

Abstract
P2-purinoceptors couple extracellular ATP to the activation of a Cl− current (ICl,ATP) in heart. We studied the molecular mechanism and intracellular signalling pathways of ICl,ATP activation in mouse heart.
Extracellular adenosine-5′-O-(3-thiotriphosphate) (ATPγS; 100 μM) activated ICl,ATP in both atrial and ventricular myocytes. A specific PKC inhibitor, bisindolylmaleimide blocked the effect of ATPγS while a PKC activator, phorbol 12,13-dibutyrate (PDBu) activated a current with identical properties to ICl,ATP. Maximal activation of ICl,ATP by ATPγS or PDBu occluded further modulation by the other agonist, suggesting that they may activate the same population of Cl− channels.
Isoprenaline increased ICl,ATP pre-activated by ATPγS or PDBu, while isoprenaline or forskolin alone failed to activate any Cl− current in these myocytes. Adenosine 3′,5′-cyclic monophosphothionate, a PKA inhibitor, prevented ATPγS or PDBu activation of ICl,ATP. Thus, ICl,ATP is regulated by dual intracellular phosphorylation pathways involving both PKA and PKC in a synergistic manner similar to cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channels.
Glibenclamide (50 μM) significantly blocked ICl,ATP activated by ATPγS or by the CFTR channel activator, levamisole.
The slope conductance of the unitary ICl,ATP in cell-attached patches was 11·8 ± 0·3 pS, resembling the known properties of CFTR Cl− channels in cardiac myocytes.
The reverse transcription polymerase chain reaction and Northern blot analysis revealed CFTR mRNA expression in mouse heart.
We conclude that ICl,ATP in mouse heart is due to activation of CFTR Cl− channels through a novel intracellular signalling pathway involving purinergic activation of PKC and PKA.
The cystic fibrosis transmembrane conductance regulator (CFTR) gene (Riordan et al. 1989) encodes a chloride (Cl−) channel (Sheppard & Welsh, 1999) that is activated by the binding of ATP to its cytoplasmic nucleotide-binding domains (NBDs) and by the phosphorylation of key serine residues in the regulatory (R) domain. Phosphorylation is mediated principally by cyclic AMP (cAMP)-dependent protein kinase A (PKA) and by protein kinase C (PKC) (Jia et al. 1997; Gadsby & Nairn, 1999; Yamazaki et al. 1999). Mutations of epithelial CFTR lead to dysfunction of the CFTR Cl− channel in cystic fibrosis (CF) patients, resulting in abnormal epithelial chloride transport (Pilewski & Frizzell, 1999).
In the heart, an isoform (exon 5-) of the epithelial CFTR gene product is responsible for cAMP-PKA-activated Cl− currents (ICl,PKA) (Levesque et al. 1992; Nagel et al. 1992; Hart et al. 1996) and PKC-activated Cl− currents (ICl,PKC) as well (Zhang et al. 1994; Collier & Hume, 1995; Middleton & Harvey, 1998). While several observations suggest that cardiac CFTR Cl− current (ICl,CFTR) may modulate resting membrane potential and action potential duration of mammalian cardiac myocytes (Hiraoka et al. 1998), the exact physiological and pathophysiological role of this channel in both normal heart and CF heart remain poorly understood (Gadsby et al. 1995; Hume et al. 2000). Since the cardiac and epithelial CFTR Cl− channel proteins are nearly identical, it is very important to know whether the cardiac CFTR Cl− channel is also defective in CF patients. In order to assess whether or not the mouse CFTR knockout model (Snouwaert et al. 1992) would be useful for determining the functional role of CFTR in heart, previous studies have attempted to characterize the properties of CFTR in mouse heart; however, these studies (Levesque & Hume, 1995; Walsh & Wang, 1996) failed to detect ICl,PKA in normal control mouse atrial and ventricular myocytes, raising doubts about whether ICl,CFTR is functionally expressed in mouse heart, and seemed to obviate the potential use of the CFTR knockout mouse model to assess the physiological and pathophysiological role of CFTR in heart.
In mouse ventricular myocytes, stimulation of purinergic receptors by extracellular nucleotides, including ATP, ADP, and ATPγS, but not adenosine nor AMP, does result in activation of a Cl− current (ICl,ATP) (Levesque & Hume, 1995) similar to the ICl,ATP described previously in guinea-pig (Matsuura & Ehara, 1992) and rat (Kaneda et al. 1994) ventricular myocytes. In both rat and mouse ventricular myocytes, the activation of ICl,ATP appeared to be coupled to P2-purinoceptor stimulation (Kaneda et al. 1994; Levesque & Hume, 1995). To date, however, the intracellular signal transduction pathways linking P2-purinoceptor stimulation to the activation of ICl,ATP in heart have not been characterized and the identity of the channel responsible for ICl,ATP is unknown. Although ICl,ATP represents the least-studied Cl− channel in heart (Hume et al. 2000), many of the known electrophysiological and pharmacological properties of ICl,ATP resemble those of ICl,CFTR (Matsuura & Ehara, 1992; Kaneda et al. 1994; Levesque & Hume, 1995). Furthermore, in epithelial cells it has been suggested that extracellular ATP may directly activate CFTR Cl− channels (Cantiello et al. 1994; Stutts et al. 1995). It seems possible, therefore, that ICl,ATP and ICl,CFTR in heart might be generated by the same protein or by proteins molecularly related to CFTR.
In the present study, we used electrophysiological, pharmacological, and molecular biological techniques to further characterize the properties of ICl,ATP in mouse heart. We first demonstrate that PKA and PKC couple purinergic receptors to the activation of ICl,ATP through dual synergistic PKC and PKA phosphorylation pathways, in a manner highly homologous to the regulation of CFTR. We then provide evidence that the properties of the unitary conductance responsible for ICl,ATP in cell-attached patches of mouse ventricular myocytes are nearly identical to the known properties of cardiac unitary CFTR Cl− channels. Finally, we present evidence using the reverse transcription-polymerase chain reaction (RT-PCR) and Northern blot analysis confirming the expression of the mouse CFTR homologue (Tata et al. 1991; Yorifuji et al. 1991) in normal mouse atrium and ventricle. These results provide evidence that purinergic receptor stimulation in mouse heart is coupled to the activation of CFTR by dual synergetic phosphorylation pathways involving both PKC and PKA, and support the potential usefulness of the CFTR knockout mouse for future functional studies of CFTR in heart.
METHODS
Preparation of single cardiac cells
Single atrial and ventricular myocytes from mouse hearts were isolated using an enzymatic dispersion technique as originally described (Levesque & Hume, 1995). Briefly, the technique involves rapidly removing the heart from mice (C-57/black inbred, male, 20 g) killed by cervical dislocation, in accordance with national guidelines, and perfusing the heart, using a modified Langendorff technique, with a physiological saline solution (PSS) warmed to 37°C. PSS contained (mM): 126 NaCl, 22 dextrose, 4·4 KCl, 5·0 MgCl2, 1·5 CaCl2, 12 Hepes, 20 taurine, 5 creatine, 5 sodium pyruvate, and 1 NaHPO4. The heart was perfused with PSS until free of blood and then with a nominally Ca2+-free PSS until the heart ceased to beat, and finally with the Ca2+-free solution containing 0·04% collagenase (CLSII, Sigma) and 1·0% bovine serum albumin (BSA) for 20–30 min. The atria and ventricles were removed and further dissected into small pieces, and cell dissociation was achieved by gentle mechanical agitation. Only rod-shaped myocytes with clear cross-striations and no blebs under isotonic conditions were used for electrophysiology and molecular biology studies.
Electrophysiological recordings
The tight-seal whole-cell patch-clamp technique was used to record whole-cell currents in isolated mouse atrial and ventricular myocytes and the cell-attached configuration of the patch-clamp technique was employed to record single Cl− channel currents from mouse ventricular myocytes (Hamill et al. 1981). Recording pipettes were prepared from borosilicate glass electrodes (1·5 mm o.d.) with tip resistance of 1–2 MΩ when filled with pipette solution. A bridge (3 M KCl in agar salt) between the bath and an Ag-AgCl reference electrode immersed in pipette solution was used to minimize changes in liquid junction potential, and junction potentials were zeroed before establishing a membrane seal. Voltage-clamp recordings were obtained using a Dagan 3900A or an Axopatch 200B patch-clamp amplifier (Axon Instruments). Single-channel currents were recorded at a gain of 500 mV pA−1, low-pass filtered with an eight-pole Bessel filter at 1 kHz, and digitized (Digidata 1200, Axon Instruments Inc.) and stored on the hard disk of an IBM PC/AT-compatible computer. Whole-cell currents were filtered at a frequency of 2 kHz and pCLAMP 6.0 or 7.0 from Axon Instruments (Clampex, Clampfit, Fetchex, Fetchan and Pstat) were used to control voltage-clamp protocols, and perform data acquisition and analysis. Voltage-clamp pulses were generated by a 12-bit digital-to-analog (D/A) convertor. To obtain whole-cell current-voltage (I–V) relations, cells were clamped from a holding potential of 0 mV to a series of test potentials from −80 to +80 mV for 400 ms in +20 mV increments at an interval of 10 s (as shown in Fig. 1). To obtain single-channel I–V relations, the membrane was clamped from a holding potential of 0 mV relative to the resting potential (RP) to a series of test potentials for 2 s at a time. The voltage of all cell-attached single-channel voltage clamps in this manuscript is expressed as RP +V, where RP is the cell membrane resting potential and V is the transpatch voltage step applied by the amplifier as would be measured at the intracellular side of the patch membrane. Hyperpolarizing and depolarizing pulses were imposed at 0·1 Hz in +20 mV increments between RP - 60 mV and RP + 120 mV. All command voltages and single-channel currents are displayed as they would be measured at the intracellular side of the membrane. Single-channel events at various voltages were manually reviewed, and the open probability of a number of channels (NPo) at any one potential was calculated as described previously (Collier & Hume, 1995; Duan et al. 1997a). The cell-attached patch configuration was checked at the end of each experiment by rupturing the patch to confirm passage from the cell-attached to the whole-cell configuration. Intracellular potentials of myocytes were immediately measured. The average intracellular potential was −83·5 ± 5·2 mV (n = 7). All experiments were performed at room temperature (22–24°C).
