

Abstract
The hyperpolarization-activated If current was recorded in inside-out macropatches from sino-atrial (SA) node myocytes during exposure of their intracellular side to pronase, in an attempt to verify if cytoplasmic f-channel domains are involved in both voltage- and cAMP-dependent gating.
Superfusion with pronase caused a quick, dramatic acceleration of channel opening upon hyperpolarization and slowing, rapidly progressing into full blockade, of channel closing upon depolarization; these changes persisted after wash off of pronase and were irreversible, indicating proteolytic cleavage of channel regions which contribute to gating.
If recorded from patches normally responding to cAMP became totally insensitive to cAMP following pronase treatment, indicating partial or total removal of channel regions involved in the cAMP-dependent activation.
The fully activated I–V relationship was not modified by pronase, indicating that internal proteolysis did not affect the f-channel conductance.
The changes in If kinetics induced by pronase were due to a large depolarizing shift of the f-channel open probability curve (56.5 ± 1.1 mV, n = 7).
These results are consistent with the hypothesis that cytoplasmic f-channel regions are implicated in dual voltage- and cAMP-dependent gating; also, since pronase does not abolish hyperpolarization-activated opening, an intrinsic voltage-dependent gating mechanism must exist which is inaccessible to proteolytic cleavage. A model scheme able to account for these data thus includes an intrinsic gating mechanism operating at depolarized voltages, and a blocking mechanism coupled to cAMP binding to the channel.
Hyperpolarization-activated channels are dually regulated by voltage and cyclic nucleotides (DiFrancesco, 1993). In cardiac pacemaker myocytes of the rabbit sino-atrial (SA) node, pacemaker f-channels open upon hyperpolarization with a threshold of about −40 to −50 mV (DiFrancesco et al. 1986); their voltage range of activation is modulated by cAMP, which binds directly to channels at their intracellular side and shifts the channel open probability curve to more positive voltages, thus increasing the fraction of available current (DiFrancesco & Tortora, 1991; DiFrancesco & Mangoni, 1994). cAMP-dependent control of f-channel activation represents the molecular basis of one of the key mechanisms involved in the autonomic regulation of pacemaker activity in the SA node and hence of heart rate (DiFrancesco, 1993).
Since the action of cAMP on the channel open probability is equivalent to a voltage shift of its voltage dependence, there may be a common mechanism by which cAMP and voltage act on f-channel kinetics. Indeed, we have recently shown that the shifting action of intracellular cAMP, and its sigmoidal concentration dependence, are accounted for by a cyclic allosteric cAMP and voltage dual-activation model, whereby both cAMP binding and voltage hyperpolarization contribute to channel opening (DiFrancesco, 1999). The allosteric model also predicts differential effects of cAMP on channel opening and closing rates, as experimentally verified. According to this interpretation, the main mechanism by which cAMP favours channel opening is by locking channels in their open configuration (DiFrancesco, 1999).
The suggestion that voltage hyperpolarization and cAMP binding stabilize open channels by a common mechanism implies that they may modify the channel conformation in similar ways. We thus considered the possibility that an intracellular channel region, sensitive to both voltage and cAMP binding, participates in channel gating.
METHODS
The methods for obtaining isolated pacemaker myocytes from rabbit SA node were as previously described (DiFrancesco et al. 1986). We anaesthesized rabbits (New Zealand White, weighing about 1 kg) by i.m. injection of xylazine 4.6 mg kg−1 and ketamine 60 mg kg−1. The animals were then killed by cervical dislocation, exsanguinated and hearts removed swiftly. We used procedures conforming with guidelines of care and use of laboratory animals as established by State (D.L. 116/1992) and European directives (86/609/CEE).
