

Abstract
Nitric oxide (NO) has been shown to modulate neuropeptide secretion from the posterior pituitary. Here we show that NO activates large-conductance Ca2+-activated K+ (BK) channels in posterior pituitary nerve terminals.
NO, generated either by the photolysis of caged-NO or with chemical donors, irreversibly enhanced the component of whole-terminal K+ current due to BK channels and increased the activity of BK channels in excised patches. NO also inhibited the transient A-current. The time courses of these effects on K+ current were very different; activation of BK channels developed slowly over several minutes whereas inhibition of A-current immediately followed NO uncaging.
Activation of BK channels by NO occurred in the presence of guanylyl cyclase inhibitors and after removal of ATP or GTP from the pipette solution, suggesting a cGMP-independent signalling pathway.
The sulfhydryl alkylating agent N-ethyl maleimide (NEM) increased BK channel activity. Pretreatment with NEM occluded NO activation.
NO activation of BK channels occurred independently of voltage and cytoplasmic Ca2+ concentration. In addition, NO removed the strict Ca2+ requirement for channel activation, rendering channels highly active even at nanomolar Ca2+ levels.
These results suggest that NO, or a reactive nitrogen byproduct, chemically modifies nerve terminal BK channels or a closely associated protein and thereby produces an increase in channel activity. Such activation is likely to inhibit impulse activity in posterior pituitary nerve terminals and this may explain the inhibitory action of NO on secretion.
Nitric oxide (NO) is recognised as an important signalling molecule in diverse physiological processes (Rees et al. 1989; Moncada et al. 1991; Garthwaite & Boulton, 1995). One important role proposed for NO is the modulation of neurosecretion, and this may be relevant to some forms of synaptic plasticity (Schuman & Madison, 1994; Garthwaite & Boulton, 1995). However, few investigations to date have directly addressed the actions of NO on nerve terminal excitability. In this study we examined the actions of NO in posterior pituitary nerve terminals. These nerve terminals are responsible for the secretion of the neuropeptides anti-diuretic hormone (ADH) and oxytocin (OT), and there is evidence that NO may regulate the secretion of these hormones. First, high levels of constitutive nitric oxide synthase (NOS) have been identified in the posterior pituitary (Bredt et al. 1990; Miyagawa et al. 1994; Pow, 1994; Kadowaki et al. 1994), and NOS activity in pituitary extracts has been reported to correlate with ADH release (Kadowaki et al. 1994). Second, agents that inhibit NOS activity, or release NO, have been shown to modulate ADH and OT release in animals (Eriksson et al. 1982; Ota et al. 1993; Summy-Long et al. 1993; Goyer et al. 1994; Kadowaki et al. 1994; Chiodera et al. 1994), hypothalamic neurons (Raber & Bloom, 1994) and isolated pituitary preparations (Lutz-Bucher & Koch, 1994). However, in the studies cited above, manipulation of NO produced variable results. Further, NO itself inhibited the stimulated release of ADH but enhanced basal secretion.
To explore the mechanisms involved in the modulation of secretion by NO we investigated the effect of NO on neurohypophysial large-conductance Ca2+-activated K+ (BK) channels (Wang et al. 1992; Bielefeldt et al. 1992). BK channels play an important role in regulating the excitability of pituitary nerve terminals. Activation of BK channels during prolonged bursts of action potentials decreases membrane excitability (Bielefeldt & Jackson, 1993, 1994) and this could lead to a reduction in secretion. Moreover, Ca2+-activated K+ channels are well characterised targets for NO signalling in other tissues; activation of these channels either directly (Bolotina et al. 1994), or via a cGMP-dependent pathway (Archer et al. 1994), contributes to relaxation of arterial smooth muscle. More recently, NO has been shown to induce a direct activation of BK channels isolated from synaptosomes (Shin et al. 1997). The present study shows a similar action of NO on neurohypophysial BK channels, which can explain some of the results regarding NO modulation of OT and ADH secretion. This cGMP-independent effect was seen in cell-free excised patches, was mimicked by sulfhydryl alkylation and occurred independently of voltage and [Ca2+]. These results suggest that interactions between NO or NO byproducts and BK channel complexes play a role in the regulation of neuropeptide release.
METHODS
Slice preparation
Experiments were carried out in accordance with the National Institutes of Health guide for the care and uses of laboratory animals. Animals were housed under 12 h light-dark cycle with free access to water and food. Posterior pituitary slices were prepared as described previously (Jackson et al. 1991; Bielefeldt et al. 1992). Male rats (220-300 g) were rendered unconscious by exposure to a rising concentration of CO2 and decapitated. The pituitary was removed and placed in ice-cold 95 % O2-5 % CO2-saturated artificial cerebrospinal fluid (ACSF) containing (mm): 125 NaCl, 4 KCl, 26 NaHCO3, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 and 10 glucose. The whole pituitary was mounted in a slicing chamber and the neurointermediate lobe was sliced at a thickness setting of 75 μm using a Vibratome. Slices were maintained for up to 2–3 h in 95 % O2-5 % CO2-saturated ACSF until recording.
Patch-clamp recording
Voltage-clamp recordings were obtained from nerve terminals in posterior pituitary slices using standard patch-clamp methods. Individual nerve terminals were located with an upright microscope (Nikon optiphot) equipped with Nomarski optics and a × 40 water-immersion objective. Recordings were made using an EPC-7 amplifier interfaced to a Macintosh Power PC running IgorPro software (Wavemetrics, Lake Oswego, OR, USA). All whole-terminal recordings were made using P/4 leak subtraction.
Patch pipettes with resistances between 2–3 MΩ were prepared from aluminosilicate glass (Garner Glass, Claremont, CA, USA). Series resistance compensation was routinely set at 50 %. Only recordings with an uncompensated series resistance of less than 10 MΩ were used for analysis. For whole-terminal recordings, the bath was continuously perfused with 95 % O2-5 % CO2-saturated ACSF at room temperature (20-24°C). For whole-terminal and outside-out patch recordings, the patch pipette contained (mm): 130 KCl, 10 NaCl, 10 Hepes, 5 EGTA, 4 MgATP and 0.1 GTP, titrated to pH 7.3 with KOH. For inside-out patches the pipette solution contained (mm): 140 KCl, 2 CaCl2, 1 MgCl2 and 10 Hepes, pH 7.3; and the bath solution contained (mm): 150 KCl, 10 Hepes, 1 MgATP, 5 EGTA, pH 7.3, with CaCl2 varied to produce the desired free [Ca2+]. Free [Ca2+] between 10−7 and 10−5 M was measured using a Ca2+-selective electrode (Fisher Scientific).
Caged-NO and photolysis
Caged-NO (potassium nitrosylpentachlororuthenate, 1–4 mm; Molecular Probes, Eugene, OR, USA) was included in the patch pipette solution in both whole-terminal and excised patch recordings. After establishment of a whole-terminal recording, we allowed at least 2–3 min for diffusion of caged-NO into the nerve terminal prior to photolysis. For photolysis, the preparation was illuminated with a 75 W Xenon arc lamp (Optiquip, Highland Mills, NY, USA) gated by an electromechanical shutter (Hsu et al. 1996). The system was modified by the addition of a capacitor in the power supply which could be discharged to generate brief periods (μ0.5 ms) of high intensity light at approximately 5–10 times the rated power of the bulb. The lamp was mounted on the microscope such that its output entered the epi-illumination pathway. For each light pulse the shutter was open for 30 ms (illuminating with low intensity) and during this time a brief high intensity light pulse was provided by discharging the capacitor.
