

Abstract
The molecular basis of the state-dependent block of L-type Ca2+ channels by dihydropyridines is still poorly understood. Therefore, we studied the enantioselectivity of Ca2+ channel block by isradipine enantiomers at three holding potentials (-80, −60 and −40 mV) in Chinese hamster ovary (CHO) cells stably expressing the rabbit lung α1C-b-subunit.
The extent of enantioselectivity did not markedly change with the holding potential (IC50 ratios of 104–138), whereas the potency of both isradipine enantiomers increased with depolarisation of the holding potential.
In addition to its block of the peak Ca2+ channel current, Ipeak, (−)-isradipine inhibited the relative current at the end of the test pulse, the so-called Ilate, normalised to Ipeak (Ilate/Ipeak). This effect was unaffected by the holding potential and revealed distinct kinetics compared to the development of conventional block of Ipeak.
When these effects were studied using an α1C-b-mutant lacking the high-affinity dihydropyridine binding site, expressed in human embryonic kidney (HEK 293) cells, both enantiomers blocked Ilate/Ipeak to a similar degree.
Our data are discussed within the framework of the ‘guarded receptor’ and the ‘modulated receptor’ hypotheses. The very different properties of the block of Ilate/Ipeak compared to those of the conventional high-affinity block of Ipeak suggest the existence of an additional mechanism possibly mediated via a second, distinct binding site.
Chiral dihydropyridine Ca2+ antagonists exert voltage-dependent and enantioselective block of L-type Ca2+ channels. The voltage-dependent action of these compounds has been demonstrated many times (Bean, 1984; Sanguinetti & Kass, 1984; Uehara & Hume, 1985; Bean et al. 1986; Mironneau et al. 1992) and has often been explained in terms of the well-accepted ‘modulated receptor’ hypothesis (Hondeghem & Katzung, 1984; Bean, 1984). Here, the drug is supposed to have a high affinity for inactivated Ca2+ channels, which are favoured at depolarised membrane potentials, and a low affinity for resting channels, which predominate at resting membrane potential. The voltage-dependent changes in dihydropyridine affinity then reflect structural rearrangements of the channel protein itself, resulting in voltage-dependent changes in the drug-receptor interaction.
The enantioselectivity of drug action is also a property that reflects the structure of the binding site and the interaction of drug and receptor, as it requires a high structural complementarity between the appropriate enantiomer and its binding site. Whereas the enantioselective action of dihydropyridine Ca2+ antagonists is well-known (e.g. Morel & Godfraind, 1987; Romanin et al. 1992), we have found no data about the voltage dependence of enantioselectivity in the literature. Knowledge about this relation could help us to understand the molecular mechanism of the voltage-dependent action of dihydropyridines. Assuming that the voltage-dependent channel gating modifies the structure of the dihydropyridine binding site, as postulated by the modulated receptor hypothesis, then it is likely that enantioselectivity of action will be affected by the voltage, as well. Thus, changes in the membrane potential that enhance the potency of dihydropyridine enantiomers would also enhance enantioselectivity. This has been shown to be the case for a pair of enantiomers blocking sodium channels (Gödicke et al. 1992).
We previously tested this hypothesis by measuring L-type Ca2+ channel block by the niguldipine enantiomers in single ventricular myocytes from guinea-pig. Interestingly, the increase in absolute potency of both enantiomers, along with the reduction in holding potential, was not accompanied by any changes in the extent of enantioselectivity (Handrock & Herzig, 1996). This result could be explained within the framework of the ‘guarded receptor’ hypothesis (Starmer et al. 1984; Handrock & Herzig, 1996).
The primary aim of this study was to investigate whether these findings could be confirmed for another splice variant of the L-type Ca2+ channel and another chiral dihydropyridine. We used Chinese hamster ovary (CHO) cells, stably expressing the rabbit lung α1C-b-subunit, and studied the voltage dependence and enantioselectivity of the effect of isradipine (PN200-110). We were able to confirm the previous principal finding of non-voltage-dependent enantioselectivity. During these experiments, we observed that, in addition to the inhibitory effect on the peak current (Ipeak), the relative current at the end of the test pulse, the so-called Ilate, normalised to Ipeak (Ilate/Ipeak), was blocked by (−)-isradipine. Analysis of the development of this effect revealed kinetics different from those of the development of conventional Ipeak block. In mutated α1C-b-channels, lacking the high-affinity dihydropyridine binding site, both enantiomers induced a similar degree of Ilate/Ipeak block.
Our data suggest different mechanisms for conventional Ipeak block and the Ilate/Ipeak block.
METHODS
Cell transfection and cell culture
Chinese hamster ovary (CHO, DG44 mutant, from L. Chasin, New York) cells were stably transfected with the α1C-b-subunit from rabbit lung as described previously, yielding the cell clone CHOCa9 (Bosse et al. 1992). No auxiliary subunits were coexpressed. Cells were grown in Dulbecco's modified Eagle's medium (Biochrom KG, Berlin, Germany) supplemented with 10% dialysed fetal bovine serum (Sigma, Deisenhofen, Germany), penicillin (30 units ml−1), streptomycin (30 μg ml−1) and non-essential amino acids (Biochrom). For electrophysiological experiments the cells were seeded in polystyrene dishes (8 cm2; Falcon, Heidelberg, Germany) at a density of between 104 and 2 × 104 cells cm−2 and used from the first to third day after plating.
For construction of the α1C-b-mutant, three amino acids of the last putative transmembrane segment IVS6 were replaced by the corresponding amino acids of the α1E-subunit (Y1485I, M1486F and I1493L; numbers according to the α1C-b sequence), yielding the chimera Ch30. The full-length cDNA encoding this chimera was cloned into the pcDNA 3 vector (Invitrogen) (Schuster et al. 1996). Human embryonic kidney cells (HEK 293, from T. Schneider, Cologne, Germany) were transiently transfected with the cDNA plasmids encoding the α1C-b-mutant, the β2a- and α2/δ-subunits and green fluorescent protein (GFP, Gibco BRL, Life Technologies) by lipofection with SuperFect (Qiagen, Hilden, Germany) at a DNA mass ratio of 1:1:1:0·1. Transfected cells were grown onto poly-L-lysine-coated glass coverslips in minimal essential medium (MEM, Biochrom) supplemented with 10% fetal bovine serum (Sigma), penicillin (30 units ml−1) and streptomycin (30 μg ml−1). Electrophysiological recordings were conducted 2 days after transfection.
Electrophysiological recordings
Currents through L-type Ca2+ channels were measured using the whole-cell configuration of the patch-clamp technique (Hamill et al. 1981). Experiments with CHOCa9 cells were performed in an external solution containing (mM): NaCl 120, BaCl2 10·8, MgCl2 1, CsCl 5·4, dextrose 10, Hepes 10 (pH 7·4). Pipettes (borosilicate glass, 6–8 MΩ) were filled with (mM): CsCl 120, MgCl2 3, MgATP 5, EGTA 10, Hepes 5 (pH 7·4). For measurement of currents through the mutated α1C-b Ca2+ channel, transfected HEK 293 cells were bathed in a solution containing (mM): NaCl 82, TEA-Cl 20, BaCl2 30, MgCl2 1, CsCl 5·4, EGTA 0·1, dextrose 10, Hepes 5 (pH 7·4). Pipettes (borosilicate glass, 2–3 MΩ) contained (mM): CsCl 102, TEACl 10, MgCl2 1, EGTA 10, Hepes 5, Na2ATP 3 (pH 7·4).
