

Abstract
Neurotransmitter release relies on a series of synaptic vesicle trafficking reactions. We have determined the molecular basis of these reactions by microinjecting, into ‘giant’ nerve terminals of squid, probes that interfere with presynaptic proteins. These probes affect neurotransmitter release and disrupt nerve terminal structure. From the nature of these lesions, it is possible to deduce the roles of individual proteins in specific vesicle trafficking reactions. This approach has revealed the function of more than a dozen presynaptic proteins and we hypothesize that neurotransmitter release requires the coordinated action of perhaps 50–100 proteins.
The classic work of Bernard Katz and colleagues, most of which was published in The The Journal of Physiology, established that quanta of neurotransmitters are released from presynaptic terminals (Katz, 1969). Further, their work showed that electrical excitation of the presynaptic terminal triggers this quantal neurotransmitter release by opening voltage-gated calcium channels. The localized elevation of calcium concentration resulting from calcium channel opening (Augustine et al. 1987) leads to the exocytotic discharge of the neurotransmitters stored within synaptic vesicles (Heuser et al. 1979). Following exocytosis, vesicular components are retrieved and recycled into new vesicles (Heuser & Reese, 1973). Thus, neurotransmitter release can be viewed as a local cycle of presynaptic vesicle trafficking with at least one reaction - the fusion of synaptic vesicles - that is greatly accelerated by calcium ions.
Currently much effort is focused on understanding the molecular basis of these synaptic vesicle trafficking reactions. While many presynaptic proteins have been identified (Südhof, 1995), their functions are largely unclear (Augustine et al. 1996). Given that synaptic vesicle trafficking involves the coordinated activity of many proteins in reactions that are regulated in both space and time, understanding this process ultimately requires study of molecular events in intact presynaptic terminals. Here we briefly summarize our efforts to meet this challenge through the use of the squid giant synapse. The large dimensions of the presynaptic terminal of this synapse make it possible to microinject molecular probes that perturb the function of individual proteins and then to evaluate the consequences of these molecular manipulations upon neurotransmitter release (Burns & Augustine, 1999). The many thousands of active zones in these terminals also allow us to use electron microscopy to identify the specific trafficking reactions that are affected by protein perturbations. In this way, we have been able to map the function of many presynaptic proteins onto defined steps of the synaptic vesicle trafficking cycle, and this review provides a general overview of these studies.
The synaptic vesicle recycling pathway
The logical underpinnings of our analysis come from studies of sequential reaction cascades. In such cascades, blockade of one reaction produces a depletion of downstream products and an accumulation of upstream precursors. By analogy, perturbation of a presynaptic protein should block the reaction in which this protein participates, causing a loss of downstream trafficking intermediates and a parallel accumulation of upstream reactants. Thus, defining the trafficking step that is blocked by disruption of a given protein should elucidate the reaction in which this protein participates. In the case of neurotransmitter release, a set of vesicle trafficking reactions has been defined, so that the goal is to empirically assign individual proteins and protein- protein interactions to specific trafficking steps. Our studies are interpreted via a modest revision (Fig. 1) of the vesicle trafficking model proposed by Heuser & Reese (1973). While there is still some debate regarding the role of endosomes in synaptic vesicle trafficking (Takei et al. 1996; Shi et al. 1998; Murthy & Stevens, 1998), the details of this particular reaction have little impact on interpreting the results presented here. We will use this model to summarize how microinjected reagents change presynaptic structure and function and how such results yield our current view of the molecular basis of synaptic vesicle trafficking.
Figure 1. Membrane trafficking reactions of the presynaptic terminal.
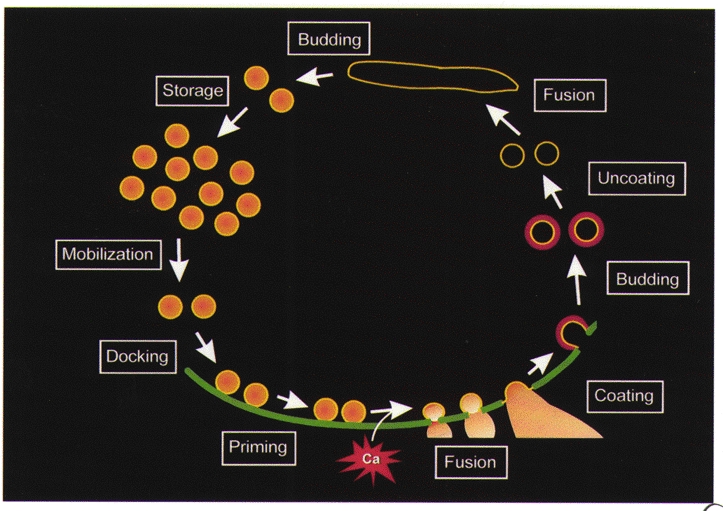
Storage of synaptic vesicles in a reserve pool
Synaptic vesicles within the interior of the presynaptic terminal are too far away from the plasma membrane to participate immediately in neurotransmitter release during an action potential. It has been proposed that these vesicles instead constitute a ‘reserve pool’ that is mobilized following the action potential to replenish vesicles that have undergone fusion (Pieribone et al. 1995). Perturbation of a protein involved in producing or maintaining this reserve pool should gradually inhibit neurotransmitter release following synaptic activity, enhance synaptic depression, and diminish the number of synaptic vesicles in the interior of the terminal. We have used these predictions to test the hypothesis that synapsins participate in keeping synaptic vesicles in this reserve pool. Synapsins are a family of proteins that are peripherally associated with the synaptic vesicle membrane (Greengard et al. 1993). All members of this family possess several conserved domains, while other domains are variable and/or subject to control by alternative splicing (Südhof et al. 1989). We have studied synapsin by concentrating on domain E, a part of the molecule that is found in synapsins from all species yet is absent in certain splice variants (type b) of vertebrate synapsins as well as invertebrate isoforms (Hilfiker et al. 1999b; Kao et al. 1999).