Figure 1. Extracellular ATPγS, but not forskolin, activates a linear membrane conductance with reversal potential near the predicted equilibrium potential of Cl− under symmetrical 150 mM Cl−.
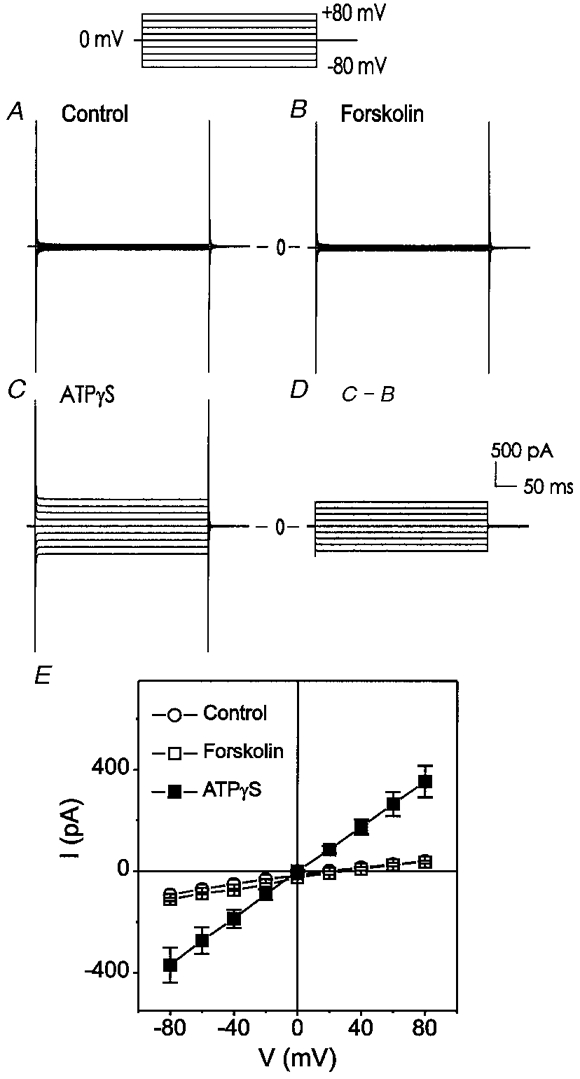
The voltage-clamp protocol is shown at the top. Cells were held at 0 mV and test potentials were applied from −80 to +80 mV in +20 mV increments at an interval of 10 s. A, negligible currents were detected under control conditions. B, subsequent exposure of the same cell to forskolin (10 μM) failed to activate any current. C, ATPγS (100 μM) caused a significant increase in membrane conductance. D, ATPγS (100 μM)-sensitive current. E, mean current-voltage (I–V) relationships of 6 different cells under control (○), forskolin (□), and ATPγS (100 μM) (▪) conditions.
Solutions and drugs
Bath and pipette solutions were chosen to facilitate Cl− current recording. Cd2+ (0·2 mM), Cs+ (5 mM), and 4-aminopyridine (2 mM) were present continuously in the bath solution to block Ca2+ and K+ currents, respectively.
The standard extracellular bath solutions used for whole-cell recordings contained (mM): 140 NaCl, 0·8 MgCl2, 1·5 CaCl2, 5 CsCl, 0·2 CdCl2, 10 Hepes, 5·5 glucose; pH 7·4; total [Cl−]o= 150 mM; 300 mosmol (kg H2O)−1 with mannitol. In experiments where the external Cl− was to be reduced, Cl− was replaced by an equimolar concentration (140 mM) of the monovalent anion iodide (I−) or aspartate (Asp−). Possible contamination from outward cation currents and non-selective cation currents was prevented by using the large impermeant cation N′-methyl-D-glucamine (NMDG) to replace the cations in the pipette solution. The standard intracellular pipette solution used for whole-cell recordings contained (mM): 150 NMDG-Cl, 5 MgATP, 0·1 NaGTP, 5 EGTA, 5 Hepes; pH 7·4; total [Cl−]i= 150 mM; 290 mosmol (kg H2O)−1 using mannitol. In experiments where the internal Cl− was to be reduced, Cl− was replaced by an equimolar concentration of Asp− (110 mM).
The standard pipette (extracellular) solutions used in cell-attached patch-clamp experiments contained (mM): 150 NMDG-Cl, 5 Hepes, 10 glucose; pH 7·4; total [Cl−]i= 150 mM. Inward cation currents such as INa, ICa, and non-selective cation currents were prevented by using the large impermeant cation NMDG to replace the cations in the pipette solution. The standard bath solutions for cell-attached configuration were the same bath (extracellular) solutions used for whole-cell recordings.
All chemicals, including isoprenaline, and forskolin were obtained from Sigma. Levamisole was purchased from Aldrich and phorbol 12,13-dibutyrate (PDBu), bisindolylmaleimide I-HCl (BIM), and adenosine-3′,5′-cyclic monophosphothioate RP-isomer (Rp-cAMP) were purchased from Calbiochem and prepared as stock solutions of 1 or 10 mM in dimethyl sulphoxide and added to a known volume of superfusion solutions to produce the desired concentrations.
RNA isolation and cDNA synthesis
Total RNA was isolated from mouse atrial and ventricular tissues using the Trizol reagent (Life Technologies), following the manufacturer's instructions. Tissues from several animals were pooled (50–100 mg) for each RNA preparation. Total RNA was incubated with RNase-free DNase (10 units) for 20 min at room temperature, followed by heat inactivation. One microgram of total RNA was reverse transcribed with Superscript II reverse transcriptase (Life Technologies) in a 20 μl reaction containing 25 ng oligo(dT) (12–18) primer, 500 μM each dNTP, 50 mM Tris-HCl, pH 8·3, 75 mM KCl, 3 mM MgCl2 and 10 mM DTT.
Reverse transcription PCR (RT-PCR)
RT-PCR was performed using primers specific for the mouse CFTR gene (Tata et al. 1991; GenBank Accession No. M69298). A 392 base pair (bp) region corresponding to nucleotides 1340–1730 was amplified with the forward primer 5′-GGGAGGAGGGATTTGGGGAA-3′ and the reverse primer 5′-GTGATGTCCTGCTGTAGTTG-3′. PCR was performed in 25 μl reactions containing Taq buffer (50 mM KCl, 10 mM Tris-HCl, 1·5 mM MgCl2, 0·1% Triton X-100, 250 μM each dNTP, 20 μM each primer, 2·5 μl cDNA and 1 unit Taq polymerase (Promega)). Amplifications were performed in a GeneAmp 2400 thermal cycler (Perkin Elmer) for 30 cycles of 94°C for 30 s; 50°C for 30 s; 72°C for 1 min, followed by a final extension at 72°C for 7 min. The RT-PCR products were resolved on a 2% agarose gel alongside a molecular weight marker. The murine CFTR 390 bp fragment was gel eluted and subcloned into the TOPO 2.1 T/A vector (Invitrogen). Plasmid DNA was prepared from overnight cultures using the QIAprep Miniprep kit (Qiagen). To confirm the presence of mouse CTFR, both strands of the plasmid DNA were sequenced using the ABI Prism cycle sequencing kit (Perkin Elmer) and analysed on a Genetic Analyser, Model 310 (Perkin Elmer).
Northern blot analysis
To confirm the expression of CFTR mRNA in mouse cardiac tissues, Northern blot analysis was performed as described previously (Duan et al. 1997b). Briefly, 20 μg of total RNA from atrial and ventricular tissue were size-fractionated on a 1% agarose-formaldehyde gel and transferred to nylon filters. Filters were baked and pre-hybridized in 50% formamide, 5 × SSC, 50 mM sodium phosphate, 5 × Denhardt's solution, sonicated salmon sperm DNA (50 μg ml−1), 0·1% SDS, 10% dextran sulphate, at 42°C overnight. The 390 bp murine CFTR cDNA probe was radiolabelled with 32P by random priming (Feinberg & Vogelstein, 1983). Hybridization was performed under the same conditions overnight. The filters were washed at high stringency (3 times in 2 × SSC at room temperature for 5 min then twice in 0·2 × SSC/0·1% SDS at 65°C for 30 min) to assure specificity of labelling. Filters were exposed to film and autoradiography was performed using a phosphoimager (BioRad).
Data analysis
All results are expressed as means ±s.e.m. Statistical comparisons were performed either by analysis of variance (ANOVA) with Scheffé contrasts for group data, or by Student's t test when only two groups were compared. A two-tailed probability of < 5% is taken to indicate statistical significance.
RESULTS
Anion-selectivity of ATPγS-activated ICl,ATP in mouse heart
In agreement with an earlier study (Levesque & Hume, 1995), the cAMP activator forskolin (10 μM) failed to activate any current in mouse ventricular myocytes; however, application of a poorly hydrolysable ATP analogue, ATPγS (100 μM), caused activation of a significant membrane conductance in 6 of 17 (35·2%) of these cells. This time-independent conductance reversed completely upon washout of ATPγS. The ATPγS-activated current had a linear current-voltage (I–V) relationship and a reversal potential of −2·3 ± 1·2 mV (n = 6), which was very close to the predicted equilibrium potential of Cl− (ECl= 0 mV) with symmetrical 150 mM Cl− (Fig. 1A). A similar ICl,ATP was also observed in 3 of 11 (27·3%) mouse atrial myocytes (data not shown).
