine-flow dose-response curves from one animal determined at PP = 40 and 100 mmHg, showing how higher pulsatility resulted in a leftward shift of the curve. Figure 2B provides the mean percentage flow change with increasing pulsatility plotted as a function of adenosine dose. Maximal augmentation of the pulsatility response (+82.6 ± 14.3%) was achieved with an adenosine administration of μ10 μg min−1. This dose by itself only raised basal flow by 66.7 ± 15%, well below the maximal adenosine response (typically 300-400%).
Figure 2.
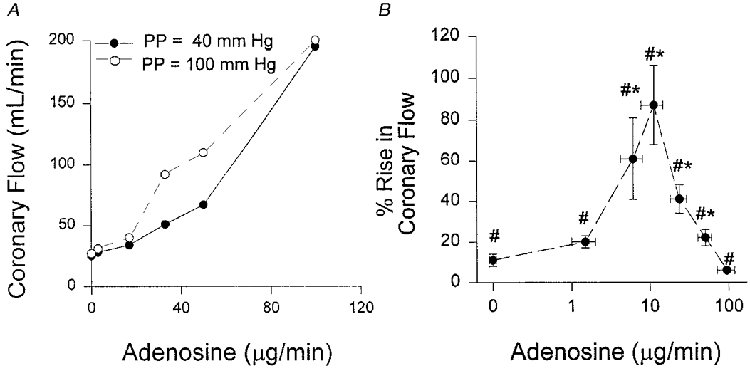
A, example of adenosine-coronary flow dose-response curve at both 40 and 100 mmHg pulse pressure from one animal. There is a leftward shift of the curve at higher PP. B, group data showing percentage increase in coronary flow due to a change in PP at different adenosine doses. Increasing PP resulted in a rise in mean coronary flow at all levels of adenosine infusion (# P < 0.05). Furthermore, at multiple doses, adenosine amplified the PP-flow response (* P < 0.05) to a maximum almost 90% above that of the control (see Methods for concentrations). This dose modestly elevated basal flow (+47%) at a resting PP of 40 mmHg.
Similar results were observed when adenosine catabolism was inhibited. Basal flow rose 2- to 3-fold, and subsequently increasing PP elevated mean flow by a further +22 ± 4%, similar to the 30 ± 3% rise obtained from equi-dilating doses of exogenously administered adenosine. Thus, it is likely that the adenosine-PP synergy did not derive from enhanced intravascular delivery of the nucleoside.
Verapamil, acetylcholine and pinacidil pulse perfusion interactions
Figure 3A (top panel) displays percentage changes in mean flow from various alternative dilators, all measured at a basal PP of 40 mmHg, with each agent administered to achieve a similar level of flow elevation (μ65%). The corresponding lower panel displays absolute mean flow change after enhancing pulsatility. As noted, adenosine amplified the PP-flow response by nearly an order of magnitude. In contrast, verapamil co-administration did not alter this response from control values (+16.6 ± 3.3%). This lack of enhancement was not due to inhibition of NO-dependent signalling, since i.c. flow increases following an acetylcholine bolus were not significantly different before or after verapamil infusion (202 ± 61% before, 183 ± 58% after).
Figure 3.
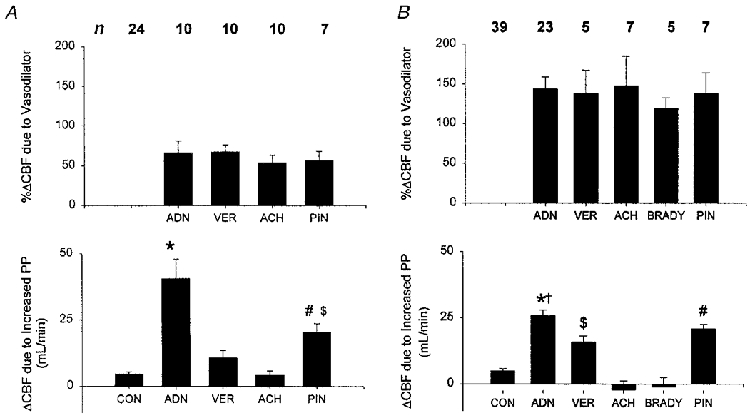
A (top), mean percentage change in coronary blood flow (CBF) induced by various vasodilators at a fixed 40 mmHg pulse pressure. ADN, adenosine; ACH, acetylcholine; VER, verapamil; PIN, pinacidil. The number of studies for each condition is shown at the top. Bottom, mean absolute change in CBF induced by raising perfusion pulsatility from 40 to 100 mmHg with and without concomitant vasodilatation. All conditions resulted in a significant rise in basal mean coronary flow at control levels of pulsatility (i.e. 40 mmHg). However, despite similar levels of basal dilatation, there were marked discrepancies in the response to augmenting pulsatility, with adenosine producing the largest interaction. * P < 0.005 vs. all other conditions; # P < 0.01 vs. CON; $ P < 0.05 vs. ACH. B, similar data presentation to A, but with a higher level of basal dilatation from each agent. BRADY, bradykinin. The magnitude of additional percentage flow elevation was smaller given the higher basal flow, but there remained marked disparities between dilators that were comparable to those observed at the lower dose. * P < 0.0001 vs. CON, BRADY, ACH; † P < 0.05 vs. VER; # P < 0.001 vs. ACH, BRADY, CON; $P = 0.01 vs. CON. With the exception of ACH and BRADY, all dilators were still associated with a significant increase in mean flow with enhanced pulsatility.
