

Abstract
A study was undertaken to examine the influence of acute renal perfusion pressure (RPP) reduction on renin release, renal renin and angiotensinogen gene expression and the role played by angiotensin II in these responses.
In chloralose-urethane anaesthetised rats, reduction of RPP to 60 mmHg for 3 h in vehicle or losartan-treated (5 days at 10 mg kg−1 bis in die (b.i.d.)) rats decreased renal blood flow by 46 and 29% (both P < 0.001), respectively, glomerular filtration rate by 45 and 57% (both P < 0.001), respectively, and sodium excretion by 96 and 98% (both P < 0.01).
Chloralose-urethane anaesthesia and surgery caused a rise in plasma renin activity but was associated with a suppression of renal renin (50%, P < 0.01) and angiotensinogen (40%, P < 0.05) gene expression. Following reduction of RPP to 60 mmHg for 3 h, plasma renin activity was increased more than 7-fold (P < 0.001) and renal renin gene expression about 2-fold (P < 0.05).
Chronic (5 days) blockade of angiotensin II receptors with losartan elevated plasma renin activity some 29-fold (P < 0.001) and caused a marked increase (30-fold, P < 0.05) in renal renin gene expression, compatible with angiotensin II exerting a negative feedback control on renin release and gene expression. Reduction of RPP to 60 mmHg for 3 h in these animals had little effect on renal renin gene expression.
From these findings it can be concluded that (a) chloralose-urethane anaesthesia and surgery had a stimulatory effect on renin release but suppressed basal levels of renal renin and angiotensinogen gene expression; (b) acute reduction of RPP for 3 h could stimulate renin gene expression in the renin producing cells; and (c) the negative feedback control of angiotensin II on renin release and synthesis which was evident following chronic losartan treatment was not apparent during short-term reduction of RPP.
Renin is the primary component of the renin-angiotensin system (RAS) which is produced mainly in the kidney by the juxtaglomerular (JG) cells, located in the walls of afferent arterioles (Johns, 1989). The renin gene produces renin mRNA which is translated to pre-prorenin by ribosomes on the endoplasmic reticulum, converted into prorenin by removal of the signal peptide, glycosylated and incorporated into the protogranules by Golgi bodies. Some of the protogranules are packaged and secreted immediately via a constitutive pathway giving rise to prorenin, or inactive renin, within the circulation, while in others, the prorenin is processed further to renin and stored in mature granules, from which it is released via a regulatory pathway as active renin on demand (Morris, 1992; Skott & Jensen, 1993; Tamur et al. 1995). Angiotensinogen (AGT) is the unique substrate for renin, and the primary source of plasma AGT is the liver where it is constitutively secreted into the circulation once synthesised by the hepatocytes. The AGT gene is expressed in many tissues, including those of the brain, spinal cord, aorta, kidney and adrenal gland where AGT appears to play a role in regulating local angiotensin II (Ang II) production (Tamura et al. 1995; Morgan et al. 1996).
A number of investigators (Goldblatt et al. 1934; Witty et al. 1971; Kirchheim et al. 1987) suggested the existence of a renal baroreceptor which stimulated renin release when renal perfusion pressure (RPP) was decreased below the autoregulatory range, and that this phenomenon was independent of the macula densa mechanism and renal nerve-mediated release. However, reduction of RPP may be accompanied by decreases in renal blood flow (RBF) and the latter may also contribute to the stimulation of renin release. In a recent study, Nafz et al. (1997), using conscious dogs, were able to dissociate alterations in RBF from changes in RPP and found that the responses of plasma renin activity (PRA) correlated with the RPP stimulus but not with the RBF changes, and concluded that RBF played no major role in renin release. There is now general agreement that a baroreceptor mechanism for renin release exists in the kidney, and probably the JG cell itself is the sensor for changes in blood pressure mediated through regulation of transmembrane Ca2+ influx (Hackenthal et al. 1990; King et al. 1993).
The balance between renin release, depletion of granular stores and renin gene expression as a consequence of short-term changes in RPP has received little attention. Applying direct mechanical stretch for 20 h to cultured rat JG cells and human Calu-6 cells resulted in a reduction in baseline renin mRNA accumulation of 26 and 46 %, respectively (Carey et al. 1997). Therefore, a relatively long-term mechanical deformation of renin producing cells decreases renin gene expression. Aortic coarctation proximal to the renal arteries in rats for 24 h increased the percentage of JG apparati containing renin and its mRNA. In addition, recruitment of renin secreting cells was observed along the afferent arterioles. Hence, 24 h of RPP reduction can enhance renin gene expression (Tufro-McReddie et al. 1993). Taken together, these data show that stimulation of the renal baroreceptor mechanism for approximately 1 day could activate renin gene expression in granular cells but the question remains as to whether shorter time periods of RPP reduction might be effective.
The aim of this study was to investigate the impact of acute (3 h) RPP reduction on renin secretion and to determine whether this could influence the level of renin gene expression. In addition, the changes in renal haemodynamic and excretory variables, and the expression of renal AGT gene were also determined. Since it has been shown that Ang II itself has a powerful negative feedback control on renin secretion and gene expression (Johns, 1992; Schunkert et al. 1992), the responses of the mentioned variables to RPP reduction were also examined in rats chronically treated with losartan to block Ang II receptors in order to remove any potential influence of Ang II.