We further examined the Cl− dependence of the current by replacing the [Cl−]o with an equimolar concentration of I− or Asp−. As shown in Fig. 2, reduction of [Cl−]o caused a decrease in the amplitude of outward currents and shifted the reversal potential (Vrev) to more positive potentials. I− and Asp− substitution of [Cl−]o shifted the Vrev of ICl,ATP from 1·4 ± 1·4 mV to 23·5 ± 1·6 mV and 58·3 ± 3·0 mV, respectively. The permeability ratios (permeability ratio of ion X with respect to Cl−, Px/PCl) were then calculated from the shifts of Vrev using the modified Goldman-Hodgkin-Katz equation (Hille, 1971). The ICl,ATP channel had a PI/PCl of 0·46 ± 0·06 (n = 3) and a PAsp/PCl of 0·13 ± 0·05 (n = 3). These results indicate that the ATPγS-activated current is indeed a Cl− current with a relative permeability of Cl− > I− >> Asp−.
Figure 2. Anion selectivity of ICl,ATP in mouse ventricular myocytes.
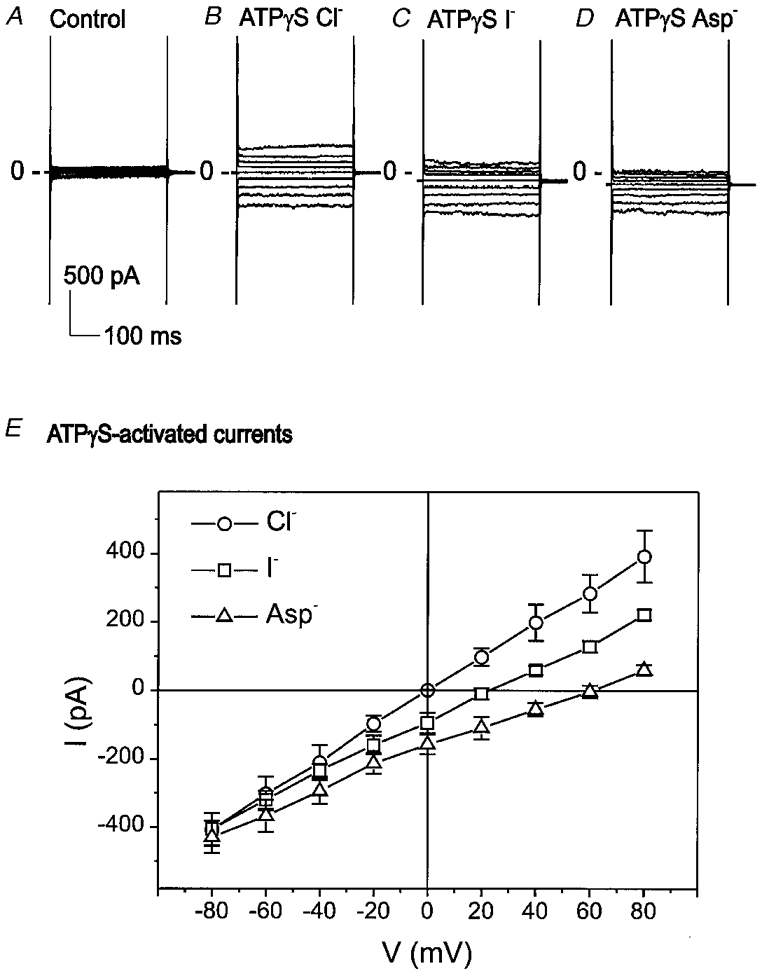
A–D, representative whole-cell currents recorded in the absence (A, control) and presence of ATPγS (100 μM). After ICl,ATP was activated by ATPγS in high extracellular Cl− ([Cl−]o= 150 mM) bath solution (B), [Cl−]o was reduced to 10 mM (ECl=+69·6 mV) by equimolar (140 mM) replacement of Cl− with I− (C) or Asp− (D). E, mean I–V relations of ATPγS-activated whole-cell currents before (○) and after [Cl−]o was substituted by I− (□) or Asp− (▵). I− and Asp− substitution of Cl− shifted the reversal potential of ICl,ATP from 1·4 ± 1·4 mV to 23·5 ± 1·6 mV and 58·3 ± 3·0 mV (n = 3), respectively.
ICl,ATP requires activation of endogenous PKC
In the presence of BIM (100 nM), a PKC inhibitor, ATPγS failed to activate any membrane conductance in mouse ventricular cells (Fig. 3A and D). After removal of BIM from superfusate, ATPγS was then able to activate a current in these cells (Fig. 3A and E). Figure 3A shows the mean I–V curves of whole-cell currents from five different cells under control (○), ATPγS + BIM (□), and ATPγS alone (▪). These results suggest that activation of ICl,ATP by purinergic receptors may be mediated by the activation of endogenous PKC. Therefore, we tested whether activation of PKC by phorbol esters might also cause activation of a similar Cl− conductance.
Figure 3. Inhibition of endogenous protein kinase C by BIM prevented the activation of ICl,ATP by ATPγS.
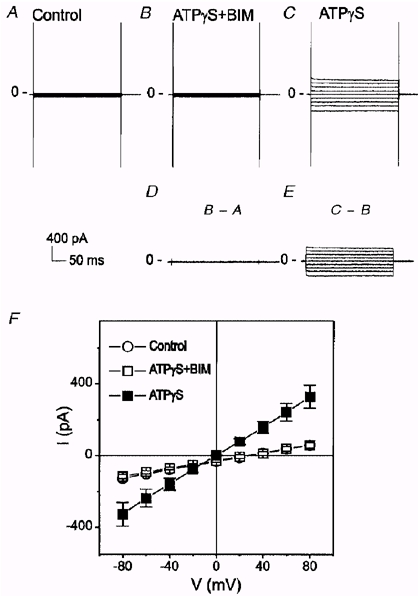
A, whole-cell current under control conditions. B, when BIM (100 nM) was applied together with ATPγS, no increase in the membrane current was observed. C, ATPγS was able to activate ICl,ATP after removal of BIM from superfusate. D, difference current between B and A. E, difference current between C and B. F, mean I–V curves from 5 different cells under control (○), ATPγS + BIM (□), and ATPγS alone (▪) conditions. Only cells which exhibited a distinct ICl,ATP following washout of BIM were included in this analysis.
As shown in Fig. 4, PDBu (100 nM) activated a current with properties (time independence, a linear I–V relationship in symmetrical Cl−) similar to those of ICl,ATP (Fig. 4B, D and F). Subsequent addition of ATPγS in the presence of PDBu failed to further increase the membrane conductance, suggesting that PDBu and ATPγS may activate the same channel. Similar results were observed from four different cells and the mean I–V curves from these cells are shown in Fig. 4F. In symmetrical Cl− gradient ([Cl−]o/[Cl−]i= 150 mM/150 mM, ECl= 0 mV), the PDBu-activated currents were linear and reversed at −3·3 ± 2·7 mV (n = 4), close to the predicted value of ECl (∼0 mV).
Figure 4. PDBu activates a Cl− current similar to ICl,ATP.
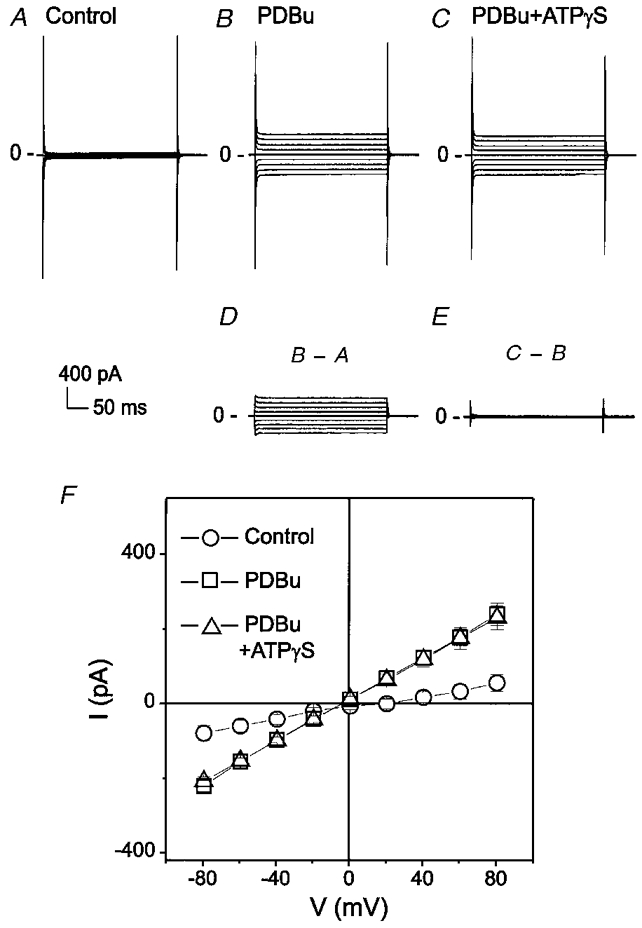
A, whole-cell recording under control conditions. B, PDBu (100 nM)-activated currents. C, subsequent addition of ATPγS in the presence of PDBu failed to further increase membrane conductance. D, PDBu-sensitive current. E, difference current between C and B. F shows mean I–V curves from 4 different cells under control (○), PDBu (100 nM) (□) and PDBu + ATPγS (100 μM) (▵) conditions.