During the experiment, cells were continuously superfused with drug-free bath solution or the isradipine-containing solution (2 ml min−1, volume of the bath chamber 2 ml). Whole-cell Ba2+ currents were elicited by depolarising voltage steps from holding potentials of −80, −60 and −40 mV in the case of CHOCa9 cells and from −80 and −40 mV in the case of transfected HEK 293 cells. For each cell a current-voltage relationship was established and the test potential at which current was maximal was applied for the rest of the protocol (+10 to +30 mV). The stimulus frequency was 0·1 Hz. For CHOCa9 cells, concentration-response curves for both isradipine enantiomers were obtained at the three holding potentials. Each cell was exposed to only one isradipine concentration and enantiomer, and the entire stimulus protocol was applied. For transfected HEK 293 cells, the isradipine enantiomers were applied at one concentration only (1 μM). All experiments were conducted at room temperature (21–24°C).
Ba2+ currents were sampled at 10 kHz and filtered (-3 dB) at 2 kHz (Axopatch 1D, Axon Instruments, Foster City, CA, USA). Leak and capacitive currents were subtracted by using a P/N pulse protocol. The pCLAMP software (version 5.5, Axon Instruments) was used for data acquisition and analysis.
The enantiomers of isradipine were a gift from Sandoz (Basle, Switzerland). They were prepared as stock solutions in absolute ethanol (final concentration ≤0·3% v/v). The purity of each enantiomer was > 99·8%.
Data analysis
Ba2+ currents were measured as the difference between the peak current within a pulse and zero (Ipeak) or between the current at the end of a pulse and zero (late current or Ilate), as indicated in the text and explained in Fig. 1C. All effects were determined after the current had reached steady state at the respective condition. Currents were analysed after normalisation, with current at −80 mV (Ipeak or Ilate) immediately before drug addition being 100%.
Figure 1. Protocol used to study Ba2+ currents in CHOCa9 cells.
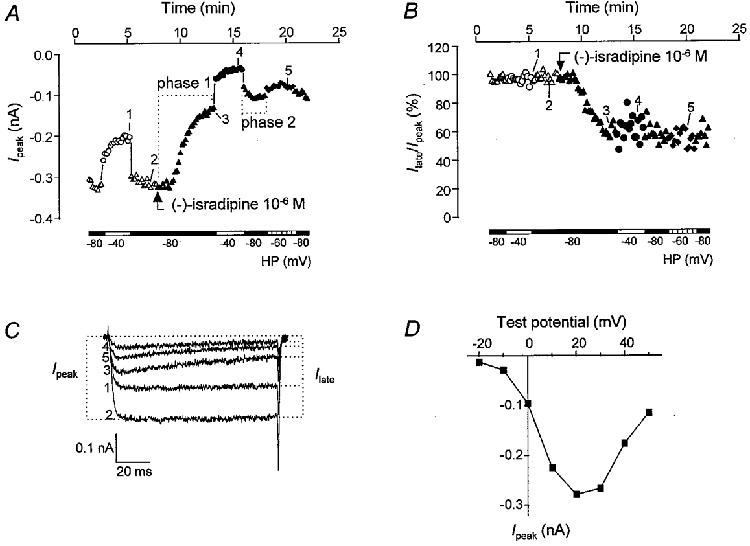
A, course of Ipeak during a single experiment with CHOCa9 cells in the absence and presence of 10−6 M (−)-isradipine. Different holding potentials (HP) were used, as indicated by the bars and the symbols: ▵, ▴, −80 mV; ○, •, −40 mV; ♦, −60 mV. Open symbols, control; filled symbols, 10−6 M (−)-isradipine. The test potential was +20 mV during the entire voltage protocol. For an explanation of phases 1 and 2, see text. B, course of Ilate/Ipeak during the same experiment as shown in A. For determination of Ilate, see C. For explanation of symbols, see A. C, original traces obtained at the time points indicated by the numbers in A and B. The determination of Ipeak and Ilate is demonstrated by the dotted lines. Test potential was +20 mV. D, current-voltage relationship at HP −80 mV under control conditions. Ba2+ current was maximal at a test potential of +20 mV.
Concentration-response curves were fitted by non-linear regression analysis using a one-to-one binding curve according to the equation E = Emax/(1 +[drug]/IC50). For the kinetic analysis of the development of both peak and late current block, the data points describing Idrug/Icontrol at the respective holding potential were fitted by an exponential association function after having multiplied them by −1. The function is given by the equation y =y0+A(1 - e-x/τ), with A describing the amplitude of the trace and τ the time constant. Association and dissociation rate constants were obtained by solving the equation kobs=kon×[drug]+koff.
Data are given as means ±s.e.m. of 3–8 experiments in the case of CHOCa9 cells and as means ±s.e.m. of 4–5 experiments in the case of Ch30-transfected HEK 293 cells.
RESULTS
Voltage dependence and enantioselectivity of the action of isradipine on Ipeak in CHOCa9 cells
The time course of the voltage protocol used to study the block of Ipeak by the isradipine enantiomers is exemplified in Fig. 1A by a single experiment. During superfusion of the cells with the bath solution, Ba2+ current was measured at holding potentials of −80 and −40 mV. In this case, the current at −40 mV was reduced by 36% compared to the current at −80 mV. This voltage-dependent inactivation ranged between 12 and 36% in all the experiments included in our analysis. After returning the holding potential to −80 mV, the dihydropyridine enantiomer - in this case 10−6 M (−)-isradipine - was added and Ba2+ current was blocked. This inhibition was markedly enhanced when the holding potential was depolarised to −40 mV. Switching the holding potential back to −80 mV resulted in an incomplete recovery from the enhanced block reached at −40 mV. Finally, current inhibition was measured at a holding potential of −60 mV, where it exhibited an intermediate level of block compared to the levels measured at −80 and −40 mV. Original traces recorded at the three holding potentials are shown in Fig. 1C. The test potential in this experiment was +20 mV, as the current was maximal under this condition (Fig. 1A).
Concentration-response curves for Ipeak block by (+)- and (−)-isradipine at the three holding potentials are depicted in Fig. 2. The half-filled symbols at the origin of the plots represent values from control experiments carried out by applying the same voltage protocol to cells superfused with bath solution containing 0·3% ethanol. In each experiment, the effect of the drug at −80 mV was measured at two time points within the protocol: at the end of phase 1, i.e. before depolarisation to −40 mV, and at the end of phase 2, i.e. after depolarisation to −40 mV (see Fig. 1A). At the latter time point, current block induced by (+)-isradipine was on average two times larger than at −80 mV before changing the holding potential to −40 mV. Thus a preceding depolarisation of the holding potential seems to favour complete binding of the (+)-enantiomer to the Ca2+ channel at −80 mV. In contrast, Ca2+ channel block by (−)-isradipine at a holding potential of −80 mV was virtually unaffected by a preceding period at −40 mV. Similar observations concerning the different kinetics of the two enantiomers were made by Morel & Godfraind (1987) and Wibo et al. (1988). This finding could be simply explained by the concentration dependence of the association process. As (−)-isradipine is the less potent enantiomer and is therefore applied at higher concentrations, its association at polarised conditions will be faster than that of (+)-isradipine at equieffective concentrations. This difference disappears in depolarised conditions. The concentration- response curves at −80 mV in Fig. 2 therefore only demonstrate the effects of the drug measured at the end of phase 2 of the protocol, where a true steady-state seems to be reached.
Figure 2. Concentration-response curves for isradipine enantiomers obtained with CHOCa9 cells.