To study the function of synapsin domain E, we synthesized peptides that mimic part of domain E of the squid synapsin homologue (Hilfiker et al. 1998). This peptide should act as a competitive inhibitor of the interaction between synapsin and any proteins that bind to it via domain E, so that injecting this peptide into the squid giant nerve terminal will impair any function that relies on these interactions. Indeed, microinjection of this peptide potently inhibits neurotransmitter release (Fig. 2A). Two lines of evidence indicate that these peptides work by reducing the reserve pool of synaptic vesicles. First, peptide injection enhances the rate and extent of synaptic depression elicited by high-frequency trains of presynaptic action potentials (Fig. 2B). Because depression is thought to reflect the balance between depletion of the releasable vesicle pool and mobilization from the reserve pool, the fact that domain E peptide enhances depression while reducing transmitter release indicates that it attenuates the supply of vesicles from the reserve pool. Second, electron microscopy reveals that domain E peptide injection reduces the number of synaptic vesicles within the nerve terminal (Fig. 2C). To quantify these structural effects, we measured the spatial distribution of synaptic vesicles. The number of vesicles immediately opposite the plasma membrane is the same both in terminals injected with an inert control peptide and those injected with the domain E peptide, but the domain E peptide reduces the number of vesicles at greater distances into the interior of the terminal (Fig. 2D). This position-dependent reduction in synaptic vesicles was characterized by calculating the ratio of vesicles in each spatial compartment of terminals injected with domain E peptide compared with controls (Fig. 2E). This analysis makes it clear that the peptide causes the selective loss of vesicles in the interior of the synapse, those that are thought to constitute the reserve pool (Pieribone et al. 1995). It is not yet known where these lost vesicles end up. Measurements of the nerve terminal perimeter indicate that the vesicular membranes are not trapped in the presynaptic plasma membrane (S. Hilfiker & G. J. Augustine, unpublished observations); instead, it is likely that these vesicles move away from the synapse because they are no longer tethered to each other and/or to other presynaptic structures, as has been proposed at lamprey synapses (Pieribone et al. 1995).
Figure 2. Tests of the presynaptic function of synapsin.

A, measurements of presynaptic (Vpre) and postsynaptic (Vpost) responses before (control), during (pepE) and after (recovery) microinjection of a peptide from domain E of squid synapsin. B, injection of peptide from domain E of rat synapsin I enhances synaptic depression, measured as the decline in the rate of rise of postsynaptic responses (EPSP) evoked by a 50 Hz train of presynaptic action potentials. EPSP slope was normalized to values measured for the first response in each train. C, electron micrographs of presynaptic terminals injected with an inert control peptide (left) or a peptide from domain E of rat synapsin I (pepE; right). D, spatial distribution of synaptic vesicles measured in 50 nm shells surrounding active zones of terminals following injection of peptide from domain E of rat synapsin I or a control peptide. E, relative spatial distribution of synaptic vesicles in terminals injected with peptide from domain E of rat synapsin I, determined from the data shown in D by dividing values for domain E-injected terminals by values measured in control terminals. Modified from Hilfiker et al. (1998).
Taken together, these results are consistent with the hypothesis that synapsin is involved in maintaining synaptic vesicles in the reserve pool and suggest that the binding of unidentified proteins to domain E plays a central role in this reaction. Because domain E peptides also prevent docked synaptic vesicles from fusing, synapsin may participate in additional reactions that are more directly involved in vesicle fusion.
Vesicle mobilization and docking
Although our work has not addressed vesicle mobilization, it has been proposed that activity-dependent mobilization of vesicles from the reserve pool is controlled by phosphorylation of synapsin by calcium-regulated kinases such as the type II calcium-calmodulin-dependent protein kinase (Llinás et al. 1991; Greengard et al. 1993). After these vesicles are mobilized, they attach to the presynaptic plasma membrane during the process of docking. Inhibiting the function of proteins needed for vesicle docking reactions should inhibit neurotransmitter release by preventing vesicles from attaching to the plasma membrane. The only treatment that we have found to produce such changes is GDPβS, a non-hydrolyzable analogue of GDP. When injected into the presynaptic terminal, GDPβS slowly inhibits neurotransmitter release and causes a selective loss of vesicles attached to the plasma membrane (P. A. Doroshenko, M. E. Burns & G. J. Augustine, unpublished observations). Conversely, injecting the GTP analogue, GTPγS, enhances the relative number of docked synaptic vesicles (Hess et al. 1993). These results suggest that a GTP-binding protein must be in its GTP-bound conformation in order for synaptic vesicles to dock. This is consistent with observations in yeast that an early and reversible stage of vacuole docking, termed tethering, is mediated by Ypt7, a small molecular weight GTP-binding protein (Ungermann et al. 1998).
While the identity of the GTP-binding protein involved in synaptic vesicle docking is presently unknown, there are suggestions that it may be rab3A, an abundant vesicular GTP-binding protein that has been postulated to serve as a negative regulator of neurotransmitter release (Geppert et al. 1998). Injection of Mss4, a protein that stimulates several rabs - including rab3A - to exchange GDP for GTP enhances neurotransmitter release (Burton et al. 1994). A peptide from rabphilin-3A, a protein that binds to the vesicular GTP-binding protein rab3A, causes a relative accumulation of vesicles that are 50-100 nm away from the plasma membrane and may be awaiting docking (Burns et al. 1998). Thus, rab3A or some other GTP-binding protein must be bound to GTP to permit vesicles to dock.