The uncaging efficiency of the light pulses was calibrated by measuring the light-induced increase in the fluorescence of CMNB-caged-fluorescein (fluorescein bis-(5-carboxymethoxy-2-nitrobenzyl) ether (Molecular Probes). CMNB-caged-fluorescein (85 μm) was introduced into terminals via the patch pipette and the pipette was then withdrawn prior to uncaging. The photoreleased fluorescein was excited by continuous illumination with a tungsten lamp in series with a 485 ± 11 nm bandpass filter. Excitation light was reflected off a 505 nm dichroic mirror treated for high UV reflectance, and focused onto the preparation with a × 40 Olympus UPlanApo water-immersion objective (NA 0.75). Emitted light passed through a 530 ± 15 bandpass filter and was measured with a photomultiplier tube. The mirror and filters were obtained from Omega (Brattleboro, VT, USA). Figure 1 shows the normalised fluorescence (means ±s.d.) from nine experiments. Note that fluorescence increased in a sigmoidal rather than monoexponential fashion because CMNB-caged-fluorescein contains two light-sensitive protecting groups, both of which must be photolysed to liberate the fluorescent dye. The data were fitted to an equation of the form:
Figure 1. Photolytic efficiency of 75 W Xenon lamp.
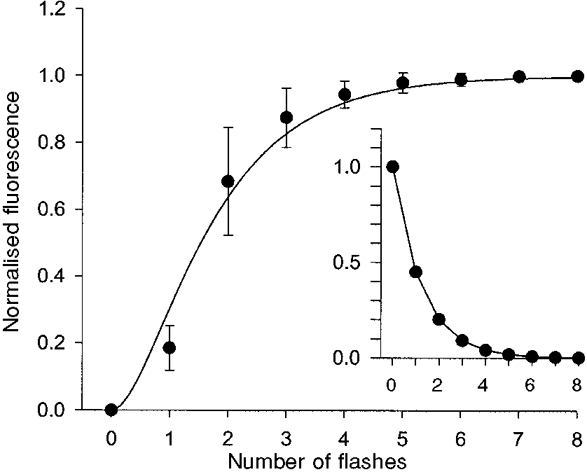
Normalised nerve terminal fluorescence (means ±s.d.) from 9 nerve terminals (loaded with 85 μm caged-fluorescein) versus the number of light flashes. The continuous curve shows the best fit to eqn (1) (see Methods) with a Δ value of 1.25. The inset shows a plot of exp(-n/Δ), or the fraction of unphotolysed caging groups remaining after each light flash. Thus each flash liberates 55 % of the caging groups. A calculation in Methods based on this result suggests that each flash will photolyse 2 % of the caged-NO.
| (1) |
where F is fluorescence, n is the number of light flashes and Δ is an uncaging constant for one cage group. The continuous curve shows the best fit with Δ= 1.25. The fraction of carboxymethoxy nitrophenyl photolysed per flash is therefore given by 1 - exp(−1/Δ), or 0.55. Thus, each flash photolyses 55 % of the caging groups. The inset to Fig. 1 shows the monoexponential decrease in intact caged groups versus number of light flashes (i.e. a plot of exp(-n/Δ), computed from the plot of fluorescein fluorescence).
The efficiency of caged-NO photolysis can then be estimated by multiplying the photolytic efficiency of CMNB-caged-fluorescein (55 %) by the ratio of the product of the quantum yield and molar extinction coefficients for both caged compounds. We determined the ratio (CMNB-caged-fluorescein/caged-NO) of molar extinction coefficients to be 10.7 by measuring their relative absorbance at 350 nm. The published quantum yield for caged-NO is 0.06 (Bettache et al. 1996). The quantum yield for caged-fluorescein is unreported but we used the value of 0.13 obtained for the structurally similar caged compound, 4,5-dimethoxy-2-nitrobenzyl)phenylephrine (Walker et al. 1993). Thus, we estimate that one light flash will convert μ2 % of the caged-NO to free NO. This value is roughly twice the value of 0.9 % estimated by Murphy et al. (1994) using 1 ms flashes with a 100 W Xenon flash lamp (70 mJ pulse−1). In a typical experiment with 4 mm caged-NO, we estimate that one flash of light will produce approximately 80 μm NO. An illumination protocol used in much of this work was five flashes, applied within 3 s to photolyse μ400 μm caged-NO. Note that because of the rapid decay of NO in solution (Moncada et al. 1991; Wink et al. 1996) these rises in [NO] will be short-lived. It is difficult to estimate the peak [NO] because of the complicated kinetics of NO decomposition (the half-life of NO ranges from 13 min at 1 μm to less than 1 s at 1 mm (Wink et al. 1996)) but it is certainly somewhat less than 400 μm, and will be indicated as such (i.e. ≤ 400 μm).
Single channel recording and analysis
Single channel data were filtered at 10 kHz and stored on videotape. Data were later replayed through a 1 kHz low-pass filter and digitised at either 2 or 5 kHz. Open probability (Po) was measured in continuous data segments of 20 s or longer with a discriminator set at half the open channel amplitude or by dividing the current integral by the maximum current amplitude (for patches containing multiple channels) using the software CHANNEL2 (Michael Smith and Peter Gage, Australian National University).
NO donors and guanylyl cyclase inhibitors
Diethylamine NONOate was from Cayman Chemicals (Ann Arbor, MI, USA), sodium nitroprusside was obtained from Sigma, and 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ) and 6-(phenylamino)-5,8-quinolinedione (LY-83,583) were obtained from Research Biochemicals International (Natick, MA, USA).
Statistics
All results are expressed as means ±s.e.m. Statistical significance was evaluated using Student's t test. P < 0.05 was considered significant.
RESULTS
NO enhances steady-state K+ current
Posterior pituitary nerve terminal K+ current was activated by a 500 ms depolarising pulse from −80 to +50 mV (Fig. 2A). When nerve terminals filled with caged-NO were illuminated, the amplitude of the K+ current increased. Figure 2A shows the K+ current recorded before and μ10 min after five successive light flashes produced a transient rise in [NO] to ≤ 400 μm (see Methods). NO prolonged the time course of the K+ current and produced an μ80 % increase in the final level of current just before the end of the voltage pulse. NO also caused a small decrease in the magnitude of the peak current.
Figure 2. NO modulation of whole-terminal K+ current.