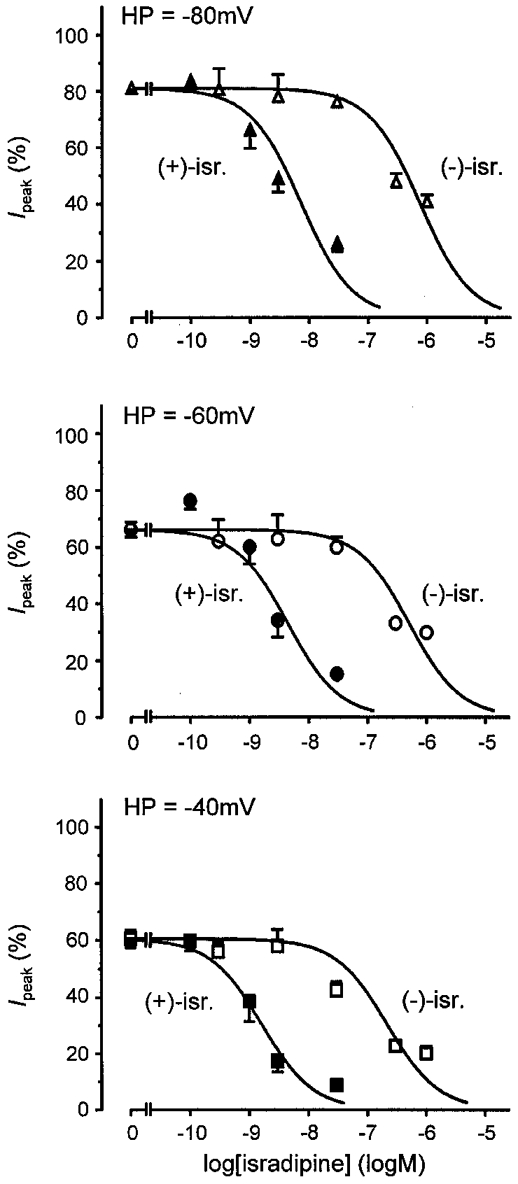
The results of Ca2+ channel block by (+)- and (−)-isradipine, obtained by analysis of Ipeak, are depicted at holding potentials (HP) of −80, −60 and −40 mV. Filled symbols, (+)-isradipine; open symbols, (−)-isradipine.
The three concentration-response curves shown in Fig. 2 illustrate that Ca2+ channel block is voltage dependent, as expected. Both enantiomers become more effective with decreasing holding potential. (+)-Isradipine is a potent Ca2+ channel blocker with an IC50 value of 7·2 × 10−9 M at −80 mV, which decreased to 4·5 × 10−9 M at −60 mV and further to 1·6 × 10−9 M at −40 mV. In the case of (−)-isradipine, the IC50 value was reduced from 7·5 × 10−7 M at −80 mV to 5·1 × 10−7 M at −60 mV and to 2·2 × 10−7 M at −40 mV (see also Table 1).
Table 1.
Summary of the IC50 values from analysis of Ipeak and Ilate in CHOCa9 cells
| IC50 values (m): analysis of Ipeak | IC50 values (m): analysis of Ilate | |||||
|---|---|---|---|---|---|---|
| Holding potential | (+)-isr. | (−)-isr. | F | (+)-isr. | (−)-isr. | F |
| −80 mV | 7.2 × 10−9 | 7.5 × 10−7 | 104.2 | 8.2 × 10−9 | 3.8 × 10−7 | 46.3 |
| −60 mV | 4.5 × 10−9 | 5.1 × 10−7 | 113.3 | 4.2 × 10−9 | 2.7 × 10−7 | 64.3 |
| −40 mV | 1.6 × 10−9 | 2.2 × 10−7 | 137.5 | 2.0 × 10−9 | 9.8 × 10−7 | 49.0 |
IC50 values for both isradipine enantiomers at the three tested holding potentials were derived from the concentration-response curves of Ipeak (Fig. 2) or Ilate (not shown), respectively. F indicates the ratio of the IC50 values of (−)- and (+)-isradipine.
As is evident from these data, current inhibition is enantioselective, with (+)-isradipine being the more potent enantiomer at the three holding potentials tested. The extent of enantioselectivity was determined as the ratio of the IC50 values of (−)- and (+)-isradipine, resulting in values of 104·2, 113·3 and 137·5 at −80, −60 and −40 mV, respectively. Thus, the potency ratios are in a rather narrow range and are virtually unaffected by the holding potentials tested.
Voltage dependence and enantioselectivity of the action of isradipine on Ilate/Ipeak in CHOCa9 cells
Closer inspection of the traces in the absence and presence of (−)-isradipine at the highest concentration applied (10−6 M) revealed that this enantiomer induced a change in current waveform during the 100 ms test pulse (see original traces in Fig. 1A). To elucidate the voltage dependence and stereoselectivity of this effect, we quantified the effect by determining Ilate (see Fig. 1A) and normalising it to Ipeak in all experiments in the absence and presence of both enantiomers. This ratio then reflects the additional drug-induced effect on Ilate, since that portion of the reduction in Ilate that is due to Ipeak block is eliminated. The time course of Ilate/Ipeak is exemplified in Fig. 1A for the same experiment shown in Fig. 1A. The control values did not depend on the holding potential, as expected. During wash-in of (−)-isradipine at −80 mV, current values considerably decreased. Once the drug had reached its steady-state concentration in the bath - approximately at the end of the pulse episode at −80 mV - Ilate/Ipeak remained nearly unchanged for the rest of the voltage protocol, showing no dependence on the holding potential. We did not observe such an effect on Ilate/Ipeak in the presence of (+)-isradipine. In Fig. 3, original and normalised current traces at holding potentials of −80 and −40 mV in the absence and presence of both enantiomers are depicted. The concentrations of (+)- and (−)-isradipine chosen for illustration at −80 mV are approximately equipotent. At −40 mV, currents in the presence of a 10-fold lower concentration of (−)-isradipine are presented, since otherwise the steady-state effect was reached instantaneously with the first pulse and development of the block would not have been demonstrated. As an equipotent concentration of (+)-isradipine was not studied, the concentration chosen for comparison was 10−9 M. Only during wash-in of (−)-isradipine at −80 mV, Ilate/Ipeak was reduced from test pulse to test pulse, as can clearly be seen after normalisation of the traces to the control peak current (Fig. 3A, top panel). At −40 mV, as well as in the presence of (+)-isradipine at both holding potentials, no drug- or voltage-dependent change in Ilate/Ipeak could be detected; the normalised traces are superimposed. Table 2 summarises the Ilate/Ipeak values before (control) and after drug addition within the same recording at the different holding potentials. In the absence of the drug, the reduction in current during the test pulse was in the range of 5–14% at both −80 and −40 mV. The presence of (−)-isradipine resulted in a concentration-dependent block of Ilate/Ipeak; the reduction in current reached 33 and 35% at 10−6 M, measured at holding potentials of −80 and −40 mV, respectively. The (+)-enantiomer did not cause any changes in the current waveform over the concentration range investigated (see the two bottom panels of Fig. 3A and B as well as Table 2). The steady-state effects of both enantiomers on Ilate/Ipeak did not depend on the time at which the measurement was taken (phase 1 or 2 of the protocol).
Figure 3. Original and normalised current traces from CHOCa9 cells.
