

Abstract
The effects of nitric oxide (NO) donors on whole-cell, TTX-sensitive sodium currents and single sodium channels in excised patches were examined in rat hippocampal neurons. The whole-cell sodium current consisted of a large transient component (INa,t) and a smaller, inactivation-resistant, persistent component (INa,p).
In acutely dissociated neurons, the amplitude of the whole-cell INa,p increased by 60–80% within a few minutes of exposure to either of two NO donors, sodium nitroprusside (SNP, 100 μm) or S-nitroso-N-acetyl-DL-penicillamine (SNAP, 100 μm).
The amplitude of INa,t was not changed significantly by the same concentrations of SNP and SNAP, indicating that NO had a selective effect on INa,p.
Both NO donors significantly increased the mean persistent current in excised inside-out patches from cultured hippocampal neurons. SNP at 10m100 μm increased average mean persistent current at a pipette potential (Vp) of +30 mV from −0.010 ± 0.014 pA (control) to −2.91 ± 1.41 pA (n= 10). SNAP at 3–100 μm increased the average mean inward current in six inside-out patches from −0.07 ± 0.02 to −0.30 ± 0.08 pA (Vp=+30 mV).
The increase in persistent Na+ channel activity recorded in inside-out patches in the presence of SNP or SNAP could be reversed by the reducing agent dithiothreitol (DTT, 2−5 mm) or by lidocaine (1–10 μm).
The average mean current recorded in the presence of SNP was 10-fold higher than that elicited by SNAP. The time delay before an increase was observed was shorter with SNP (4.0 ± 0.8 min, n= 8) than with SNAP (8.4 ± 1.6 min, n= 7).
A component of the SNP molecule added on its own, 5 mm sodium cyanide (NaCN), increased mean current in excised inside-out patches (Vp=+30 mV) from −0.06 ± 0.04 to −0.58 ± 0.21 pA (n= 19). This increase in channel activity could be blocked by 10 μm lidocaine and 2–5 mm DTT.
These results suggest that NO may directly increase the activity of neuronal persistent Na+ channels, but not transient Na+ channels, through an oxidizing action directly on the channel protein or on a closely associated regulatory protein in the plasma membrane.
Ever since the simultaneous findings by both Furchgott's and Ignarro's laboratories that nitric oxide (NO) is a potent and important vasodilator (Ignarro et al. 1987; Furchgott, 1988), many different roles for NO in various tissues have been discovered. NO is involved in the regulation of a variety of cell functions such as neurotransmission in the central nervous system and in the control of platelet aggregation. In addition, NO participates in immunological and autoimmunity reactions and, because of its cytotoxicity, overproduction is deleterious to cells. Hence, NO is a messenger in the central nervous system under normal, but also during some pathological, conditions.
NO is produced from L-arginine by the enzyme NO synthase, is highly membrane permeant and may directly influence cell function by modulating second messenger systems and ion channel activity. NO directly activates a Ca2 + -dependent K+ channel (Archer et al. 1994; Bolotina et al. 1994), stimulates a glibenclamide-sensitive K+ channel in vascular smooth muscle cells (Garland & McPhearson, 1992; Parkington et al. 1995), modulates NMDA channels (Manzoni et al. 1992; Lei et al. 1992) and has been reported to activate (Méry et al. 1993) and inhibit (Levi et al. 1994) cardiac Ca2+ channels. It is well known that NO activates guanylate cyclase and increases intracellular cGMP levels (Schulz et al. 1991) providing one possible pathway for these events. More recently it has been proposed that NO may directly influence or modulate protein structure and hence gating of various ion channels. NO, or the nitrosonium redox form of NO (NO+), has been shown to react with protein thiols resulting in S-nitrosylation (Stamler et al. 1992). Hence, NO may in this way act as an oxidizing agent by enabling disulfide bond formation between two closely located S-nitrosylated thiols in the channel protein (Lei et al. 1992).
A persistent, inactivation-resistant, TTX-sensitive Na+ current (INa,p) has been recorded in neurons (Gilly & Armstrong, 1984; French & Gage, 1985; French et al. 1990; Taylor, 1993; Crill, 1996). This persistent current may be caused by a change in the gating mode of transient Na+ channels, i.e. some channels that intermittently lose their inactivation (Alzheimer et al. 1993; Brown et al. 1994). Alternatively, INa,p may be generated by structurally different channels, perhaps following association of βγ-subunits of G-proteins (Ma et al. 1997) or some other protein, with the α-subunit of the Na+ channel. In normal healthy cells, INa,p is thought to play an important role as a pacemaker current, enhancing rythmicity and hence enabling repetitive firing of action potentials (Taylor, 1993). We have previously recorded an increase in the amplitude of INa,p but not in the transient Na+ current (INa,t) in CA1 neurons exposed to hypoxia (Hammarström & Gage, 1998), and we suggest that this may be the pathway for the prolonged and eventually damaging Na+ influx observed during hypoxia (Friedman & Haddad, 1994).
In addition to NO being a neurotransmitter in the brain (Mayer & Miller, 1990) and an important messenger released from the endothelial lining of cerebral blood vessels, NO production is also transiently increased during ischaemia (Shibata et al. 1996). Knock-out mice in which the neuronal NO synthase gene has been deleted are resistant to ischaemic brain damage (Huang et al. 1994, Panahian et al. 1996). In addition, the rate of action potential firing is altered in the presence of NO (Pehl & Schmid, 1997). These observations suggest that NO may play an important part in the neuronal cell damage and cell death occurring as a result of ischaemia.
As NO is a possible trigger for hyperactivity of INa,p during hypoxia, the aim of this study was to investigate the effect of NO donors on neuronal TTX-sensitive transient Na+ currents (INa,t) and persistent Na+ currents (INa,p).
A preliminary report of these results has appeared elsewhere (Hammarström & Gage, 1999).
METHODS
Details of experimental methods have been described previously (Premkumar et al. 1990; Ju et al. 1996; Hammarström & Gage, 1998).
Dissociation of CA1 neurons
Briefly, young adult Wistar rats (14-21 days old) were decapitated (CO2 anaesthesia) and the brain quickly transferred to ice-cold artificial cerebrospinal fluid (ACSF) (mm: 124 NaCl, 26 NaH2CO3, 3 KCl, 2.5 CaCl2, 1.3 MgSO4, 2.5 NaH2PO4 and 20 glucose) gassed with 95 % CO2-5 % O2. Brain slices approximately 500 μm thick were cut on a vibratome (Camden), in the presence of ice-cold ACSF equilibrated with 95 % CO2-5 % O2. The brain slices were enzymatically treated for 30 min with 200 U papain (Worthington Biochemicals), 1.1 mm cysteine (Sigma), 0.2 mm EDTA and 13.4 μmβ-mercaptoethanol at 35°C, following the adaptation (French et al. 1990) of the procedure originally described by Kay & Wong (1987). The CA1 region was subsequently removed and individual neurons obtained by careful mechanical trituration with a glass pipette. These methods have been approved by the Animal Experimentation Ethics Committee at the Australian National University, ACT, Australia.
