

Abstract
ATP-sensitive potassium (KATP) channels are composed of pore-forming Kir6.2 and regulatory SUR subunits. A truncated isoform of Kir6.2, Kir6.2ΔC26, forms ATP-sensitive channels in the absence of SUR1, suggesting the ATP-inhibitory site lies on Kir6.2.
Previous studies have shown that mutation of the lysine residue at position 185 (K185) in the C-terminus of Kir6.2 to glutamine, decreased the channel sensitivity to ATP without affecting the single-channel conductance or the intrinsic channel kinetics. This mutation also impaired 8-azido[32P]-ATP binding to Kir6.2.
To determine if K185 interacts directly with ATP, we made a range of mutations at this position, and examined the effect on the channel ATP sensitivity by recording macroscopic currents in membrane patches excised from Xenopus oocytes expressing wild-type or mutant Kir6.2ΔC26.
Substitution of K185 by a positively charged amino acid (arginine) had no substantial effect on the sensitivity of the channel to ATP. Mutation to a negatively charged residue markedly decreased the channel ATP sensitivity: the Ki for ATP inhibition increased from 85 μM to >30 mM when arginine was replaced with aspartic acid. Substitution of neutral residues had intermediate effects.
The inhibitory effects of ADP, ITP and GTP were also reduced when K185 was mutated to glutamine or glutamate.
The results indicate that a positively charged amino acid at position 185 is required for high-affinity ATP binding to Kir6.2. Our results demonstrate that ATP does not interact with the side-chain of K185. It remains unclear whether ATP interacts with the backbone of this residue, or whether its mutation influences ATP binding allosterically.
ATP-sensitive potassium (KATP) channels are found in pancreatic β-cells, cardiac, smooth and skeletal muscles, and certain neurones (Ashcroft & Ashcroft, 1990). They are formed by the physical association of four inwardly rectifying K+ channel subunits (Kir6.2) with four sulphonylurea receptor subunits (either SUR1, SUR2A or SUR2B) (Inagaki et al. 1995, 1996; Sakura et al. 1995; Isomoto et al. 1996; Clement et al. 1997). Kir6.2 serves as the KATP channel pore, and contains the site at which intracellular ATP and ADP bind to cause channel inhibition. SUR is a regulatory subunit that modulates the channel gating properties, enhances the apparent ATP sensitivity and acts as the target for sulphonylurea drugs, K+ channel openers and intracellular Mg-nucleotides, which modulate KATP channel activity (Aguilar-Bryan et al. 1995; Inagaki et al. 1996; Nichols et al. 1996; Gribble et al. 1997b; Tucker et al. 1997). The way in which metabolism regulates KATP activity remains unclear, but both the stimulatory effects (mediated via SUR) and inhibitory effects (mediated via Kir6.2) of intracellular adenine nucleotides are thought to be involved.
Although wild-type Kir6.2 does not express functional channels in the absence of SUR1, deletion of the last 26 amino acids (Kir6.2ΔC26) enables its independent functional expression (Tucker et al. 1997). The ability of ATP to inhibit Kir6.2ΔC26 currents demonstrates that the ATP-inhibitory site does not lie on SUR1 and suggests that it may be located on Kir6.2. Additional support for this view is provided by the fact that mutations within this subunit can severely reduce the inhibitory effects of ATP (Tucker et al. 1997, 1998; Shyng et al. 1997a; Drain et al. 1998). In addition, direct binding of the photoaffinity analogue of ATP, 8-azido ATP, to Kir6.2 has been demonstrated (Tanabe et al. 1999).
In this paper we explore the effect of mutating a C-terminal amino acid, the lysine residue at position 185 (Fig. 1), on the sensitivity of Kir6.2ΔC26 to ATP. Mutation of this residue to glutamine (K185Q) markedly decreases the channel ATP sensitivity (Tucker et al. 1997, 1998; Koster et al. 1999) and reduces the extent of photoaffinity labelling of Kir6.2 with 8-azido[32P]-ATP (Tanabe et al. 1999). Although this suggests that K185 may be involved in ATP binding, the data do not distinguish whether this residue interacts directly with ATP or whether it has an allosteric effect on ATP binding. In an attempt to resolve this issue, we have investigated the effects of substituting a range of different amino acids at position 185.
Figure 1. Putative membrane topology of Kir6.2.
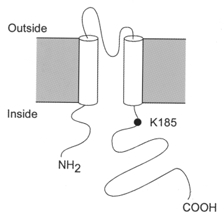
The position of residue K185, which lies in the C-terminus of the protein, is indicated.