Results with sustained acetylcholine infusion were similar to verapamil. Mean flow increased 6.3 ± 2.3% with an augmented PP, similar to control values. This dose of acetylcholine produced < 33% of the maximal dose response, so the lack of synergy could not be ascribed to saturation of acetylcholine-dependent (i.e. NO, hyperpolarization) signalling.
In contrast to verapamil and acetylcholine, pinacidil significantly amplified coronary flow responses to higher perfusion pulsatility. Flow rose 33 ± 2.6% at the higher pulse pressure, more than with acetylcholine, but still less than half that with adenosine. This result supported a role for KATP channel activation in the interaction of adenosine and pulsatility.
Dose dependence of dilator-PP interaction
To confirm that disparities in dilator-pulsatility interaction were not unique to a particular dilator response, additional studies were performed at higher doses of each agent, sufficient to raise basal flow μ120% over baseline (Fig. 3B, upper panel). As shown in Fig. 3B (lower panel), adenosine still yielded the largest amplification, significantly greater than control, acetylcholine or verapamil. At higher doses of acetylcholine or bradykinin, mean flow did not significantly change with higher pulsatility, whereas the other dilators were still associated with a flow rise at higher PP. Pinacidil again augmented PP-flow responses, and this was similar to the adenosine response at the higher dose. Lastly, the higher verapamil dose also resulted in enhanced PP-flow responses above those of the control, suggesting some vascular tone-dependent interaction at this higher level of basal relaxation.
Quinacrine and adenosine-pulse perfusion interaction
The failure of other vasodilators to reproduce the synergy between perfusion pulsatility and adenosine suggested a dependence on more than increased basal flow. To further test this, quinacrine was infused to block adenosine-mediated vasodilatation and then the interaction between adenosine and pulsatility was examined. Quinacrine almost completely blocked basal flow elevation by adenosine (Fig. 4A), from a 49.8 ± 31% flow rise over baseline to 4.3 ± 13.6% (P = 0.04). By itself, quinacrine had little effect on the flow response to enhanced pulsatility over control values (Fig. 4B). However, with the addition of adenosine, there was again augmentation of the pulsatility response (P = 0.004) to 22 ± 8%, 2.5 times the control (+8.8 ± 6.2%).
Figure 4.
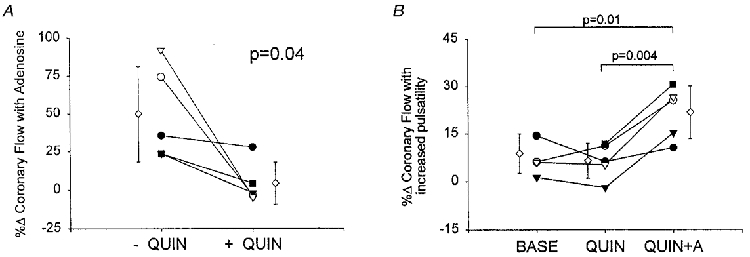
A, effect of quinacrine on adenosine-induced flow response. There was a marked decrease in coronary dilatation induced by adenosine with co-administration of quinacrine. B, percentage change in mean coronary flow with enhanced pulsatility under baseline (BASE), quinacrine alone (QUIN), and quinacrine with adenosine (QUIN+A). Despite a lack of basal flow elevation with concomitant quinacrine, adenosine still amplified the flow response to increased perfusion pulsatility. Different symbols identify individual experiments. ⋄ corresponds to mean ±s.e.m.
Regional myocardial oxygen consumption
Adenosine-mediated increases in vascular turgor can elevate regional myocardial oxygen consumption (MV̇O2) (Gregg phenomena; Gregg, 1963) and such change in metabolic demand might itself influence coronary flow. To test whether this mechanism played a role in the flow enhancement from higher pulsatility, we measured regional MV̇O2 under control conditions, and with adenosine, bradykinin or verapamil (Table 1), comparing values at 40 and 100 mmHg pulsatility. Verapamil slightly lowered MV̇O2(P < 0.05), consistent with its negative inotropic effect, while adenosine tended to raise basal MV̇O2, consistent with the Gregg phenomena. However, in none of the conditions was MV̇O2 further altered by varying PP. Thus, flow- pulsatility responses could not be ascribed to altered energetic demand.