Vesicle priming
In endocrine cells, docked vesicles (and perhaps the adjacent plasma membrane) must undergo ATP-dependent priming reactions before exocytosis can occur (Holz et al. 1989; Parsons et al. 1995; Martin et al. 1995). At least two types of ATP-dependent reactions occur during priming. First, phosphate is transferred from ATP to vesicle lipids, by a lipid kinase, to produce phosphatidylinositol 4,5-bisphosphate (Martin et al. 1995). Second, the ATPase NSF (N-ethylmaleimide-sensitive fusion protein) causes dissociation of complexed SNARE proteins, perhaps as a precursor to forming other SNARE complexes required for membrane fusion (Banerjee et al. 1996). We have used the squid giant synapse to ask whether the latter reaction occurs in presynaptic terminals. Perturbing vesicle priming should prevent docked vesicles from fusing and cause such unprimed vesicles to accumulate at the plasma membrane.
To determine whether preventing SNARE complex dissociation causes these changes, we have used several reagents that target either NSF or SNAP, the soluble NSF-attachment protein that links NSF to the SNARE proteins. To study the function of SNAP, we injected peptides from several regions of this protein (DeBello et al. 1995). Although the detailed mechanism of action of these peptides is not yet established, they prevent SNAP from supporting SNARE-mediated fusion of Golgi vesicles in vitro and presumably act by preventing SNAP from binding to other SNARE complex components. All SNAP peptides that block Golgi vesicle fusion also inhibit neurotransmitter release when microinjected into the squid presynaptic terminal, while those that are ineffective on Golgi fusion also have no effect on synaptic transmission. Conversely, microinjecting recombinant squid SNAP, or mammalian α-SNAP, causes a substantial enhancement of evoked neurotransmitter release. These results indicate that SNAP is essential for neurotransmitter release. In the case of NSF, we have injected two peptides that block the SNAP-stimulated ATPase activity of NSF, which is responsible for dissociating complexed SNARE proteins (Söllner et al. 1993). These peptides also inhibit neurotransmitter release when microinjected into the squid presynaptic terminal, while point-mutated peptides that do not prevent inhibition of the ATPase activity have no effect on release (Schweizer et al. 1998). This indicates that NSF and, more specifically, its SNAP-stimulated ATPase activity, is also important for neurotransmitter release.
Injecting either the SNAP or NSF peptides produces very similar changes in the structure of presynaptic terminals. Both peptides cause an accumulation of synaptic vesicles that are apparently docked at the plasma membrane yet cannot fuse. Thus, SNARE complex dissociation is apparently important for a step that follows vesicle docking yet precedes fusion. This is consistent with indications that these proteins must form a 20S complex that dissociates during priming of endocrine vesicles (Banerjee et al. 1996) and suggests that NSF and SNAP are involved in synaptic vesicle priming.
These results indicate that NSF, in concert with SNAP, participates in reactions that precede vesicle fusion. However, there have been suggestions that NSF works instead after fusion to prepare vesicular membrane for endocytosis (Littleton et al. 1998). Three observations indicate that this is not the primary site of action of these proteins in squid. First, injection of NSF peptides slows the kinetics of neurotransmitter release following single action potentials (Schweizer et al. 1998). While it is possible that interfering with endocytosis could also produce such kinetic effects, the simplest interpretation is that NSF acts on a priming step that precedes fusion. Second, if NSF and SNAP interfere with vesicle budding during endocytosis, then the surface area of the presynaptic terminal should increase. However, electron microscopic measurements indicate that injection of either NSF or SNAP peptides has no measurable effect on presynaptic membrane area (J. R. Morgan & G. J. Augustine, unpublished observations). Third, the speed of blockade by SNAP peptide is very rapid, with measurable inhibition of synaptic transmission occurring within 4 s (DeBello et al. 1995). Because the delay between endocytosis and subsequent refusion of retrieved vesicles is much longer - minimally 35 s (Ryan & Smith, 1995) - the peptide must block a reaction that immediately precedes fusion rather than a reaction involved in endocytosis.
Thus, in neurons it appears that NSF-mediated priming occurs after synaptic vesicles dock and serves to dissociate the membrane SNARE proteins. This runs counter to the original proposal that SNAREs work earlier to mediate membrane recognition and docking of vesicles (Rothman, 1994), and to more recent indications that NSF dissociates SNAREs prior to docking in yeast (Mayer et al. 1996). Thus far, we have microinjected nine different reagents that perturb SNARE proteins or complexes in a variety of ways. These treatments include clostridial toxins (Hunt et al. 1994; O'Connor et al. 1997) and several other reagents that prevent SNAREs from interacting (Burns, 1996; O'Connor et al. 1997; Dresbach et al. 1998; H. Tokumaru, L. L. Pelligrini, T. Ishizuka, K. Umayahara, H. Saisu, H. Betz, G. J. Augustine & T. Abe, unpublished observations). All of these treatments prevent neurotransmitter release yet none of them prevent synaptic vesicles from docking. Similar observations have been made independently in squid (Marsal et al. 1997) and in Drosophila (Broadie et al. 1995). We thus conclude that docking is SNARE independent but leads to NSF- and SNAP-dependent reactions that precede fusion. Presumably this is to dissociate membrane SNARE complexes in preparation for fusion. It is thought that the SNARE proteins are involved in the final fusion of the synaptic vesicle with the plasma membrane (Weber et al. 1998), although there are suggestions that SNAREs may not, in fact, be directly responsible for the fusion event (Ungermann et al. 1998; Coorssen et al. 1998). Further work will be needed to resolve the specific reactions that SNAREs participate in following their dissociation by NSF.