METHODS
Mouse Kir6.2 (GenbankTM D50581, Inagaki et al. 1995; Sakura et al. 1995) and rat SUR1 (GenbankTM L40624, Aguilar-Bryan et al. 1995; provided by Dr G. Bell, University of Chicago) were used in this study. A 26 amino acid C-terminal deletion of mouse Kir6.2 (Kir6.2ΔC26) was made by introduction of a stop codon at the appropriate residue (Tucker et al. 1997). Site-directed mutagenesis of Kir6.2ΔC26 was carried out by subcloning the appropriate fragments into the pALTER vector (Promega). Synthesis of capped mRNA was carried out using the mMessage mMachine in vitro transcription kit (Ambion). Amino acids are indicated by the single-letter code.
Female Xenopus laevis were anaesthetised with MS222 (2g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V-VI Xenopus oocytes were manually defolliculated, injected with ∼2 ng of mRNA encoding wild-type or mutant forms of Kir6.2ΔC26 and studied 1–4 days after injection (Gribble et al. 1997a). In some experiments, oocytes were coinjected with ∼0.04 ng full length Kir6.2 (or Kir6.2ΔC26) and ∼2 ng of SUR1 mRNAs.
Macroscopic currents were recorded from giant inside-out patches using an EPC7 patch-clamp amplifier (List Electronik) at 20–24°C (Gribble et al. 1997a). The holding potential was 0 mV and currents were evoked by repetitive 3 s voltage ramps from −110 mV to +100 mV. Currents were filtered at 0.2 kHz, digitised at 0.5 kHz using a Digidata 1200 Interface and analysed using pCLAMP software (Axon Instruments).
The pipette solution contained (mM): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 Hepes (pH 7.4 with KOH) and the internal (bath) solution contained (mM): 110 KCl, 2 MgCl2, 1 CaCl2, 30 KOH, 10 EGTA, 10 Hepes (pH 7.2 with KOH) and nucleotides as indicated. Solutions containing nucleotides were freshly made up each day and the pH was readjusted after addition of nucleotide. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
Data analysis
The slope conductance was measured by fitting a straight line to the current-voltage relation between −20 and −100 mV: the average of five consecutive ramps was calculated in each solution. ATP dose-response relationships were measured by alternating test and nucleotide-free internal (control) solutions. Currents were corrected by subtraction of the background current measured in water-injected oocytes (∼5 pA at −100 mV). Conductance (G) was expressed as a fraction of the mean of that obtained in control solution (Gc) before and after ATP application. ATP dose-response curves were fitted to the Hill equation:
where [ATP] is the ATP concentration, Ki is the ATP concentration at which inhibition is half-maximal and h is the slope factor (Hill coefficient).
All data are given as means ±s.e.m. The symbols in the figures indicate the mean and the vertical bars s.e.m. (where this is larger than the symbol). Statistical significance was tested by Student's t test or ANOVA, as appropriate. A P value of < 0.05 was assumed to be significant.
RESULTS
Previous studies have shown that mutation of the positively charged lysine residue at position 185 of Kir6.2 to glutamine (K185Q; Fig. 1) greatly decreases the inhibitory effect of ATP (Tucker et al. 1998). This residue lies within the C-terminal region of the protein and is predicted to be located intracellularly (Sakura et al. 1995). If the lysine side chain contributes directly to ATP binding, then its substitution by amino acids with altered charge, hydrophobicity and/or ability to form hydrogen bonds should have markedly different effects on the measured ATP sensitivity.
Figure 2A shows that replacement of the lysine 185 by another positively charged amino acid, arginine (K185R), had negligible effect on the extent of channel inhibition produced by 1 mM ATP, whereas substitution of the negatively charged glutamate residue (K185E) caused a dramatic reduction in ATP sensitivity.
Figure 2. Effect of ATP on wild-type and mutant (K185R or K 185E) Kir6.2ΔC26 currents.
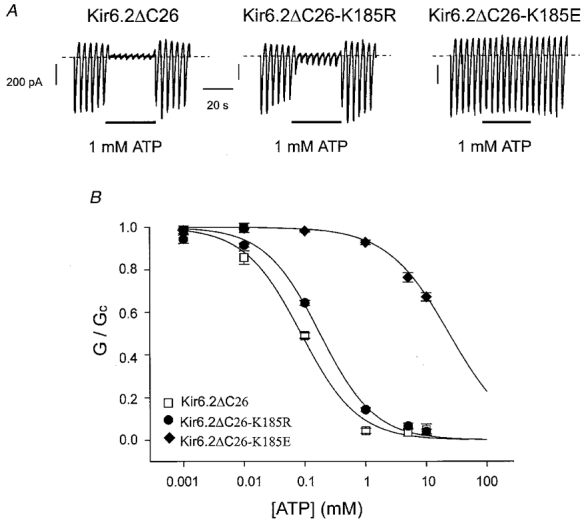
A, macroscopic currents recorded from inside-out patches excised from oocytes injected with mRNA encoding Kir6.2ΔC26, Kir6.2ΔC26-K185R or Kir6.2ΔC26-K185E. Currents were elicited in response to a series of voltage ramps from −110 to +100 mV. ATP (1 mM) was added as indicated by the bars. B, mean ATP dose-response relationships for Kir6.2ΔC26 (n = 4), Kir6.2ΔC26-K185R (n = 7) or Kir6.2ΔC26-K185E (n = 5) currents. The slope conductance (G) is expressed as a fraction of the mean of that obtained in control solution (Gc) before and after exposure to ATP. The lines are the best fit of the data to the Hill equation using the mean values for Ki and h given in Table 1.