METHODS
Animal preparation
Wistar rats, 260-390 g, were fasted overnight prior to use and were anaesthetised with 2 % fluothane in a mixture of N2O and O2 (2 and 4 l min−1, respectively). After tracheostomy, a cannula was placed into the right femoral vein, and an infusion of normal saline (150 mm NaCl) was begun at 3 ml h−1 and continued at the same rate throughout the experiment. The gaseous anaesthetic was gradually replaced (over 30 min) by 0.7–0.9 ml of an intravenous (i.v.) chloralose and urethane mixture at a concentration of 12 and 180 mg ml−1, respectively, and supplemented every 30 min with 0.05 ml of the chloralose-urethane. Another cannula was inserted into the right femoral artery, such that its tip lay within the aorta, caudal to the exit of renal arteries, for measuring RPP. The left carotid artery was also cannulated for blood pressure measurement and blood sampling. The left kidney was exposed retroperitoneally, its ureter cannulated for collection of urine, and the left renal artery was carefully cleared and a flow probe (EP 100 series probe, 2.0–2.5 mm circumference, connected to a square-wave electromagnetic flowmeter FM 501, Carolina Medical Electronic Inc., King, NC, USA) placed around it for measurement of RBF. Arterial pressure, RPP and RBF were continuously recorded on an Apple Macintosh computer using LabVIEW 2 software (National Instruments, TX, USA). The left renal nerves were dissected distal to the coeliac ganglia, and then cut. The denervation of the kidney was tested by applying silver wire electrodes to the coeliac ganglia through which pulses of 15 V, 10 Hz, 0.2 ms duration were delivered from a stimulator for 10 s. If there was a significant fall in RBF, more dissection and cutting of renal nerves were done until no change of RBF was observed during stimulation. A thread was placed around the aorta, rostral to the level of the renal arteries, and was connected to a screw device which allowed the thread to be tightened to constrict the aorta such that RPP could be reduced to the desired value. On completion of surgery, a 2 ml primer containing inulin (15 mg ml−1 in normal saline) was given i.v. and the saline infusion was changed to one of saline with inulin (15 mg ml−1). The animals were allowed 2 h equilibration and then experiments were begun.
The experimental protocol consisted of two 15 min clearance periods, taken to establish baseline levels, then RPP remained unchanged or was gradually reduced to 60 mmHg by means of suprarenal aortic constriction and once the desired pressure had been achieved, 10 min stabilisation was allowed before two 85 min clearance periods were taken to establish experimental levels. Arterial blood samples (0.35 ml) were taken at the beginning of the first control and at the end of the second control period and at the beginning and end of each experimental clearance period for determination of inulin and electrolyte concentrations. The blood was collected into cooled syringes, quickly centrifuged, and the plasma was stored at −20°C until assayed. The red blood cells were resuspended in an equal volume of normal saline and reinfused into the animal and a 5 min equilibration period elapsed before continuing the experimental protocol. At the end of the experiment a 1 ml blood sample was taken using a cooled syringe containing EDTA (5 mg ml−1) and centrifuged at 2000 g at 0–4°C. The plasma was removed and deep frozen for later estimation of PRA. During the centrifugation of the 1 ml blood sample, a further large blood sample was taken, and both kidneys were removed and their renal capsules were taken off and frozen in liquid nitrogen as quickly as possible. The animals were then killed by injecting i.v. 1 ml sodium pentobarbitone.
The rats were divided into different groups (n = 6) as follows. In Sham and 60 mmHg groups, RPP was maintained at basal levels or reduced to 60 mmHg, respectively, during the 3 h experimental period. In further groups of rats, losartan (Ang II antagonist; Merck, NJ, USA) was injected i.p. at 10 mg kg−1 b.i.d. for 5 days. One group of the losartan-treated rats was subjected to reduction of RPP to 60 mmHg for 3 h (the Los-60 mmHg group). Injection (i.v.) of 0.1 ml Ang II (2 μg ml−1), before starting the 2 h equilibration period and at the end of the experiment, increased mean blood pressure by 15-20 mmHg in non-treated rats but had no effect on mean blood pressure when given to the losartan-treated rats. There were two control groups in which losartan-treated (the Los-Cnt group) or non-treated (Cnt) rats were anaesthetised with the fluothane-O2-N2O mixture, a 1 ml blood sample was quickly taken within 5 min of surgical anaesthesia, and thereafter kidneys were immediately removed and frozen as described above.
Analyses
Inulin
Inulin in plasma and urine was assayed as previously described (Johns et al. 1976), and GFR was calculated as the clearance of inulin. Plasma and urinary sodium concentrations were measured using a flame photometer (410C; Ciba-Corning, Essex, UK).
Plasma renin activity
PRA was measured using commercially available radioimmunoassay kits (RENCTK kit, Sorin Biomedica, Saluggia, Italy). Results are expressed as nanograms Ang I generated per millilitre plasma per hour (ng Ang I ml−1 h−1).
Northern blot analysis
The extraction of total RNA from half of each kidney was carried out using the LiCl-urea precipitation method (Samani et al. 1987). RNA concentrations were determined by spectrophotometry at 260 nm and the integrity checked by running samples on agarose gels stained with ethidium bromide.
For Northern blotting, 45 μg of RNA from each kidney was electrophoresed on 1.2 % agarose gels containing 2.2 M formaldehyde and 1 × formaldehyde gel running buffer (0.04 M Mops, 8 mm sodium acetate and 1 mm EDTA), and then transferred to nylon membranes (Hybond-N; Amersham International, Buckinghamshire, UK) (Sambrook et al. 1989).
The plasmid pRRn23 containing rat renin cDNA (698 bp KpnI fragment) insertion was a gift from Dr A. Fukamizu (Tada et al. 1988) and plasmid pRAG16, which carried the rat AGT cDNA (712 bp BamHI fragment) insertion, was a gift from Professor H. Ohkubo (Ohkubo et al. 1986). The cDNA of rat glyceraldehyde-3-phosphate dehydrogenase (GAPDH) was a gift from Dr Aris Eliopoulos (Institute of Cancer Studies, University of Birmingham). The cDNAs (21 ng per reaction) were labelled radioactively with [α-32P]dCTP (International Chemical and Nuclear Pharmaceuticals Inc. (ICN), Thame, UK) using a DNA labelling kit (RadPrime DNA Labelling system; Life Technologies, Paisley, UK).