Cultured hippocampal neurons
The hippocampus was dissected from newborn Wistar rats (killed by decapitation) and the tissue triturated to dissociate cells. The cells were then grown on poly-L-lysine-coated glass coverslips and used in experiments after 5-12 days. The culture medium was minimum essential medium to which was added fetal calf serum (10 %), glucose (2 %), penicillin-streptomycin (1 %) and fungizone (1 %; Flow laboratories). Cultured cells to be used in an experiment were transferred on their coverslip to a bath filled with bath solution (for compositon see below).
Electrophysiological recording and data analysis
Tight gigaohm seal patch clamp techniques using standard glass pipettes (GC150F-15, Clarke Electromedical Instruments) were used to record currents in the whole-cell configuration and single channel activity from rat hippocampal neurons. Pipettes were pulled on a List Medical vertical pipette puller (L/M-3P-A) and had resistances of 6-10 MΩ when filled with pipette solution. In order to obtain reproducible whole-cell results, currents were not recorded for the first 10-15 min after achieving the whole-cell configuration. For single channel recordings, the pipettes (10-15 MΩ) were coated with Sylgard (Dow Corning) as close to the pipette tip as possible. Neurons that had a flat, swollen or grainy appearance were not used.
The bath solution for whole-cell recordings contained (mm): 135 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 5 CoCl2, 5 CsCl and 1 Tes adjusted to a pH of 7.4 with NaOH. The pipette solution for these experiments contained (mm): 125 CsF, 5 NaF, 10 KCl, 10 Tes and 10 EGTA, pH 7.4. The bath solution for inside-out patch recordings contained (mm): 140 potassium aspartate, 10 EGTA, 2 MgCl2, 2 CaCl2 and 10 Tes, pH adjusted with NaOH to 7.4. The pipette solution for these experiments was identical to the whole-cell bath solution.
Tetrodotoxin (TTX; Boehringer Mannheim), lidocaine, sodium nitroprusside (SNP), S-nitroso-N-acetyl-DL-penicillamine (SNAP) and DTT (all from Sigma) were applied to whole cells through fine (200 μm i.d.) tubes carefully positioned to cause a rapid change in concentration of drugs close to the patch (Ju et al. 1996). Stocks of each drug were diluted in bath solution to the required concentration immediately before an experiment. All solutions had an osmolality of 290 ± 10 mosmol kg−1.
Subtraction of whole-cell current traces recorded before and after exposure to a high concentration of TTX (0.5 μm) was used throughout to isolate TTX-sensitive Na+ currents. This subtraction isolates Na+ currents only, with a baseline of zero. The amplitude of the transient Na+ current (INa,t) was measured at its peak and the amplitude of the persistent Na+ current (INa,p) was measured at the end of a 400 ms voltage pulse from these subtracted traces. Current-voltage relationships were established by depolarizing steps from a holding potential of -100 mV (initially to a pre-pulse of -150 mV for 200 ms). With each step, the depolarizing pulse was incremented by 10 mV, to a final voltage of +40 mV. Single channel activity and whole-cell currents were activated by voltage steps from an IBM-compatible PC and digital-to-analog interface and recorded with an Axopatch-1D amplifier. Currents were analysed using ‘in-house’ computer techniques and software (Ju et al. 1992, 1996; Hammarström & Gage, 1998). Excised inside-out patches were held at a pipette potential (Vp) of +30 mV (Vm= -Vp), and a mean current calculated from 0.5-4 min recordings at 10 kHz in the absence and presence of agents. Reversal potentials were corrected for liquid junction potentials using the software program JPCalc (copyright Professor P. H. Barry, University of New South Wales, Sydney, Australia), which utilises the generalised Henderson equation for its calculations. This program gave a liquid junction potential of -16.4 mV for our excised patch solutions. This value has been added to the reversal potentials presented in the Results section.
All values are expressed as means ± 1 s.e.m. and the number of cells (n) in each group is given. Statistical analysis was performed using Student's two-tailed unpaired t test.
These techniques have been described in more detail elsewhere (Ju et al. 1992, 1996; Hammarström & Gage, 1998).
RESULTS
Whole-cell currents
NO inhibits NMDA-type glutamate receptors by oxidation of thiol groups on the channel protein (Lei et al. 1992) and this interaction has been thought to be neuroprotective during ischaemia when it may lessen the effects of the damaging glutamate overflow that occurs. To test the hypothesis that NO may also affect voltage-activated Na+ currents, we initially examined its effects on TTX-sensitive whole-cell Na+ currents in freshly dissociated CA1 neurons.
Whole-cell INa,p
Whole-cell Na+ currents were recorded first in the absence then in the presence of 0.5 μm TTX. Subtraction of the two traces revealed a larger transient and a smaller persistent Na+ current without contamination by other currents. As has been described by us and others earlier (French et al. 1990; Alzheimer et al. 1993; Hammarström & Gage, 1998), the persistent current amplitude was 0.7-1 % of the peak amplitude of the transient current, activated at more negative potentials (V½ about -50 mV) and did not show the same inactivation properties as the transient Na+ current.
When examining effects of drugs on persistent whole-cell Na+ currents that have a very small amplitude, changes in seal resistance and leak currents are an obvious problem that can produce inconsistent results. However, we have found that by TTX subtraction (see Methods) and by excluding data where significant changes in seal resistance and leak currents occurred, we can get very stable and reproducible measurements of INa,p amplitude. The graph in Fig. 1 shows the complete stability of INa,p amplitude recorded over a 15 min period (n= 8). We find that INa,p measured in this way can be very stable over long periods of time.
Figure 1. Stability of measurements of INa,p over time.

The amplitude of averaged TTX-subtracted INa,p amplitudes (from 8 hippocampal neurons) measured at the end of 400 ms depolarizing pulses relative to the amplitude of INa,p recorded when first obtaining whole-cell seal (Normalised current) plotted against time from the beginning of the whole-cell recording. There was no significant change in the amplitude for more than 15 min of recording.