Figure 2B compares the mean dose-response curves for ATP inhibition of wild-type Kir6.2ΔC26 with Kir6.2ΔC26 containing the K185R and K185E mutations. Mean data for these and other mutant channels are given in Table 1. Half-maximal inhibition (Ki) of Kir6.2ΔC26 was produced by 85 μM ATP (n = 4). The presence of an arginine at position 185 did not alter the Ki substantially (∼180 μM), but substitution of a negatively charged glutamate, or aspartate, residue caused a dramatic decrease in ATP sensitivity, shifting the Ki to >20 mM, or >30 mM, respectively (Fig. 2, Table 1). It was not possible to measure the Ki for these mutant channels accurately, because the very high ATP concentrations (>100 mM) that would be required would cause marked changes in ionic strength. Nevertheless, our results suggest that the negative charge at position 185 is not compatible with high ATP sensitivity. Single-channel recordings showed that, as is the case for K185Q (Tucker et al. 1998), mutation of K185 to glutamate did not alter the single-channel conductance or kinetics (data not shown).
Table 1.
ATP sensitivity of Kir6.2ΔC26 mutants
| Ki for ATP inhibition | h | n | Side-chain | Hydropathy | |
|---|---|---|---|---|---|
| K185 | 85 ± 8 μm | 0.95 ± 0.05 | 4 | (CH2)4NH3+ | −3.9 |
| K185R | 183 ± 9 μm | 0.95 ± 0.04 | 7 | −(CH2)3NHC(NH2)NH3+ | −4.5 |
| K185G | 164 ± 11 μm | 0.99 ± 0.04 | 6 | −H | −0.4 |
| K185S | 427 ± 38 μm | 0.88 ± 0.12 | 5 | −CH2 OH | −0.8 |
| K185N | 703 ± 141 μm | 0.79 ± 0.08 | 8 | −CH2 CONH2 | −3.5 |
| K185H | 1.17 ± 0.36 mm | 1.03 ± 0.11 | 3 | −CH2C(NHCHNCH) | −3.2 |
| K185Q | 2.25 ± 0.17 mm | 0.91 ± 0.04 | 14 | −CH2CH2CONH2 | −3.5 |
| K185A | 490 ± 29 μm | 1.00 ± 0.02 | 7 | −CH3 | 1.8 |
| K185C | 1.37 ± 0.05 mm | 0.91 ± 0.06 | 7 | −CH2SH | 2.5 |
| K185M | 2.57 ± 0.37 mm | 0.93 ± 0.06 | 7 | −CH2CH2SCH3 | 1.9 |
| K185F | 3.69 ± 0.31 mm | 0.97 ± 0.03 | 5 | −CH2C6H5 | 2.5 |
| K185L | 3.71 ± 1.58 mm | 0.79 ± 0.03 | 3 | −CH2CH(CH3)2 | 3.8 |
| K185V | 4.31 ± 0.31 mm | 0.94 ± 0.05 | 6 | −CH(CH3)2 | 4.2 |
| K185D | >30 mm | — | 2 | −CH2COO− | −3.5 |
| K185E | >20 mm | — | 6 | −CH2CH2COO− | −3.5 |
h, Hill coefficient for ATP inhibition. n, number of patches. Hydropathy values are taken from Kyte & Doolittle (1982). K1 values for all mutant channels are significantly different from wild-type.
Substitution of K185 with a neutral residue had intermediate effects on ATP sensitivity (Fig. 3, Table 1). Among the neutral residues, ATP sensitivity correlated most closely with the size of the side-chain of the amino acid at position 185, smaller side-chains, such as glycine, serine and alanine, conferring greater sensitivity to the nucleotide than larger groups such as glutamine and methionine (Fig. 3, Table 1). Asparagine had an intermediate effect. These results suggest that a bulky uncharged residue at position 185 may sterically hinder ATP binding. The Hill coefficients were not significantly different (by ANOVA) for any of the mutant channels and were close to unity, suggesting that for both wild-type and mutant channels the binding of a single ATP molecule is sufficient to cause channel inhibition.
Figure 3. Effect of ATP on wild-type and mutant (K185G, K185N or K185Q) Kir6.2ΔC26 currents.