Following enzymatic dissociation, myocytes were stored at 4 °C for the day. Aliquots were dispersed into plastic Petri dishes and superfused at a temperature of 24–26°C with a Tyrode solution containing (mM): NaCl, 140; KCl, 5.4; CaCl2, 1.8; MgCl2, 1; D-glucose, 5.5; Hepes-NaOH, 5 (pH 7.4). Selected cells were superfused by a fast-perfusion device with a high-K+ solution containing (mM): KCl, 130; NaCl, 10; MgCl2, 1; Hepes-KOH, 10; D-glucose, 5 (pH 7.4), and cell-attached patches formed with large-tipped pipettes containing (mM): NaCl, 70; KCl, 70; CaCl2, 1.8; MgCl2, 1; BaCl2, 1; MnCl2, 2; Hepes-KOH, 5 (pH 7.4). After formation of inside-out macropatches (DiFrancesco & Mangoni, 1994), we superfused their intracellular side with a solution containing (mM): potassium aspartate, 130; NaCl, 10; CaCl2, 2; EGTA, 5; Hepes-KOH, 10 (pH 7.2; pCa 7). We selected macropatches in which If recorded in the inside-out configuration showed a robust response to cAMP (DiFrancesco & Tortora, 1991), indicating free access of the perfusing solution to the cytoplasmic channel side. Pronase (Sigma P-5147, 5.1 units mg−1; 2 mg ml−1) was added to the intracellular-like solution and perfused for 1 min, after which perfusion with standard solution was resumed. We used this protocol since we noticed that modification of f-channel kinetics was completed within the first 60 s after pronase admission, and was irreversible (see Fig. 1B). Slow run-down of If in macropatches, due mostly to a negative shift of the activation curve, is known to occur in inside-out recordings (DiFrancesco & Mangoni, 1994); this process is accompanied by a negative shift of the time-constant curve (DiFrancesco, 1999), which explains why activation in macropatch measurements is normally slower than in whole-cell measurements (compare for example Fig. 4 of DiFrancesco, 1999, with Fig. 1 of DiFrancesco & Noble, 1989). In this work, we selected macropatches where the current amplitude recorded during a test pulse had stabilized before superfusion of pronase. In pilot experiments, prolonging time of exposure to pronase (up to 3 min) did not further modify channel kinetics, whereas run-down of current amplitude tended to increase (data not shown).
Figure 1. Action of pronase on macropatch If.
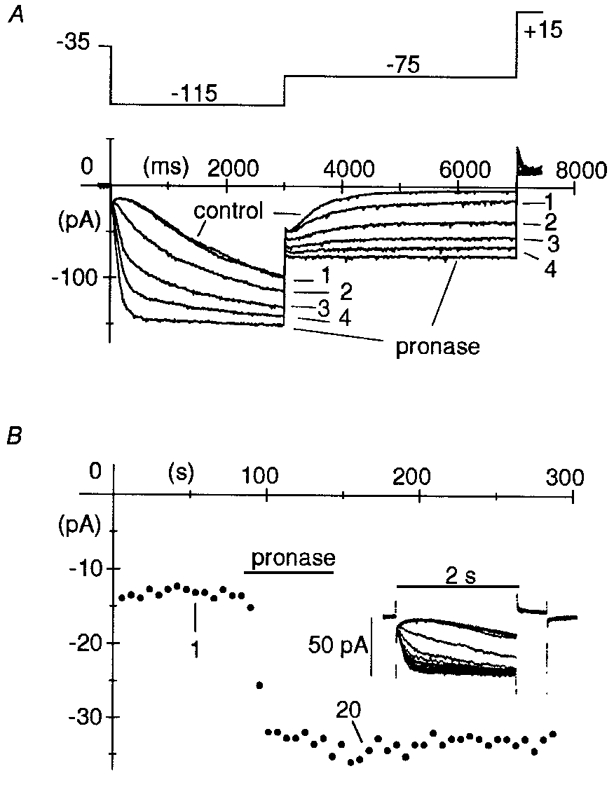
A, pronase (2 mg ml−1) was superfused on the intracellular patch side for 1 min, after which superfusion with normal intracellular-like solution was resumed, while activating/ deactivating If current traces were recorded during two-step protocols (see top panel illustrating the voltage protocol) to −115 mV (3 s) and −75 mV (4 s) applied every 10 s. Holding potential was −35 mV. A short 0.5 s step to +15 mV was applied after each pair of hyperpolarizations to fully deactivate If. Pronase induced progressive acceleration of activation at −115 mV, and slowing of deactivation at −75 mV, until after about 60 s, deactivation was fully blocked. Plotted are the first four records following switch-on of pronase superfusion and the records in control and at steady state after pronase, as indicated. B, time course of pronase action. Pronase was superfused for 1 min while applying 2 s steps to −95 mV to activate If (followed by 0.5 s steps to +15 mV to achieve full deactivation prior to subsequent activating step) every 6 s, from a holding potential of −25 mV. In the inset, 20 successive records are superimposed showing the progressive acceleration of current activation. The plot represents If amplitude at −95 mV. The amplitudes of 1st and 20th record of inset are indicated.
Figure 4. Lack of modification of the If conductance by pronase.