Effect of SNP
We initially investigated the effect of one of the most commonly studied NO donors, sodium nitroprusside (SNP), which has been used clinically to reduce blood pressure. It was found that this NO donor, when applied to the extracellular surface of CA1 neurons at a concentration of 100 μm, significantly increased the amplitude of INa,p. An example of this effect in one neuron is shown in Fig. 2A.
Figure 2. Effect of NO donors on whole-cell Na+ currents in CA1 neurons.

Currents were generated by a depolarising step to -30 mV (A) or -20 mV (B and C) from a holding potential of -100 mV. The traces shown are TTX subtracted (see Methods). A, currents recorded from a neuron before (upper trace) and after exposure to 0.1 mm SNP for 7.5 min and after 12 min of wash-out (lower traces). In this neuron, 12 min of wash-out did not reverse the effect of SNP. B, TTX-sensitive currents recorded from a neuron before (upper trace) and after 3 and 10 min of exposure to 0.1 mm SNAP (lower traces). An increase in the amplitude of INa,p was seen after 3 min and the amplitude of INa,p was doubled after 10 min of 0.1 mm SNAP perfusion. C, same neuron as in B but at a lower vertical amplification to show the lack of comparable effect of 0.1 mm SNAP on INa,t.
Before exposure to SNP, INa,p had an amplitude of 10 pA at -30 mV; 7.5 min after introduction of SNP, the amplitude of INa,p had increased to 26 pA. A similar effect was obtained in four other neurons. On average, the amplitude of INa,p was more than doubled in the presence of SNP (2.17 ± 0.28 times control, n= 5). In the CA1 neuron shown in Fig. 2A, the effect of SNP was not reversed even after 12 min of washing with fresh control bath solution. In contrast, SNP did not have a significant effect on INa,t. In five neurons exposed to 100 μm SNP, the amplitude of INa,t at -30 mV was 1.05 ± 0.15 times the amplitude of the controls, i.e. not significantly different (P= 0.7).
Effect of SNAP
A chemically quite different NO donor, S-nitroso-N-acetyl-DL-penicillamine (SNAP), was also used to ensure that the effect of SNP was due to NO. At a concentration of 0.1 mm at 25°C, SNAP has been shown to generate a stable concentration of 0.1 μm NO (Ichimori et al. 1993), which is within the physiological range.
We found that SNAP also increased the amplitude of INa,p. This is shown for one neuron in Fig. 2B. It can be seen that 100 μm SNAP increased the amplitude of INa,p by about 80 % (at -20 mV). In three similar experiments, INa,p increased in the presence of SNAP to 1.82 ± 0.14 times the amplitude recorded in control solution (at -20 mV). Again, SNAP had very little effect on INa,t, as illustrated in Fig. 2C. In five cells exposed to 100 μm SNAP, the amplitude of INa,t was 0.98 ± 0.08 times control: the difference is not significant (P= 0.65).
The current-voltage and conductance-voltage relationships for INa,p in one of the neurons before and after exposure to SNAP are shown in Fig. 3A and B. The lines through the data points are best fits to the data of the Boltzmann-type equations:
and
where Gmax is the maximum conductance, V the test potential, V0 the zero current (reversal) potential, V’ the potential at which G was 50 % of Gmax and k a slope factor.
Figure 3. Current-voltage and conductance-voltage relationships for INa,p before and after 0.1 mm SNAP.

A, current-voltage relationship of the TTX-sensitive persistent Na+ current before (^) and after a 10 min 0.1 mm SNAP exposure (•) (same cell as in Fig. 2B and C). B, conductance-voltage relationship of the TTX-sensitive persistent Na+ current before (^) and after 10 min of 0.1 mm SNAP (•) (same cell as in A). The amplitude of INa,p was divided by V - V0 to obtain the conductance (V0 is the potential at which I= 0). The lines through the data points were the best fits of the equation G = Gmax/(1 + exp((V’ - V)/k)). The line of best fit gave a Gmax of 1.5 nS before SNAP exposure and 2.4 nS in the presence of SNAP. V’ and slope factors were -41 and 12 mV in control and -42 and 13 mV in the presence of SNAP. Further details are given in the text.
It can be seen that the SNAP increased the persistent Na+ conductance at all potentials but had little effect on the voltage dependence (V’ and k). Before exposure to SNAP, the line of best fit gave a Gmax of 1.48 nS, a V’ of -41 mV and a k of 12 mV. Following a few minutes’ exposure to SNAP, Gmax had increased to 2.41 nS, but V’ and k were essentially unchanged (-42 and 13 mV, respectively).
Single channel currents in excised inside-out patches
To determine whether the effect of the NO donors on whole-cell INa,p depended on a second messenger system such as a cGMP-dependent cascade or was a direct effect on the Na+ channel protein or a closely associated regulatory protein in the plasma membrane, we examined the effect of the same NO donors, at similar concentrations to those that were used in the whole-cell experiments, on excised inside-out patches from cultured hippocampal neurons. Channel activity in the inside-out configuration was recorded by holding the patch at Vp=+30 mV (i.e. -30 mV at the inner surface of the membrane) since this inactivates INa,t but not INa,p (Saint et al. 1992; Ju et al. 1992, 1996). INa,p has been shown not to inactivate even with very prolonged depolarizations and since the whole-cell recordings showed no effect of NO on INa,t, effects of drugs on transient Na+ channels were not investigated.
The effect of SNAP
Normal INa,p activity at Vp=+30 mV consisted of infrequent 1.12 ± 0.08 pA (n= 15 patches) channel openings, often occurring in short bursts of a few openings after long quiet periods. Some patches showed no activity over prolonged periods (more than 4 min). Hence, the activity recorded before drug application (control) gave a low average mean current (-0.045 ± 0.012 pA, n= 15) recorded over long periods (3-4 min).
As for the whole-cell recordings, SNAP was diluted in bath solution immediately before application to a patch. SNAP at 3-100 μm was applied to 14 excised inside-out patches. Results from one of these experiments are shown in Fig. 4. In this patch there was some persistent Na+ channel activity in control solution (Fig. 4A). When 100 μm SNAP was applied, a large increase in persistent Na+ channel activity was observed (Fig. 4B). This increase occurred within 2-3 min and grew further over the next few minutes. In six excised patches, perfusion with 3-100 μm SNAP caused an increase in mean current. The average mean current before exposure to SNAP was -0.07 ± 0.02 pA and this increased to -0.30 ± 0.08 pA (n= 6, P= 0.02) after exposure to SNAP. The large increase in the average mean current caused by SNAP is shown in Fig. 5A. There did not seem to be a concentration dependence of the effect in the range we used: 3 μm SNAP increased mean current to -0.26 ± 0.10 pA (n= 4), which was not different from the mean current (-0.28 ± 0.13 pA, n= 3) obtained with higher concentrations (10-100 μm SNAP).