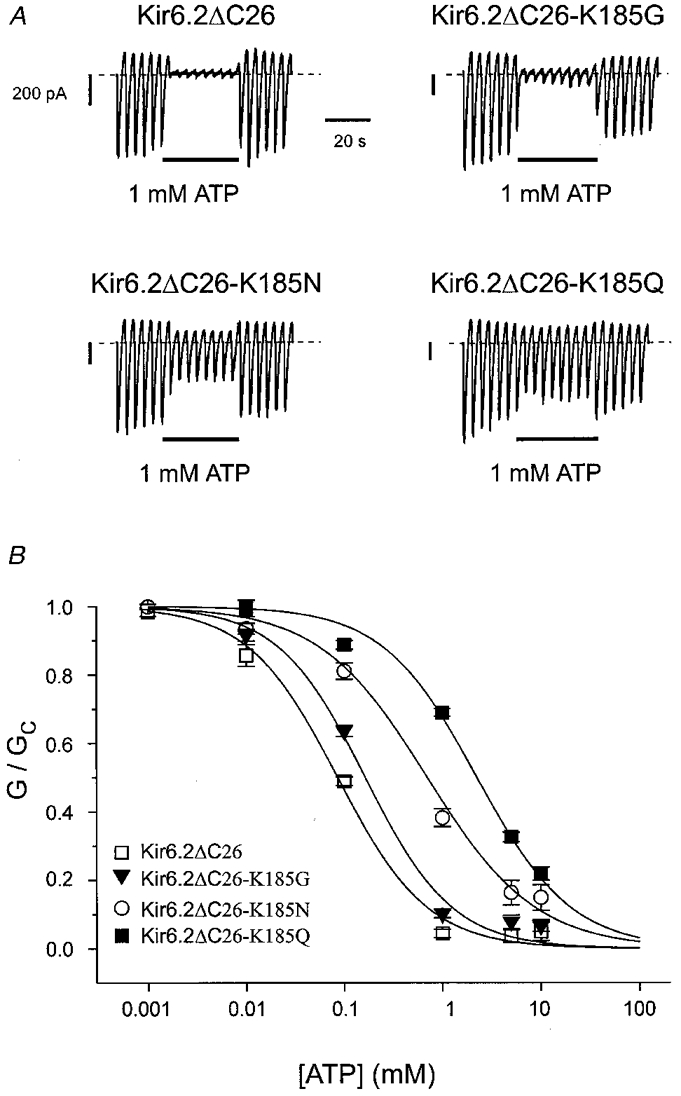
A, macroscopic currents recorded from inside-out patches excised from oocytes injected with mRNA encoding Kir6.2ΔC26, Kir6.2ΔC26-K185G, Kir6.2ΔC26-K185N or Kir6.2ΔC26-K185Q. Currents were elicited in response to a series of voltage ramps from −110 to +100 mV. ATP (1 mM) was added as indicated by the bars. B, mean ATP dose-response relationships for Kir6.2ΔC26 (n = 4), Kir6.2ΔC26-K185G (n = 6), Kir6.2ΔC26-K185N (n = 8) or Kir6.2ΔC26-K185Q (n = 14) currents. The slope conductance (G) is expressed as a fraction of the mean of that obtained in control solution (Gc) before and after exposure to ATP. The lines are the best fit of the data to the Hill equation using the mean values for Ki and h given in Table 1.
We next explored whether mutation of K185 altered the selectivity of the ATP-binding site to adenine nucleotides. Figure 4 compares ATP and ADP dose-response curves for Kir6.2ΔC26 and for Kir6.2ΔC26 containing the K185A and K185Q mutations. As is the case for ATP, both mutant channels were significantly less sensitive to ADP: the Ki values were 260 μM (Tucker et al. 1998), 1.12 ± 0.18 mM (n = 5) and 4.95 ± 0.74 mM (n = 5) for Kir6.2ΔC26, Kir6.2ΔC26-K185A and Kir6.2ΔC26-K185Q, respectively. In both cases, the sensitivity to ADP relative to that of ATP was unchanged. This result argues that K185 does not interact with the terminal phosphate group of the ATP molecule.
Figure 4. Effect of other nucleotides on mutant (K185A or K185Q) Kir6.2ΔC26 currents.