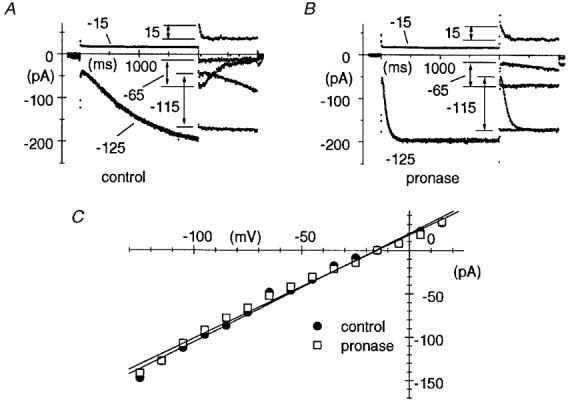
A and B, the fully activated I–V relationship of If was measured in control conditions (A) and after pronase treatment (B) according to a method previously developed (DiFrancesco et al. 1986), which consisted of two-step protocols where the current was either fully activated (by stepping to −125 mV for 2 s) or fully deactivated (by stepping to −15 mV for 2 s) before stepping to test potentials in the range −115 to +15 mV. In A and B, pairs of records shown correspond to test potentials of −115, −65 and +15 mV as indicated. Notice acceleration of current activation and slowing or block of current deactivation after pronase. C, fully activated I–V relationships in control conditions (•) and after pronase (□). The points were measured as differences between initial current values of pairs of traces corresponding to the same test potential (arrows in A and B). Linear regressions (straight lines) yielded reversal potentials of −15.7 and −15.1 mV, and conductances of 1.25 and 1.20 nS for control and pronase data, respectively.
Ramps of 60 s from −35 to −145 mV were used to measure open probability (Po) curves (normalized to channel maximum open probability), according to a previously devised protocol (DiFrancesco & Mangoni, 1994). Curves were fitted according to the square of a Boltzmann equation, such that:
where V½ is the half-activation voltage and v the inverse slope factor (DiFrancesco, 1999).
RESULTS
Recent evidence indicates expression in mammalian cardiac tissue of at least two members (HCN2, HCN4) of the hyperpolarization-activated, cyclic nucleotide-gated (HCN) channel family recently cloned (Gaußet al. 1998; Ludwig et al. 1998; Santoro et al. 1998; terminology as in Clapham, 1998). In SA node, although HCN2 and HCN4 have been reported to code for channels with different kinetics, their gating is, as in native channels, dependent upon voltage hyperpolarization and cAMP (Ludwig et al. 1999; Ishii et al. 1999).
As for other channels belonging to the cyclic nucleotide-gated (CNG) family, the cyclic nucleotide-binding site of HCN clones is located on the intracellular C-terminal domain (Gaußet al. 1998; Ludwig et al. 1998; Santoro et al. 1998). According to previous work in native f-channels, cAMP binding to its intracellular consensus site is more likely to occur when the channel is in the open configuration, and opening is favoured by cAMP since cAMP locks channels in the open state upon binding (DiFrancesco, 1999). The combined evidence from native channels and channel clones suggests therefore that the C-terminal part of the channel carrying the cAMP-binding site takes different physical conformations during voltage-dependent closed-to-open transitions, and that the cAMP consensus site is more efficiently bound in the open state. This raises the possibility that a conformational change involving the C-terminal domain participates in both voltage- and cAMP-dependent gating of f-channels.
To help understand whether f-channel gating may be associated with conformational changes of intracellular channel domains, we investigated to see if the integrity of intracellular domains affects f-channel activation and/or inactivation properties.
In Fig. 1AIf was recorded from an inside-out macropatch during a two-step activation/deactivation protocol to −115 and −75 mV applied every 10 s from a holding potential of −35 mV and pronase was superfused onto the intracellular membrane side of the patch. Within seconds from admission of pronase, a dramatic acceleration during activation and slowing during deactivation progressively occurred in the current time course. The activation time constant at −115 mV, according to square exponential fitting, varied from 1304 ms, a value similar to those obtained in control conditions (see DiFrancesco, 1999, Fig. 4C), to 119 ms after pronase, with a larger than 10-fold acceleration. The first four records following superfusion with pronase are shown in Fig. 1A along with control and steady-state pronase records. At steady state, activation was strongly accelerated and deactivation was fully blocked.
The changes induced by pronase on f-channel kinetics were not due to unspecific action on the channels, since they persisted after the 1 min period of pronase superfusion of the intracellular patch side. In fact, changes occurred quickly during superfusion of the enzyme and did not reverse upon wash out, as shown in the example of Fig. 1B, where pronase was added for 60 s while activating If by 2 s steps to −95 mV applied every 6 s. This is in agreement with the idea that pronase-induced modifications of f-channel kinetics are due to irreversible cleavage of portions of the channel protein.
The modifications illustrated in Fig. 1 were observed in all patches where If was recorded upon hyperpolarization while superfusing pronase (n = 30). In some patches we noticed a progressive decrease of the current amplitude at full activation voltages, possibly reflecting run-down due to gradual channel disruption.