Hybridisation was performed using standard protocols (Sambrook et al. 1989; Kaiser et al. 1997). For each probe, 4 h prehybridisation and 14-16 h hybridisation were carried out at 42°C in a solution containing 50 % formamide, 6 × SSPE (20 × SSPE is 3.6 M NaCl, 0.2 M NaH2PO4 and 20 mm EDTA), 5 × Denhardt's reagent (100 × Denhardt's reagent is 2 % each of Ficoll (type 400), polyvinylpyrrolidone and bovine serum albumin (fraction V)), 0.5 % SDS, 6 % polyethlene glycol and 200 μg ml−1 denatured and fragmented salmon sperm DNA. After hybridisation membranes were washed twice in a solution of 3 × SSC (20 × SSC is 3 M NaCl and 0.3 M sodium citrate) and 0.1 % SDS, and in a solution of 1 × SSC and 0.1 % SDS, each of them for 15 min at 55°C. Autoradiography was carried out at −70°C using X-ray film (Kodak X-OMAT AR-5; Sigma) and an intensifying screen. The X-ray films were then developed and the densities of the bands on autoradiographic images were measured using a densitometer (Ultrascan XL laser densitometer; Pharmacia LKB Biotechnology, Uppsala, Sweden). Before reprobing, the previous probe was stripped from the membranes by immersion in 1 l solution containing 1 mm Tris base (pH 8.0), 1 mm EDTA (pH 8.0) and 0.1 × Denhardt's reagent for 2 h at 75°C. The membrane was applied to X-ray film to check that all of the probe had been removed and then used for the subsequent hybridisation. To correct for differences in RNA loading and to determine whether the responses were gene specific, the membrane was also hybridised with a labelled GAPDH probe as a ‘house-keeping’ gene. For each lane (rat kidney) on the autoradiographic images, the ratio of renin or AGT mRNA densitometric readings to GAPDH mRNA densitometric readings was calculated as renin/GAPDH mRNA and AGT/GAPDH mRNA. The renal renin/GAPDH mRNA and AGT/GAPDH mRNA from the experimental left kidneys were expressed relative to the mean of those of the control group and were given as a percentage of Cnt. All samples to be compared were run, hybridised and autoradiographed together, at least twice. The concurrent RNA kb-ladder (Life Technologies, Paisley, UK) was used for indication of blotting quality and estimation of relative molecular masses of bands on the membrane.
Statistical analysis
The renal function parameters and blood pressure were measured by taking the mean values of the first two clearance periods as basal values, and the two clearances during which RPP was reduced to 60 mmHg or remained unchanged as experimental values, and the results were expressed as means ±s.e.m. Student's paired t test was used to analyse the differences between basal and experimental values within the groups. The difference (Δ) between experimental and basal values of renal function parameters and blood pressure was calculated in all animals. The mean Δ value for each of the parameters PRA, renin/GAPDH mRNA and AGT/GAPDH mRNA in each group was calculated and for between-group comparisons one-way analysis of variance was used.
RESULTS
Table 1 contains the results obtained from anaesthetised rats kept for 3 h during which RPP remained unchanged, no experimental manoeuvre (Sham group) being instigated. It was observed that RBF, glomerular filtration rate (GFR), absolute sodium excretion (UNaV) and fractional sodium excretion (FENa) remained stable during the 3 h experimental period, but there were small decreases in blood pressure (P < 0.05), RPP (P < 0.05) and urine flow (UV) (P < 0.05), while PRA was 33.7 ± 7.3 ng Ang I ml−1 h−1 at the end of the study. In the group in which RPP was decreased to 60 ± 1 mmHg (60 mmHg group), RBF decreased by 46 % (P < 0.001), GFR by 45 % (P < 0.001), UV by 92 % (P < 0.01), UNaVby 98 % (P < 0.01) and FENa by 96 % (P < 0.01), while there was an increase in blood pressure to 135 ± 3 mmHg (P < 0.001) and PRA to 251.5 ± 31.6 ng Ang I ml−1 h−1. Reduction of RPP to 60 ± 1 mmHg in the group subjected to chronic Ang II receptor blockade (Los-60 mmHg group) caused decreases in RBF of 29 % (P < 0.001), GFR of 57 % (P < 0.001), UV of 93 % (P < 0.01), UNaVof 98 % (P < 0.01) and FENa of 96 % (P < 0.01), while blood pressure and PRA were increased to 117 ± 3 mmHg (P < 0.001) and 345 ± 17 ng Ang I ml−1 h−1, respectively. RBF decreased less (P < 0.01) in the Los-60 mmHg group than in the 60 mmHggroup, but the magnitudes of the reductions in GFR, UV, UNaVand FENa in the two groups were not significantly different. Blockade of Ang II receptors reduced the rise in MAP occurring during the 3 h reduction of RPP to 60 mmHg in the Los-60 mmHg group compared with the 60 mmHg group (P < 0.05). Comparisons of the mean values of MAP during basal periods in the Los-60 mmHg and 60 mmHggroups showed that chronic injection of losartan resulted in a significantly (P < 0.05) lower MAP.
Table 1.
Effect of 3 h renal perfusion pressure reduction on left renal function, mean arterial pressure and plasma renin activity under chloralose-urethane (CU) anaesthesia in losartan-treated and non-treated rats
| Groups | RPP(mmHg) | RBF (ml min−1 gKw−1) | GFR (ml min−1 gKw−1) | UV (μl min−1 gKw−1) | UNaV (μmol min−1 gKw−1) | FENa (%) | MAP (mmHg) | PRA (ng AngI ml−1 h−1) |
|---|---|---|---|---|---|---|---|---|
| Sham | ||||||||
| Basal values | 103 ± 3 | 10.8 ± 0.5 | 1.16 ± 0.04 | 21.3 ± 2.8 | 4.27 ± 0.60 | 2.44 ± 0.33 | 105 ± 3 | — |
| Expt values | 100 ± 3* | 10.8 ± 0.6 | 1.17 ± 0.06 | 16.8 ± 2.7* | 4.04 ± 0.56 | 2.33 ± 0.34 | 103 ± 3* | 33.7 ± 7.3 |
| 60 mmHg | ||||||||
| Basal values | 106 ± 2 | 10.1 ± 0.4 | 1.32 ± 0.09 | 30.5 ± 5.9 | 6.47 ± 0.91 | 3.21 ± 0.56 | 108 ± 2 | — |
| Expt values | 60 ± 1‡ | 5.5 ± 0.3‡ | 0.73 ± 0.09‡ | 2.3 ± 0.2† | 0.15 ± 0.03† | 0.13 ± 0.02† | 135 ± 3‡ | 251.5 ± 31.6 |
| Los–60 mmHg | ||||||||
| Basal values | 95 ± 4 | 10.8 ± 0.3 | 1.24 ± 0.08 | 30.5 ± 5.1 | 6.85 ± 1.08 | 3.55 ± 0.52 | 97 ± 4 | — |
| Expt values | 60 ± 1‡ | 7.7 ± 0.4‡ | 0.55 ± 0.08‡ | 2.1 ± 0.4† | 0.10 ± 0.04† | 0.14 ± 0.05† | 117 ± 3‡ | 345.4 ± 17.3 |
Values are means ±s.e.m. of basal (Basal values) and experimental (Expt values) periods for renal perfusion pressure (RPP), renal blood flow (RBF), glomerular filtration rate (GFR), urine flow (UV), absolute sodium excretion (UNaV), fractional sodium excretion (FENa) and mean systemic arterial pressure (MAP). Plasma renin activity (PRA) values are means ±s.e.m. at the end of each experiment. gKw, gram kidney weight.