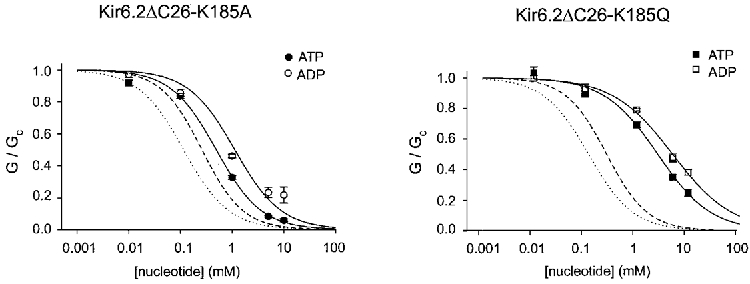
Mean ADP dose-response relationships for Kir6.2ΔC26-K185A (n = 5) and Kir6.2ΔC26-K185Q (n = 5) currents. The slope conductance (G) is expressed as a fraction of the mean of that obtained in control solution (Gc) before and after exposure to nucleotide. The lines are the best fit of the data to the Hill equation. For the data obtained in ATP solutions, the mean values for Ki and h given in Table 1. For the data obtained in ADP solutions, the mean Ki were 1.12 ± 0.18 mM (n = 5) and 4.95 ± 0.74 mM (n = 5) for Kir6.2ΔC26-K185A and Kir6.2ΔC26-K185Q, respectively, and the corresponding values of h were 0.97 ± 0.03 and 0.75 ± 0.08. The dotted and dashed lines give the mean dose-response curves obtained for wild-type Kir6.2ΔC36 for ATP and ADP, respectively (Tucker et al. 1998).
We next investigated whether there was any difference in the sensitivity of the mutant channels to the related purine nucleotide triphosphates, ITP and GTP. These nucleotides share significant structural homology to ATP. As shown in Fig. 6B, ITP is the most closely related and differs only in the nitrogen at the 1′ position, and the -NH2 group at the 6′ position, of the purine ring. In an attempt to measure the nucleotide sensitivity of the mutant channels more accurately, we coexpressed Kir6.2 with SUR1, in order to enhance the channel ATP sensitivity (Tucker et al. 1997), and thus make it possible to measure more of the dose-response curve for the mutant channels. When mutant Kir6.2 was coexpressed with wild-type SUR1, ATP enhanced, rather than reduced, the KATP current (Fig. 5). This stimulation of channel activity is mediated via the nucleotide-binding domains (NBDs) of SUR1 (Proks et al. 1998). It also occurs for wild-type channels but is masked by the inhibitory action of nucleotides on Kir6.2; when Kir6.2 is less sensitive to ATP inhibition, however, the stimulatory action of Mg-nucleotides becomes more evident. To avoid this complication, we used a mutant form of SUR1 (SUR1-KAKM) in which the lysine residues in the Walker A (WA) motifs of both NBDs have been mutated (to alanine in NBD1 and to methionine in NBD2). This has been shown to abolish the stimulatory effects of Mg-nucleotides on channel activity (Gribble et al. 1997b; Shyng et al. 1997b). As Fig. 5 also shows, mutation of the WA lysines abolished the stimulatory effect of MgATP on Kir6.2ΔC26-K185E + SUR1-KAKM currents.
Figure 6. Effects of nucleotide triphosphates on wild-type and mutant currents.
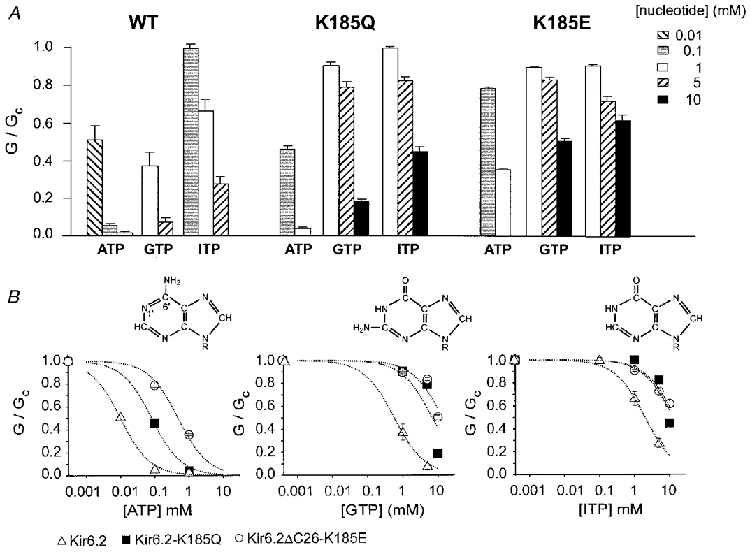
A, mean conductance recorded in the presence of the indicated nucleotide concentration, expressed relative to the mean of that recorded before and after exposure to the nucleotide, for SUR1-KAKM coexpressed with Kir6.2, Kir6.2-K185Q or Kir6.2ΔC26-K185E. The number of patches is given above each bar. B, mean concentration-inhibition relationships measured for Kir6.2 + SUR1-KAKM, Kir6.2-K185Q + SUR1-KAKM or Kir6.2ΔC26-K185E + SUR1-KAKM currents for ATP, GTP and ITP. The slope conductance (G) is expressed as a fraction of the mean (Gc) of that obtained in control solution before and after exposure to ATP. The lines are drawn through the data points by eye. Inset: chemical structures of ATP, GTP and ITP.