Table 1.
Regional myocardial arterial-venous oxygen difference and myocardial oxygen consumption at normal and increased perfusion pulse pressure
| PP (mmHg) | [Hb] (mg dl−1) | A–VO2(ml O2 ml−1) | MV̇O2 (ml O2 min−1) | |
|---|---|---|---|---|
| Control (n = 16) | 40 | 11.5 ± 2.1 | 6.1 ± 0.5 | 7.6 ± 0.5 |
| 100 | 11.6 ± 1.9 | 5.8 ± 0.6 | 7.6 ± 0.7 | |
| Adenosine (n = 7) | 40: pre-drug | 12.4 ± 1.2 | 6.4 ± 0.7 | 8.1 ± 1.0 |
| 40: post-drug | 11.9 ± 1.1 | 3.0 ± 0.5† | 10.3 ± 1.8* | |
| 100: post-drug | 11.3 ± 1.0 | 2.7 ± 0.4 | 10.45 ± 1.7 | |
| Bradykinin (n = 6) | 40: pre-drug | 12.1 ± 0.9 | 6.3 ± 0.5 | 7.5 ± 0.5 |
| 40: post-drug | 11.3 ± 1.1 | 4.1 ± 0.7† | 9.9 ± 1.7 | |
| 100: post-drug | 11.4 ± 1.1 | 4.1 ± 0.55 | 9.8 ± 1.7 | |
| Verapamil (n = 5) | 40: pre-drug | 11.6 ± 1.1 | 6.1 ± 0.5 | 8.0 ± 0.7 |
| 40: post-drug | 11.2 ± 1.2 | 2.1 ± 0.3† | 5.5 ± 1.6† | |
| 100: post-drug | 11.4 ± 1.0 | 2.6 ± 1.0 | 5.7 ± 1.4 |
PP, coronary perfusion pulse pressure; [Hb], haemoglobin concentration; A-V O2, arterial-venous oxygen difference; MV̇O2, myocardial oxygen consumption.
P = 0.06
P < 0.05 vs. pre-drug. Data are provided for the control condition and with adenosine, bradykinin or verapamil infusion. There was no significant change in regional oxygen consumption with increased pulse pressure under any of the conditions.
Systolic versus diastolic flow alteration
To further dissect the interactions between pharmacological dilators and the pulsatility of coronary flow, phasic waveforms were analysed to calculate the systolic and diastolic flow components. The mean systolic flow was determined from integrated flow waveforms spanning from the aortic pressure upstroke to the dicrotic notch. Increasing pulsatility raised the systolic driving pressure while lowering diastolic pressure. While prior studies have shown this enhances systolic flow (Saeki et al. 1995; Recchia et al. 1996), it could critically depend upon the influence of a given dilator on regional resistances and compliances. Figure 5A shows example waveforms under each condition and Fig. 5B provides group data. Systolic flow rose 50-60% at higher PPs in all conditions except with adenosine, where it rose more than twice as much (+138 ± 16%, P < 0.0001 vs. each other condition).
Figure 5.
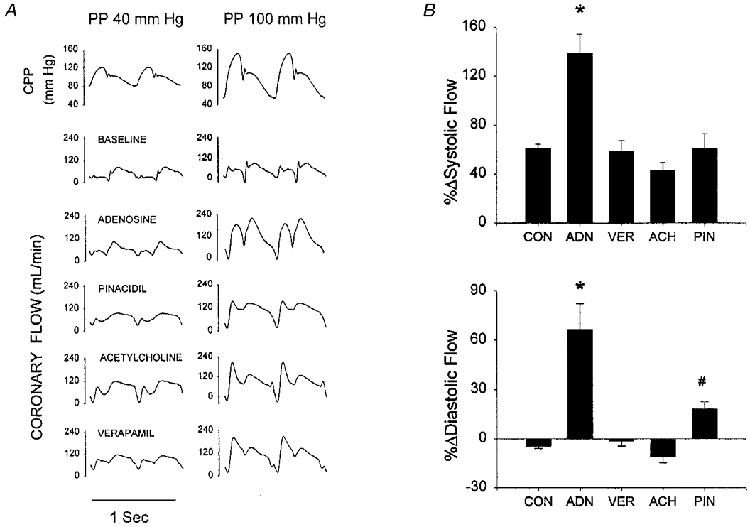
A, phasic coronary flow waveforms for 40 and 100 mmHg PP data under conditions of pre-vasodilatation with different agents. In all instances, coronary flow was predominantly diastolic at the lower PP, with phasic patterns that were generally similar. All flow waveforms displayed a marked rise in systolic flow at the higher PP. However, only adenosine and pinacidil yielded increases in diastolic flow as well, whereas mean diastolic flow declined with acetylcholine and was unchanged with verapamil. Mean diastolic pressure declined slightly from 92 to 84 mmHg at the higher PP. B, mean changes in systolic (top) and diastolic (bottom) coronary flow associated with elevation of perfusion pulsatility. All individual changes for each condition were significant (P < 0.05) with the exception of diastolic flow with verapamil. Results of multiple comparisons testing between conditions are shown by symbols: * P < 0.0001 vs. all other conditions; #P = 0.03 vs. ACH, CON.