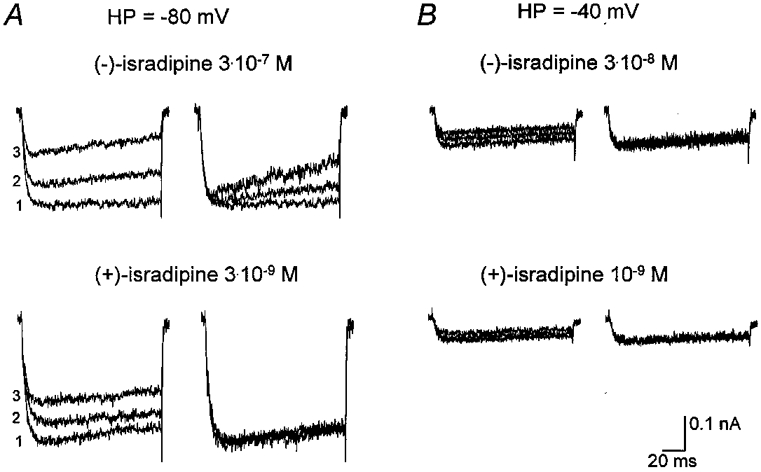
Current traces are shown in the presence of nearly equieffective concentrations of (+)- and (−)-isradipine at holding potentials (HP) of −80 and −40 mV. A, HP −80 mV: the two left panels show original traces depicting control current (1), current during wash-in of drug (2) and during the steady-state effect (3) in the presence of 3 × 10−7 M (−)-isradipine (top) or 3 × 10−9 M (+)-isradipine (bottom). In the two right panels, traces 2 and 3 were scaled to the maximal amplitude of the control trace 1 to facilitate comparison of current waveform. B, HP −40 mV: the two left panels display original traces showing the developing current block after changing the holding potential from −80 to −40 mV in the presence of 3 × 10−8 M (−)-isradipine (top) or 10−9 M (+)-isradipine (bottom). In the two right panels the two upper traces were scaled to the maximum current of the bottom trace, which was obtained immediately after the change of holding potential. The scale bar for current is only valid for the unscaled original traces.
Table 2.
Effect of isradipine enantiomers on Ilate/Ipeak in CHOCa9 cells
| Ilate/Ipeak(%) | |||
|---|---|---|---|
| −80 mV | −60 mV | −40 mV | |
| Control | 86.4 ± 1.6 | — | 85.7 ± 1.6 |
| (−)-isr. (3 × 10−10m) | 87.9 ± 1.8 | 90.5 ± 1.4 | 85.2 ± 3.3 |
| Control | 88.2 ± 3.6 | — | 88.2 ± 2.5 |
| (−)-isr. (3 × 10−9m) | 89.4 ± 3.4 | 90.2 ± 1.6 | 91.4 ± 1.7 |
| Control | 95.1 ± 1.0 | — | 95.2 ± 0.2 |
| (−)-isr. (3 × 10−8m) | 91.4 ± 2.6 | 90.5 ± 5.0 | 84.2 ± 2.0 |
| Control | 92.5 × 1.0 | — | 92.5 ± 1.2 |
| (−)-isr. (3 × 10−7m) | 72.0 ± 2.8 | 72.4 ± 5.0 | 61.7 ± 2.4 |
| Control | 94.0 ± 1.2 | — | 92.9 ± 1.7 |
| (−)-isr. (10−6m) | 67.0 ± 4.4 | 67.6 ± 6.1 | 65.3 ± 3.1 |
| Control | 92.7 ± 1.4 | — | 90.3 ± 2.2 |
| (+)-isr. (3 × 10−8m) | 91.9 ± 1.8 | 86.8 ± 2.0 | 87.5 ± 5.7 |
Ilate/Ipeak was determined before (control) and after drug addition (means ±s.e.m., n = 3–8). Control current at −60 mV was not measured, but is expected to be in the same range as for the two other holding potentials. Ilate/Ipeak values for (+)-isradipine are only given for the highest concentration applied, because at the lower concentrations, as well as at 3 × 10−8 m, no difference between values obtained in control conditions and those measured in the presence of the drug could be detected.
We next wanted to further elucidate and quantify the influence of the additional effect of (−)-isradipine on the extent of enantioselectivity of channel block and the voltage dependence of enantioselectivity. Concentration-response curves were fitted to the Ilate values (not shown) and the IC50 values were again determined (Table 1). The IC50 values for (−)-isradipine decreased from 7·5 × 10−7 M (Ipeak block) to 3·8 × 10−7 M (Ilate block) at −80 mV, from 5·1 × 10−7 M to 2·7 × 10−7 M at −60 mV and from 2·2 × 10−7 M to 9·8 × 10−8 M at −40 mV. Thus, the half-inhibitory concentrations were reduced to a comparable degree at the three holding potentials. The respective values for (+)-isradipine were unchanged. Consequently, the extent of enantioselectivity also decreased, resulting in potency ratios of 46·3 at −80 mV, 64·3 at −60 mV and 49·0 at −40 mV, but enantioselectivity was still unaffected by the holding potential.
Thus analysis of Ilate/Ipeak reveals that (−)-isradipine, but apparently not (+)-isradipine, inhibits Ilate/Ipeak and this effect is not affected by the holding potential. The lack of dependence on the holding potential and the apparent reversal in enantioselectivity found here are in contrast to the results from Ipeak analysis, where we detected voltage dependence of action and effectiveness for both enantiomers, with the (+)-enantiomer being more potent.
Kinetic analysis of development of Ipeak and Ilate/Ipeak block in CHOCa9 cells
To obtain further insight into the underlying mechanism of block of Ipeak and Ilate/Ipeak, we examined the kinetics of the development of current inhibition. For block of Ipeak, we studied the time course of inhibition at a holding potential of −40 mV for both enantiomers. For each experiment, we first normalised Ipeak in the presence of the drug to Ipeak in the absence of the drug for each test pulse at −40 mV by calculating the ratios of the respective current amplitudes obtained at corresponding time points after changing the holding potential from −80 to −40 mV. This was necessary to eliminate that component of current reduction which is due to the development of voltage-dependent inactivation. The data points could be fitted by monoexponential functions, and time constants for each experiment were determined. An example is shown in Fig. 4A. The corresponding rate constants, kobs, were then plotted against drug concentration, as shown in Fig. 4A for both enantiomers. Data points were fitted by linear regression. The derived association rate constant for (+)-isradipine, 4·5 × 106 M−1 s−1, is about 60 times higher than the respective value for (−)-isradipine, 7·7 × 104 M−1 s−1, whereas the dissociation rate constant of the (+)-enantiomer, 0·020 s−1, is about 3 times lower than the respective value of the (−)-enantiomer, 0·069 s−1. The calculated values for the rate constants kon and koff can be taken as a rough estimate only of the kinetics of the binding process, as in the case of (−)-isradipine the data points are not as convincingly fitted by linear regression. Nevertheless, we consider it appropriate to perform this sort of analysis, which requires a one-to-one interaction between drug and binding site, as it has been shown many times in the literature that binding of isradipine is mediated via a homogeneous receptor population and obeys the law of mass action (Kokubun et al. 1986; Morel & Godfraind, 1987, 1988; Kamp et al. 1988; Wibo et al. 1988; Bosse et al. 1992). Our data indicate that the enantioselectivity of block of Ipeak at −40 mV is mainly due to the different association kinetics of the enantiomers. The dissociation constants (KD) deduced from the kinetic analysis are 4·5 × 10−9 M for (+)-isradipine and 9·0 × 10−7 M for (−)-isradipine and they are quite similar to the experimentally determined steady-state IC50 values of 1·6 × 10−9 M and 2·2 × 10−7 M, respectively.
Figure 4. Kinetic analysis of development of Ipeak block by isradipine enantiomers in CHOCa9 cells.