P < 0.05
P < 0.01
P < 0.001 for comparison between basal and expermental values.
Figure 1 shows that the PRA level in the 60 mmHg group was increased significantly in comparison with that of the Sham group (P < 0.001). The mean value of PRA in theSham group (33.7 ± 7.3 ng Ang I ml−1 h−1) was higher (P < 0.001) than that of the Cnt group (2.8 ± 0.3 ng Ang I ml−1 h−1), which indicates an excitatory effect of chloralose-urethane anaesthesia and/or surgery on renin secretion. The 3 h reduction of RPP to 60 mmHg in the Los-60 mmHg group increased PRA to higher values than that of immediately killed losartan-treated rats (Los-Cnt group; 80.4 ± 6.2 ng Ang I ml−1 h−1, P < 0.01). The higher levels of PRA in the Los-Cnt group compared with theCnt group (P < 0.001) indicate that inactivation of Ang II receptors had an excitatory effect on basal renin secretion.
Figure 1. Changes in plasma renin activity after sham operation, 3 h reduction of RPP, losartan injection or 3 h reduction of RPP in losartan-treated rats.
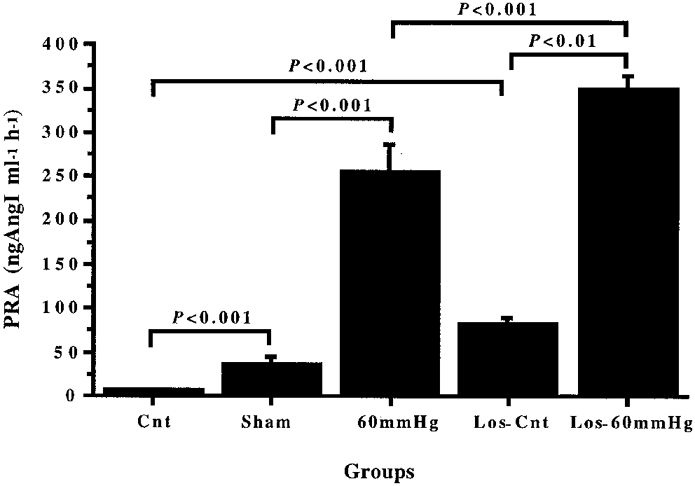
Effect of renal perfusion pressure reduction to 60 mmHg for 3 h in losartan-treated, 10 mg kg−1 b.i.d. for 5 days i.p., (Los-60 mmHg) or non-treated (60 mmHg) rats on plasma renin activity (PRA). In the Sham group, rats had undergone 3 h of sham stimulation. Control non-treated (Cnt) and losartan-treated (Los-Cnt) rats were immediately killed.
The autoradiograms of the Northern blots for total RNA of left kidneys taken from Cnt, Los-Cnt and Los-60 mmHg groups which were probed for renin, AGT and GAPDH mRNAs are presented in Fig. 2.
Figure 2. Northern blot autoradiographs for renin (top), angiotensinogen (AGT; middle), and glyceraldehyde-3-phosphate dehydrogenase (GAPDH; bottom) mRNAs.
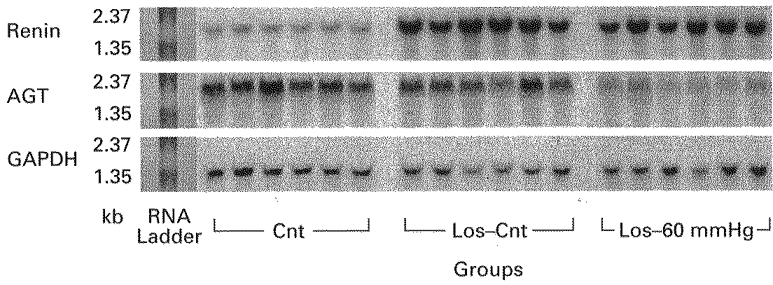
Comparisons were made between left kidneys taken from rats either losartan treated, 10 mg kg−1 b.i.d. for 5 days i.p., (Los-Cnt) or non-treated (Cnt) which were immediately killed, and losartan-treated rats in which RPP was reduced to 60 mmHg for 3 h (Los-60 mmHg).
The left renal renin/GAPDH mRNA (Fig. 3A) and AGT/GAPDH mRNA (Fig. 4) ratios in theSham group were about 51 % (P < 0.01) and 42 % (P < 0.05), respectively, lower than those of the Cnt group. This implies that chloralose-urethane anaesthesia and surgery had a depressant action on both renal renin and Ang gene expression. Reduction of RPP to 60 mmHg for 3 h (60 mmHg group) increased the renal renin/GAPDH mRNA ratio by about 40 % (P < 0.05) in comparison with that of the Sham group, while the renal AGT/GAPDH mRNA ratio of the 60 mmHg group was similar to that of theSham group.