Even greater discrepancies were observed in diastolic flow. Diastolic flow was modestly reduced (−4.5 ± 1.5% in controls, −11.1 ± 3.5% with ACh, both P < 0.05) and unchanged with verapamil at higher PPs. However, flow increased 67 ± 16% with adenosine, far more than with the other dilators. Pinacidil yielded a modest 18% increase, higher than control or acetylcholine (P = 0.03), but still one-third that from adenosine (P < 0.0001). Since diastolic pressures were identical in all cases, this difference in flow reflects a greater fall in net resistance from the adenosine- PP interaction.
DISCUSSION
The major novel finding of this study is that a combination of low dose adenosine and enhanced perfusion pulsatility markedly augments mean coronary flow in a manner largely specific to the modes of action of adenosine. While this may in part be due to KATP activation, as pinacidil also augmented flow elevation with higher pulsatility, quantitative discrepancies between adenosine and pinacidil responses suggested that additional mechanisms were involved. This was probably not due to increased basal shear, as neither acetylcholine or verapamil duplicated the adenosine-PP interaction, and inhibition of adenosine-induced dilatation by quinacrine still yielded amplified flow responses at higher pulsatility. A potentiating role of low levels of adenosine on coronary flow when pulsatility is simultaneously increased is intriguing, as it suggests a plausible and novel mechanism for flow modulation during exercise or other high output states, when both stimuli are present.
Pulse pressure modulation of coronary flow
Increasing perfusion pulsatility exposes the coronary vascular bed to both increased phasic shear and cyclic distension. Both stimuli individually increase NO release and nitric oxide synthase (NOS) gene expression in vitro (Kuo et al. 1991; Awolesi et al. 1995). There are fewer studies of this behaviour in vivo, but data do support a role for NO release, as coronary flow elevation from increased pulsatility is partially inhibited by L-NMMA (Recchia et al. 1996). Recent studies have also demonstrated that pulsatile perfusion can elicit NOS-independent relaxation via smooth muscle hyperpolarization (Popp et al. 1998; Busse & Fleming, 1998). This response may be due to arachidonic acid metabolites linked to cytochrome P450 oxidase (Hecker et al. 1994; Campbell et al. 1996) or, as suggested by others, to an endogenous cannabinoid (Randall & Kendall, 1997) or K+ that effluxes via charybdotoxin-apamin-sensitive endothelial K+ channels (Edwards et al. 1998).
For any given PP, cyclic stretch signalling should be enhanced if the tone of vessels exposed to pulsatility declines. Some evidence supporting this interaction was observed with verapamil, which at low doses had no synergistic effect, but at higher doses enhanced PP responses. It is intriguing that both low- and high-dose acetylcholine (and high-dose bradykinin) failed to augment PP-flow responses. However, acetylcholine and bradykinin induce vasorelaxation via NO release (Smith & Canty, 1993; Kuo et al. 1995; Kuga et al. 1997) and hyperpolarization (Hayabuchi et al. 1998), perhaps triggering the same pathways central to PP-flow signalling and thus blunting synergistic interaction.
Mechanisms of potentiation of PP response
Adenosine is thought to primarily relax coronary arteries by binding to A2 receptors, resulting in increased cAMP and PKA (Kusachi et al. 1983; White & Angus, 1987), which is in turn linked to a non-voltage-dependent K+ current and intracellular hyperpolarization (Quayle & Standen, 1994; Daut et al. 1994; Kleppisch & Nelson, 1995). The latter is blocked by glibenclamide, implicating KATP channel activation (Dart & Standen, 1993). Adenosine also acts on A1 receptors, which couple to an array of intracellular signalling pathways (Ralevic & Burnstock, 1998) and contribute to vasodilatation (Merkel et al. 1992). Intracellular hyperpolarization enhances Ca2+ entry, which could contribute to NOS activation, thereby further enhancing a vasodilator response (Illiano et al. 1992; Fukao et al. 1997). The extent to which adenosine dilatation is NO dependent remains controversial, and may be species dependent, with greater interactions in porcine vessels. In canine coronary arteries, we have not observed significant inhibition of adenosine dilatation by NOS inhibition with L-NMMA at basal pulsatility (Recchia et al. 1996). However, L-NMMA inhibited coronary flow at higher pulsatility, indicating that adenosine-NO interaction may require the synergistic effect of cyclic distension in this setting.