Vesicle fusion
After priming, calcium triggers exocytosis by participating in one or more reactions that catalyse vesicle fusion. Given that fusion occurs within a few hundred microseconds of calcium entering the nerve terminal (Llinás et al. 1981; Augustine et al. 1985) or of calcium accumulating within the presynaptic cytoplasm (Adler et al. 1991; Heidelberger et al. 1994), these reactions must be small in number and relatively rapid (Almers, 1994).
Although lipids may serve as a binding site for calcium (see Zimmerberg et al., this volume), it is more widely assumed that calcium acts by binding to a presynaptic protein. Among the many calcium-binding proteins found in neurons, the leading candidate for this presynaptic calcium receptor is synaptotagmin, a calcium-binding protein that is an integral part of the synaptic vesicle membrane (Südhof & Rizo, 1996). Many of the properties of synaptotagmin are in line with the expected properties of the presynaptic calcium receptor (DeBello et al. 1993). Thus, it is thought that synaptotagmin may be involved in transducing a presynaptic calcium rise into the signal for membrane fusion.
Disrupting the function of a protein involved in coupling calcium to membrane fusion should block fusion and cause primed synaptic vesicles to accumulate at the plasma membrane. These predictions have been tested by using a variety of experimental approaches to perturb synaptotagmin function (Broadie et al. 1994; Littleton et al. 1994; Geppert et al. 1994; Thomas & Elferink, 1998). Our approach was based on the use of binding site peptides to prevent the interaction of synaptotagmin with other presynaptic proteins. Most of this work relied on Pep20, a peptide that mimics a conserved motif within the C2B domain of synaptotagmin (Bommert et al. 1993). Recent biochemical studies suggest that this motif is important for a number of protein-protein interactions, including oligomerization of synaptotagmin, binding of the adaptor protein AP-2, and binding of voltage-gated calcium channels (Chapman et al. 1998). This motif also participates in the binding of inositol polyphosphates (Fukuda et al. 1995a). Microinjecting Pep20 into the squid terminal is therefore expected to disrupt one or more of these interactions.
Pep20 inhibits neurotransmitter release when injected into the squid giant presynaptic terminal. Blockade is dose dependent, requiring a few hundred micromolar Pep20 for maximal inhibition. Imaging experiments indicate that Pep20 does not interfere with calcium entry into the presynaptic terminal but instead reduces the efficacy of calcium in triggering transmitter release. Electron microscopy reveals that Pep20 injection causes a selective accumulation of docked synaptic vesicles but has little effect on vesicles in other spatial compartments. These results show that synaptotagmin is important for a reaction that occurs after synaptic vesicles dock but before they fuse, consistent with the hypothesis that synaptotagmin is the calcium receptor that regulates transmitter release. More recent genetic evidence shows that knock-out of synaptotagmin genes selectively impairs a fast component of evoked neurotransmitter release in mice (Geppert et al. 1994) and changes the slope of the relationship between external calcium concentration and transmitter release in Drosophila (Littleton et al. 1994). These experiments offer additional support for the hypothesis that synaptotagmin is the calcium receptor protein. However, it is not yet clear how vesicle fusion is triggered by calcium-bound synaptotagmin, and it is possible that one of the several proposed interactions with SNARE proteins could be important (Hilfiker et al. 1999a).
Vesicle budding
After fusion, synaptic vesicle membrane is recovered via endocytosis. Heuser & Reese (1973) proposed that endocytosis in nerve terminals is based on a membrane budding process that requires the formation of coated pits and coated vesicles (Fig. 1). This seems to involve some sort of coating protein that is widely assumed to be clathrin. However, this model of endocytosis has been difficult to establish in neurons and has led to decades of debate (Cremona & DeCamilli, 1997; Palfrey & Artalejo, 1998).
We have studied the mechanism of membrane budding by focusing on the proteins that assemble clathrin into ‘cages’. If clathrin cages are important for endocytosis, then inhibition of cage formation should reduce the number of coated pits and vesicles and should trap vesicular membrane in the plasma membrane. Based on studies in non-neuronal cells, it is thought that AP-2 is needed for assembly of clathrin on the plasma membrane (Schmid, 1997). AP-2 is a ubiquitous and multimeric protein complex that binds to clathrin and assembles it into cages. AP-2 also binds to other proteins on the plasma membrane and is thought to serve as an ‘adaptor’ which links clathrin cages to regions of the plasma membrane that undergo budding. Neurons also contain AP180, a monomeric protein that can assemble clathrin (Zhou et al. 1992). Our approach to defining the role of clathrin in endocytosis has been to perturb the function of these two potential clathrin assembly proteins in the nerve terminal.
For this purpose, we have made peptides that mimic a conserved motif found in multiple copies in both proteins, which apparently allows clathrin binding (Morgan et al. 1999; J. R. Morgan, X. Zhao, K. Prasad, G. J. Augustine & E. M. Lafer, unpublished observations). In vitro, these peptides prevent both AP-2 and AP180 from assembling clathrin into cages, and thus are good tools for studying the physiological function of clathrin assembly. When injected into neurons, these clathrin assembly inhibitor peptides dramatically impair endocytosis. This conclusion is based on several observations. First, the peptides gradually inhibit neurotransmitter release, as would be expected if endocytosis were blocked and caused a loss of synaptic vesicles. Second, electron microscopy reveals the predicted loss of synaptic vesicles in peptide-injected terminals. This loss of synaptic vesicles is due to impairment of budding from the plasma membrane, because the surface area of the presynaptic terminal is increased. Further, the number of coated vesicles within the terminal is drastically reduced. All of these changes are as predicted for blockade of membrane coating and budding from the plasma membrane. Thus, we conclude that assembly of clathrin by AP180 and AP-2 is necessary for endocytosis. These results are consistent with the functional and structural changes produced by null mutation of the Drosophila AP180 gene (Zhang et al. 1998) and with studies of the function of clathrin at a lamprey synapse (Shupliakov et al. 1997; Brodin et al. 1997). Our results are the first evidence that neurons operating under physiological conditions require clathrin and its adaptor proteins for endocytosis.