Figure 4. Effect of SNAP on INa,p activity in an excised inside-out patch.
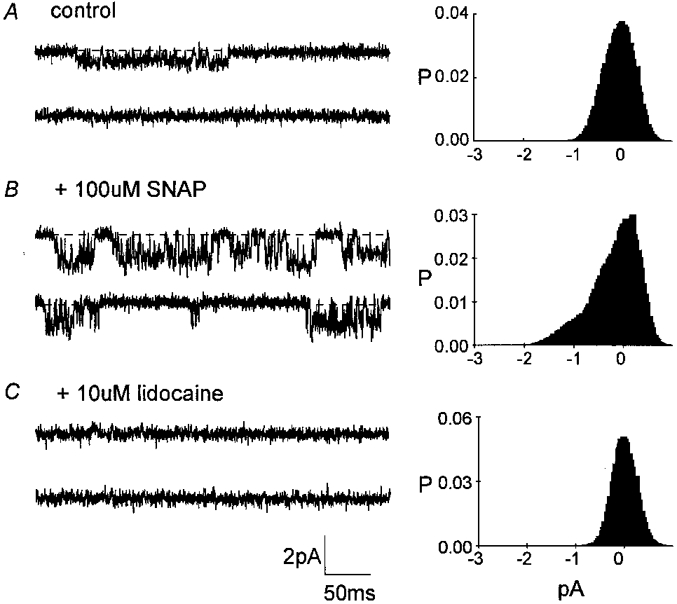
The representative 400 ms traces shown were obtained from 3-4 min recordings from a patch held at Vp=+30 mV. The accompanying all-points histograms were calculated from the full 3-4 min current record sampled at 10 kHz. A, control activity. B, increased activity after 5 min exposure to 0.1 mm SNAP. C, SNAP-induced activity was completely blocked after 4-5 min exposure to 10 μm lidocaine and 0.1 mm SNAP.
Figure 5. Increases in INa,p produced by SNP and SNAP in inside-out patches.
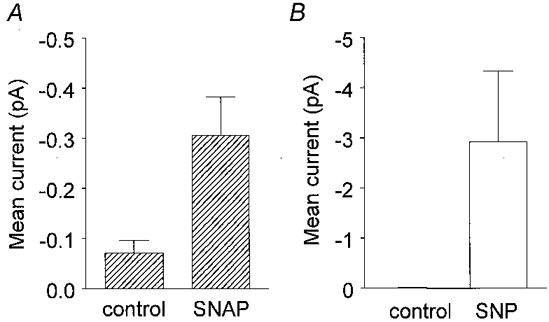
The histograms show mean current amplitudes. The vertical lines denote ± 1 s.e.m.A, mean data from 6 patches before (control, -0.07 ± 0.02 pA) and after 3-100 μm SNAP (-0.30 ± 0.08 pA, P= 0.02). B, mean data from 10 patches before (control, -0.010 ± 0.014 pA, n= 10) and after 10-100 μm SNP (-2.91 ± 1.41 pA, n= 10, P= 0.05).
To confirm that the increased channel activity was due to Na+ channels, lidocaine was applied to inside-out patches after a few minutes of exposure to SNAP. Complete block by lidocaine of the increased channel activity produced by SNAP is illustrated in Fig. 4C. The concentration of lidocaine we used is within the range that blocks neuronal Na+ channels (see Hammarström & Gage, 1998).
Effect of SNP
In 10 out of 12 inside-out patches, 10-100 μm SNP increased persistent Na+ channel activity. This effect is illustrated in one patch shown in Fig. 6B and D. An increase in channel activity was normally seen after a delay of about 1 min and then the activity kept increasing over the next few minutes. In 2 out of the 12 patches, the activity initially increased then subsided even in the continued presence of 10 μm SNP. The reason for this is unknown. In the 10 patches that showed a sustained increase in channel activity, the mean current at Vp=+30 mV increased from -0.010 ± 0.014 to -2.91 ± 1.41 pA (P= 0.05, Fig. 5B).
Figure 6. Effects of SNP, DTT and lidocaine on an inside-out patch.
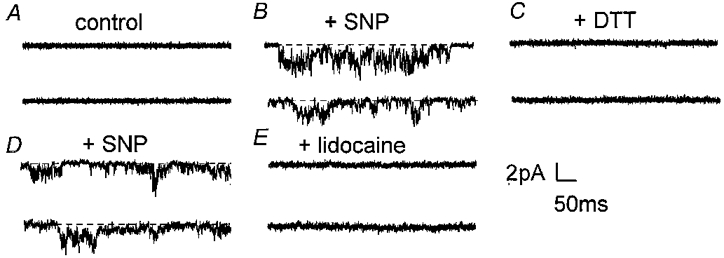
Current traces were obtained at a holding potential of -30 mV (Vp=+30 mV) and are all from the same patch. A, control current records before exposure to SNP. B, after approximately 4 min in the presence of 10 μm SNP. C, inhibition of channel activity after 2-3 min of perfusion with 2 mm DTT (with 10 μm SNP). D, the return of activity after 1.5 min of washing out DTT with a solution containing 10 μm SNP. E, abolition of SNP-elicited channel activity by 10 μm lidocaine (with 10 μm SNP, 10 min).
The increase in channel activity caused by SNP was blocked by 10 μm lidocaine, as illustrated in Fig. 6. Lidocaine reduced the average mean current (Vp=+30 mV) recorded in the presence of SNP or SNAP (n= 5) to -0.07 ± 0.04 pA (Fig. 7A). This confirms that the increase in channel activity recorded in the presence of the two NO donors was due to Na+ channel activity.
Figure 7. Average effects of DTT and lidocaine on current activity produced by SNP or SNAP.
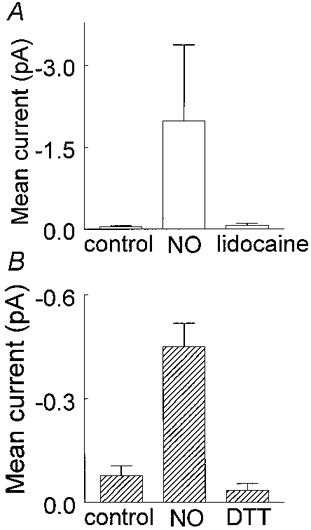
Histograms show averaged data and the vertical bars denote 1 s.e.m.A, mean current was measured from 5 patches before (-0.04 ± 0.02 pA), and after exposure to 3-100 μm SNP or SNAP (≈5.5 min exposure, -2.46 ± 1.70 pA) and then in the presence of 10 μm lidocaine plus NO donor (-0.07 ± 0.04 pA). B, mean current was measured in 4 excised patches before (-0.03 ± 0.02 pA), and after approximately 7.5 min exposure to SNP or SNAP (-0.45 ± 0.07 pA) and then after 2-3 min perfusion with 2-5 mm DTT plus NO donor (-0.03 ± 0.04 pA).