The Cl− dependence of the PDBu-activated current was further examined using different intracellular Cl− concentrations ([Cl−]i). As shown in Fig. 5, when [Cl−]i was partially substituted with equimolar Asp− (110 mM; [Cl−]i= 40 mM), PDBu reversibly activated a time-independent outwardly rectifying current with a reversal potential (-34·6 ± 1·2 mV, n = 4) very close to the predicted ECl (-33·9 mV). These results strongly suggest that PDBu-activated current in mouse ventricular myocytes is a Cl−-dependent current and that the activation of ICl,ATP by ATPγS may be due to purinoceptor-coupled activation of endogenous PKC.
Figure 5. Cl− sensitivity of PDBu-activated whole-cell currents in mouse ventricular myocytes.
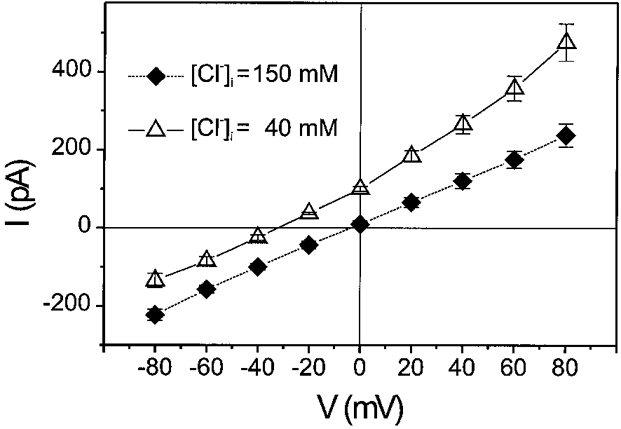
Mean I–V curves of PDBu-activated currents in asymmetrical (▵, n = 4), and symmetrical Cl− gradient (♦, n = 4). With an asymmetrical Cl− gradient ([Cl−]o/[Cl−]i= 150 mM/40 mM, ECl=−33·9 mV), PDBu-activated currents were outwardly rectifying and reversed at −34·6 ± 1·2 mV (n = 4). With a symmetrical Cl− gradient ([Cl−]o/[Cl−]i= 150 mM/150 mM, ECl= 0 mV), PDBu-activated currents were linear and reversed at −3·3 ± 2·7 mV (n = 4).
Isoprenaline enhances ICl,ATP pre-activated by ATPγS or PDBu
The anion selectivity, I–V relations and rectification properties of ICl,ATP activated by either ATPγS or PDBu in mouse heart (also see Levesque & Hume, 1995) strongly resemble those of cardiac and epithelial ICl,CFTR in other species (Anderson et al. 1991; Gadsby et al. 1995). It has been reported that both PKC and PKA activate ICl,CFTR in heart and their effects are additive (Zhang et al. 1994; Collier & Hume, 1995; Yamazaki et al. 1999). It is very interesting that Jia et al. have recently found that PKA-mediated phosphorylation alone is not a sufficient stimulus to open the CFTR Cl− channel in the presence of MgATP; initial, constitutive PKC phosphorylation may be required for acute activation of the CFTR channel by PKA (Jia et al. 1997). It is possible, therefore, that a low level of basal PKC activity in mouse cardiac myocytes is responsible for the failure of acute elevation of intracellular cAMP to activate Cl− currents in these cells (Levesque & Hume, 1995; cf. Fig. 1). If this is true, then the ICl,ATP pre-activated by ATPγS or PDBu should be further enhanced by subsequent application of cAMP stimulators. To test this hypothesis, we examined the effects of isoprenaline on ICl,ATP pre-activated by ATPγS or PDBu. As shown in Fig. 6, while isoprenaline alone failed to activate any current in ventricular myocytes (A and B), addition of isoprenaline (1 μM) caused a further significant increase in the ATPγS-activated ICl,ATP in these myocytes (D). Removal of ATPγS only partially reduced ICl,ATP (E). Further removal of isoprenaline completely reduced the current (F). Similar results were obtained from four different cells under identical conditions and the mean I–V curves from these cells are shown in Fig. 6G.
Figure 6. Synergistic activation of ICl,ATP by ATPγS and isoprenaline in mouse ventricular myocytes.
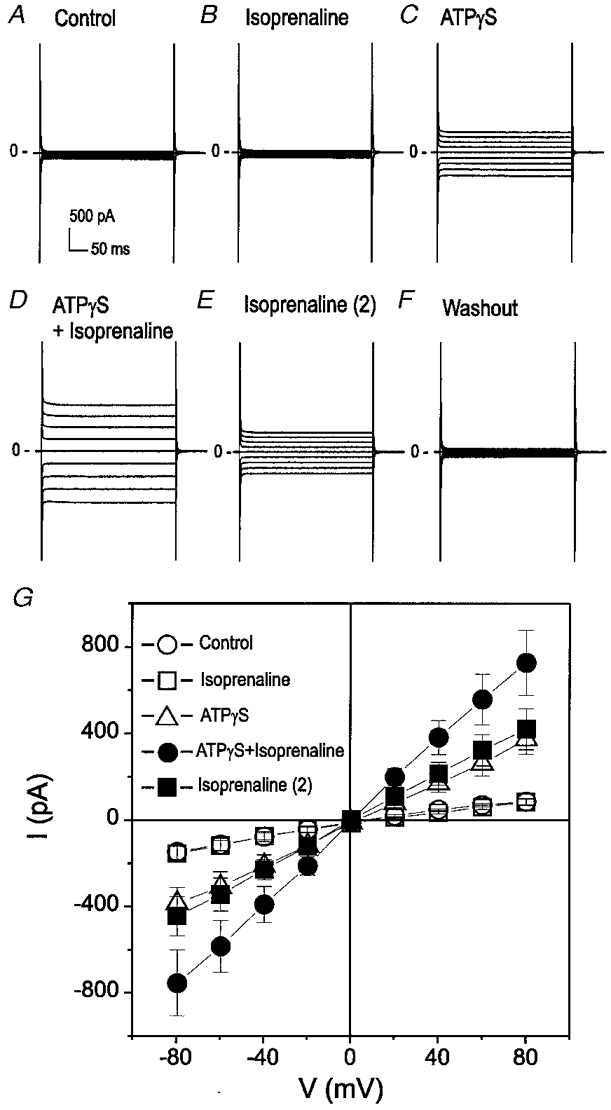
A, whole-cell recording under control conditions. B, isoprenaline alone failed to activate ICl,ATP. C, ATPγS (100 μM) activated ICl,ATP. D, subsequent addition of isoprenaline (1 μM) in the presence of ATPγS caused a further increase in the membrane conductance. E, removal of ATPγS partially reduced ICl,ATP. F, further removal of isoprenaline completely reduced the current. G, mean I–V curves from 4 different cells under control (○), isoprenaline (1 μM) (□), ATPγS (100 μM) (▵), isoprenaline + ATPγS (•), and isoprenaline alone (▪) conditions.
Similarly, as shown in Fig. 7, the PDBu pre-activated ICl,ATP in mouse ventricular myocytes was also enhanced by isoprenaline in mouse ventricular myocytes. These results suggest that activation of PKC by ATPγS or PDBu may be required for the modulation of ICl,ATP by intracellular cAMP-PKA.
Figure 7. Synergistic activation of ICl,ATP by PDBu and isoprenaline in mouse ventricular myocytes.
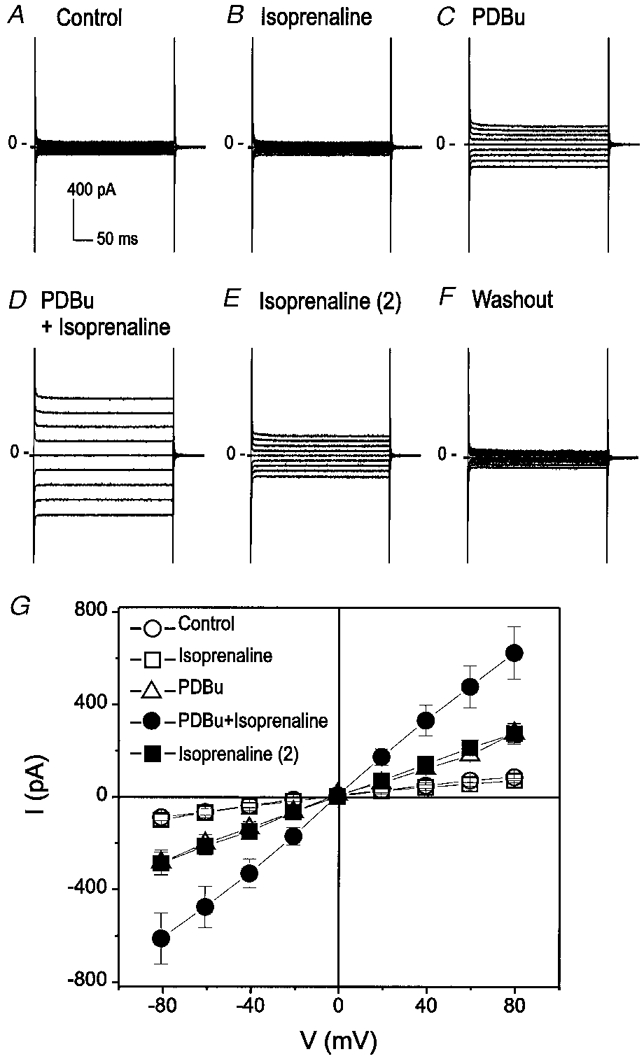
A, whole-cell recording under control conditions. B, isoprenaline alone failed to activate ICl,ATP. C, PDBu (100 nM) activated ICl,ATP. D, subsequent addition of isoprenaline (1 μM) in the presence of PDBu caused a further increase in the membrane conductance. E, removal of PDBu partially reduced ICl,ATP. F, further removal of isoprenaline completely reduced the current. G, mean I–V curves from 3 different cells under control (○), isoprenaline (1 μM) (□), PDBu (100 nM) (▵), isoprenaline + PDBu (•), and isoprenaline alone (▪) conditions.