Surprisingly, similar lesions in vesicle budding are produced by a fragment of rabphilin-3A (Burns et al. 1998). Presynaptic microinjection of the N-terminal portion of this protein, which includes the binding site for rab3A, inhibits transmitter release. This fragment also reduces the number of synaptic vesicles and coated vesicles and increases the surface area of the presynaptic plasma membrane. This indicates that rabphilin-3A may play a role in membrane budding during endocytosis. Given the evidence that rabphilin-3A may also be involved in vesicle docking (see above), it appears that this protein has multiple functions in synaptic vesicle trafficking. In this regard it is interesting that other C2 domain proteins, such as synaptotagmin and protein kinase C, have also been implicated in more than one vesicle trafficking reaction within the nerve terminal (Robinson et al. 1993; Fukuda et al. 1995b; Jorgensen et al. 1995; Gillis et al. 1996). Thus, the C2 domain protein family may serve as molecular coordinators of presynaptic vesicle trafficking reactions.
Endosomal fusion
Heuser & Reese (1973) proposed that recycled vesicular membrane transits through endosomes and other intermediates following membrane budding (Fig. 1). Impairment of these endocytotic reactions should reduce the number of synaptic vesicles and endosomes without increasing the surface area of the presynaptic plasma membrane. Peptides from both SNAP and NSF have such effects; they decrease the number of synaptic vesicles (DeBello et al. 1995; Schweizer et al. 1998) and, as mentioned above, do not increase presynaptic plasma membrane area. These results indicate that NSF and SNAP are important within the endocytotic pathway, at a reaction somewhere downstream of vesicle budding. While it is not yet clear which reaction within the endocytotic pathway is affected by these perturbations, we suggest that these peptides block a SNARE-dependent mechanism that ordinarily allows retrieved vesicles to fuse with endosomes. Disruption of the membrane SNARE proteins does not prevent endocytosis (Hunt et al. 1994; Burns, 1996; O'Connor et al. 1997; Dresbach et al. 1998). The differential involvement of SNAREs and NSF/SNAP in endocytosis is in agreement with the SNARE hypothesis, which predicts that NSF and SNAP are involved in many fusion reactions, while membrane SNAREs are involved in compartment-specific fusion reactions (Rothman, 1994). Examination of endosomes in nerve terminals with impaired NSF and/or SNAP should provide a definitive test of the role of these proteins - and endosomes - in synaptic vesicle recycling.
Conclusions
A summary of the findings described here is sh own in Fig. 3. This figure emphasizes that one or more specific proteins have been shown to participate in almost all of the known reactions of the synaptic vesicle trafficking cycle. Further work will be necessary to identify the many other presynaptic proteins likely to be important and to clarify how all these proteins act. But compared with the paucity of information that was available only a couple of years ago, there are reasons to be optimistic that a molecular understanding of neurotransmitter release is achievable, although perhaps not within immediate reach.
Figure 3. A model for the control of membrane trafficking reactions by presynaptic proteins.

While we are at the early stages of a molecular understanding of neurotransmitter release, it is still worthwhile to search for general themes within the currently known proteins and reactions. It is clear that neurons have taken advantage of some of the same molecular mechanisms employed for membrane trafficking in all cells, such as SNARE proteins, GTP-binding proteins, and clathrin. It is also clear that neurons have embellished these conserved reactions by using other proteins, such as synapsins, synaptotagmins, and others, to incorporate other reactions that confer high speed and calcium regulation on the trafficking of synaptic vesicle constituents. In this way, neurons are able to modify the basic mechanisms of membrane trafficking to secrete neurotransmitters very rapidly and for extended periods of time.
Acknowledgments
The work summarized in this review arises from our collaboration with many other people, including T. Abe, H. Betz, K. Bommert, M. Charlton, A. Czernik, P. Doroshenko, T. Dresbach, P. Greengard, C. Heuss, J. Hunt, T. Ishizuka, H.-T. Kao, E. Lafer, V. O'Connor, L. Pelligrini, K. Prasad, J. Rothman, H. Saisu, T. Schaeffer, S. Wang, M. Womack and W. Whiteheart. We thank them for their many valuable contributions to this work. This work was supported by NIH grant NS-21624.