Figure 5. Effect of ATP on Kir6.2ΔC26-K185E + SUR1-KAKM currents.
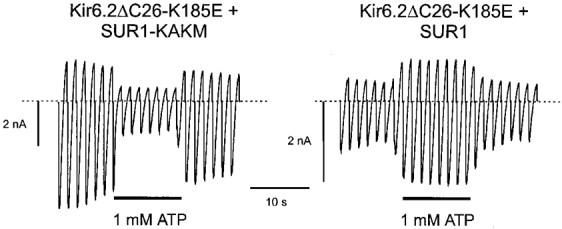
Macroscopic currents recorded from inside-out patches excised from oocytes injected with mRNA encoding Kir6.2ΔC26-K185E and either SUR1 or SUR1-KAKM. Currents were elicited in response to a series of voltage ramps from −110 to +100 mV. ATP (1 mM) was added as indicated by the bars.
Figure 6A shows the effect of different concentrations of ATP, ITP and GTP on the relative conductance of patches excised from oocytes coexpressing SUR1-KAKM plus either Kir6.2 or Kir6.2 containing the K185Q or K185E mutations. The data are also plotted on a semilogarithmic scale in Fig. 6B. Kir6.2 + SUR1-KAKM currents were significantly less sensitive to ITP and GTP than to ATP, with half-maximal inhibition being produced by around 10 μM ATP, 500 μM GTP and 2 mM ITP. The values for ATP and GTP are similar to those reported previously for Kir6.2 + SUR1 (Trapp et al. 1997; Tucker et al. 1997). Even in the presence of SUR1-KAKM, it was still not possible to obtain an accurate value for Ki for nucleotide inhibition of the mutant channels, because of the very high nucleotide concentrations that would be required (as discussed earlier). Nevertheless, it is clear that both mutations cause a marked shift in sensitivity to GTP and ITP, as well as to ATP.
DISCUSSION
Our results demonstrate that residue 185 has a marked influence on the sensitivity of Kir6.2ΔC26 currents to ATP, the greatest ATP sensitivity being found when this residue is positively charged and the smallest when it is negatively charged.
One mechanism by which a mutation may indirectly alter the channel ATP sensitivity is by impairing the ability of the channel to close (Shyng et al. 1997a; Trapp et al. 1998). However, as previously reported (Tucker et al. 1998), the properties of single-channel Kir6.2ΔC26-K185Q currents were not different from those of wild-type Kir6.2ΔC26 currents. This argues that the reduced ATP sensitivity is not a consequence of an impaired ability of the channel to close, and thus that the mutation is likely to interfere either with ATP binding or with the mechanism by which binding is linked to channel closure. In addition, because mutation of K185 does not modify the single-channel current amplitude, or the extent of rectification, this residue probably does not contribute functionally to the KATP channel pore. Earlier studies have also suggested that the K185Q mutation impairs binding of 8-azido[32P]-ATP to Kir6.2 (Tanabe et al. 1999). If we assume that the structural requirements for ATP binding are similar to those for 8-azido ATP, this suggests that ATP binding is impaired in Kir6.2-K185Q channels. As discussed earlier, we can exclude the possibility that changes in the channel open state contribute to the reduced binding of 8-azido ATP.
It seems clear, therefore, that the K185Q mutation decreases ATP sensitivity by impairing ATP binding. The simplest hypothesis for this finding is that the negatively charged phosphate tail of ATP interacts with the positively charged lysine at position 185. Our experiments do not support this idea. If this were the case, then replacement of K185 with a neutral or negatively charged group would be expected to disrupt nucleotide binding. For example, if K185 interacted with the terminal (γ) phosphate of ATP, then such mutations should lead to a reduction in ATP, but not in ADP, sensitivity. However, ADP sensitivity was also shifted when K185 was neutralised to alanine or glutamine. This makes it unlikely that K185 addresses the γ-phosphate of ATP.
To determine if the β-phosphate of ATP might be involved in high-affinity nucleotide inhibition, it would be desirable to compare the AMP sensitivity of channels in which K185 had been mutated. Unfortunately, we observed very little block of Kir6.2ΔC26 currents by AMP (Ki≡ 9 mM, Tucker et al. 1998), which makes this experiment impractical: at the high nucleotide concentrations that would be required, it would be hard to measure any shift in the Ki of the mutant channels accurately. If the β-phosphate of ATP does indeed interact with K185, then one might expect that when K185 was neutralised the ATP sensitivity would fall towards, but not exceed, that of AMP. This is in fact what happened. However, the fact that the K185G mutant still exhibits high ATP sensitivity, despite the fact that glycine cannot participate in charge-charge interactions, argues that interaction of lysine 185 with the phosphate tail of nucleotide triphosphates is not involved in high-affinity inhibition.