To ascertain that the action of pronase on f-channel kinetics was accompanied by cleavage of regions implicated in the cAMP-induced activation, we checked the response of pronase-treated macropatches to cAMP. We found that patches responding normally to cAMP prior to pronase superfusion became totally insensitive to cAMP. In the patch shown in Fig. 2, for example, a full response to cAMP (10 μM, a concentration yielding near-saturation effects, DiFrancesco & Tortora, 1991) was obtained before pronase superfusion (Fig. 2A); following pronase treatment, however, If activation was strongly accelerated and deactivation slowed until it was fully blocked, but the current was completely insensitive to further addition of cAMP (Fig. 2B). In all patches where cAMP (10 μM) was tested, exposure to pronase led to full loss of responsiveness (n = 23).
Figure 2. Lack of action of cAMP on pronase-modified If.
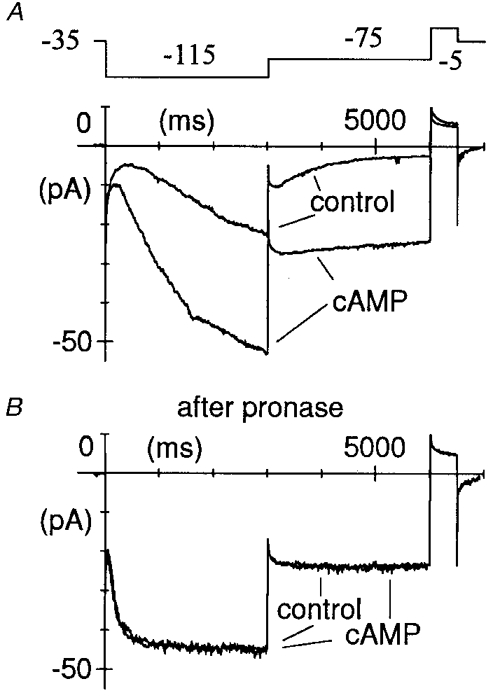
Macropatch If was recorded during two-step protocols (as shown in the top panel in A) from a holding potential of −35 mV to −115 and to −75 mV before (A) and after pronase (B). If responded normally to cAMP (10 μM) in control conditions (A); following pronase treatment, however, If activation was accelerated and deactivation blocked, and the current became completely insensitive to cAMP (B).
The strong acceleration during If activation and slowing during If deactivation, eventually leading to block of deactivation, suggests that pronase acts by shifting the current kinetics to more positive voltages. To verify this, and evaluate the size of the shift induced by pronase on If gating kinetics, we measured f-channel open probability curves (normalized to maximum channel open probability) by applying slow ramps, where patches were hyperpolarized from −35 mV to −145 mV in 60 s (DiFrancesco & Mangoni, 1994; Accili & DiFrancesco, 1996). In the patch shown in Fig. 3, the midpoint of the open probability curve and the inverse slope coefficient were −107.7 and 5.5 mV, respectively, in control conditions. The rather negative position of the midpoint of the open probability curve, a feature typical of inside-out macropatch recordings, was in the range reported previously (DiFrancesco & Mangoni, 1994). After pronase, the open probability curve midpoint shifted to −55.2 mV (by 52.5 mV) and the inverse slope coefficient was 8.0 mV (Fig. 3A, bottom panel). Notice that the fully activated current at −145 mV measured during ramp application (middle panel of Fig. 3A) was little changed by pronase, indicating that pronase did not affect the maximal If conductance. Although the patch If was normally responsive to cAMP (10 μM) in control conditions (not shown), after pronase the cAMP sensitivity was fully lost, as apparent from the ramp analysis (Fig. 3A). The shift induced by pronase was much larger than shifts caused by saturating concentrations of cAMP. To allow for a direct comparison, in Fig. 3B typical open probability curves of If in control conditions and in the presence of 10 μM cAMP are shown from a pronase-untreated macropatch. The midpoint of the open probability curve and the inverse slope coefficient were −96.8 and 8.4 mV for the control, and −79.9 and 8.6 mV, respectively, for the cAMP curve (shift, 16.9 mV). In n = 7 macropatches, pronase caused a shift of 56.5 ± 1.1 mV in the f-channel open probability curve. The inverse slope factor did not change significantly (from 6.78 ± 0.72 to 7.18 ± 0.44 mV, n = 7, P > 0.05).
Figure 3. Large depolarizing shift of f-channel open probability curve induced by pronase.