References
- Adler EM, Augustine GJ, Duffy SN, Charlton MP. Alien intracellular calcium chelators attenuate neurotransmitter release at the squid giant synapse. Journal of Neuroscience. 1991;11:1496–1507. doi: 10.1523/JNEUROSCI.11-06-01496.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almers W. Synapses. How fast can you get? Nature. 1994;367:682–683. doi: 10.1038/367682a0. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Burns ME, DeBello WM, Pettit DL, Schweizer FE. Exocytosis: Proteins and perturbations. Annual Review of Pharmacology and Toxicology. 1996;36:659–701. doi: 10.1146/annurev.pa.36.040196.003303. [DOI] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium entry and transmitter release at voltage-clamped nerve terminals of squid. The Journal of Physiology. 1985;367:163–181. doi: 10.1113/jphysiol.1985.sp015819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Charlton MP, Smith SJ. Calcium action in synaptic transmitter release. Annual Review of Neuroscience. 1987;10:633–693. doi: 10.1146/annurev.ne.10.030187.003221. [DOI] [PubMed] [Google Scholar]
- Banerjee A, Barry VA, DasGupta BR, Martin TFJ. N-Ethylmaleimide-sensitive factor acts at a prefusion ATP-dependent step in Ca2+-activated exocytosis. Journal of Biological Chemistry. 1996;271:20223–20226. doi: 10.1074/jbc.271.34.20223. [DOI] [PubMed] [Google Scholar]
- Bommert K, Charlton MP, DeBello WM, Chin GJ, Betz H, Augustine GJ. Inhibition of neurotransmitter release by C2-domain peptides implicates synaptotagmin in exocytosis. Nature. 1993;363:163–165. doi: 10.1038/363163a0. [DOI] [PubMed] [Google Scholar]
- Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proceedings of the National Academy of Sciences of the USA. 1994;91:10727–10731. doi: 10.1073/pnas.91.22.10727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broadie K, Prokop A, Bellen HJ, O'Kane CJ, Schulze KL, Sweeney ST. Syntaxin and synaptobrevin function downstream of vesicle docking in Drosophila. Neuron. 1995;15:663–673. doi: 10.1016/0896-6273(95)90154-x. [DOI] [PubMed] [Google Scholar]
- Brodin L, Low P, Gad H, Gustafsson J, Pieribone VA, Shupliakov O. Sustained neurotransmitter release: new molecular clues. European Journal of Neuroscience. 1997;9:2503–2511. doi: 10.1111/j.1460-9568.1997.tb01679.x. [DOI] [PubMed] [Google Scholar]
- Burns ME. Durham, NC, USA: Duke University; 1996. Molecular mechanisms of neurotransmitter release: the functions of Rab3A, Rabphilin, and SNAP-25. PhD Thesis. [Google Scholar]
- Burns ME, Augustine GJ. Functional studies of presynaptic proteins at the squid giant synapse. In: Bellen H, editor. Neurotransmitter Release: Frontiers in Molecular Biology. Oxford University Press; 1999. pp. 237–264. [Google Scholar]
- Burns ME, Sasaki T, Takai Y, Augustine GJ. Rabphilin-3A: a multifunctional regulator of synaptic vesicle traffic. Journal of General Physiology. 1998;111:243–255. doi: 10.1085/jgp.111.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton JL, Burns ME, Gatti E, Augustine GJ, De Camilli P. Specific interactions of Mss4 with members of the Rab GTPase subfamily. EMBO Journal. 1994;13:5547–5558. doi: 10.1002/j.1460-2075.1994.tb06892.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman ER, Desai RC, Davis AF, Tornehl CK. Delineation of the oligomerization, AP-2 binding, and synprint binding region of the C2B domain of synaptotagmin. Journal of Biological Chemistry. 1998;273:32966–32972. doi: 10.1074/jbc.273.49.32966. [DOI] [PubMed] [Google Scholar]
- Coorssen JR, Blank PS, Tahara M, Zimmerberg J. Biochemical and functional studies of cortical vesicle fusion: the SNARE complex and Ca2+ sensitivity. Journal of Cell Biology. 1998;143:1845–1857. doi: 10.1083/jcb.143.7.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremona O, DeCamilli P. Synaptic vesicle endocytosis. Current Opinion in Neurobiology. 1997;7:323–330. doi: 10.1016/s0959-4388(97)80059-1. [DOI] [PubMed] [Google Scholar]
- DeBello WM, Betz H, Augustine GJ. Synaptotagmin and neurotransmitter release. Cell. 1993;74:947–950. doi: 10.1016/0092-8674(93)90716-4. [DOI] [PubMed] [Google Scholar]
- DeBello WM, O'Connor V, Dresbach T, Whiteheart SW, Wang SS, Schweizer FE, Betz H, Rothman JE, Augustine GJ. SNAP-mediated protein-protein interactions essential for neurotransmitter release. Nature. 1995;373:626–630. doi: 10.1038/373626a0. [DOI] [PubMed] [Google Scholar]
- Dresbach T, Burns ME, O'Connor V, DeBello WM, Betz H, Augustine GJ. A neuronal Sec1 homolog regulates neurotransmitter release at the squid giant synapse. Journal of Neuroscience. 1998;18:2923–2932. doi: 10.1523/JNEUROSCI.18-08-02923.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukuda M, Kojima T, Aruga J, Niinobe M, Mikoshiba K. Functional diversity of C2 domains of synaptotagmin family. Mutational analysis of inositol high polyphosphate binding domain. Journal of Biological Chemistry. 1995a;270:26523–26527. doi: 10.1074/jbc.270.44.26523. [DOI] [PubMed] [Google Scholar]
- Fukuda M, Moreira JE, Lewis FM, Sugimori M, Niinobe M, Mikoshiba K, Llinás R. Role of the C2B domain of synaptotagmin in vesicular release and recycling as determined by specific antibody injection into the squid giant synapse preterminal. Proceedings of the National Academy of Sciences of the USA. 1995b;92:10708–10712. doi: 10.1073/pnas.92.23.10708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, Südhof TC. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79:717–27. doi: 10.1016/0092-8674(94)90556-8. [DOI] [PubMed] [Google Scholar]