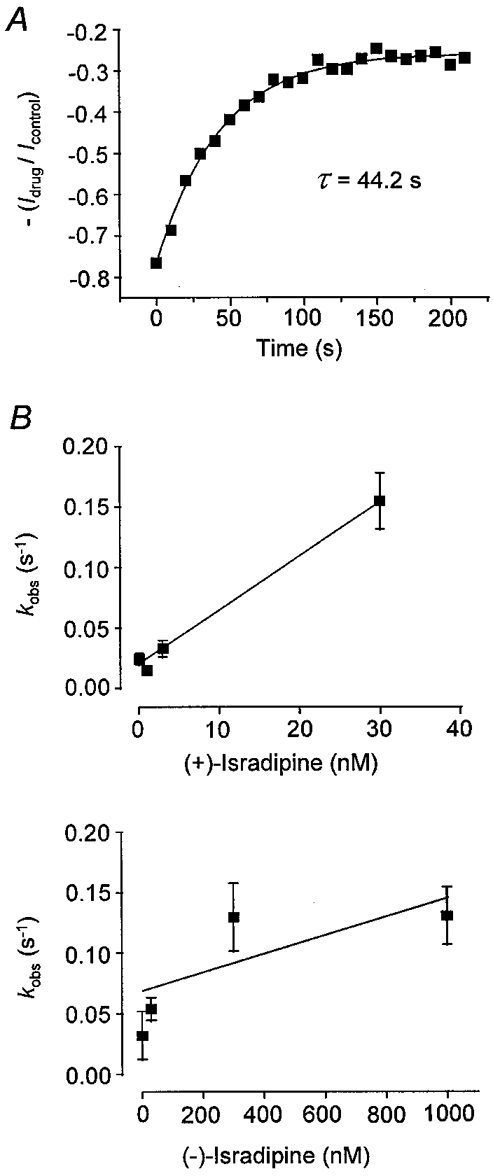
Development of Ipeak block was analysed at a holding potential of −40 mV. A, the analysis is exemplified by a single experiment. Ipeak in the presence of 3 × 10−9 M (+)-isradipine at HP −40 mV was normalised to control current at HP −40 mV; Ipeak was obtained at corresponding time points after the change of holding potential. The first value refers to the steady-state value at −80 mV immediately before changing the holding potential. Data points were fitted by a monoexponential function, yielding the time constant (τ) of 44·2 s. B, the observed rate constant, kobs (the reciprocal value of τ), plotted against drug concentration. Points were fitted by linear regression. Calculated rate constants: for (+)-isradipine, kon= 4·5 × 106 M−1 s−1, koff= 0·020 s−1; for (−)-isradipine: kon= 7·7 × 104 M−1 s−1, koff= 0·069 s−1.
The kinetics of development of Ilate/Ipeak block were analysed for (−)-isradipine at holding potentials of −80 and −40 mV. The current in the presence of the drug was normalised to the control current in order to eliminate from the analysis the current decay that took place in the absence of drug during the test pulse. Therefore, current traces which showed a stable effect of the drug, normally the last few traces for the respective experimental condition, were averaged and divided by the mean control current obtained by averaging stable control traces at the same holding potential in the same cell. An example is illustrated in Fig. 5A. The trace resulting from the ratio of the averaged drug and control currents was then fitted by a monoexponential function, as shown in Fig. 5A. The resulting rate constants were again plotted against drug concentration at the two holding potentials and fitted by linear regression (Fig. 5A). The determination of association and dissociation rate constants (kon and koff) yielded valuesfor kon of 3·1 × 107 and 1·8 × 107 M−1 s−1 and for koff of 28·7 s−1 and 22·0 s−1 at −80 and −40 mV, respectively. A comparison of the values at the two holding potentials reveals that the association and dissociation kinetics of Ilate/Ipeak block are obviously not affected by the holding potentials investigated. The dissociation constants derived from this kinetic analysis are 9·3 × 10−7 and 1·2 × 10−6 M at −80 and −40 mV, respectively. As we could not directly derive IC50 values for the block of Ilate/Ipeak from our experiments because we used only three effective (−)-isradipine concentrations, a further evaluation of the kinetically derived KD values was not possible. From the values given in Table 2 we can roughly estimate, assuming one-to-one interaction of the drug with the binding site, that the IC50 values would be in the range of 10−6 M at both holding potentials.
Figure 5. Kinetic analysis of development of Ilate/Ipeak block by (−)-isradipine in CHOCa9 cells.
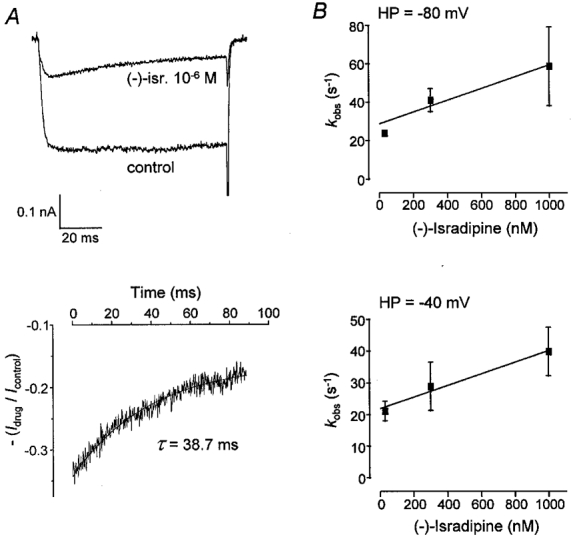
Development of Ilate/Ipeak block was analysed at holding potentials of −80 and −40 mV. A, the analysis is exemplified by a single experiment at HP −80 mV. The mean current from several control traces and that from several traces in the presence of 10−6 M (−)-isradipine under steady-state conditions (upper panel) were used for kinetic analysis. A monoexponential function was fitted to the ratio of the mean currents starting from the time point at which peak current was recorded (lower panel). B, the reciprocal values of the observed time constants (τ), kobs, were plotted against drug concentration. At HP −80 mV: kon= 3·1 × 107 M−1 s−1, koff= 28·7 s−1; at HP −40 mV: kon= 1·8 × 107 M−1 s−1, koff= 22·0 s−1.
A comparison of the kinetic data of (−)-isradipine from the two analyses at −40 mV reveals that both association and dissociation take place much quicker in the case of Ilate/Ipeak block than in the case of Ipeak block. The derived KD values reveal a similar potency of (−)-isradipine for inducing Ipeak and Ilate/Ipeak block.
Voltage dependence and enantioselectivity of the action of isradipine on Ilate/Ipeak in HEK 293 cells transfected with the chimera Ch30
The analysis of Ilate/Ipeak block in CHOCa9 cells reveals that Ilate/Ipeak was reduced only in the presence of (−)-isradipine. The apparent lack of effect of (+)-isradipine could be due to its strong block of Ipeak, which might mask the effect on Ilate/Ipeak. To test this hypothesis, we transiently transfected HEK 293 cells with a mutated α1C-b-subunit, in which three amino acids in segment IVS6 were replaced by the respective amino acids of the dihydropyridine-insensitive α1E-subunit, yielding the chimera Ch30. This resulted in an ∼100-fold reduction in the extent of Ipeak block by (+)-isradipine (Schuster et al. 1996). It was assumed that the use of the Ch30 chimera would reveal a possible effect of the otherwise more potent (+)-enantiomer on Ilate/Ipeak. The same voltage protocol as for the CHOCa9 cells was applied (see Fig. 1A), except that the current inhibition at a holding potential of −60 mV was not checked at the end of the protocol. A single concentration (10−6 M) of each enantiomer was studied. Both enantiomers still inhibited Ipeak in a voltage-dependent manner, but the effects were considerably weakened compared to those obtained with the wild-type α1C-b-subunit. The presence of 10−6 M (+)-isradipine led to a decrease in peak current to 80·0 ± 7·4% at −80 mV and 56·9 ± 2·7% at −40 mV. In the case of the (−)-enantiomer, the current was reduced to 86·6 ± 4·3% at −80 mV and 68·6 ± 3·9% at −40 mV. Enantioselectivity seemed to be nearly abolished. This is not surprising, as the removal of the amino acids essential for high affinity binding in the α1C-b-subunit will consequently lead to a loss of the high complementarity between the (+)-enantiomer and its binding site.