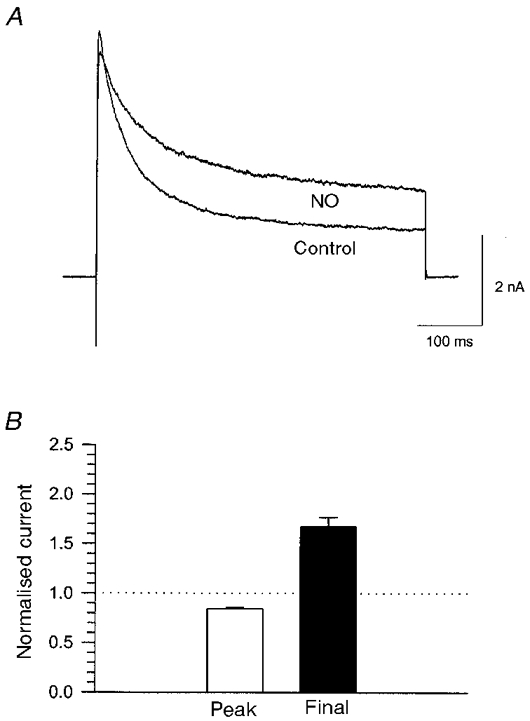
A, whole-terminal K+ current evoked by a 500 ms pulse from −80 to +50 mV before and approximately 10 min after uncaging of NO with 5 light flashes in 3 s (raising [NO] to < = 400 μm). NO increased the current at the end of the pulse by 82 % and caused a small reduction of 8 % in the peak current. B, mean (±s.e.m.) changes in peak and final current from 25 terminals, expressed as a fraction of control. In each terminal NO was released with 5 successive light flashes as in A. Current was recorded 5–10 min later to allow for the slow time course of NO action (see Fig. 3).
Illumination without caged-NO had no effect other than a 30 ms transient increase in current (Hsu et al. 1999), indicating that the changes in K+ current were not an artifact of illumination. In addition, there were no changes in the K+ current when the caged-NO solution was illuminated with UV light prior to filling the patch pipette, thus ruling out a non-specific action of other products of photolysis.
Photorelease of NO had similar effects on K+ current in 25 nerve terminals although the magnitude of these effects varied. The mean increase in the final current after five rapid UV flashes was 70 ± 8 %, while the mean decrease in the peak current was 15 ± 2 % (Fig. 2B). With further uncaging of NO (up to 10 light flashes) the final current increased by as much as 2- to 3-fold (n = 4), and thereafter decreased with further illumination. Note that the concentration of caged-NO in the pipette solution was 4 mm in these experiments so that further illumination caused the release of more NO. However, since NO decays rapidly (Moncada et al. 1991), [NO] was unlikely to rise above the 400 μm level generated by the five rapidly applied light flashes (< 3 s). Thus, the further enhancement of K+ current by additional uncaging of NO most probably reflects the cumulative effect of successive [NO] rises.
Effects of NO on different kinetic components of K+ current
Macroscopic K+ current in pituitary nerve terminals inactivates with biexponential kinetics. Previous studies have shown that the rapidly inactivating component is an A-current while the slowly inactivating and sustained components are primarily carried by a BK current (Bielefeldt et al. 1992). A delayed rectifier channel also makes a small contribution to the sustained current but is less well characterised. Figure 3A illustrates the effect of NO on the individual kinetic components of macroscopic K+ current. Prior to uncaging of NO, the K+ current evoked by a 400 ms depolarisation from −120 to +50 mV could be well fitted with a double exponential function (see legend to Fig. 3) yielding rapidly inactivating, slowly inactivating, and sustained components. The continuous curve (Fig. 3A) shows the best fit to this function. Similar analysis from eight experiments showed that NO (≤ 400 μm) increased the amplitude of the sustained component, A0, by 83 ± 28 %, which is consistent with the action of NO on final current described above. NO also reduced the amplitude of the rapidly inactivating component, A1, by 35 ± 7 % (Fig. 3B) and slowed its rate of decay by 60 ± 23 %. NO slightly decreased the amplitude of the slowly inactivating component, A2 (by 22 ± 8 %), and its rate of inactivation by 124 ± 36 %.
Figure 3. Effect of NO on the different kinetic components of whole-terminal K+ current.
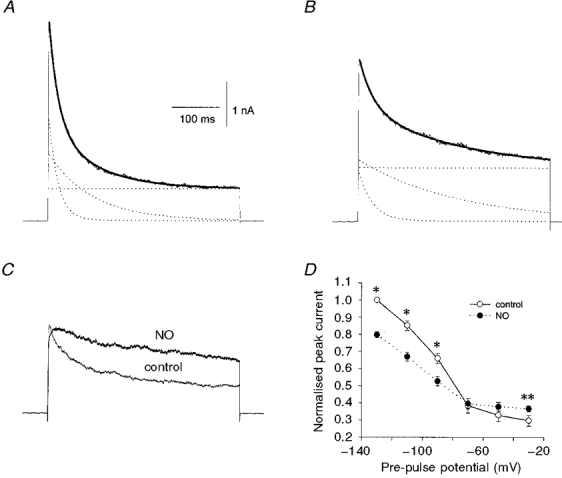
The reduction in peak K+ current by NO was dependent on the holding potential. Much greater effects were seen on K+ currents evoked following pre-pulses to −120 mV (as in Fig. 3A and B), than following more positive pre-pulses (Fig. 3C). Peak current was unchanged (−70 and −50 mV) or enhanced (−30 mV, P < 0.05) with more positive pre-pulses (Fig. 3C and D). This effect is probably due to voltage-dependent inactivation of the rapidly inactivating A-current (Bielefeldt et al. 1992) (50 % inactivation at μ-85 mV). At potentials more positive than −60 mV the A-current makes little contribution to the overall K+ current (μ6 % of peak current at −30 mV; Bielefeldt et al. 1992) so that the inhibitory action of NO on this channel is mostly occluded. As a result, when nerve terminals are held near the resting potential the most prominent effect of NO is the enhancement of BK current (note that the effect of NO on the voltage dependence of final current is described below, Fig. 8).
Figure 8. NO activation of final K+ current is independent of voltage.
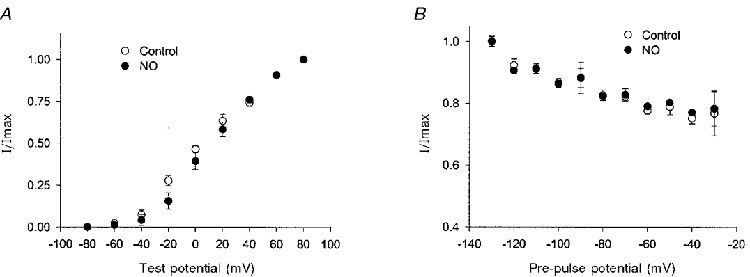
The final K+ current reflects BK current with essentially no contribution from A-current. A, amplitude of final K+ current following 500 ms voltage steps from −120 mV to various test potentials, control (n = 3 terminals), and after (n = 5 terminals) NO (< = 400 μm). B, plot of final current elicited by a 500 ms pulse to +50 mV versus pre-pulse potential, control (n = 6 terminals), and after (n = 4 terminals) NO. In A and B, current amplitudes were normalised to the maximum value.