Effects of SNP and SNAP on single channel conductance and reversal potential
There was no obvious change in the amplitude of single Na+ channel currents during exposure of patches to SNP or SNAP. Average single channel current amplitude in 15 patches (Vp=+30 mV) was -1.12 ± 0.08 pA before exposure to the NO donors and -1.16 ± 0.08 pA (n= 19) in the presence of the NO donors (P= 0.68). Hence, the increase in mean current is most likely to be due to an increase in channel open probability. The fraction of time channels were open (NPo, where N is the number of channels) increased from 0.005 ± 0.002 to 0.137 ± 0.050 (n= 6, P= 0.02) following exposure to SNP. Similarly, NPo increased from 0.060 ± 0.040 to 0.147 ± 0.076 (n= 4) following exposure to SNAP.
Both in the absence and presence of the NO donors, the reversal potential for persistent channels before and after exposure to the NO donors was Vp= -29 ± 3 mV (n= 4), which was more positive than expected from the Na+ equilibrium potential of Vp= -79 mV (see solutions in Methods). From this reversal potential, the conductance of these channels was about 20 pS both in the absence and presence of NO donors. The measured reversal potential would suggest that the channels are permeable also to K+. The Goldman-Hodgkin-Katz equation gives a PNa/PK ratio of about 4.
Effect of DTT
The increase in channel activity elicited by NO could not be readily reversed by simply washing with perfusing solution (see Fig. 2A). If this was due to NO oxidizing or forming adducts with free SH groups such as cysteine residues on the Na+ channel protein (or a closely associated regulatory protein) and enabling disulfide-bond formation, it should be possible to reverse this effect with a reducing agent (sulfhydryl-regenerating agent) such as dithiothreitol (DTT), as was found for non-selective cation channels recorded from brown adipose tissue by Koivisto & Nedergaard (1995).
The ability of DTT to reverse the effects of the NO donors was examined in four inside-out patches. It can be seen in Fig. 6C that exposure to 2 mm DTT for 2-3 min completely abolished channel activity elicited in the presence of 10 μm SNP (Fig. 6B). When DTT was washed out with a solution containing 10 μm SNP, the channel activity returned (Fig. 6D) and could later be inhibited by lidocaine (Fig. 6E). DTT reversibly reduced the mean current of -0.45 ± 0.07 pA (n= 4) recorded in the presence of SNP or SNAP to -0.03 ± 0.04 pA (P= 0.001, Fig. 7B). The channel activity in the presence of DTT appeared slightly less than the control level suggesting that disulfide bond formation may be associated with persistent Na+ channel activity. However, this difference was not statistically significant (P= 0.25) and needs to be examined further.
Differences in the effects of SNP and SNAP
The time delay before an increase in channel activity was observed appeared to be longer with SNAP (8.4 ± 1.6 min, n= 7) than with SNP (4.0 ± 0.8 min, n= 8) and the relative increase in mean current caused by similar concentrations of SNAP and SNP was greater with SNP (see Fig. 5, note a 10-fold difference in y-axis scale). It has been reported that SNP-elicited responses may have two components, one due to NO release and a second component caused by cyanide accumulation (Padgett & Whorton, 1995). It was therefore thought that the difference seen in the effects of SNAP and SNP could be due to SNP decomposition being accompanied by cyanide release (a maximum of five equivalents of CN− per molecule of SNP) (Feelisch, 1998). Hence, we investigated the effects of sodium cyanide (NaCN) on INa,p.
Effect of NaCN
Cyanide has been suggested to interact directly with N-methyl-D-aspartate receptors to alter their conformation and gating (Arden et al. 1998). However, direct effects of CN− on Na+ channels in excised patches have not been reported previously.
Application of 5 mm NaCN to inside-out patches held at Vp=+30 mV caused an increase in persistent Na+ channel activity in a similar manner to SNP and SNAP. An example of the effect of NaCN in an inside-out patch after 5 min exposure to the drug is shown in Fig. 8. The patch was essentially silent in control solution (Fig. 8A) but in the presence of 5 mm NaCN there was a significant increase in channel activity (Fig. 8B) which could be abolished with 10 μm lidocaine (Fig. 8C). The average change in mean current recorded from 19 patches exposed to 5 mm NaCN is shown in Fig. 9. After an average of 4.5 ± 1.0 min of cyanide perfusion, the mean current increased from -0.06 ± 0.04 to -0.58 ± 0.21 pA (n= 19, P= 0.02). Lidocaine reduced the average mean current recorded in the presence of NaCN (n= 4) to -0.01 ± 0.01 pA (see Fig. 9). The effect of NaCN could also be reversed by DTT (-0.03 ± 0.02 pA, n= 4).
Figure 8. Effect of NaCN on Na+ channel activity.

Representative traces are from an inside-out patch held at Vp=+30 mV. A, traces before drug application. B, after a 5 min application of 5 mm NaCN. C, inhibition of activity by 10 μm lidocaine.
Figure 9. The effect of 5 mm NaCN on mean persistent Na+ current.

Mean current recorded from 19 inside-out patches in control (-0.06 ± 0.04 pA), after approximately 5 min of exposure to 5 mm NaCN (-0.58 ± 0.21 pA) and after lidocaine exposure (-0.01 ± 0.01 pA, n= 4). NaCN significantly increased inward persistent channel activity which could be abolished by 10 μm lidocaine. The difference between control and 5 mm NaCN is statistically significant (n= 19, P= 0.02) as is the difference between 5 mm NaCN and 10 μm lidocaine (n= 4, P= 0.01).
Effect of DTNB
We also examined the effect of another oxidising but membrane-impermeant agent, 5,5′-dithiobis-2-nitrobenzoic acid (DTNB) (Sucher et al. 1990). Traces shown in Fig. 10 illustrate the effect of adding 1 mm DTNB to an inside-out patch excised from a hippocampal neuron. Figure 10A shows a typical quiet patch in control solution. After 5-6 min of perfusing with DTNB (Fig. 10B) channel activity had increased significantly. DTNB (1 mm) increased channel activity in a similar manner to SNP and SNAP in all three excised patches examined.