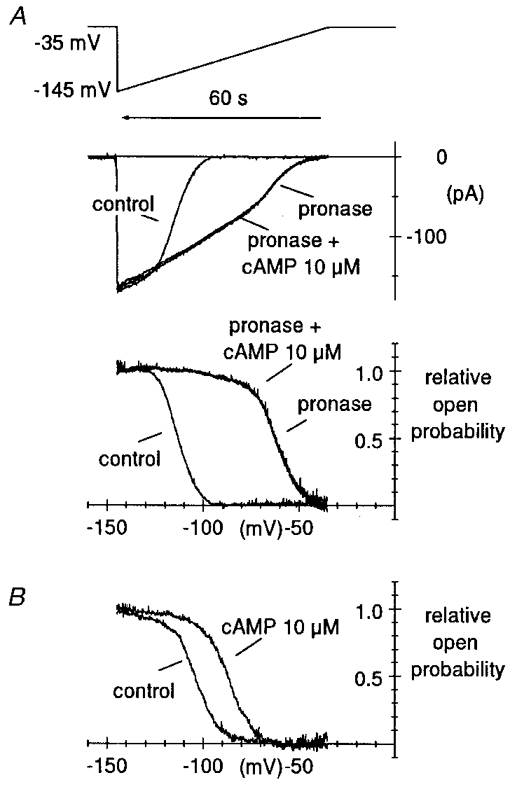
A, curves of the relative channel open probability (Po), normalized to maximum channel open probability, were measured by a voltage-ramp method previously developed (DiFrancesco & Mangoni, 1994) in control condition and after pronase treatment, as indicated. Ramps were applied from a holding potential of −35 mV to −145 mV and lasted 60 s (top panel). Notice that time runs backward for comparison with the open probability curves in the bottom panel. Ramp currents recorded in control conditions and after pronase, the latter with and without cAMP, are shown in the middle panel. Fitting open probability curves by the second power of the Boltzmann equation (see Methods) yielded V½ = −107.7 mV, v = 5.48 mV and V½ = −55.2 mV, v = 8.02 mV for the control and pronase curve, respectively (shift = 52.5 mV). The Po curve was not modified by 10 μM cAMP after pronase treatment. B, the same protocol was applied to determine, for comparison, the action of cAMP in a pronase-untreated macropatch. Best fitting parameters were V½ = −96.8 mV, v = 8.44 mV and V½ = −79.9 mV, v = 8.61 mV for the control and cAMP curve, respectively (shift = 16.9 mV).
To confirm the lack of action of pronase on the fully activated If, we compared the IfI–V relationship measured after the 1 min pronase superfusion with that in control conditions. As shown in Fig. 4, despite the marked modification of activation/deactivation kinetics, the fully activated current was unaltered by pronase treatment. In the macropatch of Fig. 4, linear regression yielded reversal potentials of −15.7 and −15.1 mV, and conductances of 1.25 and 1.20 nS from the control and pronase curves, respectively. In n = 8 patches, the mean If conductance change measured within 60–120 s after pronase treatment was −2.0 ± 1.2 % relative to control conditions.
DISCUSSION
These data show that superfusion of the intracellular side of SA node cell macropatches by pronase led to: (1) a large depolarizing shift of the f-channel open probability curve and (2) abolition of the shifting action of cAMP. The latter action implies that the pronase-induced cleavage of portions of the channel intracellular domain is likely to include, partially or totally, the cAMP-binding site.
On the other hand, our data do not allow identification of the part of the channel protein whose cleavage leads to the observed shift of the channel open probability curve in Fig. 3. However, our model of cAMP-induced activation (DiFrancesco, 1999) is consistent with opening of the channel as a consequence of a conformational change involving the cAMP-binding site (located at the C-terminus in HCN clones; Gaußet al. 1998; Ludwig et al. 1998; Santoro et al. 1998), which acquires in the open state a configuration with higher cAMP affinity. Binding of cAMP then allosterically stabilizes open channels mainly by locking subunits in their open state. This means that both voltage hyperpolarization and cAMP binding concur to activate channels by favouring the same open configuration, according to a dual cyclic allosteric scheme (DiFrancesco, 1999). A consequence of this arrangement is that the action of cAMP on open probability is equivalent to a depolarizing voltage shift.
The results presented here agree with the hypothesis that f-channel opening involves a conformational change of a pronase-sensitive domain, which simultaneously exposes the cAMP-binding site. Interestingly, a conformational change favouring cyclic nucleotide binding is known to occur in CNG channels (Tibbs et al. 1998). We may assume for example that a gate physically occluding the channel mouth is formed by part of the same C-terminus, or by a portion of the channel which is coupled to the cAMP-induced conformational change. In this case, opening would result from removal of a blocking gate located on (or coupled to) the cAMP-binding domain, with simultaneous exposure of the cAMP consensus site to free access of cAMP molecules.