Time course of NO action
The increase in the BK current usually followed uncaging after a delay of more than 30-60 s and the current reached a final plateau several minutes later. In contrast, the action of NO on the A-current was rapid. Figure 4 shows an example of the time course of NO action. Immediately after raising [NO] to approximately 80 μm (Fig. 4A, arrow), the peak current was reduced but there was no change in the amplitude of the final current. Figure 4B shows K+ current following two and three flashes of light. The time courses of peak and final K+ currents are shown in Fig. 4C. After two further light flashes each liberating 80 μm NO (Fig. 4B and C), the final current increased gradually with time. Even after the third light flash the rise in the final current took μ10 min to stabilise (Fig. 4C). In general, we found that the increase in final current was maintained for the lifetime of a whole-terminal recording (μ15-30 min), showing no signs of recovery. In contrast, the peak current decreased rapidly after uncaging but often showed a slight increase later in the experiment (for instance after the first and second flashes in Fig. 4C). This is probably not a recovery in A-current but rather is likely to be due to a compensatory increase in BK current (see Fig. 3). The observation that peak current, but not late current, is modified after a single light flash suggests that NO has kinetically distinct actions on the two major channels responsible for K+ current in this preparation. In the rest of this study, we focused our attention on the BK channel.
Figure 4. Time course of NO modification of K+ current.
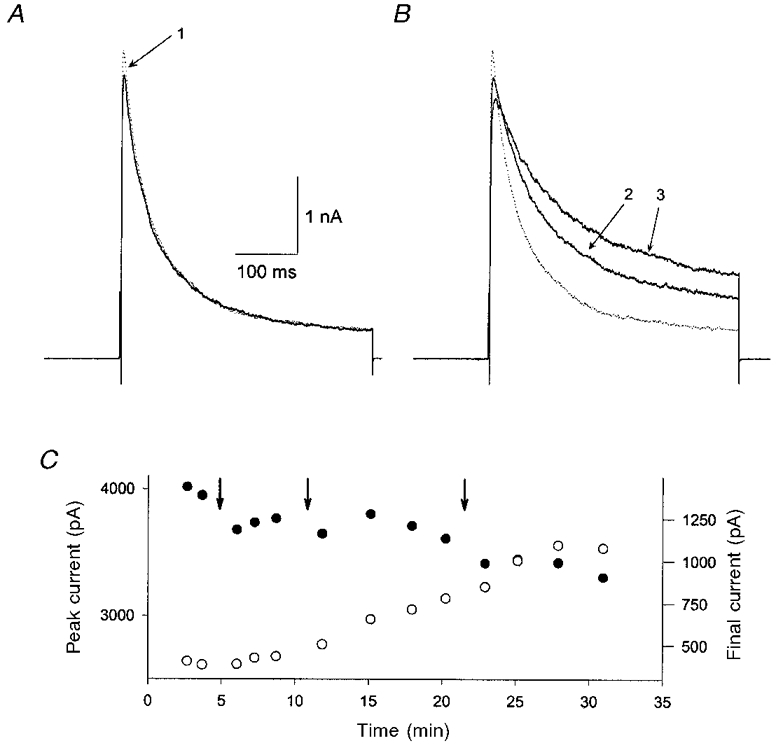
A and B, whole-terminal K+ currents evoked by a 400 ms depolarisation to +50 mV from a pre-pulse potential of −120 mV. A shows the response immediately after the first light flash (1) used to raise [NO] to 80 μm. B shows the current after a second (2) and third (3) light flash. C, plot showing the amplitudes of the peak (•) and final (○) K+ currents versus time for the same experiment shown in A. Arrows indicate illumination with a single flash. Note that the final current increased slowly and continued to rise for about 10 min after the third light flash whereas the peak current was inhibited immediately after the first light flash.
NO activates single BK channels
The analysis of whole-terminal K+ current above suggests that current through BK channels is enhanced by NO. Since the BK channel is readily identified by its large conductance, we tested the effects of NO on single K+ channel activity in excised inside-out patches.
In 150 mm/140 mm (bath/pipette) K+ solutions, BK channels exhibited outward rectification with slope conductances of 162 and 257 pS at negative and positive holding potentials, respectively (Fig. 5B). As shown in Fig. 5A, these channels were sensitive to free [Ca2+], and were reversibly inhibited when [Ca2+] was decreased from 100 μm to 1 nM (nominal). A plot of Poversus[Ca2+] averaged from nine patches revealed a half-maximal response of μ0.7 μm at a holding potential of +30 mV. (This value is higher than a previously reported value of μ250 nM at +40 mV (Bielefeldt & Jackson, 1993), because here [Ca2+] was determined using a Ca2+-selective electrode. We found a 2- to 3-fold difference in measured and theoretically predicted calcium over the range 10−7 to 10−5 M.)
Figure 5. Single channel properties of nerve terminal BK channels.
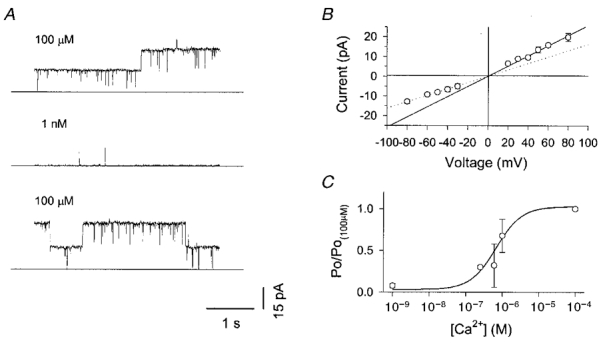
A, Ca2+-sensitive BK channel activity in an inside-out patch held at +60 mV. The free [Ca2+] in the bath solution was sequentially changed between 100 μm and 1 nM as indicated, by perfusion with Ca2+-EGTA-buffered solutions. B, single channel current-voltage relationship from 10 patches with 150 mm/140 mm (bath/pipette) K+ and > 1 μm cytoplasmic free Ca2+. The channels exhibited modest outward rectification. Slope conductances at negative and positive potentials were 162 and 257 pS, respectively. C, normalised open probability versus free [Ca2+] obtained from 9 patches. The continuous curve represents the best fit to a Hill equation with a KA of 680 nM and a Hill coefficient of 1.2.
We also observed large conductance channels with little to no Ca2+ sensitivity in at least 40 % of inside-out patches (data not shown). These channels exhibited a similar conductance (μ250 pS at positive potentials) to the Ca2+-sensitive channels. Whether this channel is a separate class of channel or the result of modification of normally Ca2+-sensitive channels is unclear. The results described below suggest that these Ca2+-insensitive channels might be the result of endogenous NO-induced modification.
Figure 6A shows the result of raising [NO] to ≤ 240 μm (3 flashes instead of the usual 5) in an inside-out patch containing a single BK channel (holding potential +30 mV and < 1 nM free Ca2+). NO increased channel activity in a time-dependent fashion: the initial Po was 0.004 but increased to 0.11 and 0.97, 30 and 90 s after uncaging, respectively. Amplitude histograms of data before and after uncaging show that NO did not alter the amplitude of the single channel current (Fig. 6B). Figure 6D shows that NO (80 to ≤ 400 μm) activated BK channels in four of four inside-out patches (P < 0.05) and in five of five outside-out patches (P < 0.05) with < 1 nM free Ca2+. Note that in these experiments caged-NO was photolysed in the pipette solution. This demonstrates that NO is active when released from either side of the membrane and is consistent with NO being a highly membrane-permeant gas. In general, maximal activation of BK channels occurred 1–3 min after uncaging of NO, as in Fig. 6A. Thus, the time course of single channel activation in excised patches was similar, if somewhat faster, to that of the late component of K+ current in whole-terminal recordings (Fig. 4). In some patches photorelease of NO was immediately followed by the activation of previously quiescent channels. An example of this is shown in Fig. 6C, where a second BK channel with high Po was activated within seconds of the UV pulse (arrow).