We next determined Ilate/Ipeak. The results are depicted in Fig. 6. In contrast to the results with the CHOCa9 cells, (+)-isradipine induced a strong reduction in Ilate/Ipeak, which showed no clear dependence on the two holding potentials tested. (−)-Isradipine also inhibited Ilate/Ipeak independently of the holding potential used, but was slightly less potent than the (+)-enantiomer. Thus, the enantioselectivity of Ilate/Ipeak block in Ch30 is strongly reduced compared to that found with CHOCa9 cells for Ipeak inhibition.
Figure 6. Effect of isradipine enantiomers on Ilate/Ipeak in cells transfected with the chimera Ch30.
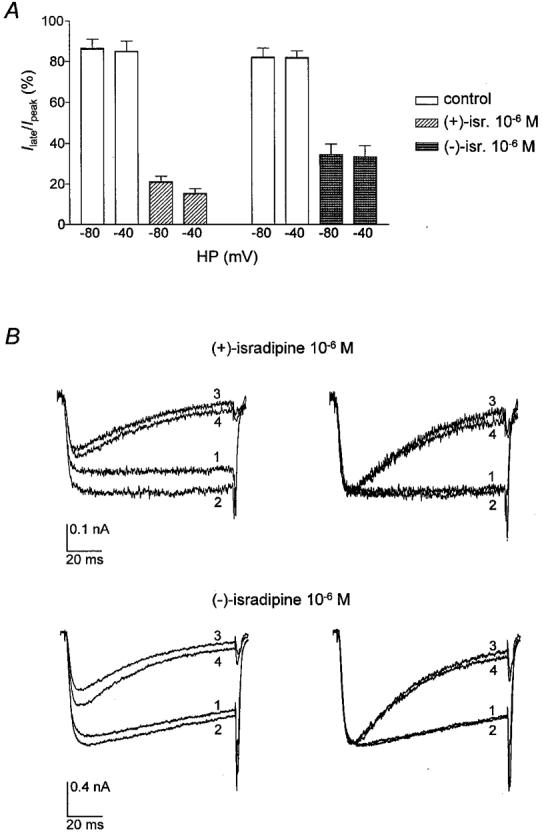
Ilate/Ipeak at holding potentials of −80 and −40 mV was determined in HEK 293 cells transfected with the chimera Ch30. A, Ilate/Ipeak during 100 ms test pulses under control conditions and in the presence of 10−6 M (+)- or (−)-isradipine at HP −80 and −40 mV. The control columns represent the values before drug application in the same experiments. B, original (left) and normalised (right) traces in the absence and presence of (+)- or (−)-isradipine at both holding potentials. For normalisation, traces 1, 3 and 4 were scaled to the maximum current (trace 2). 1: HP −40 mV, control; 2: HP −80 mV, control; 3: HP −40 mV, drug; 4: HP −80 mV, drug. Test potential was +30 mV. Scale bars for current are only valid for the unscaled original traces.
The experiments with Ch30-transfected cells support our hypothesis that the apparent lack of effect of (+)-isradipine on Ilate/Ipeak in the native α1C-b-channel was occluded by its strong effect on Ipeak, although we cannot exclude the possibility that (+)-isradipine has no effect at all on Ilate/Ipeak in CHOCa9 cells. The lack of influence of the holding potential on Ilate/Ipeak found in Ch30-transfected cells is in accordance with the results for the wild-type α1C-b-channel.
DISCUSSION
Our results demonstrate that the isradipine enantiomers exhibit Ca2+ channel block in two different ways: by the conventional, well-known inhibition of Ipeak and by inhibition of Ilate/Ipeak, with both effects displaying very different characteristics. Our data indicate that these two effects are mediated by two different mechanisms and possibly operate at two different binding sites for the following reasons. First of all, the inhibition of Ipeak clearly displayed a high degree of enantioselectivity in CHOCa9 cells, whereas block of Ilate/Ipeak did not (in both Ch30-transfected and CHOCa9 cells). The latter issue will be discussed later. Secondly, the mutations in Ch30-transfected cells markedly reduced the Ipeak effect without affecting the action of (−)-isradipine on Ilate/Ipeak. Thirdly, inhibition of Ipeak by both enantiomers depended on the holding potential, whereas the extent of inhibition of Ilate/Ipeak (in both Ch30-transfected and CHOCa9 cells) did not. This points to different molecular mechanisms, e.g. interaction of the enantiomers with different channel states with different dependencies on holding potential. Fourthly, both effects markedly differ in their kinetics of association and dissociation. This suggests that the two conformational states or possibly separate binding sites are accessed differently by the dihydropyridine.