Kuo & Chancellor (1995) first suggested an interaction between adenosine and NO-dependent vasodilatation in a study of isolated porcine coronary epicardial vessels (50-150 μm diameter). In their study, adenosine potentiated laminar flow-induced dilatation by stimulating further NO release, and this effect was blocked by glibenclamide but not indomethacin, supporting a role for KATP channel activation. While pinacidil amplified the pulsatile perfusion effect on mean flow in the present study, the response was still less than with an equi-dilating dose of adenosine. Thus, an action other than KATP stimulation appears to contribute. The pinacidil data also suggest that the synergy was not solely related to a specific vascular site of action, as the actions of both adenosine (Kanatsuka et al. 1989) and KATP agonist (Sato et al. 1994) are principally localized to distal (< 150 μm) arterioles.
Our hypothesis of flow-independent synergistic action of adenosine and perfusion pulsatility was further corroborated by data obtained with the PLA2 inhibitor quinacrine. Quinacrine inhibits Ca2+-dependent K+ channels (Denson et al. 1996; Hayabuchi et al. 1998) and glibenclamide-sensitive K+ channels (Sakuta & Yoneda, 1994), and markedly inhibits coronary flow augmentation from adenosine (Kimura & Satoh, 1985). The fact that moderate adenosine-PP synergy persisted despite quinacrine suggests additional mechanisms. There is no evidence that quinacrine directly inhibits agonist binding to A2 or A1 receptors, so receptor-coupled responses that are primarily independent of PLA2 could still contribute. For example, A1 receptors couple through PKC and PLC pathways (Ralevic & Burnstock, 1998), resulting in increased intracellular Ca2+ and Ca2+-dependent hyperpolarization and/or NOS activation. Further studies are needed using selective agonists and inhibitors of adenosine receptor subtypes to clarify their respective signal transduction roles. Another factor may be stretch-activated Ca2+ channels (Busse & Fleming, 1998). It remains difficult to determine the in vivo role of cytochrome P450-dependent arachidonic acid metabolites, since the majority of inhibitors are complex agents that do not solely or specifically block their synthesis. For example, quinacrine also blocks glibenclamide-sensitive K+ currents by a mechanism independent of PLA2 or cytochrome P450 (Sakuta & Yoneda, 1994).
Systolic versus diastolic flow alterations
Increased systolic pressure with a concomitant fall in mean diastolic pressure is a hallmark of increased pulsatility. While this has been considered a mechanism for compromising coronary perfusion, we have previously shown (Saeki et al. 1995; Recchia et al. 1996) and confirm in the present study that pulsatility elevates systolic flow while maintaining diastolic flow, thereby augmenting mean flow. The rise in systolic flow probably reflects capacitative effects related to enhanced stretch of arterial vessels (Goto et al. 1996). Interestingly, the magnitude of systolic flow rise at the higher PP with verapamil, acetylcholine or pinacidil co-infusion was very similar, suggesting a dependence on the mechanical stimulus itself rather than on the specific mechanisms by which vascular tone is reduced. However, adenosine plus enhanced pulsatility led to a much larger rise in systolic flow, indicating either a greater rise in capacitance, or facilitation of flow despite myocardial contraction. During diastole, this same combination uniquely elevated mean flow despite the decline in mean pressure consistent with more pronounced interactive effects of both stimuli on lowering coronary resistance. Simultaneous direct measurements of microvascular distension (Kuo et al. 1995) combined with varying PP and adenosine stimulation may further clarify the mechanisms for these interactions
Limitations
A limitation of our in vivo preparation is that signals specifically related to phasic shear cannot be separated from those associated with cyclic distension of the arteries. As it proved difficult to establish constant flow in the in vivo coronary bed while varying pulsatility, our conclusions about the role of shear per se were based on comparisons with other dilator agents at matched levels of mean flow. The limitation is that each agent does not exactly produce an identical anatomic distribution of dilatation within the vascular bed. Even so, the results with quinacrine further support mechanisms other than basal flow itself. In vitro studies provide more control over the mechanical stimuli, but may miss in vivo mechanisms linked to the particular distribution of capacitances and resistances and dilator sensitivities and interactions in the intact cardiac vasculature.