- Geppert M, Südhof TC. RAB3 and synaptotagmin: the yin and yang of synaptic membrane fusion. Annual Review of Neuroscience. 1998;21:75–95. doi: 10.1146/annurev.neuro.21.1.75. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Mossner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Greengard P, Valtorta F, Czernik AJ, Benfenati F. Synaptic vesicle phosphoproteins and regulation of synaptic function. Science. 1993;259:780–785. doi: 10.1126/science.8430330. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Heinemann C, Neher E, Matthews G. Calcium dependence of the rate of exocytosis in a synaptic terminal. Nature. 1994;371:513–515. doi: 10.1038/371513a0. [DOI] [PubMed] [Google Scholar]
- Hess SD, Doroshenko PA, Augustine GJ. A functional role for GTP-binding proteins in synaptic vesicle cycling. Science. 1993;259:1169–1172. doi: 10.1126/science.8438167. [DOI] [PubMed] [Google Scholar]
- Heuser JE, Reese TS. Evidence for recycling of synaptic vesicle membrane during transmitter release at the frog neuromuscular junction. Journal of Cell Biology. 1973;57:315–344. doi: 10.1083/jcb.57.2.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser JE, Reese TS, Dennis MJ, Jan Y, Jan L, Evans L. Synaptic vesicle exocytosis captured by quick freezing and correlated with quantal transmitter release. Journal of Cell Biology. 1979;81:275–300. doi: 10.1083/jcb.81.2.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Greengard P, Augustine GJ. Coupling calcium to SNARE-mediated synaptic vesicle fusion. Nature Neuroscience. 1999a;2:104–106. doi: 10.1038/5659. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao H-T, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philosophical Transactions of the Royal Society of London B. 1999b;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilfiker S, Schweizer FE, Kao H-T, Czernik AJ, Greengard P, Augustine GJ. Two sites of action for synapsin domain E in regulating neurotransmitter release. Nature Neuroscience. 1998;1:29–35. doi: 10.1038/229. [DOI] [PubMed] [Google Scholar]
- Holz RW, Bittner MA, Peppers SC, Senter RA, Eberhard DA. MgATP-independent and MgATP-dependent exocytosis. Evidence that MgATP primes adrenal chromaffin cells to undergo exocytosis. Journal of Biological Chemistry. 1989;264:5412–5419. [PubMed] [Google Scholar]
- Hunt JM, Bommert K, Charlton MP, Kistner A, Habermann E, Augustine GJ, Betz H. A post-docking role for synaptobrevin in synaptic vesicle fusion. Neuron. 1994;12:1269–1279. doi: 10.1016/0896-6273(94)90443-x. [DOI] [PubMed] [Google Scholar]
- Jorgensen EM, Hartwieg E, Schuske K, Nonet ML, Jin Y, Horvitz HR. Defective recycling of synaptic vesicles in synaptotagmin mutants of. Caenorhabditis elegans. Nature. 1995;378:196–199. doi: 10.1038/378196a0. [DOI] [PubMed] [Google Scholar]
- Kao H-T, Porton B, Hilfiker S, Stefani G, Pieribone VA, Greengard P. Molecular evolution of the synapsin gene family. Journal of Experimental Zoology. 1999 in the Press. [PubMed] [Google Scholar]
- Katz B. The Release of Neural Transmitter Substances. Liverpool University Press; 1969. [Google Scholar]
- Littleton JT, Chapman ER, Kreber R, Garment MB, Carlson SD, Ganetzky B. Temperature-sensitive paralytic mutations demonstrate that synaptic exocytosis requires SNARE complex assembly and disassembly. Neuron. 1998;21:401–413. doi: 10.1016/s0896-6273(00)80549-8. [DOI] [PubMed] [Google Scholar]
- Littleton JT, Stern M, Perin M, Bellen HJ. Calcium dependence of neurotransmitter release and rate of spontaneous vesicle fusions are altered in Drosophila synaptotagmin mutants. Proceedings of the National Academy of Sciences of the USA. 1994;91:10888–10892. doi: 10.1073/pnas.91.23.10888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llinás R, Gruner JA, Sugimori M, McGuinness TL, Greengard P. Regulation by synapsin I and Ca2+-calmodulin-dependent protein kinase II of transmitter release in squid giant synapse. Journal of Physiology. 1991;436:257–282. doi: 10.1113/jphysiol.1991.sp018549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsal J, Ruiz-Montasell B, Blasi J, Moreira JE, Contreras D, Sugimori M, Llinás R. Block of transmitter release by botulinum C1 action on syntaxin at the squid giant synapse. Proceedings of the National Academy of Sciences of the USA. 1997;94:14871–14876. doi: 10.1073/pnas.94.26.14871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin TF, Hay JC, Banerjee A, Barry VA, Ann K, Yom HC, Porter BW, Kowalchyk JA. Late ATP-dependent and Ca2+-activated steps of dense core granule exocytosis. Cold Spring Harbor Symposium on Quantitative Biology. 1995;60:197–204. doi: 10.1101/sqb.1995.060.01.022. [DOI] [PubMed] [Google Scholar]
- Mayer A, Wickner W, Haas A. Sec18p (NSF)-driven release of Sec17p (alpha-SNAP) can precede docking and fusion of yeast vacuoles. Cell. 1996;85:83–94. doi: 10.1016/s0092-8674(00)81084-3. [DOI] [PubMed] [Google Scholar]
- Morgan JR, Zhao X, Womack M, Prasad K, Augustine GJ, Lafer EM. A role for the clathrin assembly domain of AP180 in synaptic vesicle endocytosis. Journal of Neuroscience. 1999 doi: 10.1523/JNEUROSCI.19-23-10201.1999. in the Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy VN, Stevens CF. Synaptic vesicles retain their identity through the endocytic cycle. Nature. 1998;392:497–501. doi: 10.1038/33152. [DOI] [PubMed] [Google Scholar]