We next considered the possibility that K185 might interact with the purine moiety of ATP. It is clear from our experiments that ATP is a very much more potent blocker of the wild-type channel than GTP. Similar results have been observed previously for both native β-cell KATP channels and for Kir6.2 + SUR1 (Ashcroft & Rorsman, 1989; Trapp et al. 1997; Tucker et al. 1998). This suggests that high-affinity ATP binding requires the NH2 group at position 6 of the purine ring, and the nitrogen at the 1′ position. Because lysine serves as a hydrogen donor, it is unlikely that K185 interacts with the NH2 group (also a hydrogen donor). However, K185 could potentially interact with the nitrogen at position 1 of the adenine ring (a hydrogen acceptor). This group is replaced by a hydrogen donor, NH, in both ITP and GTP, which might explain their lower potency. To examine this possibility, we mutated K185 to a glutamate, an amino acid that serves as a hydrogen acceptor, and to glutamine which may serve as both an H+ acceptor and an H+ donor. If interaction of K185 with the residue at position 1′ of the purine ring is important for ATP sensitivity, and for discriminating between different purine nucleotide triphosphates, we would expect these mutations to decrease the channel ATP sensitivity and concomitantly enhance its sensitivity to ITP and GTP. However, we observed that sensitivity to all three nucleotides was reduced when K185 was mutated to glutamate or glutamine, and that the potency of ITP and GTP relative to that of ATP was unchanged. This suggests that K185 does not address the nitrogen at position 1′ of the adenine ring. Indeed, the participation of the side-chain of K185 in hydrogen bonding to any part of the ATP molecule cannot be involved in high-affinity ATP binding, because the channel still retained high ATP sensitivity when lysine was replaced by glycine, and glycine cannot function as a hydrogen donor.
To determine which side-chains were compatible with ATP inhibition, we made a large range of mutations at position 185. The data indicate that a positive charge at K185 is preferred for high-affinity ATP inhibition. Replacement of lysine with arginine produced only a small effect on ATP sensitivity, while replacement with a negatively charged glutamate or aspartate resulted in a dramatic decrease in the inhibitory effect of ATP. Neutralisation of K185 also reduced ATP sensitivity, to differing extents, but not as markedly as substituting a negative charge. Differences in size, hydrophobicity and structure of the amino acid side-chain probably account for the variable effects of charge neutralization. Taken together, these data suggest that lysine 185 forms a charge-charge interaction with another residue that is important for nucleotide binding.
As discussed earlier, our results demonstrate that K185 is unlikely to bind to the phosphate tail of ATP. They also indicate that this lysine does not interact with the adenine ring of ATP. This is because any interaction with the purine ring is expected to involve hydrogen bonding, and we found no clear correlation between the ability of the amino acid at position 185 to form hydrogen bonds and its effect on ATP sensitivity. Furthermore, neutralization of K185, or substitution of a negative charge, did not alter the selectivity to purine nucleotide triphosphates. The data therefore suggest that the side-chain of lysine 185 does not interact directly with the ATP molecule. We can offer three explanations for the critical importance of K185 on ATP binding. First, K185 might influence ATP binding electrostatically, increasing the ATP concentration in the vicinity of the ATP-binding site. Secondly, ATP may interact with the backbone of the lysine residue. Thirdly, K185 may not interact directly with ATP, but instead exert an allosteric effect on ATP binding. In the last two cases, the fact that a positive charge is preferred suggests that K185 forms an ionic bond with another region of the channel protein.
Mutations in both the N and C termini of Kir6.2 have been shown to influence ATP sensitivity (Tucker et al. 1997; 1998; Shyng et al. 1997a; Drain et al. 1998). One of these mutations, in the N-terminus of Kir6.2 (R50G), reduces the channel ATP sensitivity without affecting the single channel kinetics, by a mechanism that appears to involve an allosteric effect on ATP binding rather than a direct interaction with the nucleotide (Proks et al. 1998; Tanabe et al. 1999). In addition, there is unpublished evidence that the N and C termini of Kir6.2 interact (Tucker & Ashcroft, 1999). This suggests that mutation of R50 (or indeed K185) may affect the ATP binding site by influencing the allosteric interaction between the N and C termini of Kir6.2. The precise contribution of the N and C termini to the ATP binding site remains unresolved.