We will discuss the properties of block of Ipeak and Ilate/Ipeak separately. The main finding concerning the conventional Ipeak block of wild-type α1C-b-channels by the isradipine enantiomers is that, at the holding potentials tested, there was no voltage dependence of enantioselectivity but the absolute potency of the enantiomers was modulated. The potency ratios are in a narrow range between 104 and 138 at the three holding potentials and are close to the value known from radioligand binding studies (∼100, e.g. Bosse et al. 1992). The high extent of enantioselectivity in our study is mainly due to the faster association process for (+)-isradipine than for (−)-isradipine, as derived from the kinetic analysis. No comparable electrophysiological data concerning the voltage dependence of enantioselectivity are available from the literature, as usually only (+)-isradipine is the subject of study. Morel & Godfraind (1987) investigated the binding of both isradipine enantiomers to intact vessels at high and low potassium concentrations and found potency ratios of 56 at resting membrane potentials and of 40 at depolarised membrane potentials. Thus, enantioselectivity of isradipine binding did not increase with depolarisation of the membrane, in agreement with our data. It might be argued that our voltage protocol was inappropriate for revealing a dramatic increase in enantioselectivity, since the most depolarised holding potential we used, −40 mV, led to a steady-state inactivation of 12–36% under control conditions. We have already addressed this problem using model calculations based on the modulated receptor hypothesis (Handrock & Herzig, 1996). We showed that within the range of conditions covering about 0–30% steady-state inactivation, the most dramatic changes in enantioselectivity would be expected. How can our data now be interpreted within the framework of the two models, the modulated receptor hypothesis and the guarded receptor hypothesis, which offer different explanations for the mechanism of state-dependent drug interaction with the channel? Our results are in accordance with the data we first obtained on this subject (Handrock & Herzig, 1996), which were interpreted in terms of the guarded receptor hypothesis (Starmer et al. 1984; see also Gödicke et al. 1992). Here, it was postulated that the affinity of a drug for its binding site is constant, and voltage-dependent gating of the channel modulates the access and/or escape of the drug from the immediate vicinity of the binding site and therefore the drug's potency. In the case of two enantiomers possessing identical physicochemical properties, their access to or escape from the site is expected to be influenced to the same extent by channel gating and consequently, enantioselectivity should be unaffected. This is exactly what we found. Our results do not exclude a modulated receptor mechanism, but this would imply that the state-dependent conformational changes of the binding site do not affect enantioselectivity. We consider this to be unlikely, as enantioselectivity of action is a reflection of high structural complementarity between drug and receptor, but we cannot exclude this from our data. A guarded receptor mechanism would imply that different regions of the channel protein determine enantioselectivity, i.e. the primary binding site, and voltage dependence of dihydropyridine action. The primary dihydropyridine binding site has been quite well characterised. The most critical high-affinity determinants are located in the transmembrane segments IIIS5, IIIS6 and IVS6 of the α1-subunit (for review see Striessnig et al. 1998). It is still unclear which regions of the channel protein determine the voltage dependence of dihydropyridines. Schuster et al. (1996) demonstrated that the Ch30 mutant, while displaying an ∼100-fold lower dihydropyridine affinity, still has the full voltage dependence of dihydropyridine action of the wild-type α1C-b-channel. Our results on Ipeak block with the Ch30 mutant also support this finding. In particular, the mutations in the Ch30 channel did not affect the gating behaviour in the absence of the drug (Schuster et al. 1996; Lacinová & Hofmann, 1998). Thus, the altered pharmacology of this mutant is not secondary to, for example, an altered distribution of channel states. However, other studies suggest a role for primary dihydropyridine attachment points in mediating voltage dependence of action (Ito et al. 1997; Bodi et al. 1997). Considering the results for Ch30-transfected cells (this study and Schuster et al. 1996), it should be noted that the mutations in Ch30 affected only part of the dihydropyridine interaction site. Nevertheless, stereoselectivity of Ipeak block was almost abolished, pointing to the importance of these three amino acids for high-affinity binding. Note that, in contrast to the wild-type α1C-b-subunit, the mutant α1C-b-subunit was coexpressed with β2a- and α2/δ-subunits, which may have effects on enantioselectivity. However, Mitterdorfer et al. (1994) demonstrated a 5-fold increase in isradipine enantioselectivity when a β-subunit was coexpressed with a wild-type α1C-subunit. Therefore, it seems unlikely that the diminished enantioselectivity in Ch30-transfected cells is due to the coexistence of auxiliary subunits. Summarising our own data and those from the literature, it seems that amino acids remote from the putative primary dihydropyridine binding site, as well as those which are part of the binding site, except for the critical attachment points in IVS6, affect the voltage dependence of potency.
During our study with the CHOCa9 cells, we detected an inhibition of Ilate/Ipeak by (−)-isradipine. The finding, already discussed, that enantioselectivity was not voltage dependent was not affected by incorporating the concentration-dependent reduction of Ilate/Ipeak by (−)-isradipine into the analysis by determining Ilate instead of Ipeak. We will now consider the properties of Ilate/Ipeak block in more detail. Our experiments with Ch30-transfected cells were guided by the idea that the apparent lack of effect of (+)-isradipine on Ilate/Ipeak in CHOCa9 cells could be occluded by its strong effect on Ipeak at the higher concentrations applied, especially at 30 nM. The fact that block of Ilate/Ipeak by (+)-isradipine is never observed in CHOCa9 cells, not even at 300 nM (Lacinová & Hofmann, 1998), may be interpreted in four ways: (1) (+)-isradipine inhibits Ilate/Ipeak in CHOCa9 cells in the same concentration range as in Ch30-transfected cells, where the threshold concentration is 30 nM (see Lacinová & Hofmann, 1998), (2) (+)-isradipine induces this effect in CHOCa9 cells at concentrations above 300 nM, (3) (+)-isradipine has no effect at all on Ilate/Ipeak in CHOCa9 cells or (4) Ilate/Ipeak block was simply missed at low (+)-isradipine concentrations in CHOCa9 cells because the kinetics of reaction are different: assuming that a 100-fold higher potency of (+)-isradipine than (−)-isradipine for block of Ilate/Ipeak is exclusively due to a 100-fold lower koff, then block by (+)-isradipine at low concentrations would be too slow (τ≥3·5 or 4·5 s at −80 or −40 mV) to be detected within the 100 ms test pulse. If we consider that enantioselectivity, at least, of Ipeak block (Fig. 4) is mainly based on different kon values, case 4 is unlikely. The possibilities 1–3 imply that the enantioselectivity of Ilate/Ipeak block in CHOCa9 cells is different from that of Ipeak, being either abolished (1) or reversed (2, and 3 as an extreme case of 2). As 1 μM (+)-isradipine clearly blocks Ilate/Ipeak in Ch30-transfected cells, it is tempting to speculate that case 1 holds true. Lacinová & Hofmann (1998) showed that this effect occurs concentration dependently. (−)-Isradipine at 1 μM also induces inhibition of Ilate/Ipeak in Ch30-transfected cells, which was similar in magnitude to that produced by the (+)-enantiomer, indicating a lack of enantioselectivity of Ilate/Ipeak block in these cells. The effects of both enantiomers were unaffected by the holding potential. When comparing the (−)-isradipine-induced effect on Ilate/Ipeak in Ch30-transfected cells with the respective effect in CHOCa9 cells, this enantiomer was found to be slightly more potent in the former. The similar characteristics of Ilate/Ipeak block by (−)-isradipine in Ch30-transfected and CHOCa9 cells suggest that the binding site mediating this effect is not grossly modified by the three point mutations.
The observation that a dihydropyridine alters channel kinetics is not new in principle. It has been shown for nisoldipine, nitrendipine, nifedipine and nimodipine, mostly with Ba2+ as the charge carrier (Lee & Tsien, 1983; Sanguinetti & Kass, 1984; Gurney et al. 1985; Cohen & McCarthy, 1987), but has neither been described for isradipine nor studied for its two enantiomers. The fact that no such observations have been made for isradipine before might be due to the use of Ca2+ as charge carrier in most whole-cell studies. Therefore, a possible change in current kinetics might have been obscured by the fast Ca2+-dependent inactivation in previous studies. It has often been suggested that a block of open channels underlies this effect. We purposely avoided using this term instead of ‘Ilate/Ipeak block’, because it has mechanistic implications which our results did not prove. Our data do not enable us to clarify the underlying mechanism. The very rapid kinetics of association and dissociation, which allowed the drug to bind during the test pulse and to fully dissociate between the pulses, support the idea of binding to open channels. However, one could also imagine an interaction with pre-open closed states that prevail during test pulses. Alternatively, drug-bound channels might still be able to open, but might then inactivate rapidly. We dismiss this possibility for several reasons: no potency difference in the effect on block of Ilate/Ipeak is observed between (+)- and (−)-isradipine, or between depolarised and polarised holding potentials, or between cells with wild-type subunits and those expressing Ch30 mutants, i.e. Ilate/Ipeak block is not favoured by any means predicted to enhance channel inactivation before the test pulse. Finally, and most importantly, hastened inactivation of channels that have bound the drug beforehand should lead to an enhanced fraction, but not rate of current decay, with increasing drug concentration (see Fig. 5A). To discriminate between other possibilities, e.g. binding to open or pre-open closed channels, single-channel analysis will be necessary. To clearly elucidate whether this low-affinity interaction takes place at its own distinct site, further mutagenesis and structure-activity relationship studies will be helpful.