Figure 10. Effect of 1 mm DTNB on inside-out hippocampal patch.
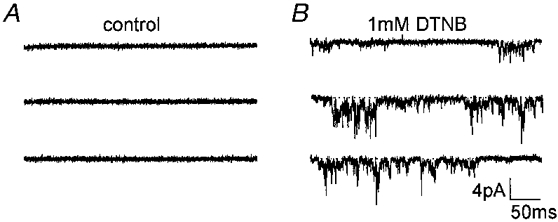
A sulfhydryl specific reagent, DTNB, caused an increase in persistent Na+ channel activity in a patch showing no channel activity over a previous 4 min recording period. A, before 1 mm DTNB was perfused. B, after about 6 min of exposure to 1 mm DTNB.
DISCUSSION
Our results show that NO increases INa,p, a current believed to be important for pacemaker activity in spontaneously firing neurons (Taylor, 1993). The amplitude of whole-cell INa,p increased by 60-80 % within a few minutes of SNAP or SNP exposure. No significant effect on INa,t was observed. NO may, by increasing INa,p, alter the firing rate and the ionic balance in these cells and eventually cause cell damage.
We were interested in determining how NO was increasing INa,p. It is well known that NO donors activate guanylate cyclase and cGMP-dependent pathways in most cells but it is now becoming recognised that direct effects of NO on membrane proteins involving S-nitrosylation and disulfide bond formation may also be important. A regulatory mechanism of ion channel activity involving sulfhydryl to disulfide conversion has been described previously and ‘regulatory thiols’ have been demonstrated in K+ channels (Ruppersberg et al. 1991), N-methyl-D-aspartate-receptor channels (Lei et al. 1992), inositol(1,4,5)trisphosphate (IP3) receptors (Kaplin et al. 1994) and ryanodine receptor calcium release channels (Zaidi et al. 1989; Salama et al. 1992). To explore the way in which SNP and SNAP affect neuronal Na+ channels, we examined their effects in excised inside-out patches from hippocampal neurons, where we could assume that the cGMP pathway would not be available. Both NO donors significantly increased the mean INa,p in excised patches at concentrations similar to those used in the whole-cell experiments. The channels we observed in the excised patches were Na+ channels and the evidence for this was (1) the pipette solution for single channel recordings contained cobalt and caesium to block Ca2+ and K+ channels, (2) currents were blocked by lidocaine, and (3) currents reversed at +30 mV (Vp= -30 mV), consistent with a Na+ channel with a PNa/PK ratio of 4 but excluding the possibility that these channels were Cl− channels. The effect of SNP and SNAP in excised inside-out patches suggests that NO is acting directly on the Na+ channel protein, or a closely associated regulatory protein, in the plasma membrane, thereby directly regulating the activity of persistent Na+ channels. Modulation of protein thiols can be induced by biologically active nitrosothiol compounds (R-SNOs, e.g. SNAP) via direct interaction (transnitrosation) or indirectly following decomposition to nitrosonium cation (NO+), nitroxyl anion (NO−), and/or nitric oxide (NO.). The process of transferring an NO-group to a protein is termed S-nitrosylation. The effect we describe probably occurred as a result of a conformational change in the Na+ channel protein due to disulfide bond formation between two closely located S-nitrosylated thiols in the protein, since single nitrosothiols are very labile (Girard & Potier, 1993) but the change in INa,p was robust. The change in conformation may convert a transient to a persistent channel by removing inactivation.
The hypothesis that the effect of NO is due to disulfide bond formation was supported by similar increases in channel activity elicited by the more sulfhydryl-specific agent, DTNB, and also the reversal of the effect of the NO donors with the sulfhydryl reducing agent DTT. DTT has been reported to reduce several thiol oxidation states, among them mixed disulfides, intra- and intermolecular disulfides and sulfenic acids (Padgett & Whorton, 1995). The fact that DTT sometimes appeared to reduce the mean current below the control level (Fig. 7B) may indicate that the channel is often at least partially oxidised. In agreement with this, inactivation of axonal Na+ channels can be altered by glutathione. It was shown (Strupp et al. 1992) that 5 mm glutathione suppressed long-lasting single Na+ channel currents in excised patches from rat dorsal spinal root ganglia, suggesting that cysteine-based disulfide bridges contribute to channel kinetics even under control conditions. Our experiments also suggest, since both DTT and DTNB are membrane impermeant, that this regulation of persistent sodium channel activity involves an intracellular site of the channel protein.
The time taken to elicit an increase in mean current and the magnitude of the increase were different for SNAP (3-100 μm) (an S-nitrosothiol, RSNO) and SNP (10-100 μm) (an inorganic iron nitrosyl compound) (see Results and Fig. 5). The differences in the magnitude of the increase and the time course of action of the two NO donors may be related to transnitrosation reactions, the kinetics of NO release and/or whether the NO formation is via an enzymatic or non-enzymatic pathway. Such differences between NO donors, including SNP and SNAP, have been described previously (Feelisch, 1998). Nevertheless, both NO donors elicited the same effect, i.e. they both increased INa,p activity. We wanted, however, to investigate this point further and examined the effect of a vital component of the SNP molecule, cyanide, which is released in maybe even larger amounts than NO (5 FeCN moieties per molecule of SNP), on INa,p activity. We found that cyanide added by itself increased the persistent sodium channel activity, activity which could be reversed by DTT or lidocaine. These results suggest that the observed difference between SNP and SNAP may be due, at least in part, to cyanide release from the SNP complex. Interestingly, the effect of cyanide, which has been suggested to act as a reducing agent in some preparations (Karnik & Khorana, 1990), could be blocked by a reducing agent, DTT. It may be that cyanide is affecting Na+ channels in a similar manner to N-methyl-D-aspartate receptors where it was reported to have subunit-specific interactions similar to those of the oxidizing agent DTNB (Arden et al. 1998). Alternatively, CN− inhibits a Ca2+-dependent K+ current in excised patches from central neurons (Jiang & Haddad, 1994), an interaction believed to involve an iron-containing moiety since it can be mimicked by iron chelators such as 1,10-phenantroline. It is well established that both CN− and NO interact with haem-containing moieties, e.g. guanylate cyclase and cytochrome a3 of the mitochondrial electron transfer chain. Therefore, another explanation may be that CN− (and possibly NO) elicits its effects via a haem-protein interaction that increases the levels of free radicals in the immediate vicinity of the sodium channel. This issue is presently under investigation in our laboratory.