Activation of ICl,ATP is dependent upon endogenous PKA
The above results indicate that extracellular ATP activates ICl,ATP through purinoceptor-coupled activation of PKC, which may permit synergistic activation of ICl,ATP by PKA phosphorylation. It has recently been reported that ATPγS may also stimulate intracellular cAMP through purinoceptor-coupled activation of adenylyl cyclase V (Puceat et al. 1998). On the other hand, Middleton & Harvey reported that in guinea-pig ventricular myocytes phorbol esters alone failed to elicit CFTR current although they were able to potentiate subsequent CFTR channel activation by PKA (Middleton & Harvey, 1998). Therefore, we next asked the question whether purinoceptor-activated PKA is also involved in the process of ICl,ATP activation. We initially observed that the phosphatase inhibitor, calyculin A (10 nM), was able to increase the current amplitude of ICl,ATP pre-activated by ATPγS or PDBu (data not shown). We next examined the effects of a specific inhibitor of PKA, Rp-cAMP, on ICl,ATP pre-activated by ATPγS. Figure 8A shows the time course of changes in whole-cell currents at +80 mV and −80 mV monitored continuously when the cell was consecutively exposed to control solution for 10 min, ATPγS (100 μM) until the changes in current amplitudes reached steady state (∼15 min), and then ATPγS + Rp-cAMP (100 μM) until the changes in currents reached steady state. Rp-cAMP eventually abolished the ATPγS-activated current. Similar results were observed in three other cells under the same conditions and Rp-cAMP caused a 78·3 ± 6·8% inhibition of ICl,ATP after application of the drug for 10 min. Furthermore, in cells (n = 9) exposed to Rp-cAMP (100 μM), ATPγS (100 μM) failed to activate ICl,ATP (Fig. 8A). Rp-cAMP also prevented the activation of ICl,ATP by PDBu (data not shown), suggesting that PKA is involved in the activation of ICl,ATP by ATPγS or PDBu.
Figure 8. Activation of ICl,ATP by ATPγS is prevented by inhibition of endogenous PKA by Rp-cAMP in mouse ventricular myocytes.
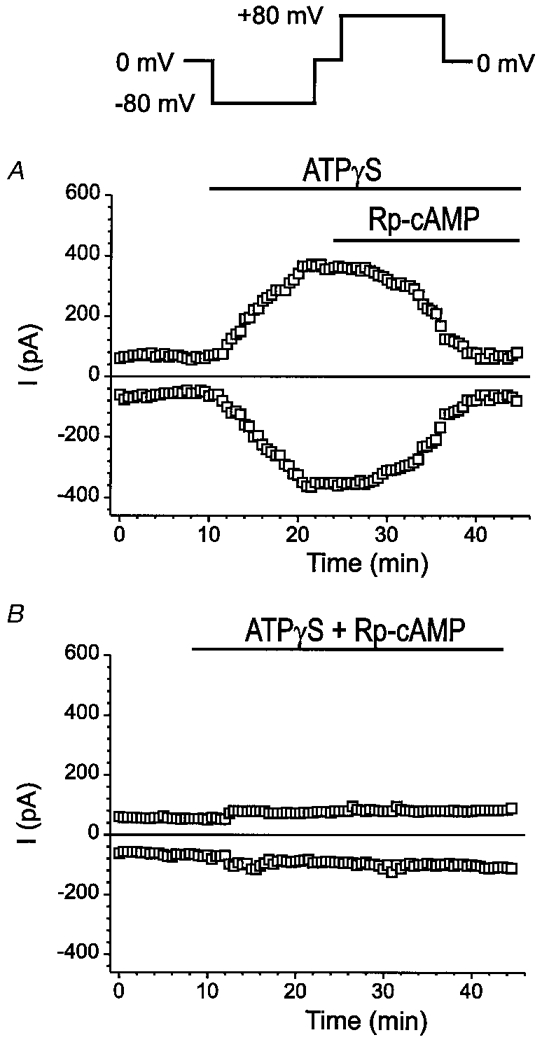
A, time course of changes in whole-cell currents at +80 mV and −80 mV continuously monitored (voltage-clamp protocol is shown on the top) when the cell was consecutively exposed to control solution for 10 min, ATPγS (100 μM) until the changes in current amplitudes reached steady state (≈15 min), and ATPγS + Rp-cAMP (100 μM) until the changes in currents reached steady state. Rp-cAMP eventually abolished the activation of the current even in the presence of ATPγS. B, in the presence of Rp-cAMP (100 μM) ATPγS (100 μM) failed to activate ICl,ATP. Same protocol as in A was used to monitor the time course of whole-cell currents.
Glibenclamide blocks ICl,ATP activated by ATPγS and levamisole
Glibenclamide, a KATP channel inhibitor, is also an effective blocker of both epithelial and cardiac CFTR Cl− channels (Yamazaki & Hume, 1997; Sheppard & Robinson, 1997). As shown in Fig. 9A, 50 μM glibenclamide blocked ICl,ATP activated by 100 μM ATPγS. The inhibition was not voltage dependent (69·8 ± 3·4% inhibition at −80 mV and 71·8 ± 3·2% inhibition at +80 mV, n = 3, P = n.s.).
Figure 9. Glibenclamide blocks ICl,ATP activated by ATPγS and levamisole.
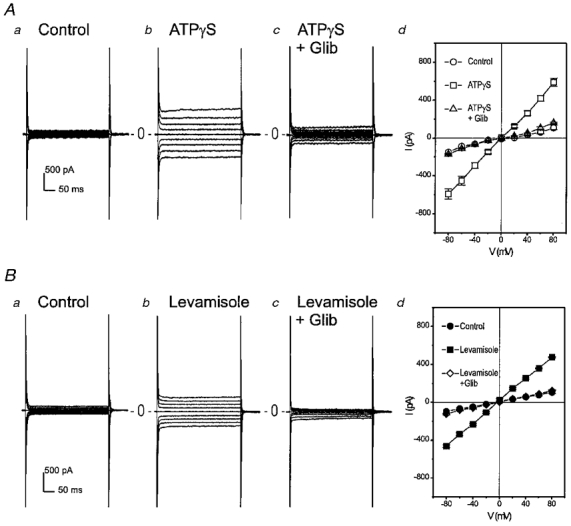
A, ATPγS (100 μM)-activated ICl,ATP (b) in mouse ventricular myocytes was significantly inhibited by 50 μM glibenclamide (c). Panel d shows mean I–V curves of whole-cell currents recorded from 3 different myocytes in the absence (○) and presence (□) of ATPγS (100 μM) and in the presence (▵) of ATPγS and glibenclamide (50 μM). B, levamisole, a reported CFTR Cl− channel activator, activated a linear time-independent current in mouse ventricular myocytes (b and d) and this levamisole-activated current was also significantly inhibited by 50 μM glibenclamide (c and d). Panel d shows mean I–V curves of whole-cell currents recorded from 4 different myocytes in the absence (•) and presence (▪) of levamisole (500 μM) and in the presence (⋄) of levamisole and glibenclamide (50 μM).
It was recently shown that phenylimidazothiazole compounds, especially levamisole, are able to activate CFTR Cl− channels through a cAMP-independent pathway (Becq et al. 1996). Therefore we tested whether levamisole also activates a Cl− current in mouse cardiac myocytes. As shown in Fig. 9A, 1 mM levamisole activated a linear time-independent current which was also inhibited by 50 μM glibenclamide in four of eight (50%) mouse ventricular myocytes tested. The glibenclamide inhibition of the levamisole-activated current was identical to the inhibition of ICl,ATP activated by ATPγS in terms of the voltage-independence and the sensitivity of the channel to the blocker (73·9 ± 2·5% inhibition at −80 mV and 75·5 ± 1·5% inhibition at +80 mV, n = 4), suggesting that levamisole may activate the same ICl,ATP in mouse ventricular myocytes. Although it is not clear at this point whether levamisole activates the current through an inhibition of membrane-associated alkaline phosphatases (Becq et al. 1996) or a second target other than phosphatase inhibition (Mun et al. 1998).
Single-channel properties of ICl,ATP in mouse ventricular myocytes
The cell-attached patch-clamp mode was used to record single-channel currents from cells exposed to ATPγS. Pipette (external) solutions contained 150 mM Cl−. Figure 10A shows representative currents recorded (2 min) when the patch was clamped at −40 mV relative to the resting membrane potential (RP - 40 mV) under control conditions and after exposure of the cell to ATPγS (100 μM). Corresponding amplitude histograms are shown on the right of each panel. No channel opening was detected under control conditions (Fig. 10Aa). Subsequent exposure of the same cell to ATPγS activated inward currents with an amplitude of ∼2·24 pA and an open probability (NPo) of 0·34 (Fig. 10Ab). Similar single-channel activity activated by ATPγS was observed in 5 of 16 (31%) ventricular myocytes. Mean NPo of the single-channel ICl,ATP at RP - 40 mV was 0·25 ± 0·12 (n = 5).
Figure 10. Single-channel properties of extracellular ATPγS-activated Cl− currents in mouse ventricular myocytes.