Our hypothesis that different mechanisms underly the inhibition of Ipeak and Ilate/Ipeak are strongly supported by the results of Lacinová & Hofmann (1998): they demonstrated that (i) (+)-isradipine-induced current decay (calculated as 1 - (Ilate/Ipeak)) in Ch30-transfected cells does not depend on the holding potential, consistent with our own data, (ii) current decay depends on the test potential, (iii) the extent of current decay is not related to the extent of Ipeak inhibition, and (iv) the recovery from inactivation in the presence of (+)-isradipine is considerably accelerated in Ch30 compared to wild-type channels. They suggest an open channel block as the underlying mechanism.
To summarise our data, we demonstrated that Ipeak block of the wild-type α1C-b-channel by the isradipine enantiomers is characterised by a high degree of enantioselectivity and dependence of absolute potency, but not enantioselectivity, on the holding potential. These data are compatible with the idea of a guarded receptor. In contrast with the properties of Ipeak block, inhibition of Ilate/Ipeak displays an altered enantioselectivity and no dependence on the holding potential (in both CHOCa9 and Ch30-transfected cells). We therefore conclude that two different mechanisms are involved: a high-affinity interaction mediating inhibition of Ipeak and a low-affinity interaction mediating block of Ilate/Ipeak.
Acknowledgments
The excellent technical help of Elke Schröder and Elke Hippauf is gratefully acknowledged.
References
- Bean BP. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proceedings of the National Academy of Sciences of the USA. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bean BP, Sturek M, Puga A, Hermsmeyer K. Calcium channels in muscle cells isolated from rat mesenteric arteries: Modulation by dihydropyridine drugs. Circulation Research. 1986;59:229–235. doi: 10.1161/01.res.59.2.229. [DOI] [PubMed] [Google Scholar]
- Bodi I, Yamaguchi H, Hara M, He M, Schwartz A, Varadi G. Molecular studies on the voltage dependence of dihydropyridine action on L-type Ca2+ channels. Journal of Biological Chemistry. 1997;272:24952–24960. doi: 10.1074/jbc.272.40.24952. [DOI] [PubMed] [Google Scholar]
- Bosse E, Bottlender R, Kleppisch T, Hescheler J, Welling A, Hofmann F, Flockerzi V. Stable and functional expression of the calcium channel α1 subunit from smooth muscle in somatic cell lines. EMBO Journal. 1992;11:2033–2038. doi: 10.1002/j.1460-2075.1992.tb05260.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen CJ, McCarthy RT. Nimodipine block of calcium channels in rat anterior pituitary cells. The Journal of Physiology. 1987;387:195–225. doi: 10.1113/jphysiol.1987.sp016570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gödicke J, Herzig S, Mescheder A, Mohr K, Steinke F. Enantioselectivity of asocainol studied at different conditions: a novel approach to check the feasibility of molecular models of antiarrhythmic drug action. Naunyn-Schmiedeberg's Archives of Pharmacology. 1992;346:345–351. doi: 10.1007/BF00173550. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Nerbonne JM, Lester HA. Photoinduced removal of nifedipine reveals mechanisms of calcium antagonist action on single heart cells. Journal of General Physiology. 1985;86:353–379. doi: 10.1085/jgp.86.3.353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Handrock R, Herzig S. Stereoselectivity of Ca2+ channel block by dihydropyridines: no modulation by the voltage protocol. European Journal of Pharmacology. 1996;309:317–321. doi: 10.1016/0014-2999(96)00465-7. [DOI] [PubMed] [Google Scholar]
- Hondeghem LM, Katzung BG. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel-blocking drugs. Annual Review of Pharmacology and Toxicology. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- Ito H, Klugbauer N, Hofmann F. Transfer of the high-affinity dihydropyridine sensitivity from L-type to non-L-type calcium channel. Molecular Pharmacology. 1997;52:735–740. doi: 10.1124/mol.52.4.735. [DOI] [PubMed] [Google Scholar]
- Kamp TJ, Sanguinetti MC, Miller RJ. Voltage-dependent binding of dihydropyridine calcium channel blockers to guinea pig ventricular myocytes. Journal of Pharmacology and Experimental Therapeutics. 1988;247:1240–1247. [PubMed] [Google Scholar]
- Kokubun S, Prod'hom B, Becker C, Porzig H, Reuter H. Studies on Ca channels in intact cardiac cells: voltage-dependent effects and cooperative interactions of dihydropyridine enantiomers. Molecular Pharmacology. 1986;30:571–584. [PubMed] [Google Scholar]
- Lacinová L, Hofmann F. Isradipine interacts with the open state of the L-type calcium channel at high concentrations. Receptors and Channels. 1998;6:153–164. [PubMed] [Google Scholar]
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialysed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- Mironneau J, Yamamoto T, Sayet I, Arnaudeau S, Rakotoarisoa L, Mironneau C. Effect of dihydropyridines on calcium channels in isolated smooth muscle cells from rat vena cava. British Journal of Pharmacology. 1992;105:321–328. doi: 10.1111/j.1476-5381.1992.tb14253.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitterdorfer J, Froschmayr M, Grabner M, Striessnig J, Glossmann H. Calcium channels: the β-subunit increases the affinity of dihydropyridine and Ca2+ binding sites of the α1-subunit. FEBS Letters. 1994;352:141–145. doi: 10.1016/0014-5793(94)00938-4. [DOI] [PubMed] [Google Scholar]
- Morel N, Godfraind T. Prolonged depolarization increases the pharmacological effect of dihydropyridines and their binding affinity for calcium channels of vascular smooth muscle. Journal of Pharmacology and Experimental Therapeutics. 1987;243:711–715. [PubMed] [Google Scholar]
- Morel N, Godfraind T. Selective modulation by membrane potential of the interaction of some calcium entry blockers with calcium channels in rat mesenteric artery. British Journal of Pharmacology. 1988;95:252–258. doi: 10.1111/j.1476-5381.1988.tb16571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanin C, Seydl K, Glossmann H, Schindler H. The dihydropyridine niguldipine inhibits T-type Ca2+ currents in atrial myocytes. Pflügers Archiv. 1992;420:410–412. doi: 10.1007/BF00374478. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Kass RS. Voltage-dependent block of calcium channel current in the calf cardiac Purkinje fiber by dihydropyridine calcium channel antagonists. Circulation Research. 1984;55:336–348. doi: 10.1161/01.res.55.3.336. [DOI] [PubMed] [Google Scholar]
- Schuster A, Lacinová L, Klugbauer N, Ito H, Birnbaumer L, Hofmann F. The IVS6 segment of the L-type calcium channel is critical for the action of dihydropyridines and phenylalkylamines. EMBO Journal. 1996;15:2365–2370. [PMC free article] [PubMed] [Google Scholar]
- Starmer CF, Grant AO, Strauss HC. Mechanisms of use-dependent block of sodium channels in excitable membranes by local anesthetics. Biophysical Journal. 1984;46:15–27. doi: 10.1016/S0006-3495(84)83994-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Striessnig J, Grabner M, Mitterdorfer J, Hering S, Sinegger MJ, Glossmann H. Structural basis of drug binding to L Ca2+ channels. Trends in Pharmacological Sciences. 1998;19:108–115. doi: 10.1016/s0165-6147(98)01171-7. [DOI] [PubMed] [Google Scholar]
- Uehara A, Hume J R. Interactions of organic calcium channel antagonists with calcium channels in single frog atrial cells. Journal of General Physiology. 1985;85:621–647. doi: 10.1085/jgp.85.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wibo M, Deroth L, Godfraind T. Pharmacologic relevance of dihydropyridine binding sites in membranes from rat aorta: Kinetic and equilibrium studies. Circulation Research. 1988;62:91–96. doi: 10.1161/01.res.62.1.91. [DOI] [PubMed] [Google Scholar]