A cartoon model depicting this scheme is illustrated in Fig. 5. In the top panel (control), channels are thought to switch from open bound (OB) to open (O) configurations following state transitions which involve displacement of a gate (depicted as a ball for representative purposes only) physically occluding the channel mouth. The blocking region could for example be located on the C-terminal end, but this is not essential for the validity of the proposed model: any other location (such as for example on the N-terminal) would be equally valid, as long as the processes of gating and cAMP binding are coupled. Displacement of the occluding gate results in a new configuration (O) which is more easily accessible to cAMP binding, a process leading to stabilization of open channels due to a locking action of cAMP (DiFrancesco, 1999).
Figure 5. Model interpretation of the action of pronase on f-channel gating.
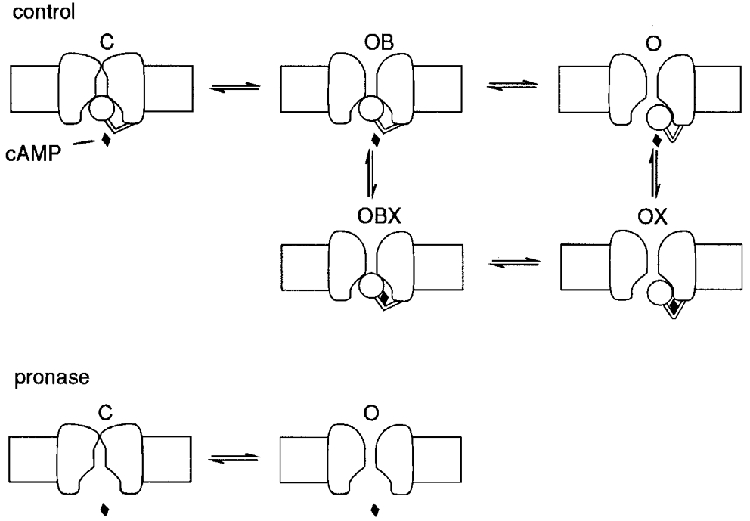
Upper panel (control), a terminal ball region is assumed to switch on voltage hyperpolarization from an open-blocked (OB) configuration, where it blocks the internal channel mouth, to an open one (O), the latter binding one cAMP molecule (OX) with higher affinity than the former (OBX). cAMP binding locks the ball in the open state. Symbols OB, O, OBX and OX refer to the allosteric model of DiFrancesco (1999, Fig. 2; replacing O for R, relaxed and OB for T, tense). Lower panel (pronase), protease cleavage of the terminal ball region abolishes the corresponding gating mechanism and the cAMP dependence. A second, voltage-dependent gating mechanism inaccessible to pronase is assumed to underly voltage-dependent kinetics between states C (closed) and O in the absence of the ‘ball’-dependent gating. Further explanation in text.
According to this scheme, removal by proteolysis of the channel protein terminal regions (Fig. 5, bottom panel, pronase) should have a twofold effect: (1) an expectedly large depolarizing shift of the Po curve, since removal of the blocking region abolishes ball-dependent closed states and, like cAMP, prevents channels blocking (the O configuration after pronase in the bottom panel of Fig. 5 corresponds to the four states OB, O, OBX and OX in control conditions); (2) abolition of the cAMP action. Both these effects were found when the intracellular side of macropatches was superfused with pronase (Figs 2 and 3).
The model scheme corresponding to configurations OB, O, OBX and OX coincides with the allosteric model previously used to describe the dual voltage and cAMP dependence of f-channel gating (symbols OB for open blocked and O for open, replacing T for tense and R for relaxed as used by DiFrancesco, 1999). According to the allosteric scheme, on the assumption that each subunit has a single cAMP-binding site, the probability P that each channel (gating) subunit is in the open state (consisting of states O and OX) is:
| (1) |
where X is cAMP concentration, KOB and KO are dissociation constants for agonist binding to blocked and open configurations, respectively, and where the equilibrium constant L = [OB]/[O] of open/blocked transitions in the absence of agonist depends on voltage according to the Boltzmann relation:
| (2) |
where V½ is the half-activation voltage and v the inverse slope coefficient (DiFrancesco, 1999). For channels obeying square exponential kinetics, the probability of channel opening will be Po = P2.
If, as experimentally verified, the action of pronase is to remove by cleavage an inhibitory channel domain upon which cAMP normally exerts its action, then in terms of the above scheme, pronase can be assumed to fully abolish this same inhibition. Pronase would thus behave as a cAMP agonist with infinite affinity, a condition which in eqn (1) can be expressed by setting KO = 0. Substituting into eqn (1), this yields the relation P = 1 for all cAMP concentrations.