Figure 6. NO and NO donors activate single BK channels in excised patches.
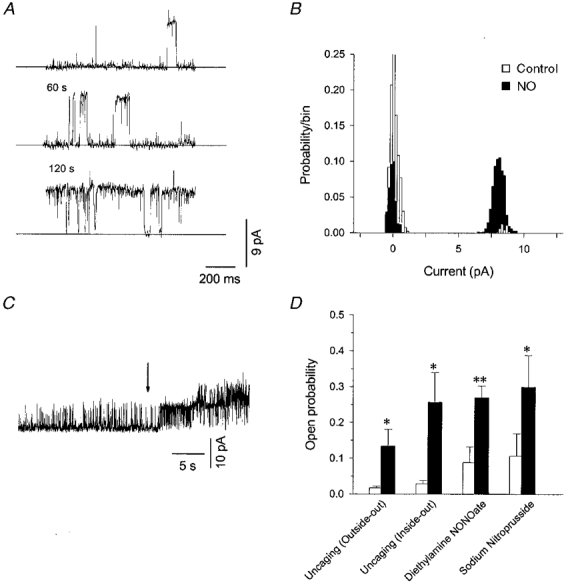
A, activity from a single BK channel in an excised (inside-out) patch before, and 60 and 120 s after uncaging of NO (< = 240 μm). The holding potential was +30 mV and free [Ca2+] was nominally 1 nM. B, amplitude histogram for the same channel as in A showing that the NO-induced modification did not affect the open channel amplitude (≈8 pA). C, continuous record of activity from an excised patch containing at least two BK channels showing that one channel with a high Po was activated shortly after uncaging (arrow). The patch was held at +30 mV and the bath contained 100 μm free Ca2+. D, summary of the effects of caged-NO and NO donors on BK channel activity (▪); *P < 0.05, **P < 0.01, compared with control (□). The differences in control Po are due to the different [Ca2+] used. The free [Ca2+] on the cytosolic face of the membrane was 1 and 160 nM for caged-NO and donor experiments, respectively. The full ionic composition is given in the Methods.
Effects similar to those seen with caged-NO were observed with NO donors. Both diethylamine NONOate (230 μm to 1 mm, 5 patches, P < 0.01) and sodium nitroprusside (5–8 mm, 5 patches, P < 0.05) increased channel Po in inside-out patches with μ160 nM free Ca2+ in the bath. Note that control channel activity was greater in these NO donor experiments compared with NO-uncaging experiments due to the higher free [Ca2+]. In addition, ATP and GTP were omitted from the bath solution in these experiments, indicating that NO activation of channels did not depend on channel phosphorylation or G-protein activation. We also tested another commonly used NO donor, S-nitrosocysteine (SNC) (Broillet & Firestein, 1997). Although SNC activated BK channels in three of four experiments, we also found that it caused a partial, reversible inhibition of whole-terminal and single-channel K+ currents. This inhibitory action of SNC was not likely to involve NO and may be attributed to the end-product L-cysteine, since it persisted long after the NO-releasing lifetime of the donor (μ3 h). Due to the complex actions of SNC, these data were not included in this study.
NO activation is independent of Ca2+
One mechanism by which NO may activate BK channels is by altering their Ca2+ sensitivity. Figure 7A shows representative BK channel activity from an inside-out patch at three different Ca2+ concentrations both before and after NO uncaging. Under control conditions, most of the activity was due to a single BK channel, although occasional superimposed activity from a second channel could also be seen at 100 μm Ca2+. The activity of the main channel was very Ca2+ dependent, with Po close to 0 and 1 when the free [Ca2+] in the bath solution was 1 nM and 100 μm, respectively. However, after uncaging, the channel was highly active at all [Ca2+]. A plot of Poversus[Ca2+] for this channel (Fig. 7B) shows that NO increased Po to close to unity at all [Ca2+] and essentially removed the Ca2+ dependence of activation. This effect was seen in three of five patches which appeared to have a single channel. Figure 7C shows a plot of normalised Poversus[Ca2+] from nine patches containing either single or multiple BK channels. NO produced a significant increase in activity at all [Ca2+]. Unlike the single channel shown in Fig. 7A and B, NO increased Po at 100 μm due to activation of quiescent channels and because Po under control conditions was often less than unity. The Ca2+ sensitivity apparent in the averaged post-NO data probably reflects the fact that a fraction of channels were not modified by NO. These results indicate that NO activates channels independently of their Ca2+-sensitive properties, and further shows that NO modification nearly eliminates the Ca2+ requirement for channel activation.
Figure 7. NO activation of BK channels is Ca2+ independent.
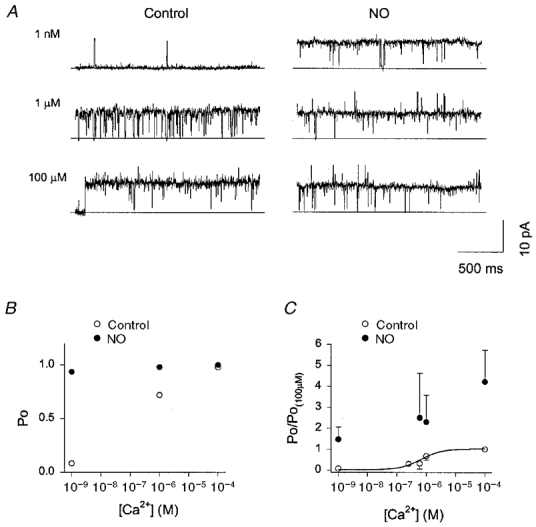
A, activity from an inside-out patch with varying bath [Ca2+], before (Control) and after uncaging of NO. The majority of activity was due to a single channel which exhibited a strong Ca2+ dependence under control conditions. Occasional superimposed activity from a second channel can be seen at 100 μm Ca2+. After NO uncaging the first channel was highly active at all [Ca2+]. B, Poversus[Ca2+] plot for the main channel in A showing that NO removed the Ca2+ dependence of activity and increased Po to close to unity. C, normalised Poversus[Ca2+] from 9 inside-out patches showing that NO increased activity at all [Ca2+]. Note that, unlike the single channel shown in A and B, NO increased channel activity at 100 μm Ca2+. This reflected both an increase in the number of active channels and an increase in Po (which was less than unity under control conditions).
NO does not affect voltage-dependent activation and inactivation
Previous studies have shown that the sustained component of the whole-terminal K+ current exhibits voltage-dependent activation and inactivation (Bielefeldt et al. 1992). Therefore, we considered the possibility that NO modulated the BK channel by altering its voltage dependence. Figure 8A shows a plot of the final K+ current versus voltage, before and after NO. The amplitudes were normalised to the maximum value in order to focus on the voltage dependence. This plot shows that NO had little effect on the voltage-dependent activation of the K+ current. Similarly, NO had little effect on the relative extent of inactivation observed in the final current following pre-pulses ranging from −130 to −30 mV (Fig. 8B). Thus, the actions of NO appear to be independent of the voltage-sensitive properties of the BK channel.