Physiological considerations
The present study may have potential implications regarding coronary flow regulation during exercise or high output states, and in individuals with widened pulse perfusion, such as the elderly. Arterial PP rises with exercise, with central aortic PP increasing nearly 2-fold and with greater changes in femoral or brachial arteries (Rowell et al. 1968; Gerstenblith et al. 1987). Aged individuals display progressive increases in arterial pulse pressure at rest (Franklin et al. 1997) and even higher PPs during exercise (Rowell et al. 1968; Gerstenblith et al. 1987). Although there is controversy regarding its precise role in physiological flow regulation (Van Bibber et al. 1997; Yada et al. 1999), adenosine may rise with both dobutamine ( van Wylen et al. 1990) and exercise (Bacchus et al. 1982; McKenzie et al. 1982). The present study shows that even modest levels of adenosine agonist activity can be far more effective in augmenting flow if pulsatility is simultaneously increased. It is intriguing to speculate that agents such as dipyridamole, which inhibits adenosine uptake, might be administrable at a dose that improved exercise perfusion in patients with wide pulse pressures far more than would be observed at rest. Further studies are needed to test this hypothesis.
Acknowledgments
This work was supported by an NIH grant HL-47511 (D.A.K.), an American Heart Association Fellowship Grant (F.A.R.) and by institutional support from the Università di Torino (P.P.), the Università di Perugia (N.P.) and the University of Tokyo (T.I.). The authors also gratefully acknowledge the expert technical assistance of Richard Tunin.
References
- Akatsuka Y, Egashira K, Katsuda Y. ATP sensitive potassium channels are involved in adenosine A2 receptor mediated coronary vasodilation in the dog. Cardiovascular Research. 1994;28:906–911. doi: 10.1093/cvr/28.6.906. [DOI] [PubMed] [Google Scholar]
- Awolesi MA, Sessa WC, Sumpio BE. Cyclic strain upregulates nitric oxide synthase in cultured bovine aortic endothelial cells. Journal of Clinical Investigation. 1995;96:1449–1454. doi: 10.1172/JCI118181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacchus AN, Wly SW, Knabb RM, Rubio R, Berne RM. Adenosine and coronary blood flow in conscious dogs during normal physiological stimuli. American Journal of Physiology. 1982;243:H628–633. doi: 10.1152/ajpheart.1982.243.4.H628. [DOI] [PubMed] [Google Scholar]
- Busse R, Fleming I. Pulsatile stretch and shear stress: physical stimuli determining the production of endothelium-derived relaxing factors. Journal of Vascular Research. 1998;35:73–84. doi: 10.1159/000025568. [DOI] [PubMed] [Google Scholar]
- Campbell WB, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatreinoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:699–709. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Dart C, Standen NB. Adenosine-activated potassium current in smooth muscle cells isolated from the pig coronary artery. The Journal of Physiology. 1993;471:767–786. doi: 10.1113/jphysiol.1993.sp019927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daut J, Standen NB, Nelson MT. The role of the membrane potential of endothelial and smooth muscle cells in the regulation of coronary blood flow. Journal of Cardiovascular Electrophysiology. 1994;5:154–181. doi: 10.1111/j.1540-8167.1994.tb01156.x. [DOI] [PubMed] [Google Scholar]
- Denson DD, Worrell RT, Eaton DC. A possible role for phospholipase A2 in the action of general anesthetics. American Journal of Physiology. 1996;270:C636–644. doi: 10.1152/ajpcell.1996.270.2.C636. [DOI] [PubMed] [Google Scholar]
- Edwards G, Dora KA, Gardner MJ, Garland CJ, Weston AH. K+ is an endothelium-derived hyperpolarizing factor in rat arteries. Nature. 1998;396:269–272. doi: 10.1038/24388. [DOI] [PubMed] [Google Scholar]
- Ely SW, Matherne GP, Coleman SD, Berne RM. Inhibition of adenosine metabolism increases myocardial interstitial adenosine concentrations and coronary flow. Journal of Molecular and Cellular Cardiology. 1992;24:1321–1332. doi: 10.1016/0022-2828(92)93097-4. [DOI] [PubMed] [Google Scholar]
- Franklin SS, Gustin W, Wong ND, Larson MG, Weber MA, Kannel WB, Levy D. Hemodynamic patterns of age-related changes in blood pressure. Circulation. 1997;96:308–315. doi: 10.1161/01.cir.96.1.308. [DOI] [PubMed] [Google Scholar]
- Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Sources of Ca2+ in relation to generation of acetylcholine-induced endothelium-dependent hyperpolarization in rat mesenteric artery. British Journal of Pharmacology. 1997;120:1328–1334. doi: 10.1038/sj.bjp.0701027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstenblith G, Renlund DG, Lakatta EG. Cardiovascular response to exercise in younger and older men. Federation Proceedings. 1987;46:1834–1839. [PubMed] [Google Scholar]
- Goto M, Vanbavel E, Giezeman MJMM, Spaan JAE. Vasodilator effect of pulsatile pressure on coronary resistance vessels. Circulation Research. 1996;79:1039–1045. doi: 10.1161/01.res.79.5.1039. [DOI] [PubMed] [Google Scholar]
- Gregg DE. Effect of coronary perfusion pressure or coronary flow on oxygen usage of the myocardium. Circulation Research. 1963;13:497–500. doi: 10.1161/01.res.13.6.497. [DOI] [PubMed] [Google Scholar]
- Hartley SA, Kozlowski RZ. ATP increases Ca(2+)-activated K+ channel activity in isolated rat arterial smooth muscle cells. Biochimica et Biophysica Acta. 1996;1283:192–198. doi: 10.1016/0005-2736(96)00094-6. [DOI] [PubMed] [Google Scholar]
- Hayabuchi Y, Nakaya Y, Matsuoka S, Kuroda Y. Endothelium-derived hyperpolarizing factor activates Ca2+-activated K+ channels in porcine coronary artery smooth muscle cells. Journal of Cardiovascular Pharmacology. 1998;32:642–649. doi: 10.1097/00005344-199810000-00018. [DOI] [PubMed] [Google Scholar]
- Hecker M, Bara AT, Bauersachs J, Busse R. Characterization of endothelium-derived hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. The Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Illiano S, Nagao T, Vanhoutte PM. Calmidazolium, a calmodulin inhibitor, inhibits endothelium-dependent relaxations resistant to nitro-L-arginine in the canine coronary artery. British Journal of Pharmacology. 1992;107:387–392. doi: 10.1111/j.1476-5381.1992.tb12756.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanatsuka H, Lamping KG, Eastham CL, Dellsperger KC, Marcus ML. Comparison of the effects of increased myocardial oxygen consumption and adenosine on the coronary microvascular resistance. Circulation Research. 1989;65:1296–1305. doi: 10.1161/01.res.65.5.1296. [DOI] [PubMed] [Google Scholar]
- Kimura T, Satoh S. Inhibitory effect of quinacrine on myocardial reactive hyperemia in the dog. Journal of Pharmacology and Experimental Therapeutics. 1985;232:269–274. [PubMed] [Google Scholar]
- Kleppisch T, Nelson MT. Adenosine activates ATP-sensitive potassium channels in arterial myocytes via A2 receptors and cAMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1995;92:12441–12445. doi: 10.1073/pnas.92.26.12441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuga T, Mohri M, Egashira K, Hirakawa Y, Tagawa T, Shimokawa H, Takeshita A. Bradykinin-induced vasodilation of human coronary arteries in vivo: role of nitric oxide and angiotensin-converting enzyme. Journal of the American College of Cardiology. 1997;30:108–112. doi: 10.1016/s0735-1097(97)00112-5. [DOI] [PubMed] [Google Scholar]
- Kung CF, Tschudi MR, Noll G, Clozel JP, Luscher TF. Differential effects of the calcium antagonist mibefradil in epicardial and intramyocardial coronary arteries. Journal of Cardiovascular Pharmacology. 1995;26:312–318. doi: 10.1097/00005344-199508000-00018. [DOI] [PubMed] [Google Scholar]
- Kuo L, Chancellor JD. Adenosine potentiates flow-induced dilation of coronary arterioles by activating KATP channels in endothelium. American Journal of Physiology. 1995;269:H541–549. doi: 10.1152/ajpheart.1995.269.2.H541. [DOI] [PubMed] [Google Scholar]
- Kuo L, Chilian WM, Davis MJ. Interaction of pressure- and flow-induced responses in porcine coronary resistance vessels. American Journal of Physiology. 1991;261:H1706–1715. doi: 10.1152/ajpheart.1991.261.6.H1706. [DOI] [PubMed] [Google Scholar]
- Kuo L, Davis MJ, Chilian WM. Longitudinal gradient for endothelium-dependent and independent vascular responses in the coronary microcirculation. Circulation. 1995;92:518–525. doi: 10.1161/01.cir.92.3.518. [DOI] [PubMed] [Google Scholar]
- Kusachi S, Thompson RD, Olsson RA. Ligand selectivity of dog coronary adenosine receptor resembles that of adenylate cyclase stimulatory (Ra) receptors. Journal of Pharmacology and Experimental Therapeutics. 1983;227:316–321. [PubMed] [Google Scholar]