Since the treatment with pronase, however, does not lead to constitutively open channels but rather to a very large positive shift of the open probability curve, it might seem logical to assume that pronase is equivalent to a cAMP agonist with very high, but finite affinity. Although this possibility cannot be excluded, we do not favour it, since it would imply that the proteolytic action of pronase is incomplete, or that a subset of f-channels is not accessible to pronase. This contrasts with the evidence we find that: (1) the action of pronase reaches a steady state within tens of seconds (Fig. 1B), and no extra acceleration of activation occurs when pronase superfusion is mantained for several minutes; and (2) channel kinetics become totally insensitive to cAMP, suggesting full removal of channel regions interacting with cAMP.
An alternative possibility to explain the ability of channels to close after pronase, though at much more depolarized voltages, is to assume the existence of an independent, intrinsic (i.e. not interacting with cAMP) gating mechanism inaccessible to cleavage and distinct from the blocking mechanism. To represent this, a state C (for closed) has been included in the model scheme of Fig. 5. The large shift of open probability curve after pronase implies that normally, intrinsic gating is non-functional. Thus, the intrinsic gate activates much more rapidly and deactivates much more slowly than the putative blocking gate (Figs 1 and 2), such that the latter can be assumed to govern f-channel open/close kinetics under normal conditions. In other words, at voltages at which the blocking mechanism operates, the state C will be essentially empty.
It is worth noting that the scheme presented in Fig. 5 has features reminiscent of kinetic models proposed to hold for K+ channels. Shaker K+ channels, for example, open upon depolarization and then inactivate according to two distinct mechanisms: N-type inactivation, due to N-terminal-dependent block, and C-type inactivation, mainly located at the outer channel mouth (Baukrowitz & Yellen, 1995). Assuming, on the basis of purely descriptive purposes, that the intrinsic gating mechanism of f-channels is analogous to the K+ channel C-type inactivation, and that the blocking mechanism is analogous to N-type inactivation, opening of f-channels upon hyperpolarization could then be considered as consisting of the sequential removal of two inactivation mechanisms (rather than a closed-to-open transition), similar to the removal of C-type and N-type inactivation states in K+ channels, also occurring upon membrane hyperpolarization (compare with Baukrowitz & Yellen, 1995, Fig. 6). The hyperpolarization-induced activation process of f-channels would then be regarded as analogous to removal of the inactivation process of K+ channels.
A limit on this comparison is that, unlike K+ channels, f-channels do not possess an inactivated state at hyperpolarized voltages. On the other hand, the physiological voltage range of If activation is not dissimilar from the inactivation range of some A-type K+ channels (compare for example DiFrancesco et al. 1986, Fig. 5 with Covarrubias et al. 1991, Fig. 5 and Thompson, 1977 as reported in Hille, 1992, p. 119). Further, it is known that the voltage dependence of activation and inactivation processes can be modified by several amino acid substitutions in K+ channel clones (Liman et al. 1991; Lopez et al. 1991; Tytgat et al. 1993; Baukrowitz & Yellen, 1995; Zou et al. 1998).
Finally, there are previous indications that an inactivation process can be converted into an activation process via mutations altering the voltage dependence of gating. For example, the delayed outward rectifying behaviour of Shaker K+ channels can be converted into that of an inward rectifier (such as the KAT1 channel) by amino acid substitutions in the S4 segment, which remove positively charged residues and are able to shift the activation curve to highly negative voltages (Miller & Aldrich, 1996).
In conclusion, the action of pronase superfusion on the intracellular side of f-channels can be interpreted to indicate that: (1) the changes in gating due to proteolysis are accounted for by an extension of the allosteric model previously proposed to describe the dual voltage and cAMP dependence of f-channel gating (DiFrancesco, 1999); (2) there is a proteolysis-sensitive domain that inhibits opening and is partly overcome by cAMP; (3) cleavage of this domain fully removes the cAMP-dependent channel inhibition and acts as an irreversible agonist with very high efficacy; and (4) an intrinsic gating mechanism inaccessible to pronase can account for the residual voltage dependence after proteolysis.
Acknowledgments
This work was supported by the Ministero dell Università e della Ricerca Scientifica e Tecnologica (MURST) and by Telethon. We wish to thank A. Ferroni and A. Malgaroli for discussion.