NO signalling pathway
Many of the cellular effects of NO are mediated by soluble guanylyl cyclase (GC) and cGMP (Moncada et al. 1991; Garthwaite & Boulton, 1995). These signalling pathways may underlie the NO-mediated activation of BK current in whole-terminal recordings since both ATP and GTP were included in the pipette solution. However, NO was still able to activate single channels in excised patches in the absence of ATP and GTP, suggesting a GC-independent action. As a further test of cGMP signalling in the activation of whole-terminal K+ current by NO, we measured the response to NO in the presence of specific GC inhibitors or with no ATP and GTP in the patch pipette. Figure 9 shows that NO activation of whole-terminal K+ current was unaffected by removal of ATP and GTP (7 terminals) from the pipette solution or by replacement of ATP with the non-hydrolysable ATP analogue, AMP-PNP (β,γ-imido adenosine triphosphate; 4 mm, 2 terminals). In addition, the specific GC inhibitors ODQ (4 terminals) and LY-83,583 (4 terminals) did not affect the response to NO (Fig. 9). Thus, both whole-terminal and single-channel experiments are consistent with the hypothesis that NO actions on BK channels are independent of GC and cGMP signalling pathways.
Figure 9. NO activation is independent of cGMP and phosphorylation.
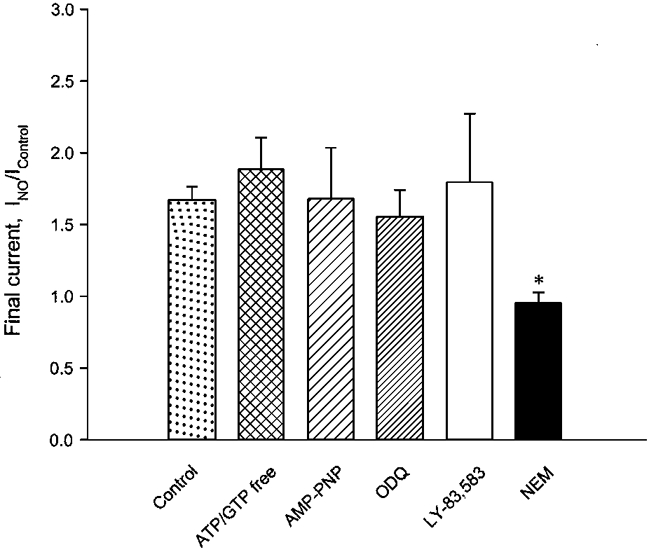
Mean (±s.e.m.) fractional increase in final K+ current after NO uncaging under various conditions: control (25 terminals; from Fig. 2B), omission of ATP and GTP from the pipette solution (7 terminals), replacement of ATP with the non-hydrolysable analogue AMP-PNP (4 mm, 2 terminals), inclusion of the specific guanylyl cyclase inhibitors ODQ (16 μm, 4 terminals) or LY-83,583 (17 μm, 4 terminals) in the pipette solution, and bath application or inclusion of NEM (0.5–5 mm, 6 terminals) in the pipette solution. Data are the maximal response obtained after photorelease of < = 400 μm NO. NEM treatment alone prevented NO activation (*P < 0.001, compared with control). Note that the control measurement in the NEM experiment is current after NEM enhancement (see Fig. 10), and this result means that NO caused no further enhancement beyond that caused by NEM.
NEM mimics and occludes the effect of NO
Because NO actions on non-haemoproteins are thought to depend on sulfhydryl groups (Arnelle & Stamler, 1995), we tested the effect of the sulfhydryl alkylating agent N-ethylmaleimide (NEM). Pre-treatment with NEM (0.5–5 mm, 6 terminals) in either the pipette or bath solution fully prevented activation by NO (Figs 9 and 10, P < 0.001). Furthermore, bath application of NEM alone (0.5–5 mm) produced an approximately 2-fold increase in the final K+ current (Fig. 10A and C) with a mean increase of 93 ± 2 % (4 terminals, P < 0.05), without altering the peak current. NEM also increased the activity of BK channels in excised patches (Fig. 10B), with a mean increase of 53 ± 13 % (Fig. 10C, 3 patches, P < 0.05). Taken together, these results strongly suggest that NO or NO byproducts act via modification of sulfhydryl groups on BK channels or on a closely associated molecule. Interestingly, the enhancement in channel activity produced by NEM alone contrasts with the result seen in smooth muscle, where NEM reduced channel activity (Bolotina et al. 1994).
Figure 10. NEM increases whole-terminal K+ current and single BK channel activity.
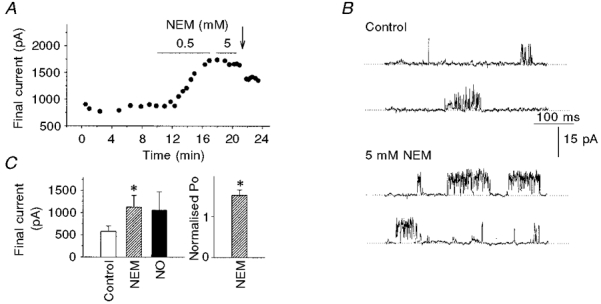
A, plot of final current versus time from a single experiment. Perfusion of 0.5 mm NEM into the bath solution produced a nearly 2-fold increase in current. No further activation was observed with 5 mm NEM, suggesting that the effect was already saturated. NEM also prevented subsequent activation by NO; in this case photolysis of caged-NO (arrow) reduced the final K+ current. B, single channel activity of a BK channel in an inside-out patch held at −60 mV. NEM (5 mm) increased Po from 0.16 to 0.26. C, left panel: summary of effects of bath perfusion of NEM (0.5–5 mm). NEM increased the final current from 580 ± 120 to 1120 ± 270 pA (4 terminals, *P < 0.05, compared with Control). In two of these experiments, further NO treatment had little effect. Right panel, mean Po (normalised to control) for 3 patches after treatment with 5 mm NEM (*P < 0.05, compared with control).
DISCUSSION
These results show that NO enhances BK channel activity in the peptidergic terminals of the posterior pituitary. Photolysis of caged-NO increased the sustained component and slowed the rate of decay of whole-terminal K+ current. NO also increased single BK channel activity in excised patches. In contrast, the transient A-current was reduced by NO, although at physiological voltages the reduction in this current was mostly compensated for by the increase in BK current, to leave the peak only slighty reduced (Fig. 3C). The enhancement of this current is consistent with recent studies describing NO activation of BK channels in brain synaptosomes (Shin et al. 1997) and smooth muscle (Bolotina et al. 1994), although the latter study reported different effects with NEM. Collectively, these results suggest that BK channels may be important neuronal targets for NO and may contribute to NO regulation of neurosecretion.