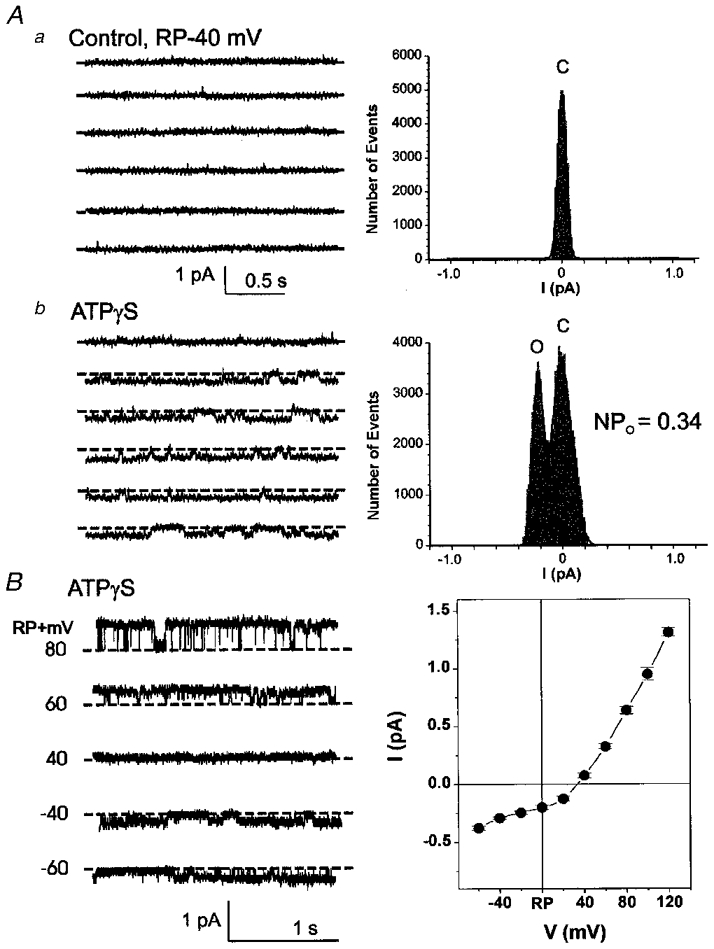
Cell-attached patch-clamp mode was used to record unitary currents before and after the cells were exposed to ATPγS (100 μM). Pipette (external) solutions contained 150 mM Cl−. A, representative current tracings (2 min recording) when the cell-attached patch was clamped at −40 mV relative to the resting membrane potential (RP - 40 mV) under control conditions (a) and in the presence of ATPγS (100 μM) (b); displayed currents low-pass filtered at 150 Hz. Corresponding amplitude histogram is shown on the right of each panel. Data were fitted to a Gaussian distribution (Fetchan) and the continuous line indicates the results. Aa, no channel opening was detected under control conditions. Ab, subsequent exposure of the same cell to ATPγS activated small inward currents with an amplitude of ≈2·24 pA and an open probability (NPo) of 0·34. B, current-voltage (I–V) relationship of ATPγS-activated unitary Cl− currents. Representative current tracings from cell-attached patches when the patch was clamped from a holding potential of 0 mV relative to the resting membrane potential (RP) to various potentials from RP - 60 mV to RP + 80 mV for 2 s in increments of +20 mV; displayed currents low-pass filtered at 1 kHz. Mean I–V relationship (±s.e.m.) from 4 different cells studied under the same conditions is shown in the right panel. Potentials are expressed as a change from RP as measured from the inside of the cell. The mean reversal potential was RP + 28·1 ± 2·6 mV, and the slope conductance (from RP + 20 mV to RP + 120 mV) was 11·8 ± 0·3 pS when the pipette (external) solution contained 150 mM NMDG-Cl.
The I–V relationship of ATPγS-activated unitary ICl,ATP was also determined (Fig. 10A). The mean I–V curves from four different cells studied under the same conditions were outwardly rectifying with a slope conductance of (RP + 20 mV to RP + 120 mV) 11·8 ± 0·3 pS (n = 4) when the pipette (external) solution contained 150 mM NMDG-Cl. The reversal potential was RP + 28·1 ± 2·6 mV (n = 4).
CFTR is transcriptionally expressed in both atrium and ventricle of mouse heart
The results described thus far on ICl,ATP anion-selectivity, rectification, single-channel conductance, PKC- and PKA-dependent activation mechanisms, and levamisole activation and glibenclamide inhibition, suggest that ICl,ATP and ICl,CFTR may be one and the same in mouse heart. Therefore, we performed experiments to verify molecular expression of CFTR in mouse heart.
Figure 11A shows an agarose gel depicting a CFTR-specific RT-PCR product generated from RNA derived from mouse atrial and ventricular tissue. The RT-PCR reaction of total RNA prepared from both atrial and ventricular tissue with specific primers designed to amplify a 392-nucleotide product of mouse CFTR (nucleotide positions 1340–1730) confirmed the transcriptional expression of CFTR in both atrium and ventricle. The RT-PCR product was sequenced and determined to be identical to the previously cloned murine CFTR (Tata et al. 1991; Yorifuji et al. 1991).
Figure 11. Molecular expression of CFTR in mouse heart.
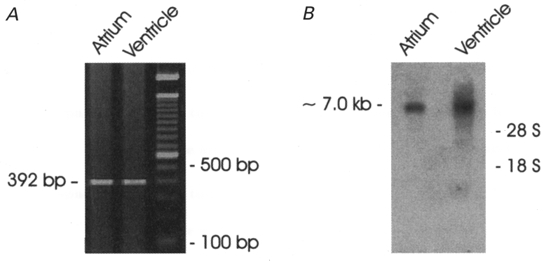
A, agarose gel depicting CFTR-specific RT-PCR product from mouse atrial and ventricular tissue. The CFTR-specific primers amplified a 392-nucleotide product of mouse CFTR (nucleotide positions 1340–1730) which confirmed transcriptional expression of CFTR in both atrial and ventricular cells. B, Northern blot analysis of CFTR expression in mouse cardiac tissues. Total RNA from atrial (9 μg) and ventricular (20 μg) tissue was hybridized with a 32P-labelled mouse CFTR probe (392 bp) as described in Methods. The mouse CFTR transcript was detected at ≈7·0 kb.
Northern blot analysis also indicated that CFTR messenger RNA is expressed in both atrium and ventricle of mouse heart. Figure 11A shows hybridization to a transcript of ∼7·0 kb, which is similar in size to CFTR mRNA (Riordan et al. 1989; Levesque et al. 1992; Nagel et al. 1992).
DISCUSSION
A previous study from this laboratory demonstrated that extracellular ATP activates ICl,ATP through stimulation of P2-receptors in mouse ventricular myocytes (Levesque & Hume, 1995). In the present study, we further examined the intracellular signal transduction mechanisms responsible for purinergic activation of ICl,ATP, identified the single channels responsible for ICl,ATP, and demonstrated transcriptional expression of CFTR in mouse heart. Our data strongly suggest that ICl,ATP in mouse heart may be attributed to the activation of CFTR Cl− channels.
In the past decade at least seven different types of Cl− currents have been described in the heart (Hiraoka et al. 1998; Hume et al. 2000). These include Cl− currents activated by intracellular cAMP-PKA (ICl,PKA), PKC (ICl,PKC), Ca2+ (ICl,Ca), extracellular ATP (ICl,ATP), cell swelling (ICl,swell), hyperpolarization, and a basally active Cl− current (ICl,b). Functional and molecular studies indicate that ICl,PKA, and ICl,PKC as well, are encoded by an isoform of CFTR (Levesque et al. 1992; Nagel et al. 1992; Hart et al. 1996; Yamazaki et al. 1999), whereas the volume-regulated outwardly rectifying Cl− channel (ICl,vol), including ICl,b, may be encoded by a member of the ClC Cl− channel family, ClC-3 (Duan et al. 1997b, 1999b). Another member of the ClC family, ClC-2, may encode the volume-regulated inwardly rectifying Cl− channel recently found in heart (Furukawa et al. 1998; Duan et al. 1999a). At this point, the molecular identity of the proteins responsible for ICl,ATP and ICl,Ca remain unknown (Hume et al. 2000).
The electrophysiological properties of ICl,ATP reported here and by other authors (Matsuura & Ehara, 1992; Kaneda et al. 1994; Levesque & Hume, 1995) are indistinguishable from ICl,CFTR in terms of their anion selectivity, rectification, time independence, and single-channel conductance (Gadsby et al. 1995; Hume et al. 2000). Furthermore, in the present study, we found that a specific PKC inhibitor prevented the activation of ICl,ATP, while the phorbol ester, PDBu, activated a Cl− current identical to that activated by ATPγS, strongly indicating that activation of intracellular PKC may play a major role in the coupling of P2-receptor stimulation to channel activation. Direct evidence for extracellular ATP-induced increase in PKC activity in cardiac myocytes has been obtained (Legssyer et al. 1988; Kunapuli & Daniel, 1998), suggesting that PKC may be a common second messenger for P2-receptor regulation of Cl− channels in other species as well (Matsuura & Ehara, 1992; Kaneda et al. 1994). It has been well established that PKC and PKA activate CFTR Cl− channels in heart (Zhang et al. 1994; Collier & Hume, 1995; Yamazaki et al. 1999) and other tissues (Bajnath et al. 1993; Winpenny et al. 1995; Jia et al. 1997; Lansdell et al. 1998). Potentiation of cAMP-PKA activation of CFTR by PKC has been observed in many other cells as well (Bajnath et al. 1993; Winpenny et al. 1995; Jia et al. 1997; Lansdell et al. 1998; Gadsby & Nairn, 1999). Therefore, it is not too surprising that purinergic receptor stimulation in mouse cardiac myocytes may be linked to activation of CFTR Cl− channels through a mechanism involving both PKC and PKA phosphorylation. Future studies are planned to test this hypothesis further using cardiac myocytes isolated from homozygous CFTR knockout mice (which were not readily available from commercial sources for the present experiments).