Figure 7 presents a speculative model that accounts for the effects of different substitutions at position 185 on ATP inhibition. We hypothesise that in the wild-type channel the side-chain of the lysine 185 points into a hydrophobic pocket in the protein, where it co-ordinates a negatively charged residue and thereby stabilizes the three-dimensional structure of Kir6.2. Mutations that destabilize this interaction disrupt ATP inhibition, by allosterically altering the binding pocket for ATP. Replacement of K185 by arginine has little effect, because this residue is also positively charged and is of similar size. Glycine, which is small and hydrophilic, does not alter ATP binding because it does not approach the negative charge. This allows water and/or ions to enter the hydrophobic pocket and stabilize the negative charge. As the size of the side-chain of an uncharged hydrophilic residue at position 185 is increased there is increasingly greater charge destabilization and gradual loss of ATP sensitivity (serine, asparagine, histamine, glutamine). This is because residue 185 approaches more closely to the negative charge, thereby excluding water and decreasing the extent of shielding. Hydrophobic residues exclude water and prevent stabilisation of the negative charge even more effectively which explains why alanine, cysteine, methionine, phenylalanine, leucine and valine reduce ATP sensitivity. Again, the smaller side-chains have less severe effects. The dramatic reduction in ATP sensitivity associated with a negatively charged residue at position 185 are explained by severe disruption of the hydrophobic pocket due to charge repulsion. This model holds whether ATP interacts directly with the backbone of the lysine 185 or whether it binds to different residue. In either case, destabilisation of the hydrophobic pocket produces a conformational change in Kir6.2 that disrupts the ATP-binding site.
Figure 7. Putative model for the role of K185 in ATP inhibition.
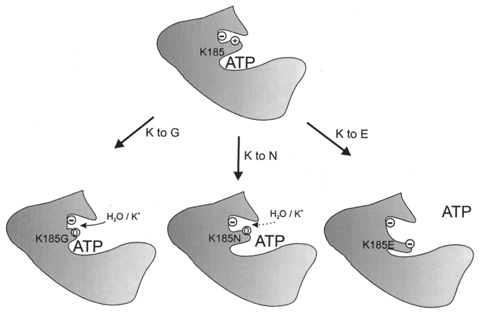
See text for explanation.
Acknowledgments
We thank Yamanouchi Research International, the Wellcome Trust and the British Diabetic Association for support.
References
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, Boyd AE, González G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β-cell high-affinity sulphonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Ashcroft SJH. Properties and functions of ATP-sensitive K-channels. Cellular Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Rorsman P. Electrophysiology of the pancreatic β-cell. Progress in Biophysics and Molecular Biology. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- Clement JP, IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Drain P, Li LH, Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM. Properties of cloned ATP-sensitive K-currents expressed in Xenopus oocytes. The Journal of Physiology. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by MgADP and diazoxide. EMBO Journal. 1997b;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulphonylurea receptors determines the sensitivity of the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulphonylurea receptor forms with BIR (Kir6.2) a smooth muscle type of ATP-sensitive K+ channel. Journal of Biological Chemistry. 1996;271:24321–24325. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- Koster JC, Sha Q, Shyng S-L, Nichols CG. ATP inhibition of KATP channels: control of nucleotide sensitivity by the N-terminal domain of the Kir6.2 subunit. The Journal of Physiology. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyte J, Doolittle RF. A simple method for displaying the hydropathic character of a protein. Journal of Molecular Biology. 1982;157:105–132. doi: 10.1016/0022-2836(82)90515-0. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng S-L, Nestorowicz A, Glasser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Proks P, Gribble FM, Adhikari R, Tucker SJ, Ashcroft FM. Involvement of the N-terminus of Kir6.2 in the inhibition of the KATP channel by ATP. The Journal of Physiology. 1998;514:19–25. doi: 10.1111/j.1469-7793.1999.019af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Letters. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Control of rectification and gating of KATP channels by the Kir6.2 subunit. Journal of General Physiology. 1997a;110:141–153. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP. Journal of General Physiology. 1997b;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanabe K, Tucker SJ, Matsuo M, Proks P, Ashcroft FM, Seino S, Amachi T, Ueda K. Direct photoaffinity labeling of the Kir6.2 subunit of the ATP-sensitive K+ channel by 8-Azido-ATP. Journal of Biological Chemistry. 1999;274:3931–3933. doi: 10.1074/jbc.274.7.3931. [DOI] [PubMed] [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of KATP channel gating and implications for channel inhibition by ATP. Journal of General Physiology. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapp S, Tucker SJ, Ashcroft FM. Activation and inhibition of K-ATP currents by guanine nucleotides is mediated by different channel subunits. Proceedings of the National Academy of Sciences of the USA. 1997;94:8872–8877. doi: 10.1073/pnas.94.16.8872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Ashcroft FM. Mapping the physical interaction between the intracellular domains of an inwardly rectifying potassium current, Kir6.2. Journal of Biological Chemistry. 1999. in the Press. [DOI] [PubMed]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO Journal. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+-channels in the absence of the sulphonylurea receptor. Nature. 1997;378:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]