References
- Accili EA, DiFrancesco D. Inhibition of the hyperpolarization-activated current (if) of rabbit SA node myocytes by niflumic acid. Pflügers Archiv. 1996;431:757–762. doi: 10.1007/BF02253840. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: a tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Clapham D. Not so funny anymore: pacing channels are cloned. Neuron. 1998;21:5–7. doi: 10.1016/s0896-6273(00)80508-5. [DOI] [PubMed] [Google Scholar]
- Covarrubias M, Wei A, Salkoff L. Shaker, Shal, Shab and Shaw express independent K+ current systems. Neuron. 1991;7:763–773. doi: 10.1016/0896-6273(91)90279-9. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. Pacemaker mechanisms in cardiac tissue. Annual Reviews of Physiology. 1993;55:451–467. doi: 10.1146/annurev.ph.55.030193.002323. [DOI] [PubMed] [Google Scholar]
- DiFrancesco D. Dual allosteric modulation of pacemaker (f) channels by cAMP and voltage in rabbit SA node. The Journal of Physiology. 1999;515:367–376. doi: 10.1111/j.1469-7793.1999.367ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D, Ferroni A, Mazzanti M, Tromba C. Properties of the hyperpolarizing-activated current (if) in cells isolated from the rabbit sino-atrial node. The Journal of Physiology. 1986;377:61–88. doi: 10.1113/jphysiol.1986.sp016177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D, Mangoni M. The modulation of single hyperpolarization-activated (if) channels by cyclic AMP in the rabbit SA node. The Journal of Physiology. 1994;474:473–482. doi: 10.1113/jphysiol.1994.sp020038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiFrancesco D, Noble D. Current if and its contribution to cardiac pacemaking. In: Jacklet Jw., editor. Neuronal and Cellular Oscillators. New York: Dekker; 1989. pp. 31–57. [Google Scholar]
- DiFrancesco D, Tortora P. Direct activation of cardiac pacemaker channels by intracellular cyclic AMP. Nature. 1991;351:145–147. doi: 10.1038/351145a0. [DOI] [PubMed] [Google Scholar]
- Gauß R, Seifert R, Kaupp BU. Molecular identification of a hyperpolarization-activated channel in sea urchin sperm. Nature. 1998;393:583–587. doi: 10.1038/31248. [DOI] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. 2. Sunderland: Sinauer; 1992. [Google Scholar]
- Ishii TM, Takano M, Xie L-H, Noma A, Ohmori H. Molecular characterization of the hyperpolarization-activated cation channel in rabbit sinoatrial node. Journal of Biological Chemistry. 1999;274:12835–12839. doi: 10.1074/jbc.274.18.12835. [DOI] [PubMed] [Google Scholar]
- Liman ER, Hess P, Weaver F, Koren G. Voltage sensing residues in the S4 region of a mammalian K+ channel. Nature. 1991;353:752–756. doi: 10.1038/353752a0. [DOI] [PubMed] [Google Scholar]
- Lopez GA, Jan JN, Jan LY. Hydrophobic substitution mutations in the S4 sequence alter voltage-dependent gating in Shaker K+ channels. Neuron. 1991;7:327–336. doi: 10.1016/0896-6273(91)90271-z. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Jeglitsch M, Hofmann F, Biel M. A family of hyperpolarization-activated mammalian cation channels. Nature. 1998;393:587–591. doi: 10.1038/31255. [DOI] [PubMed] [Google Scholar]
- Ludwig A, Zong X, Stieber J, Hullin R, Hofmann F, Biel M. Two pacemaker channels from human heart with profoundly different activation kinetics. EMBO Journal. 1999;18:2323–2339. doi: 10.1093/emboj/18.9.2323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller AG, Aldrich RW. Conversion of a delayed rectifier K+ channel to a voltage-gated inward rectifier K+ channel by three amino acid substitutions. Neuron. 1996;16:853–858. doi: 10.1016/s0896-6273(00)80105-1. [DOI] [PubMed] [Google Scholar]
- Santoro B, Liu DT, Yao H, Bartsch D, Kandel ER, Siegelbaum SA, Tibbs GR. Identification of a gene encoding a hyperpolarization-activated pacemaker channel of brain. Cell. 1998;93:717–729. doi: 10.1016/s0092-8674(00)81434-8. [DOI] [PubMed] [Google Scholar]
- Tibbs GR, Liu DT, Leypold BG, Siegelbaum SA. A state-independent interaction between ligand and a conserved arginine residue in cyclic-nucleotide-gated channels reveals a functional polarity of the cyclic-nucleotide binding site. Journal of Biological Chemistry. 1998;273:4497–4505. doi: 10.1074/jbc.273.8.4497. [DOI] [PubMed] [Google Scholar]
- Thompson SH. Three pharmacologically distinct potassium channels in molluscan neurons. The Journal of Physiology. 1977;265:465–488. doi: 10.1113/jphysiol.1977.sp011725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tytgat J, Nakazawa K, Gross A, Hess P. Pursuing the voltage sensor of a voltage-gated mammalian potassium channel. Journal of Biological Chemistry. 1993;268:23777–23779. [PubMed] [Google Scholar]
- Zou A, Xu QP, Sanguinetti MC. A mutation in the pore region of HERG K+ channels expressed in Xenopus oocytes reduces rectification by shifting the voltage dependence of inactivation. The Journal of Physiology. 1998;509:129–137. doi: 10.1111/j.1469-7793.1998.129bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]