- McKenzie JE, Steffen RP, Haddy FJ. Relationship between adenosine and coronary resistance in conscious exercising dogs. American Journal of Physiology. 1982;242:H24–29. doi: 10.1152/ajpheart.1982.242.1.H24. [DOI] [PubMed] [Google Scholar]
- Merkel LA, Lappe RW, Rivera LM, Cox BF, Perrone MH. Demonstration of vasorelaxant activity with an A1-selective adenosine agonist in porcine coronary artery: involvement of potassium channels. Journal of Pharmacology and Experimental Therapeutics. 1992;260:437–443. [PubMed] [Google Scholar]
- Popp R, Fleming I, Busse R. Pulsatile stretch in coronary arteries elicits release of endothelium-derived hyperpolarizing factor. Circulation Research. 1998;82:696–703. doi: 10.1161/01.res.82.6.696. [DOI] [PubMed] [Google Scholar]
- Quayle JM, Standen NB. KATP channels in vascular smooth muscle. Cardiovascular Research. 1994;28:797–804. doi: 10.1093/cvr/28.6.797. [DOI] [PubMed] [Google Scholar]
- Ralevic V, Burnstock G. Receptors for purines and pyrimidines. Pharmacology Reviews. 1998;50:413–492. [PubMed] [Google Scholar]
- Randall MD, Kendall DA. Involvement of a cannabinoid in endothelium-derived hyperpolarizing factor-mediated coronary vasorelaxation. European Journal of Pharmacology. 1997;335:205–209. doi: 10.1016/s0014-2999(97)01237-5. [DOI] [PubMed] [Google Scholar]
- Recchia FA, Senzaki H, Saeki A, Byrne BJ, Kass DA. Pulse pressure-related changes in coronary flow in vivo are modulated by nitric oxide and adenosine. Circulation Research. 1996;79:849–856. doi: 10.1161/01.res.79.4.849. [DOI] [PubMed] [Google Scholar]
- Rowell LB, Brengelmann GB, Blackmon JR, Bruce RA, Murray JA. Disparities between aortic and peripheral pulse pressure induced by upright exercise and vasomotor changes in man. Circulation. 1968;37:954–964. doi: 10.1161/01.cir.37.6.954. [DOI] [PubMed] [Google Scholar]
- Saeki A, Recchia F, Kass DA. Systolic flow augmentation in hearts ejecting into a model of stiff aging vasculature: influence on myocardial perfusion-demand balance. Circulation Research. 1995;76:132–141. doi: 10.1161/01.res.76.1.132. [DOI] [PubMed] [Google Scholar]
- Sakuta H, Yoneda I. Inhibition by SKF 525A and quinacrine of endogenous glibenclamide-sensitive K+ channels in follicle-enclosed Xenopus oocytes. European Journal of Pharmacology. 1994;252:117–121. doi: 10.1016/0014-2999(94)90583-5. [DOI] [PubMed] [Google Scholar]
- Sato K, Kanatsuka H, Sekiguchi N, Akai K, Want Y, Sugimura A, Kamagai T, Komaru T, Shirato K. Effect of an ATP sensitive potassium channel opener, levcromakalim, on coronary arterial microvessels in the beating canine heart. Cardiovascular Research. 1994;28:1780–1786. doi: 10.1093/cvr/28.12.1780. [DOI] [PubMed] [Google Scholar]
- Shinoda M, Toki Y, Murase K, Mokuno S, Okumura K, Ito T. Types of potassium channels involved in coronary reactive hyperemia depend on duration of preceding ischemia in rat hearts. Life Sciences. 1997;61:997–1007. doi: 10.1016/s0024-3205(97)00604-8. [DOI] [PubMed] [Google Scholar]
- Smith TP, Canty JMJ. Modulation of coronary autoregulatory responses by nitric oxide. Evidence for flow-dependent resistance adjustments in conscious dogs. Circulation Research. 1993;73:232–240. doi: 10.1161/01.res.73.2.232. [DOI] [PubMed] [Google Scholar]
- Stepp DW, van Bibber R, Kroll K, Feigl EO. Quantitative relation between interstitial adenosine concentration and coronary blood flow. Circulation Research. 1996;79:601–610. doi: 10.1161/01.res.79.3.601. [DOI] [PubMed] [Google Scholar]
- van Bibber R, Stepp DW, Kroll K, Feigl EO. Role of adenosine in norepinephrine-induced coronary vasodilation. American Journal of Physiology. 1997;273:H557–565. doi: 10.1152/ajpheart.1997.273.2.H557. [DOI] [PubMed] [Google Scholar]
- van Wylen DGL, Willis J, Sodhi J, Weiss RJ, Lasley RD, Mentzer RM. Cardiac microdialysis to estimate interstitial adenosine and coronary blood flow. American Journal of Physiology. 1990;258:H1642–1649. doi: 10.1152/ajpheart.1990.258.6.H1642. [DOI] [PubMed] [Google Scholar]
- White TD, Angus JA. Relaxant effects of ATP and adenosine on canine large and small coronary arteries in vitro. European Journal of Pharmacology. 1987;143:119–126. doi: 10.1016/0014-2999(87)90741-2. [DOI] [PubMed] [Google Scholar]
- Yada T, Richmond KN, van Bibber R, Kroll K, Feigl EO. Role of adenosine in local metabolic coronary vasodilation. American Journal of Physiology. 1999;276:H1425–1433. doi: 10.1152/ajpheart.1999.276.5.H1425. [DOI] [PubMed] [Google Scholar]