Figure 3. Changes in renal renin gene expression after 3 h reduction of RPP or sham operation, losartan injection or 3 h reduction of RPP in losartan-treated rats.
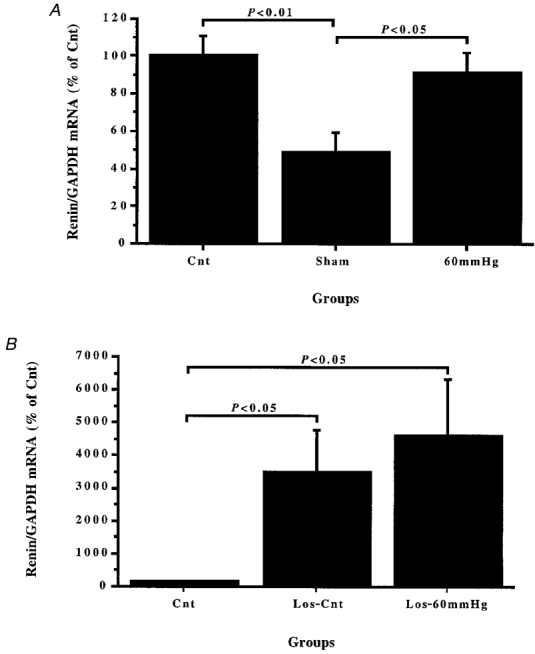
Comparisons were made between left kidneys taken from groups of rats which were immediately killed (Cnt) and in which RPP was reduced for 3 h to 60 mmHg (60 mmHg), or remained unchanged (Sham) (A), and left kidneys taken from losartan-treated rats, 10 mg kg−1 b.i.d. for 5 days i.p. (Los-Cnt), and losartan-treated rats in which RPP was reduced to 60 mmHg for 3 h (Los-60 mmHg) (B). The ratio of left renal renin per glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA densitometric readings (renin/GAPDH mRNA) was expressed relative to the mean of those of the control group (Cnt) and was given as a percentage of Cnt.
Figure 4. Changes in renal angiotensinogen gene expression after 3 h reduction of RPP or sham operation, losartan injection or 3 h reduction of RPP in losartan-treated rats.
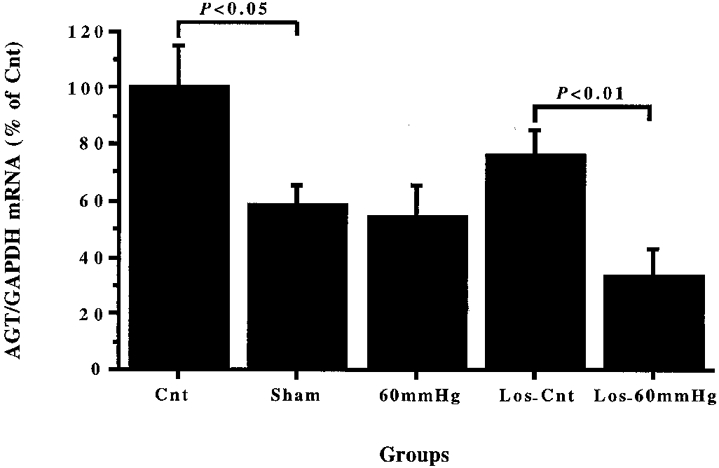
Comparisons were made between left kidneys taken from groups of rats which were immediately killed (Cnt) and in which RPP was reduced for 3 h to 60 mmHg (60 mmHg), or remained unchanged (Sham), losartan-treated rats, 10 mg kg−1 b.i.d. for 5 days i.p., (Los-Cnt) and losartan-treated rats in which RPP was reduced to 60 mmHg for 3 h (Los-60 mmHg). The ratio of left renal angiotensinogen (AGT) per glyceraldehyde-3-phosphate dehydrogenase (GAPDH) mRNA densitometric readings (AGT/GAPDH mRNA) was expressed relative to the mean of those of the control group (Cnt) and is given as a percentage of Cnt.
Comparison of the left renal renin/GAPDH mRNA ratios (Fig. 3B) in the Cnt and Los-Cnt groups revealed that 5 days administration of losartan caused about a 30-fold increase in renal renin mRNA levels. This figure also shows that there was no significant difference between left renal renin/GAPDH mRNA in the Los-Cnt and Los-60 mmHg groups. The left renal AGT/GAPDH mRNA ratio (Fig. 4) in the Los-Cnt group was not statistically different from that of the Cnt group, but it was more than 2-fold higher (P < 0.01) than that of the Los-60 mmHg group, while the left renal AGT/GAPDH in the Los-60 mmHg and 60 mmHg groups were not statistically different. These data indicated that chronic blockade of Ang II receptors had minimal effects on renal AGT gene expression, either under control conditions or after prolonged chloralose-urethane anaesthesia and surgical stress.
DISCUSSION
The present study investigated the impact of 3 h of RPP reduction on renin release, renal renin and AGT gene expression and the interaction with circulating Ang II. It is important to recognise that reduction of RPP may activate both renal baroreceptor- and macula densa-mediated renin release, but the former mechanism is likely to be the primary mediator which determines renin secretion through the regulation of transmembrane Ca2+ influx at the granular cells (Hackenthal et al. 1990; King et al. 1993). Reduction in intracellular Ca2+ concentration and its subsequent dissociation from calmodulin activates the KCl-H+ exchanger at the granular membrane to release renin from prepackaged renin granules (Skott & Jensen, 1993; King & Fray, 1994). Moreover, Ca2+-calmodulin also inhibits renin gene expression, and it has been suggested that this occurs through inhibition of cAMP-responsive element binding protein (CREB) transactivation (Ying et al. 1997).