An important and novel aspect of this report is that NO activates BK channels independently of their voltage- and Ca2+-dependent properties. Instead, NO increases the activity of channels at all voltages and [Ca2+], and activates previously quiescent channels. Interestingly, NO modification abolishes the Ca2+ requirement for channel activity. This raises the possibility that NO modification in vivo will activate BK channels independently of the high frequency stimulation normally needed to raise cytosolic Ca2+. Thus, NO-modified BK channels could generally inhibit action potential propagation in terminals rather than merely terminating bursts of activity (Bielefeldt & Jackson, 1993, 1994).
Mechanism of action
Although NO is known to activate the cGMP signalling cascade (Moncada et al. 1991; Garthwaite & Boulton, 1995), this pathway does not appear to be involved in the NO activation of BK channels. The NO action persisted in the presence of GC inhibitors and under MgATP-GTP free conditions. NO also activated BK channels in excised patches in the absence of ATP and GTP. Moreover, NEM, a reagent that specifically alkylates free sulfhydryl groups, mimicked the effects of NO and occluded subsequent NO activation. These results suggest that NO, or a related reactive nitrogen species, modifies sulfhydryl groups on the BK channel protein or a closely associated protein. Further evidence in favour of chemical modification is the dramatic alteration in channel Ca2+ sensitivity following NO treatment. This suggests that NO can induce a conformational change in the protein either directly or allosterically in a way which mimics the action of Ca2+ binding. Although our results support a direct action of NO rather than via cGMP signalling, it remains possible that the latter pathway could contribute to regulation of BK channels in vivo since soluble guanylyl cyclase may have been washed out in our experiments.
It is unclear whether the reactive species responsible for the chemical modification is NO itself or a byproduct. In aqueous solution, NO auto-oxidises to form a number of biologically active species including NO2, N2O3 and N2O4 (Wink et al. 1996). The involvement of a NO byproduct is suggested by the short lifetime of NO (μ5 s in biological solutions), which is difficult to reconcile with the slow time course of activation seen in both whole-terminal and single-channel experiments. However, the results are also consistent with an alternative hypothesis that the rate-determining step in channel activation may occur subsequent to protein nitrosylation. While NO is known to oxidise vicinal thiols to form disulfides (Pryor et al. 1982), the oxidation reaction is believed to occur via formation of an intermediate S-nitrosothiol (Arnelle & Stamler, 1995). Furthermore, it is unclear whether S-nitrosothiol can be formed from NO alone or from a reaction with another NO byproduct such as N2O3 (Kharitonov et al. 1995). Whatever the case, it is likely that one or more reactive nitrogen intermediates are involved in a process that culminates in the chemical modification of a protein.
Interestingly, the activation of BK channels by NEM reported here is opposite to the results obtained by Bolotina et al. (1994). They found that NEM inhibited BK channel activity whereas we found that NEM enhanced BK channel activity. The different results seen here may be related to tissue-specific differences in BK channel structure, or differential effects of NO on distinct sulfhydryl groups. On the other hand, our results are consistent with recent reports in other channels. For example, Broillet & Firestein (1996) have reported that NO can activate cyclic nucleotide-gated channels. As in the present study, this effect could be reproduced by NEM treatment with occlusion of subsequent NO activation. Ryanodine receptor channels are also activated by S-nitrosothiols and NEM (Xu et al. 1998), and this effect is associated with a reduction in the number of free thiol groups on the channel protein. Thus, modification of protein sulfhydryl groups by reactive nitrogen species may represent a widely used pathway for ion channel modulation.
Implications for secretion
Our results suggest a mechanism by which NO reduces evoked secretion of the neuropeptides ADH and OT. Activation of BK channels even at low [Ca2+] and the overall increase in K+ current should depress the excitability of the terminals either by inducing a hyperpolarisation or by providing a current shunt. Other manipulations that enhance BK channel activity in this preparation, including enhanced calcium entry and enhanced protein phosphorylation, lead to action potential failure (Bielefeldt & Jackson, 1993, 1994), and presumably to a reduction in secretion as well. Thus, NO should reduce action potential invasion of the terminal arborisation. Interestingly, the bimodal and biphasic action of NO on the different K+ currents raises the possibility of a more complex regulation. The slow time course of activation of the BK channel by NO would imply that the inhibition of evoked release will occur after a delay of a few minutes. In contrast, the reduction of the A-current by NO occurred much more rapidly (Fig. 3). The A-current has been shown to inactivate during high frequency electrical activity, to broaden action potentials and facilitate calcium entry (Jackson et al. 1991). Reduction of the A-current by NO would therefore enhance secretion by broadening action potentials. Because the action of NO on the A-current developed within a few seconds, the initial effect of NO production would be to enhance release. The blockade of action potentials produced by BK channel opening would terminate this effect minutes later. Thus, the two opposing effects of NO on the two principal K+ channels in neurohypophysial membranes may reflect initiation and termination components of essentially one response. The previous reports that NO inhibits secretion of neurohypophysial peptides may reflect the fact that these assays were done without taking a time course, so that early and delayed effects could not be resolved.
Acknowledgments
We thank Dr Jeff Walker for helpful comments on the chemistry of caged compounds and Dr Ed Chapman for use of a Ca2+-selective electrode. This research was supported by NIH grant NS 30016 to M.B.J.