The effect of NO described here may occur during hypoxia and may be a critical link between anoxia and neuronal cell injury. It has been proposed that NO donors may be useful for preventing neuronal injury, since chronic administration of e.g. nitroglycerine would downregulate the NMDA-receptor channel. The effect of NO described here would increase the excitability of neurons and disrupt the normal oscillations in membrane potential and rhythmic firing of action potentials (Alonso & Llinás, 1989; Amitai, 1994) and hence would not support the use of NO donors clinically as neuroprotective agents. It may be that the effect of NO donors on INa,p observed may be related to the increase in INa,p amplitude that occurs during hypoxia in these cells (Hammarström & Gage, 1998), but more experiments are needed to confirm this.
Protecting the human brain from the spread of ischaemic damage is a challenging clinical problem and identifying the cascade of events involved in the neuronal cell damage is crucial for development of better treatments. An increase in INa,p would cause membrane depolarization and initially an increase in the rate of firing of action potentials. A sustained influx of Na+ may reverse the Na+-Ca2+ exchanger and thereby increase intracellular free Ca2+ levels as has been proposed in white matter where TTX protected axons against anoxic injury (Stys et al. 1992). It is of interest that mice in which the neuronal NO synthase gene has been deleted are resistant to focal and transient global ischaemia (Huang et al. 1994; Panahian et al. 1996). This observation strengthens the argument for a pathological significance of the neuronal NO pathway in the cascade initiated by ischaemia in vivo. One new link proposed here is via the oxidation of intracellular regulatory thiols of the Na+ channel protein.
Acknowledgments
A.K.M.H. is a Postdoctoral Fellow supported by the National Heart Foundation of Australia. We would like to thank John Curmi for assistance with cultured hippocampal neurons.
References
- Alonso A, Llinás RR. Subthreshold Na+-dependent theta-like rythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:1175–1177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. Journal of Neuroscience. 1993;13:660–673. doi: 10.1523/JNEUROSCI.13-02-00660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai Y. Membrane potential oscillations underlying firing patterns in neocortical neurons. Neuroscience. 1994;63:151–161. doi: 10.1016/0306-4522(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Archer SL, Huang JM, Hampl V, Nelson DP, Shultz PJ, Weir EK. Nitric oxide and cGMP cause vasorelaxation by activation of a charybdotoxin-sensitive K channel by cGMP-dependent protein kinase. Proceedings of the National Academy of Sciences of the USA. 1994;91:7583–7587. doi: 10.1073/pnas.91.16.7583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arden SR, Sinor JD, Potthoff WK, Aizenman E. Subunit-specific interactions of cyanide with the N-methyl-D-aspartate receptor. Journal of Biological Chemistry. 1998;273:21505–21511. doi: 10.1074/jbc.273.34.21505. [DOI] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Brown AM, Schwindt PC, Crill WE. Different voltage dependence of transient and persistent Na+ currents is compatible with modal-gating hypothesis for sodium channels. Journal of Neurophysiology. 1994;71:2562–2565. doi: 10.1152/jn.1994.71.6.2562. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium currents in mammalian central neurons. Annual Review of Physiology. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Feelisch M. The use of nitric oxide donors in pharmacological studies. Naunyn-Schmiedeberg's Archives of Pharmacology. 1998;358:113–122. doi: 10.1007/pl00005231. [DOI] [PubMed] [Google Scholar]
- French CR, Gage PW. A threshold sodium current in pyramidal cells in rat hippocampus. Neuroscience Letters. 1985;56:289–293. doi: 10.1016/0304-3940(85)90257-5. [DOI] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. Journal of General Physiology. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Anoxia induced an increase in intracellular sodium in rat cortical neurons in vitro. Brain Research. 1994;663:329–334. doi: 10.1016/0006-8993(94)91281-5. [DOI] [PubMed] [Google Scholar]
- Furchgott RF. Studies on relaxation of rabbit aorta by sodium nitrite: the basis for the proposal that the acid-activatable inhibitory factor from bovine retractor penis is inorganic nitrite and the endothelium-derived relaxing factor is nitric oxide. In: Vanhoutte PM, editor. Vasodilation: Vascular Smooth Muscle, Peptides, Autonomic Nerves and Endothelium. New York: Raven Press; 1988. pp. 401–414. [Google Scholar]
- Garland JG, McPherson GA. Evidence that nitric oxide does not mediate the hyperpolarization and relaxation to acetylcholine in the rat small mesenteric artery. British Journal of Pharmacology. 1992;105:429–435. doi: 10.1111/j.1476-5381.1992.tb14270.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilly WF, Armstrong CM. Threshold channels - a novel type of sodium channel in squid giant axon. Nature. 1984;309:448–450. doi: 10.1038/309448a0. [DOI] [PubMed] [Google Scholar]
- Girard P, Potter P. NO, thiols and disulfides. FEBS Letters. 1993;320:7–8. doi: 10.1016/0014-5793(93)81645-g. [DOI] [PubMed] [Google Scholar]
- Hammarström AKM, Gage PW. Inhibition of oxidative metabolism increases persistent sodium current in rat hippocampal neurons. The Journal of Physiology. 1998;510:735–741. doi: 10.1111/j.1469-7793.1998.735bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarström AKM, Gage PW. The effect of nitric oxide donors on persistent sodium current in hippocampal neurons. Biophysical Journal. 1999;76:A195. doi: 10.1111/j.1469-7793.1999.t01-1-00451.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- Ichimori K, Arroyo CM, Pronai L, Fukahori M, Nakazawa H. The reactions of 3,5-dibromo-4-nitrosobenzenesulfonate and its biological applications. Free Radical Research Communications. 1993;19:S129–139. doi: 10.3109/10715769309056s129. [DOI] [PubMed] [Google Scholar]