ICl,ATP is unlikely to be due to the activation of ICl,Ca, because the electrophysiological properties of ICl,ATP and ICl,Ca are very different and ATP and its analogue ATPγS activate ICl,ATP in the presence of Ca2+ channel blockers and high intracellular EGTA (10 mM) or BAPTA (20 mM) (Levesque & Hume, 1995). It is also unlikely that ICl,ATP is associated with ICl,vol, because ICl,ATP is activated by extracellular nucleotide and intracellular PKC and PKA while ICl,vol is inhibited by these manoeuvres (Duan et al. 1997b; Clemo & Baumgarten, 1998; Duan et al. 1999b). The properties of ICl,ATP are also very different from those of ICl,vol: ICl,ATP has a linear I–V with symmetrical Cl−, a small single-channel conductance (∼11 pS), and an anion selectivity of Cl− > I− while ICl,vol has an outwardly rectifying I–V with symmetrical Cl−, an intermediate single-channel conductance (∼40 pS), and an anion selectivity of I− > Cl− (Vandenberg et al. 1994; Duan et al. 1997b).
The pharmacological properties of ICl,ATP examined are also similar to those of ICl,CFTR. ICl,ATP was significantly inhibited by 50 μM glibenclamide (72% at +80 mV), consistent with the reported lower IC50 value (12·5 μM at +50 mV) for glibenclamide block of cardiac ICl,CFTR, compared to reported IC50 values for glibenclamide block of cardiac ICl,vol and ICl,Ca (193 μM and 62 μM, respectively; Sheppard & Robinson, 1997). In addition, the CFTR Cl− channel activator, levamisole, was found to activate macroscopic Cl− currents similar to ICl,ATP in mouse myocytes, which were also potently inhibited by 50 μM glibenclamide.
The single-channel conductance and reversal potential of unitary ICl,ATP recorded in the cell-attached membrane patches from mouse ventricular myocytes (Fig. 10A) are very similar to those reported for CFTR Cl− channels in cell-attached membrane patches activated by either PKA (Ehara & Ishihara, 1990; Sheppard & Welsh, 1999) or PKC (Bajnath et al. 1993; Collier & Hume, 1995). Although the NPo of unitary ICl,ATP in mouse heart is lower than that reported for CFTR Cl− channels in guinea-pig heart, it is close to the NPo of murine epithelial CFTR Cl− channels expressed in Chinese hamster ovary cells (Lansdell et al. 1998), which were incidentally also found to be activated by PDBu but somewhat refractory to stimulation by activators of human epithelial CFTR Cl− channels.
We also observed that the activation of ICl,ATP by ATPγS or phorbol esters enabled a stimulatory effect of isoprenaline on ICl,ATP, indicating that initial PKC phosphorylation of the channel is a prerequisite for activation of ICl,ATP by cAMP-PKA. This is very similar to the results of a recent study from Jia et al. on PKA and PKC regulation of epithelial CFTR Cl− channel function, in which they found that constitutive PKC phosphorylation is essential for acute activation of CFTR by PKA (Jia et al. 1997). They suggest that PKA phosphorylation alone is not a sufficient stimulus to open CFTR chloride channels and initial phosphorylation of the channel by PKC has a permissive role in priming the channel for acute activation by PKA. Furthermore, Liedtke & Cole also found that PKC inhibitors as well as antisense oligonucleotide to PKC-ε prevented the stimulatory effects of forskolin + CPT-cAMP or adrenaline (epinephrine) on CFTR-mediated Cl− secretion (Liedtke & Cole, 1998). Similar synergistic effects of PKA and PKC phosphorylation on cardiac CFTR channels have also been described (Middleton & Harvey, 1998; Yamazaki et al. 1999). Differences in basal PKC activity may contribute to the variable cAMP responsiveness of CFTR channels in different cell types. After pretreatment of cells with PKC inhibitors native CFTR channels in cardiac myocytes are almost completely refractory to PKA stimulation by elevation of intracellular cAMP (Middleton & Harvey, 1998). Therefore, purinergic receptor stimulation, which causes an activation of PKC and possibly PKA as well (Puceat et al. 1998), may represent a novel, alternative pathway for CFTR regulation (see Fig. 12). This may explain why stimulation of P2-purinoceptors is able to activate a Cl− current in mouse cardiac myocytes, but stimulation of intracellular cAMP by isoprenaline or forskolin alone fails to, even though molecular evidence clearly shows that CFTR mRNA is expressed in these cells. This may also explain why electrophysiological studies on human atrial and ventricular myocytes have failed to detect the activation of ICl,PKA by isoprenaline or forskolin (Oz & Sorota, 1995) while molecular evidence shows that the CFTR message is indeed expressed in both atrial and ventricular human myocardium (Levesque et al. 1992; Warth et al. 1996). Since CFTR Cl− channel function is regulated through a multitude of receptor-signal transduction pathways, assessment of functional expression of CFTR Cl− channels in heart should not be limited to only testing the cAMP-PKA response of the channel.
Figure 12. Schematic representation of proposed novel intracellular signal transduction pathway for regulation of CFTR chloride channel function through purinergic receptors.
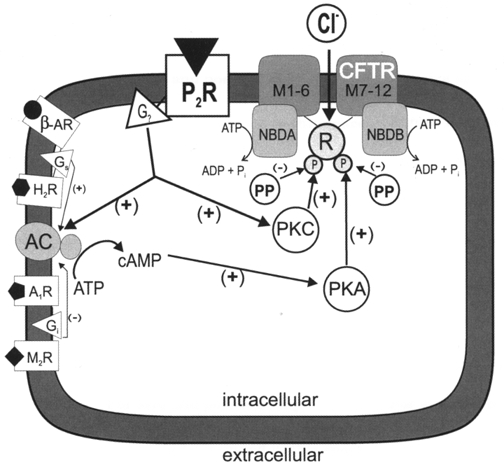
Stimulation of P2-purinergic receptors (P2R) increases intracellular PKC and adenylyl cyclase (AC)-cAMP-PKA activities through an unidentified heterotrimeric G protein (G?) (Legssyer et al. 1988; Kunapuli & Daniel, 1998; Puceat et al. 1998). Initial PKC phosphorylation permits subsequent PKA phosphorylation of the regulatory domain (R) of the CFTR channel (Jia et al. 1997), which causes activation of the two nucleotide-binding domains (NBDA and NBDB). ATP hydrolysis at NBDA and NBDB opens the channel (Sheppard & Welsh, 1999; Gadsby & Nairn, 1999). Other reported regulatory pathways for CFTR regulation are included on the left part of the model. Other abbreviations include: M1-6, the predicted CFTR membrane-spanning segments 1–6; M7-12, the predicted CFTR membrane-spanning segments 7–12; PP, phosphatase; β-AR, β-adrenergic receptor; Gs, heterotrimeric stimulatory G protein; H2R, histamine type 2 receptor; A1R, adenosine type 1 receptor; Gi, heterotrimeric inhibitory G protein; M2R, muscarinic type 2 receptor; (+), stimulatory effect; (−), inhibitory effect.
It is well known that ATP is an important neurotransmitter released from sympathetic nerves and the concentration of ATP can be significantly increased in the coronary circulation during a variety of normal or diseased states (Gordon, 1986; Kunapuli & Daniel, 1998). Therefore, the finding that ICl,ATP is due to purinergic regulation of CFTR Cl− channel function may have significant physiological and pathophysiological importance and potential clinical relevance. Although our study did not specifically address which P2-purinoceptor subtype is linked to activation of CFTR in mouse heart, the fact that activation of ICl,ATP was prevented by specific PKA or PKC inhibitors, argues against involvement of P2X-purinoceptors, since these are usually associated with ligand-gated channels (Kunapuli & Daniel, 1998). Finally, our results resurrect consideration of the use of CFTR knockout mice as a potentially useful animal model for future investigations of CFTR function in the heart.
Acknowledgments
This study was supported by NIH grant HL52803. D.D. was supported by the Medical Research Council of Canada. F.B. was supported by the Western States Affiliate of the American Heart Association.