It is necessary to emphasise that the studies herein were undertaken in anaesthetised surgically stressed animals, which would raise the degree of activation of the RAS (Hackenthal et al. 1990) and could affect the responses to RPP reduction. Indeed, this was apparent in that the PRA level in the sham-operated group was higher than in the control group, in which the animals were subjected to minimal stimulation of anaesthesia and surgery (Fig. 1). However, in the sham-operated rats both renal renin and AGT mRNA were lower compared with those of the control group (Figs 3A and 4), which suggested that chloralose- urethane anaesthesia was having an overall action to decrease the expression of both genes under basal conditions. The reason for such a suppression could either be a direct effect of chloralose-urethane on renin and AGT producing cells, possibly to cause reductions in renin and AGT gene transcription rate and/or increases in their mRNA degradation, or alternatively, PRA, and hence Ang II, which was elevated by the 8 h chloralose-urethane infusion (i.e. the total duration of the experiment) and could have acted in negative feedback fashion (Johns, 1992; Schunkert et al. 1992) to attenuate further generation of renin at the gene level. However, because most other reports have indicated an absence (Nakamura et al. 1990; Samani et al. 1994) or a stimulatory effect (Schunkert et al. 1992) of Ang II on renal AGT mRNA level, the inhibition of renal AGT gene expression through a direct negative feedback of Ang II was less probable.
Reduction of RPP for 3 h to 60 mmHg (60 mmHg group) caused marked falls in RBF, GFR and water and sodium excretions (Table 1). The falls in RPP and Na+ load to the tubules would have activated renal baroreceptor and macula densa mechanisms, and would explain the more than 7-fold increase in PRA in comparison with the sham-operated group (Fig. 1). Importantly, the higher level of renal renin mRNA in the 60 mmHg group than in theSham group (Fig. 3A) indicated that a 3 h reduction of RPP to 60 mmHg was of sufficient duration to raise renal renin gene expression. On the other hand, 3 h of RPP reduction had no impact on renal AGT gene expression (Fig. 4), which was consistent with other reports which showed that in 2K-1C rats renal AGT mRNA levels were not changed even during the chronic phases following constriction of the renal artery (ElDahr et al. 1993; Von-Thun et al. 1994).
It has been reported that Ang II via the mediation of AT1 receptors and through mobilisation of intracellular Ca2+ could have an inhibitory action on renin release (Hackenthal et al. 1990; Skott & Jensen, 1993) and also renin gene expression (Johns, 1992) at the JG cells. The elevations in Ang II induced by the experimental challenge could act in a negative feedback fashion to inhibit not only renal renin release but also renin gene expression. To test this possibility, the effect of 5 days AT1 blockade on renal renin release and gene expression, renal AGT gene expression and renal function responses to reduction in RPP were investigated. The initial study compared the PRA (Fig. 1) and renal renin mRNA levels (Fig. 3B) in losartan-treated and non-treated rats killed immediately, and showed that 5 days of AT1 blockade caused an approximate 30-fold increase in both variables. In a simplistic sense, these observations can be interpreted as indicating a removal of basal Ang II influences, releasing a negative feedback control and hence stimulation of renal renin release and gene expression. These data are consistent with other observations which suggested an inhibitory effect of endogenous Ang II on renin secretion and gene expression (Geary et al. 1992; Samani et al. 1994; Doughty et al. 1995). An important point that arose was that the renin secretion and gene transcription increased in equal proportions, which would suggest that the increased renin secretion was due to an elevated transcription rate. Importantly Kawamura et al. (1988) showed that a constitutive pathway exists for active renin secretion which is responsible for newly synthesised renin and is different from the secretion via mature granules. Thus, the present findings would support the viewpoint of Dzau et al. (1988) that a change in transcription rate controls the constitutive pathway of renin secretion and is a more economic and direct pathway of secretion that may assume a more important role during a phase of chronic stimulation.
The level of PRA (Fig. 1) achieved after RPP reduction to 60 mmHg in the losartan-treated rats was greater than that observed in the non-treated rats, although the increase in absolute terms compared with their relative controls was similar in the two groups. A number of reports (Dzau et al. 1988; Morris, 1992) have indicated that acute stimuli induce renin secretion only via the regulatory pathway, and it is likely that under the present experimental conditions, RPP reduction and chloralose-urethane anaesthesia acted via this pathway to increase renin secretion. In terms of the losartan-treated group in which basal PRA and renal renin mRNA levels were much higher, the acute anaesthesia and reduced RPP induced renin release through the regulatory pathway which was additive to the chronic losartan-induced basal renin release. Moreover, the challenge to the renal baroreceptor mechanism was not sufficient to raise renal renin mRNA beyond the already marked level present in the losartan-treated control group (Fig. 3B). This suggests that the potential direct negative feedback effect of Ang II on JG cells had only a minor influence on renin release and gene expression during the acute challenge. These data are compatible with the suggestion of Ehmke et al. (1996) that even though the Ang II short-loop feedback on JG cells has a strong tonic negative modulation on renin release and synthesis, it does not participate in the acute control of renin release by a pressure-dependent mechanism.
Interestingly, the 5 days administration of losartan, which had a marked stimulatory effect on renin gene expression, did not change the renal AGT mRNA level (Fig. 4). This finding would suggest that endogenous Ang II was having no effect on renal AGT gene expression, which is consistent with the reports of Nakamura et al. (1990) and Samani et al. (1994). However, it is necessary to mention that Schunkert et al. (1992) found that after 3 days infusion of exogenous Ang II there was a suppression of plasma renin concentration and renal renin mRNA and an elevation of renal AGT mRNA. Therefore, in spite of no effect of endogenous Ang II on renal expression of AGT, chronically increasing the level of Ang II can upregulate renal AGT gene expression and downregulate renal renin gene expression. Reducing RPP to 60 mmHg for 3 h in losartan-treated rats under chloralose-urethane anaesthesia decreased renal AGT mRNA levels in comparison with the immediately killed losartan-treated group (Fig. 4). As discussed above, the 8 h exposure to chloralose-urethane anaesthesia in sham-operated rats actually resulted in a suppression of renal AGT gene expression, and the 3 h RPP reduction to 60 mmHg did not affect this depressed value. The levels of renal AGT mRNA in the Los-60 mmHg and 60 mmHg groups were similar, which can be taken to indicate that Ang II did not play a role in the chloralose- urethane anaesthesia-induced depression of renal AGT gene expression.