- Ignarro LJ, Buga GM, Wood KS, Byrns RE, Chaudhuri G. Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proceedings of the National Academy of Sciences of the USA. 1987;84:9265–9269. doi: 10.1073/pnas.84.24.9265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang C, Haddad GG. A direct mechanism for sensing low oxygen levels by central neurons. Proceedings of the National Academy of Sciences of the USA. 1994;91:7198–7201. doi: 10.1073/pnas.91.15.7198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y, Saint DA, Gage PW. Effects of lignocaine and quinidine on the persistent sodium current in rat ventricular myocytes. British Journal of Pharmacology. 1992;107:311–316. doi: 10.1111/j.1476-5381.1992.tb12743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. The Journal of Physiology. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplin AI, Ferris CD, Voglmaier SM, Snyder SM. Purified reconstituted inositol 1,4,5-triphosphate receptors: thiol reagents act directly on the receptor protein. Journal of Biological Chemistry. 1994;269:28972–28978. [PubMed] [Google Scholar]
- Karnik SS, Khorana HG. Assembly of functional rhodopsin requires a disulfide bond between cysteine residues 110 and 187. Journal of Biological Chemistry. 1990;265:17520–17524. [PubMed] [Google Scholar]
- Kovisto A, Nedergaard J. Modulation of calcium-activated non-selective cation channel activity by nitric oxide in rat brown adipose tissue. The Journal of Physiology. 1995;486:59–65. doi: 10.1113/jphysiol.1995.sp020790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei SZ, Pan ZH, Aggarwal SK, Chen HS, Hartman J, Sucher NJ, Lipton SA. Effect of nitric oxide production on the redox modulatory site of the NMDA receptor-channel complex. Neuron. 1992;8:1087–1099. doi: 10.1016/0896-6273(92)90130-6. [DOI] [PubMed] [Google Scholar]
- Levi RC, Alloatti G, Penna C, Gallo MP. Guanylate-cyclase-mediated inhibition of cardiac ICa by carbachol and sodium nitroprusside. Pflügers Archiv. 1994;426:419–426. doi: 10.1007/BF00388305. [DOI] [PubMed] [Google Scholar]
- Ma JY, Catterall WA, Scheuer T. Persistent sodium currents through brain sodium channels induced by G protein betagamma subunits. Neuron. 1997;19:443–452. doi: 10.1016/s0896-6273(00)80952-6. [DOI] [PubMed] [Google Scholar]
- Manzoni O, Prezeau L, Marin P, Deshager S, Bockaert J, Fagni L. Nitric oxide-induced blockade of NMDA receptors. Neuron. 1992;8:653–662. doi: 10.1016/0896-6273(92)90087-t. [DOI] [PubMed] [Google Scholar]
- Mayer ML, Miller RJ. Excitatory amino acid receptors, second messengers and regulation of intracellular Ca2+ in mammalian neurons. Trends in Physiological Sciences. 1990;11:254–260. doi: 10.1016/0165-6147(90)90254-6. [DOI] [PubMed] [Google Scholar]
- Méry PF, Pavoine C, Belhassen L, Pecker F, Fischmeister R. Nitric oxide regulates cardiac Ca2+ current. Involvement of cGMP-inhibited and cGMP-stimulated phosphodiesterases through guanylyl cyclase activation. Journal of Biological Chemistry. 1993;268:26286–26295. [PubMed] [Google Scholar]
- Padgett CM, Whorton AR. S-nitrosoglutathione reversibly inhibits GAPDH by S-nitrosylation. American Journal of Physiology. 1995;269:C739–749. doi: 10.1152/ajpcell.1995.269.3.C739. [DOI] [PubMed] [Google Scholar]
- Panahian N, Yoshida T, Huang PL, Hedley-Whyte ET, Dalkara T, Fishman MC, Moskowitz MA. Attenuated hippocampal damage after global cerebral ischemia in mice mutant in neuronal nitric oxide synthase. Neuroscience. 1996;72:343–354. doi: 10.1016/0306-4522(95)00563-3. [DOI] [PubMed] [Google Scholar]
- Parkington HC, Tonta MA, Coleman HA, Tare M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. The Journal of Physiology. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehl U, Schmid HA. Electrophysiological responses of neurons in the rat spinal cord to nitric oxide. Neuroscience. 1997;77:563–573. doi: 10.1016/s0306-4522(96)00495-2. [DOI] [PubMed] [Google Scholar]
- Premkumar LS, Chung S-H, Gage PW. GABA-induced potassium channels in cultured neurons. Proceedings of the Royal Society B. 1990;241:153–158. doi: 10.1098/rspb.1990.0079. [DOI] [PubMed] [Google Scholar]
- Ruppersberg PJ, Stocker M, Pongs O, Heinemann SH, Frank R, Koenen M. Regulation of fast inactivation of cloned mammalian Ik(A) channels by cysteine oxidation. Nature. 1991;352:711–714. doi: 10.1038/352711a0. [DOI] [PubMed] [Google Scholar]
- Saint DA, Ju YK, Gage PW. A persistent sodium current in rat ventricular myocytes. The Journal of Physiology. 1992;453:219–231. doi: 10.1113/jphysiol.1992.sp019225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salama G, Abramson JJ, Pike GK. Sulphydryl reagents trigger Ca2+ release from the sarcoplasmic reticulum of skinned rabbit psoas fibres. The Journal of Physiology. 1992;454:389–420. doi: 10.1113/jphysiol.1992.sp019270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz S, Yuen PST, Garbers DL. The expanding family of guanylyl cyclases. Trends in Physiological Sciences. 1991;12:116–120. doi: 10.1016/0165-6147(91)90519-x. [DOI] [PubMed] [Google Scholar]
- Shibata M, Araki N, Hamada J, Sasaki T, Shimazu K, Fukuuchi Y. Brain nitrite production during global ischemia and reperfusion: an in vivo microdialysis study. Brain Research. 1996;734:86–90. [PubMed] [Google Scholar]
- Stamler JS, Simon DI, Osborne JA, Mullins ME, Jaraki O, Michel T, Singel D, Loscalzo J. S-nitrosylation of proteins with nitric oxide: synthesis and characterization of biologically active compounds. Proceedings of the National Academy of Sciences of the USA. 1992;89:444–448. doi: 10.1073/pnas.89.1.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strupp M, Quasthoff S, Mitrovic N, Grafe P. Glutathione accelerates sodium channel inactivation in excised rat axonal membrane patches. Pflügers Archiv. 1992;421:283–285. doi: 10.1007/BF00374840. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Ionic mechanism of anoxic injury in mammalian CNS white matter: role of Na+ channels and Na+-Ca2+ exchanger. Journal of Neuroscience. 1992;12:430–439. doi: 10.1523/JNEUROSCI.12-02-00430.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sucher NJ, Wong LA, Lipton SA. Redox modulation of NMDA receptor-mediated Ca2+ flux in mammalian central neurons. NeuroReport. 1990;1:29–32. doi: 10.1097/00001756-199009000-00009. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Na+ currents that fail to inactivate. Trends in Neurosciences. 1993;16:455–460. doi: 10.1016/0166-2236(93)90077-y. [DOI] [PubMed] [Google Scholar]
- Zaidi NF, Lagenaur CF, Abramson JJ, Pessah IN, Salama G. Reactive disulfides trigger Ca2+ release from sarcoplasmic reticulum via an oxidation reaction. Journal of Biological Chemistry. 1989;264:21725–21736. [PubMed] [Google Scholar]