References
- Anderson MP, Gregory RJ, Thompson S, Souza DW, Paul S, Mulligan RC, Smith AE, Welsh MJ. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Bajnath RB, Groot JA, De Jonge HR, Kansen M, Bijman J. Synergistic activation of non-rectifying small-conductance chloride channels by forskolin and phorbol esters in cell-attached patches of the human colon carcinoma cell line HT-29 cl.19A. Pflügers Archiv. 1993;425:100–108. doi: 10.1007/BF00374509. [DOI] [PubMed] [Google Scholar]
- Becq F, Verrier B, Chang XB, Riordan JR, Hanrahan JW. cAMP- and Ca2+-independent activation of cystic fibrosis transmembrane conductance regulator channels by phenylimidazothiazole drugs. Journal of Biological Chemistry. 1996;271:16171–16179. doi: 10.1074/jbc.271.27.16171. [DOI] [PubMed] [Google Scholar]
- Cantiello HF, Prat AG, Reisin IL, Ercole LB, Abraham EH, Amara JF, Gregory RJ, Ausiello DA. External ATP and its analogs activate the cystic fibrosis transmembrane conductance regulator by a cyclic AMP-independent mechanism. Journal of Biological Chemistry. 1994;269:11224–11232. [PubMed] [Google Scholar]
- Clemo HF, Baumgarten CM. Protein kinase C activation blocks ICl(swell) and causes myocyte swelling in a rabbit congestive heart failure model. Circulation. 1998;98:I-695. [Google Scholar]
- Collier ML, Hume JR. Unitary chloride channels activated by protein kinase C in guinea pig ventricular myocytes. Circulation Research. 1995;76:317–324. doi: 10.1161/01.res.76.2.317. [DOI] [PubMed] [Google Scholar]
- Duan D, Britton F, Ye L, Horowitz B, Hume JR. A novel anionic inward rectifier in cardiac myocytes encoded by ClC-2. Biophysical Journal. 1999a;76:A147. [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. Journal of General Physiology. 1999b;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Hume JR, Nattel S. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circulation Research. 1997a;80:103–113. doi: 10.1161/01.res.80.1.103. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997b;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Ehara T, Ishihara K. Anion channels activated by adrenaline in cardiac myocytes. Nature. 1990;347:284–286. doi: 10.1038/347284a0. [DOI] [PubMed] [Google Scholar]
- Feinberg AP, Vogelstein B. A technique for radiolabeling DNA restriction endonuclease fragments to high specific activity. Analytical Biochemistry. 1983;132:6–13. doi: 10.1016/0003-2697(83)90418-9. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Ogura T, Katayama Y, Hiraoka M. Characteristics of rabbit ClC-2 current expressed in Xenopus oocytes and its contribution to volume regulation. American Journal of Physiology. 1998;274:C500–512. doi: 10.1152/ajpcell.1998.274.2.C500. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nagel G, Hwang TC. The CFTR chloride channel of mammalian heart. Annual Review of Physiology. 1995;57:387–416. doi: 10.1146/annurev.ph.57.030195.002131. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nairn AC. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiological Reviews. 1999;79:S77–107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- Gordon JL. Extracellular ATP: effects, sources and fate. Biochemical Journal. 1986;233:309–319. doi: 10.1042/bj2330309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hart P, Warth JD, Levesque PC, Collier ML, Geary Y, Horowitz B, Hume JR. Cystic fibrosis gene encodes a cAMP-dependent chloride channel in heart. Proceedings of the National Academy of Sciences of the USA. 1996;93:6343–6348. doi: 10.1073/pnas.93.13.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. The permeability of the sodium channel to organic cations in myelinated nerve. Journal of General Physiology. 1971;58:599–619. doi: 10.1085/jgp.58.6.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiraoka M, Kawano S, Hirano Y, Furukawa T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovascular Research. 1998;40:23–33. doi: 10.1016/s0008-6363(98)00173-4. [DOI] [PubMed] [Google Scholar]
- Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiological Reviews. 2000. in the Press. [DOI] [PubMed]
- Jia Y, Mathews CJ, Hanrahan JW. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. Journal of Biological Chemistry. 1997;272:4978–4984. doi: 10.1074/jbc.272.8.4978. [DOI] [PubMed] [Google Scholar]
- Kaneda M, Fukui K, Doi K. Activation of chloride current by P2-purinoceptors in rat ventricular myocytes. British Journal of Pharmacology. 1994;111:1355–1360. doi: 10.1111/j.1476-5381.1994.tb14894.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunapuli SP, Daniel JL. P2 receptor subtypes in the cardiovascular system. Biochemical Journal. 1998;336:513–523. doi: 10.1042/bj3360513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansdell KA, Delaney SJ, Lunn DP, Thomson SA, Sheppard DN, Wainwright BJ. Comparison of the gating behaviour of human and murine cystic fibrosis transmembrane conductance regulator Cl− channels expressed in mammalian cells. The Journal of Physiology. 1998;508:379–392. doi: 10.1111/j.1469-7793.1998.379bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legssyer A, Poggioli J, Renard D, Vassort G. ATP and other adenine compounds increase mechanical activity and inositol trisphosphate production in rat heart. The Journal of Physiology. 1988;401:185–199. doi: 10.1113/jphysiol.1988.sp017157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levesque PC, Hart PJ, Hume JR, Kenyon JL, Horowitz B. Expression of cystic fibrosis transmembrane regulator Cl− channels in heart. Circulation Research. 1992;71:1002–1007. doi: 10.1161/01.res.71.4.1002. [DOI] [PubMed] [Google Scholar]
- Levesque PC, Hume JR. ATPo but not cAMPi activates a chloride conductance in mouse ventricular myocytes. Cardiovascular Research. 1995;29:336–343. [PubMed] [Google Scholar]
- Liedtke CM, Cole TS. Antisense oligonucleotide to PKC-epsilon alters cAMP-dependent stimulation of CFTR in Calu-3 cells. American Journal of Physiology. 1998;275:C1357–1364. doi: 10.1152/ajpcell.1998.275.5.C1357. [DOI] [PubMed] [Google Scholar]
- Matsuura H, Ehara T. Activation of chloride current by purinergic stimulation in guinea pig heart cells. Circulation Research. 1992;70:851–855. doi: 10.1161/01.res.70.4.851. [DOI] [PubMed] [Google Scholar]
- Middleton LM, Harvey RD. PKC regulation of cardiac CFTR Cl− channel function in guinea pig ventricular myocytes. American Journal of Physiology. 1998;275:C293–302. doi: 10.1152/ajpcell.1998.275.1.C293. [DOI] [PubMed] [Google Scholar]
- Mun EC, Mayol JM, Riegler M, O'Brien TC, Farokhzad OC, Song JC, Pothoulakis C, Hrnjez BJ, Matthews JB. Levamisole inhibits intestinal Cl− secretion via basolateral K+ channel blockade. Gastroenterology. 1998;114:1257–1267. doi: 10.1016/s0016-5085(98)70432-9. [DOI] [PubMed] [Google Scholar]
- Nagel G, Hwang TC, Nastiuk KL, Nairn AC, Gadsby DC. The protein kinase A-regulated cardiac Cl− channel resembles the cystic fibrosis transmembrane conductance regulator. Nature. 1992;360:81–84. doi: 10.1038/360081a0. [DOI] [PubMed] [Google Scholar]
- Oz MC, Sorota S. Forskolin stimulates swelling-induced chloride current, not cardiac cystic fibrosis transmembrane-conductance regulator current, in human cardiac myocytes. Circulation Research. 1995;76:1063–1070. doi: 10.1161/01.res.76.6.1063. [DOI] [PubMed] [Google Scholar]
- Pilewski JM, Frizzell RA. Role of CFTR in airway disease. Physiological Reviews. 1999;79:S215–S255. doi: 10.1152/physrev.1999.79.1.S215. [DOI] [PubMed] [Google Scholar]
- Puceat M, Bony C, Jaconi M, Vassort G. Specific activation of adenylyl cyclase V by a purinergic agonist. FEBS Letters. 1998;431:189–194. doi: 10.1016/s0014-5793(98)00747-9. [DOI] [PubMed] [Google Scholar]
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, Zielenski J, Lok S, Plavsic N, Chou JL. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. Published erratum appears in Science245 1437 1989. [DOI] [PubMed] [Google Scholar]
- Sheppard DN, Robinson KA. Mechanism of glibenclamide inhibition of cystic fibrosis transmembrane conductance regulator Cl− channels expressed in a murine cell line. The Journal of Physiology. 1997;503:333–346. doi: 10.1111/j.1469-7793.1997.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard DN, Welsh MJ. Structure and function of the CFTR chloride channel. Physiological Reviews. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Snouwaert JN, Brigman KK, Latour AM, Malouf NN, Boucher RC, Smithies O, Koller BH. An animal model for cystic fibrosis made by gene targeting. Science. 1992;257:1083–1088. doi: 10.1126/science.257.5073.1083. [DOI] [PubMed] [Google Scholar]
- Stutts MJ, Lazarowski ER, Paradiso AM, Boucher RC. Activation of CFTR Cl− conductance in polarized T84 cells by luminal extracellular ATP. American Journal of Physiology. 1995;268:C425–433. doi: 10.1152/ajpcell.1995.268.2.C425. [DOI] [PubMed] [Google Scholar]
- Tata F, Stanier P, Wicking C, Halford S, Kruyer H, Lench NJ, Scambler PJ, Hansen C, Braman JC, Williamson R. Cloning the mouse homolog of the human cystic fibrosis transmembrane conductance regulator gene. Genomics. 1991;10:301–307. doi: 10.1016/0888-7543(91)90312-3. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Yoshida A, Kirk K, Powell T. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. Journal of General Physiology. 1994;104:997–1017. doi: 10.1085/jgp.104.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh KB, Wang C. Effect of chloride channel blockers on the cardiac CFTR chloride and L-type calcium currents. Cardiovascular Research. 1996;32:391–399. doi: 10.1016/0008-6363(96)00075-2. [DOI] [PubMed] [Google Scholar]
- Warth JD, Collier ML, Hart P, Geary Y, Gelband CH, Chapman T, Horowitz B, Hume JR. CFTR chloride channels in human and simian heart. Cardiovascular Research. 1996;31:615–624. [PubMed] [Google Scholar]
- Winpenny JP, McAlroy HL, Gray MA, Argent BE. Protein kinase C regulates the magnitude and stability of CFTR currents in pancreatic duct cells. American Journal of Physiology. 1995;268:C823–828. doi: 10.1152/ajpcell.1995.268.4.C823. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Britton F, Collier ML, Horowitz B, Hume JR. Regulation of recombinant cardiac cystic fibrosis transmembrane conductance regulator chloride channels by protein kinase C. Biophysical Journal. 1999;76:1972–1987. doi: 10.1016/S0006-3495(99)77356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca(2+)-activated Cl− channels in mammalian cardiac myocytes. Circulation Research. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]
- Yorifuji T, Lemna WK, Ballard CF, Rosenbloom CL, Rozmahel R, Plavsic N, Tsui LC, Beaudet AL. Molecular cloning and sequence analysis of the murine cDNA for the cystic fibrosis transmembrane conductance regulator. Genomics. 1991;10:547–550. doi: 10.1016/0888-7543(91)90434-g. [DOI] [PubMed] [Google Scholar]
- Zhang K, Barrington PL, Martin RL, Teneick R. Protein kinase-dependent Cl− currents in feline ventricular myocytes. Circulation Research. 1994;75:133–143. doi: 10.1161/01.res.75.1.133. [DOI] [PubMed] [Google Scholar]