The importance of Ang II in causing an elevation in blood pressure during the chronic studies became clear as the basal blood pressure in losartan-treated rats was lower than in non-treated rats (Table 1), which confirmed that the tonic actions of Ang II can be an important contributor in determining blood pressure. In addition, Ang II was also involved in increasing systemic blood pressure during the period of reduced RPP, since the magnitude of increases in blood pressure due to RPP reduction to 60 mmHg in losartan-treated rats was lower than in non-treated rats. The reductions in RBF in the Los-60 mmHg group were smaller than in the 60 mmHg group, while the falls in GFR in the two groups were similar (Table 1). Therefore, during RPP reduction to 60 mmHg, Ang II, even under the conditions of maximum vasodilatory influences of the RBF autoregulation mechanisms, was still able to constrict renal arterioles and decrease RBF, but since there was no accompanying reduction of GFR, it indicated that Ang II was able to affect both afferent and efferent arterioles. The support of GFR under these conditions most likely reflected a preferential action of Ang II at the efferent arteriole as has been established in other studies (Abdi & Johns, 1997).
Together, these findings provide further insight into the interactions between physiological challenges to cause renin release and renin gene expression. They show that reduction of renal perfusion pressure below the autoregulatory limit for 3 h was sufficient to cause renin release and to activate renin gene expression. This suggests that the granular stores act as a powerful buffering mechanism such that under more modest conditions renin gene expression is unlikely to be altered to any great extent. However, the results indicate that when necessary, renin gene expression can be activated over a relatively short 3 h time frame. The data show that the chloralose-urethane anaesthesia caused renin release which was associated with a reduction in both renal renin and AGT gene expression, which may be partly due to a raised circulating angiotensin II, but also may in part be a direct action of the anaesthetic itself. A final important observation was that although chronic blockade of Ang II receptors had little effect on renal AGT gene expression, PRA was raised along with a very marked elevation of renal renin gene expression. This would be consistent with the suggestion that Ang II has an important long-term negative feedback control, not only on renin release, but also on renin gene expression.
Acknowledgments
We thank Professor H. Ohkubo for the gift of plasmid pRAG16 containing rat Ang cDNA, Dr A. Fukamizu for the plasmid pRRn23 containing rat renin cDNA and Dr Aris Eliopoulos for the cDNA of rat GAPDH. We acknowledge the contribution of Professor N. J. Samani and Dr M. Kaiser in the Northern blotting analysis. S.M.S.M. was in receipt of a scholarship from Shiraz University of Medical Sciences, Shiraz, The Islamic Republic of Iran.
References
- Abdi A, Johns EJ. The effect of angiotensin II receptor antagonists on kidney function in two-kidney, two-clip Goldblatt hypertensive rats. European Journal of Pharmacology. 1997;331:185–192. doi: 10.1016/s0014-2999(97)01038-8. [DOI] [PubMed] [Google Scholar]
- Carey RM, McGrath HE, Pentz ES, Gomez RA, Barrett PQ. Biomechanical coupling in renin-releasing cells. Journal of Clinical Investigation. 1997;100:1566–1574. doi: 10.1172/JCI119680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doughty SE, Ferrier RK, Hillan KJ, Jackson DG. The effects of Zeneca ZD8731, an angiotensin II antagonist, on renin expression by juxtaglomerular cells in the rat: Comparison of protein and mRNA expression as detected by immunohistochemistry and in-situ hybridization. Toxicologic Pathology. 1995;23:256–261. doi: 10.1177/019262339502300303. [DOI] [PubMed] [Google Scholar]
- Dzau VJ, Burt DW, Pratt RE. Molecular biology of the renin-angiotensin system. American Journal of Physiology. 1988;255:F563–573. doi: 10.1152/ajprenal.1988.255.4.F563. [DOI] [PubMed] [Google Scholar]
- Ehmke H, Berthold H, Just A, Hackenthal E, Kirchheim HR. Acute short-loop feedback inhibition of renin release by angiotensin II does not play a physiological role in the conscious dog. Kidney International. 1996;50:1801. [Google Scholar]
- ElDahr SS, Dipp S, Guan S, Navar LG. Renin, angiotensinogen, and kallikrein gene-expression in 2-kidney Goldblatt hypertensive rats. American Journal of Hypertension. 1993;6:914–919. doi: 10.1093/ajh/6.11.914. [DOI] [PubMed] [Google Scholar]
- Geary KM, Hunt MK, Peach MJ, Gomez RA, Carey RM. Effects of angiotensin converting enzyme inhibition, sodium depletion, calcium, isoproterenol, and angiotensin II on renin secretion by individual renocortical cells. Endocrinology. 1992;131:1588–1594. doi: 10.1210/endo.131.4.1396304. [DOI] [PubMed] [Google Scholar]
- Goldblatt H, Lynch J, Hanzel RF, Summerville WW. The production of persistent elevation of systolic blood pressure by means of renal ischemia. Journal of Experimental Medicine. 1934;59:347–380. doi: 10.1084/jem.59.3.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackenthal E, Paul M, Ganten D, Taugner R. Morphology, physiology, and molecular biology of renin secretion. Physiological Reviews. 1990;70:1067–1116. doi: 10.1152/physrev.1990.70.4.1067. [DOI] [PubMed] [Google Scholar]
- Johns DW. Angiotensin II inhibits renin gene expression by a direct action on the AT1 receptor of rat juxtaglomerular cells. Circulation. 1992;86:I601. [Google Scholar]