References
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnelle DR, Stamler JS. NO+, NO, and NO− donation by S-nitrosothiols: implications for regulation of physiological functions by S-nitrosylation and acceleration of disulfide formation. Archives of Biochemistry and Biophysics. 1995;318:279–285. doi: 10.1006/abbi.1995.1231. [DOI] [PubMed] [Google Scholar]
- Bettache N, Carter T, Corrie JE, Ogden D, Trentham DR. Photolabile donors of nitric oxide: ruthenium nitrosyl chlorides as caged nitric oxide. Methods in Enzymology. 1996;268:266–281. doi: 10.1016/s0076-6879(96)68029-x. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. A calcium-activated potassium channel causes frequency-dependent action-potential failures in a mammalian nerve terminal. Journal of Neurophysiology. 1993;70:284–298. doi: 10.1152/jn.1993.70.1.284. [DOI] [PubMed] [Google Scholar]
- Bielefeldt K, Jackson MB. Phosphorylation and dephosphorylation modulate a Ca2+-activated K+ channel in rat peptidergic nerve terminals. The Journal of Physiology. 1994;475:241–254. doi: 10.1113/jphysiol.1994.sp020065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielefeldt K, Rotter JL, Jackson MB. Three potassium channels in rat posterior pituitary nerve terminals. The Journal of Physiology. 1992;458:41–67. doi: 10.1113/jphysiol.1992.sp019405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Bredt DS, Hwang PM, Snyder SH. Localization of nitric oxide synthase indicating a neural role for nitric oxide. Nature. 1990;347:768–770. doi: 10.1038/347768a0. [DOI] [PubMed] [Google Scholar]
- Broillet MC, Firestein S. Direct activation of the olfactory cyclic nucleotide-gated channel through modification of sulfhydryl groups by NO compounds. Neuron. 1996;16:377–385. doi: 10.1016/s0896-6273(00)80055-0. [DOI] [PubMed] [Google Scholar]
- Broillet MC, Firestein S. β subunits of the olfactory cyclic nucleotide-gated channel form a nitric oxide activated Ca2+ channel. Neuron. 1997;18:951–958. doi: 10.1016/s0896-6273(00)80334-7. [DOI] [PubMed] [Google Scholar]
- Chiodera P, Volpi R, Coiro V. Inhibitory control of nitric oxide on the arginine-vasopressin and oxytocin response to hypoglycaemia in normal men. NeuroReport. 1994;5:1822–1824. doi: 10.1097/00001756-199409080-00034. [DOI] [PubMed] [Google Scholar]
- Eriksson S, Appelgren B, Rundgren M, Andersson B. Vasopressin release in response to intracerebroventricular L-alanine and L-arginine, and its dependence upon CSF NaCl concentration. Acta Physiologica Scandinavica. 1982;116:75–81. doi: 10.1111/j.1748-1716.1982.tb10601.x. [DOI] [PubMed] [Google Scholar]
- Garthwaite J, Boulton CL. Nitric oxide signaling in the central nervous system. Annual Review of Physiology. 1995;57:683–706. doi: 10.1146/annurev.ph.57.030195.003343. [DOI] [PubMed] [Google Scholar]
- Goyer M, Bui H, Chou L, Evans J, Keil LC, Reid IA. Effect of inhibition of nitric oxide synthesis on vasopressin secretion in conscious rabbits. American Journal of Physiology. 1994;266:H822–828. doi: 10.1152/ajpheart.1994.266.2.H822. [DOI] [PubMed] [Google Scholar]
- Hsu S-F, Ahern GP, Jackson MB. Ultra-violet light-induced changes in membrane properties in secretory cells. Journal of Neuroscience Methods. 1999 doi: 10.1016/s0165-0270(99)00071-0. in the Press. [DOI] [PubMed] [Google Scholar]
- Hsu S-F, Augustine GJ, Jackson MB. Adaptation of Ca(2+)-triggered exocytosis in presynaptic terminals. Neuron. 1996;17:501–512. doi: 10.1016/s0896-6273(00)80182-8. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Konnerth A, Augustine GJ. Action potential broadening and frequency-dependent facilitation of calcium signals in pituitary nerve terminals. Proceedings of the National Academy of Sciences of the USA. 1991;88:380–384. doi: 10.1073/pnas.88.2.380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadowaki K, Kishimoto J, Leng G, Emson PC. Upregulation of nitric oxide synthase (NOS) gene expression together with NOS activity in the rat hypothalamo-hypophysial system after chronic salt loading: evidence of a neuromodulatory role of nitric oxide in arginine vasopressin and oxytocin secretion. Endocrinology. 1994;134:1011–1017. doi: 10.1210/endo.134.3.7509733. [DOI] [PubMed] [Google Scholar]
- Kharitonov VG, Sundquist AR, Sharma VS. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. Journal of Biological Chemistry. 1995;270:28158–28164. doi: 10.1074/jbc.270.47.28158. [DOI] [PubMed] [Google Scholar]
- Lutz-Bucher B, Koch B. Evidence for an inhibitory effect of nitric oxides on neuropeptide secretion from isolated neural lobe of the rat pituitary gland. Neuroscience Letters. 1994;165:48–50. doi: 10.1016/0304-3940(94)90706-4. [DOI] [PubMed] [Google Scholar]
- Miyagawa A, Okamura H, Ibata Y. Coexistence of oxytocin and NADPH-diaphorase in magnocellular neurons of the paraventricular and the supraoptic nuclei of the rat hypothalamus. Neuroscience Letters. 1994;171:13–16. doi: 10.1016/0304-3940(94)90592-4. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Murphy KP, Williams JH, Bettache N, Bliss TV. Photolytic release of nitric oxide modulates NMDA receptor-mediated transmission but does not induce long-term potentiation at hippocampal synapses. Neuropharmacology. 1994;33:1375–1385. doi: 10.1016/0028-3908(94)90039-6. [DOI] [PubMed] [Google Scholar]
- Ota M, Crofton JT, Festavan GT, Share L. Evidence that nitric oxide can act centrally to stimulate vasopressin release. Neuroendocrinology. 1993;57:955–959. doi: 10.1159/000126459. [DOI] [PubMed] [Google Scholar]
- Pow DV. Immunocytochemical evidence for a glial localisation of arginine, and a neuronal localisation of citrulline in the rat neurohypophysis: implications for nitrergic transmission. Neuroscience Letters. 1994;181:141–144. doi: 10.1016/0304-3940(94)90579-7. [DOI] [PubMed] [Google Scholar]
- Pryor W, Church D, Govindan C, Crank G. Oxidation of thiols by nitric oxide and nitrogen dioxide: Synthetic utility and toxicological implications. Journal of Organic Chemistry. 1982;47:156–159. [Google Scholar]
- Raber J, Bloom FE. IL-2 induces vasopressin release from the hypothalamus and the amygdala: role of nitric oxide-mediated signaling. Journal of Neuroscience. 1994;14:6187–6195. doi: 10.1523/JNEUROSCI.14-10-06187.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rees DD, Palmer RM, Moncada S. Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proceedings of the National Academy of Sciences of the USA. 1989;86:3375–3378. doi: 10.1073/pnas.86.9.3375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuman EM, Madison DV. Nitric oxide and synaptic function. Annual Review of Neuroscience. 1994;17:153–183. doi: 10.1146/annurev.ne.17.030194.001101. [DOI] [PubMed] [Google Scholar]
- Shin JH, Chung S, Park EJ, Uhm DY, Suh CK. Nitric oxide directly activates calcium-activated potassium channels from rat brain reconstituted into planar lipid bilayer. FEBS Letters. 1997;415:299–302. doi: 10.1016/s0014-5793(97)01144-7. [DOI] [PubMed] [Google Scholar]
- Summy-Long JY, Bui V, Mantz S, Koehler E, Weisz J, Kadekaro M. Central inhibition of nitric oxide synthase preferentially augments release of oxytocin during dehydration. Neuroscience Letters. 1993;152:190–193. doi: 10.1016/0304-3940(93)90515-m. [DOI] [PubMed] [Google Scholar]
- Walker JW, Martin H, Schmitt FR, Barsotti RJ. Rapid release of an alpha-adrenergic receptor ligand from photolabile analogues. Biochemistry. 1993;32:1338–1345. doi: 10.1021/bi00056a020. [DOI] [PubMed] [Google Scholar]
- Wang G, Thorn P, Lemos JR. A novel large-conductance Ca(2+)-activated potassium channel and current in nerve terminals of the rat neurohypophysis. The Journal of Physiology. 1992;457:47–74. doi: 10.1113/jphysiol.1992.sp019364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wink DA, Grisham MB, Mitchell JB, Ford PC. Direct and indirect effects of nitric oxide in chemical reactions relevant to biology. Methods in Enzymology. 1996;268:12–31. doi: 10.1016/s0076-6879(96)68006-9. [DOI] [PubMed] [Google Scholar]
- Xu L, Eu JP, Meissner G, Stamler JS. Activation of the cardiac calcium release channel (ryanodine receptor) by poly-S-nitrosylation. Science. 1998;279:234–237. doi: 10.1126/science.279.5348.234. [DOI] [PubMed] [Google Scholar]