- O'Connor V, Heuss C, De Bello WM, Dresbach T, Charlton MP, Hunt JH, Pellegrini LL, Hodel A, Burger MM, Betz H, Augustine GJ, Schäfer T. Disruption of syntaxin-mediated protein interactions blocks neurotransmitter secretion. Proceedings of the National Academy of Sciences of the USA. 1997;94:12186–12191. doi: 10.1073/pnas.94.22.12186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palfrey HC, Artalejo CR. Vesicle recycling revisited: rapid endocytosis may be the first step. Neuroscience. 1998;83:969–989. doi: 10.1016/s0306-4522(97)00453-3. [DOI] [PubMed] [Google Scholar]
- Parsons TD, Coorssen JR, Horstmann H, Almers W. Docked granules, the exocytic burst and the need for ATP hydrolysis in endocrine cells. Neuron. 1995;15:1085–1096. doi: 10.1016/0896-6273(95)90097-7. [DOI] [PubMed] [Google Scholar]
- Pieribone VA, Shupliakov O, Brodin L, Hilfiker-Rothenfluh S, Czernik AJ, Greengard P. Distinct pools of synaptic vesicles in neurotransmitter release. Nature. 1995;375:493–497. doi: 10.1038/375493a0. [DOI] [PubMed] [Google Scholar]
- Robinson PJ, Sontag JM, Liu JP, Fyske EM, Slaughter C, McMahon H, Südhof TC. Dynamin GTPase regulated by protein kinase C phosphorylation in nerve terminals. Nature. 1993;365:163–166. doi: 10.1038/365163a0. [DOI] [PubMed] [Google Scholar]
- Rothman JE. Mechanisms of intracellular protein transport. Nature. 1994;372:55–63. doi: 10.1038/372055a0. [DOI] [PubMed] [Google Scholar]
- Ryan TA, Smith SJ. Vesicle pool mobilization during action potential firing at hippocampal synapses. Neuron. 1995;14:983–989. doi: 10.1016/0896-6273(95)90336-4. [DOI] [PubMed] [Google Scholar]
- Schmid SL. Clathrin-coated vesicle formation and protein sorting: an integrated process. Annual Review of Biochemistry. 1997;66:511–548. doi: 10.1146/annurev.biochem.66.1.511. [DOI] [PubMed] [Google Scholar]
- Schweizer FE, Dresbach T, DeBello WM, O'Connor V, Augustine GJ, Betz H. Regulation of neurotransmitter release kinetics by NSF. Science. 1998;279:1203–1206. doi: 10.1126/science.279.5354.1203. [DOI] [PubMed] [Google Scholar]
- Shi G, Faundez V, Roos J, Dell'Angelica EC, Kelly RB. Neuroendocrine synaptic vesicles are formed in vitro by both clathrin-dependent and clathrin-independent pathways. Journal of Cell Biology. 1998;143:947–955. doi: 10.1083/jcb.143.4.947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shupliakov O, Low P, Grabs D, Gad H, Chen H, David C, Takei K, DeCamilli P, Brodin L. Synaptic vesicle endocytosis impaired by disruption of dynamin-SH3 domain interactions. Science. 1997;276:259–263. doi: 10.1126/science.276.5310.259. [DOI] [PubMed] [Google Scholar]
- Söllner T, Bennett MK, Whiteheart SW, Scheller RH, Rothman JE. A protein assembly-disassembly pathway in vitro that may correspond to sequential steps of synaptic vesicle docking, activation, and fusion. Cell. 1993;75:409–418. doi: 10.1016/0092-8674(93)90376-2. [DOI] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle: a cascade of protein-protein interactions. Nature. 1995;375:645–653. doi: 10.1038/375645a0. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Czernik AJ, Kao HT, Takei K, Johnston PA, Horiuchi A, Kanazir SD, Wagner MA, Perin MS, De Camilli P, Greengard P. Synapsins: mosaics of shared and individual domains in a family of synaptic vesicle phosphoproteins. Science. 1989;245:1474–1480. doi: 10.1126/science.2506642. [DOI] [PubMed] [Google Scholar]
- Südhof TC, Rizo J. Synaptotagmins: C2 domain proteins that regulate synaptic vesicle traffic. Neuron. 1996;17:379–388. doi: 10.1016/s0896-6273(00)80171-3. [DOI] [PubMed] [Google Scholar]
- Takei K, Mundigl O, Daniell L, DeCamilli P. The synaptic vesicle cycle: a single vesicle budding step involving clathrin and dynamin. Journal of Cell Biology. 1996;133:1237–1250. doi: 10.1083/jcb.133.6.1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas DM, Elferink LA. Functional analysis of the C2A domain of synaptotagmin 1: implications for calcium-regulated secretion. Journal of Neuroscience. 1998;18:3511–3520. doi: 10.1523/JNEUROSCI.18-10-03511.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungermann C, Sato K, Wickner W. Defining the function of trans-SNARE pairs. Nature. 1998;396:543–548. doi: 10.1038/25069. [DOI] [PubMed] [Google Scholar]
- Weber T, Zemelman BV, McNew JA, Westermann B, Gmachl M, Parlati F, Söllner TH, Rothman JE. SNAREpins: Minimal machinery for membrane fusion. Cell. 1998;92:759–772. doi: 10.1016/s0092-8674(00)81404-x. [DOI] [PubMed] [Google Scholar]
- Zhang B, Koh YH, Beckstead RB, Budnik V, Ganetsky B, Bellen HJ. Synaptic vesicle size and number are regulated by a clathrin adaptor protein required for endocytosis. Neuron. 1998;21:1465–1475. doi: 10.1016/s0896-6273(00)80664-9. [DOI] [PubMed] [Google Scholar]
- Zhou S, Sousa R, Tannery HN, Lafer EM. Characterization of a novel synapse-specific protein. II. cDNA cloning and sequence analysis of the F1–20 protein. Journal of Neuroscience. 1992;12:2144–2155. doi: 10.1523/JNEUROSCI.12-06-02144.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zimmerberg J, Coorssen JR, Vogel SS, Blank PS. The Journal of Physiology. 1999;520:15–22. doi: 10.1111/j.1469-7793.1999.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]