- Johns EJ. Role of angiotensin II and the sympathetic nervous system in the control of renal function. Journal of Hypertension. 1989;7:695–701. [PubMed] [Google Scholar]
- Johns EJ, Lewis BA, Singer B. The sodium retaining effect of renal nerve activity in the cat: Role of angiotensin II formation. Clinical Science. 1976;51:93–102. doi: 10.1042/cs0510093. [DOI] [PubMed] [Google Scholar]
- Kaiser M, Vincent M, Kenyon CJ, Gomez-Sanchez CE, Cumin F, Lodwick D, Sassard J, Samani NJ. Analysis of phenotypic consequences of renin gene polymorphism in Lyon rats. Journal of Hypertension. 1997;15:365–372. doi: 10.1097/00004872-199715040-00007. [DOI] [PubMed] [Google Scholar]
- Kawamura M, Parmentier M, Inagami T. Localization and secretion of newly synthesized and stored renin: two compartments and secretory mechanisms. American Journal of Physiology. 1988;255:F100–107. doi: 10.1152/ajprenal.1988.255.1.F100. [DOI] [PubMed] [Google Scholar]
- King JA, Fray JCS. Hydrogen and potassium regulation of (pro)renin processing and secretion. American Journal of Physiology. 1994;267:F1–12. doi: 10.1152/ajprenal.1994.267.1.F1. [DOI] [PubMed] [Google Scholar]
- King JA, Lush DJ, Fray JCS. Regulation of renin processing and secretion: Chemiosmotic control and novel secretory pathway. American Journal of Physiology. 1993;265:C305–320. doi: 10.1152/ajpcell.1993.265.2.C305. [DOI] [PubMed] [Google Scholar]
- Kirchheim HR, Ehmke H, Hackenthal E, Lowe W, Persson P. Autoregulation of renal blood flow, glomerular filtration rate and renin release in conscious dogs. Pflügers Archiv. 1987;410:441–449. doi: 10.1007/BF00586523. [DOI] [PubMed] [Google Scholar]
- Morgan L, Pipkin FB, Kalsheker N. Angiotensinogen: Molecular biology, biochemistry and physiology. International Journal of Biochemistry and Cell Biology. 1996;28:1211–1222. doi: 10.1016/s1357-2725(96)00086-6. [DOI] [PubMed] [Google Scholar]
- Morris BJ. Molecular biology of renin. I: Gene and protein structure, synthesis and processing. Journal of Hypertension. 1992;10:209–214. doi: 10.1097/00004872-199203000-00002. [DOI] [PubMed] [Google Scholar]
- Nafz B, Berthold H, Ehmke H, Hackenthal E, Kirchheim HR, Persson PB. Flow versus pressure in the control of renin release in conscious dogs. American Journal of Physiology. 1997;273:F200–205. doi: 10.1152/ajprenal.1997.273.2.F200. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Iwao H, Fukui K, Kimura S, Tamaki T, Nakanishi S, Abe Y. Regulation of liver angiotensinogen and kidney renin mRNA levels by angiotensin II. American Journal of Physiology. 1990;258:E1–6. doi: 10.1152/ajpendo.1990.258.1.E1. [DOI] [PubMed] [Google Scholar]
- Ohkubo H, Nakayama K, Tanaka T, Nakanishi S. Tissue distribution of rat angiotensinogen mRNA and structural analysis of its heterogeneity. Journal of Biological Chemistry. 1986;261:319–323. [PubMed] [Google Scholar]
- Samani NJ, Cumin F, Kelly M, Wood JM. Expression of components of the RAS during prolonged blockade at different levels in primates. American Journal of Physiology. 1994;267:E612–619. doi: 10.1152/ajpendo.1994.267.4.E612. [DOI] [PubMed] [Google Scholar]
- Samani NJ, Morgan K, Brammar WJ, Swales JD. Detection of renin messenger RNA in rat-tissues: Increased sensitivity using an RNAse protection technique. Journal of Hypertension. 1987;5:S19–S21. doi: 10.1097/00004872-198707002-00005. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning - A Laboratory Manual. New York: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Schunkert H, Ingelfinger JR, Jacob H, Jackson B, Bouyounes B, Dzau VJ. Reciprocal feedback regulation of kidney angiotensinogen and renin messenger RNA expressions by angiotensin II. American Journal of Physiology. 1992;263:E863–869. doi: 10.1152/ajpendo.1992.263.5.E863. [DOI] [PubMed] [Google Scholar]
- Skott O, Jensen BL. Cellular and intrarenal control of renin secretion. Clinical Science. 1993;84:1–10. doi: 10.1042/cs0840001. [DOI] [PubMed] [Google Scholar]
- Tada M, Fukamizu A, Seo MS, Takahashi S, Murakami K. Nucleotide sequence of rat renin cDNA. Nucleic Acids Research. 1988;16:3576. doi: 10.1093/nar/16.8.3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K, Umemura S, Fukamizu A, Ishii M, Murakami K. Recent advances in the study of renin and angiotensinogen genes: From molecules to the whole body. Hypertension Research. 1995;18:7–18. doi: 10.1291/hypres.18.7. [DOI] [PubMed] [Google Scholar]
- Tufro-McReddie A, Chevalier RL, Everett AD, Gomez RA. Decreased perfusion pressure modulates renin and Ang II type 1 receptor gene expression in the rat kidney. American Journal of Physiology. 1993;264:R696–702. doi: 10.1152/ajpregu.1993.264.4.R696. [DOI] [PubMed] [Google Scholar]
- Von-Thun AM, ElDahr SS, Vari RC, Navar LG. Modulation of renin-angiotensin and kallikrein gene expression in experimental hypertension. Hypertension. 1994;23:I131–136. doi: 10.1161/01.hyp.23.1_suppl.i131. [DOI] [PubMed] [Google Scholar]
- Witty RT, Davis JO, Johnson JA, Prewitt RL. Effects of papaverin and hemorrhage on renin secretion in the non-filtering kidney. American Journal of Physiology. 1971;221:1666–1671. doi: 10.1152/ajplegacy.1971.221.6.1666. [DOI] [PubMed] [Google Scholar]
- Ying LH, Morris BJ, Sigmund CD. Transactivation of the human renin promoter by the cyclic AMP/protein kinase A pathway is mediated by both cAMP-responsive element binding protein-1 (CREB)-dependent and CREB-independent mechanisms in Calu-6 cells. Journal of Biological Chemistry. 1997;272:2412–2420. doi: 10.1074/jbc.272.4.2412. [DOI] [PubMed] [Google Scholar]