

Abstract
We measured membrane capacitance (Cm) in cultured rat melanotrophs pretreated with Clostridium spiroforme toxin (CST), which specifically depolymerises cortical filamentous actin (F-actin). Phalloidin staining confirmed that CST treatment depolymerised the F-actin.
In control cells, cytosol dialysis with 1 μm increased Cm by 23 ± 4% (n = 11) relative to the resting Cm 400 s after the start of patch rupture. In CST-treated cells the increase in Cm was 32 ± 5% (n = 15), not significantly different from controls. The rate of Cm increase was affected transiently by CST treatment, peaking at 1 min after patch rupture. The maximal rate of Cm increase was 4.27 ± 0.85 fF s−1 (n = 12; measured 200 s after the start of patch rupture) in controls and 8.0 ± 1.35 fF s−1 (n = 23; measured 75 s after the start of patch rupture) in CST-treated cells (P < 0.01).
In control cells cytosol dialysis with 0 μm decreased Cm by 9 ± 3% (n = 7), in CST-treated cells Cm increased by 11 ± 3% (n = 7) relative to resting Cm 400 s after the start of cytosol dialysis. The rate of change in Cm remained constant (controls: -1 to -2 fF s−1; CST treatment: 1-2 fF s−1).
Transient and sustained effects of CST treatment on changes in Cm at high or low [Ca2+]i, respectively, suggest a distinct role of cytoskeleton in Ca2+-dependent and Ca2+-independent changes in Cm. Transient enhancement of the rate of Cm by CST is consistent with a barrier role of cytoskeleton in regulated exocytosis. The sustained effect of CST on Ca2+-independent changes in Cm suggests cytoskeletal involvement in endocytosis.
The actin cytoskeleton is thought to play an important role in a myriad of cellular processes and also in exocytosis (Burgoyne & Morgan, 1993; Trifaró & Vitale, 1993). There are two views on how actin cytoskeleton may affect exocytosis. One view is that evidence obtained during the past decade has suggested that the cortical cell cytoskeleton might act as a barrier in regulated secretion. Cortical filamentous actin (F-actin) was found to form a subplasmalemmal network, which is thought to prevent the translocation of secretory granules from cytoplasm to the plasmalemma. When cells are stimulated to secrete, an accompanying transient disruption of the cortical F-actin barrier was observed in chromaffin cells (Cheek & Burgoyne, 1986; Vitale et al. 1995). In addition to a barrier role, a positive essential role of F-actin in regulated exocytosis is suggested by other evidence. Whereas actin monomer-binding proteins promoted spontaneous exocytosis in permeabilized acinar cells, greater degrees of F-actin disassembly had a negative effect on exocytosis (Muallem et al. 1995). In mast cells, stimulation of secretory activity triggers not only the disassembly of cortical actin but also the appearance of actin filaments that seem to provide a structural support for degranulation (Norman et al. 1996).
To address the role of actin cytoskeleton in exocytosis we studied melanotrophs from the rat pituitary pars intermedia, which secrete a number of peptides deriving from post-translational processing of pro-opiomelanocortin, including β-endorphin, α-melanocyte stimulating hormone and adrenocorticotrophin (Mains & Eipper, 1979). Changes in cytosolic calcium and the activity of GTP-binding proteins appear important regulators of hormone secretion in these excitable cells (see Zorec, 1996). However, whether these regulatory mechanisms act through the cytoskeleton or not, is not known.
It was shown that the peripheral cytoplasm of anterior pituitary cells contains patches of subcortical actin filaments (Senda et al. 1994). If this peripheral cytoskeleton acts as a barrier for secretory granules, then the disaggregation of subcortical actin should increase the rate of secretion (Burgoyne, 1990). To study this hypothesis at cellular level, we monitored the secretory activity of single rat melanotrophs by the patch-clamp membrane capacitance (Cm) measurements. Capacitance increases are a direct measure of increases in the area of the plasma membrane, and a rise in Cm over several minutes is indicative of net exocytosis (Neher & Marty, 1982). Secretory activity of melanotrophs was stimulated by cytosol dialysis with a solution containing high calcium. Actin cytoskeleton was disaggregated by pretreating cells with Clostridium spiroforme toxin (CST), which specifically ADP ribosylates cellular actin (Popoff & Boquet, 1988). The extent of cytoskeleton disaggregation was monitored by phalloidin immunostaining. Phalloidin binds to and stabilises F-actin as well as accelerates actin polymerisation by reducing the rate constant for depolymerisation of the newly formed filaments (Estes et al. 1981).
We found that pretreatment of cells by CST reduces the amount of phalloidin stained subcortical actin network, indicating a depolymerisation of F-actin. The maximal rate of membrane capacitance rise increased twofold in CST-treated cells, which is consistent with the view that the subcortical actin acts as a barrier for secretory activity.
METHODS
Cell culture
Cell culture of melanotrophs from the rat pars intermedia (male Wistar rats, 200-300 g) was prepared by standard methods (Rupnik & Zorec, 1992, 1995). Animals were killed by exposing them to an inflow of 100% CO2 atmosphere followed by decapitation. This procedure was approved by the Veterinary Administration of the Slovenian Ministry for Agriculture and Forestry according to the Law for Animal Health Protection and Instructions for Granting Permits for Animal Experimentation for Scientific Purposes. Cells plated on poly-l-lysine-covered glass coverslips were kept in an incubator at 36°C, in 95% humidity and 5% CO2 in a cell culture medium (a mixture of: α-minimal essential medium (αMEM), DMEM (Dulbecco's modified Eagle's medium) and F-12 medium GIBCO, GB) for 1-7 days before experimentation. Before beginning, cell-covered coverslips were transferred to the recording chamber mounted on an inverted microscope (Nikon TMS, Japan). The recording medium in the chamber consisted of (mm): NaCl, 131.8; CaCl2, 1.8; KCl, 5; MgCl2, 2; Hepes/NaOH, 10; D-glucose, 10; NaH2PO4, 0.5; NaHCO3, 5; pH at 7.2. All chemicals were obtained from Sigma (USA) unless otherwise stated.
Electrophysiological recording
Using the whole-cell patch-clamp technique, the cells were voltage clamped at a holding potential of -70 mV. Membrane capacitance (Cm) was recorded using a two-phase lock-in amplifier (1600 Hz, 1 mV peak to peak) incorporated into a patch-clamp amplifier (SWAM Cell, Henigman, Slovenia (see Zorec et al. 1991)). A DC current (low pass, 1-10 Hz, -3 dB), holding potential and real and imaginary admittance signals (low pass, 1 Hz, -3 dB) were used in the calculations (Zorec et al. 1991). Reversal potential for cell membrane current used in the calculation was -50 mV, which did not change during a recording. The plots of the passive cell parameters, access conductance (Ga), parallel combination of leak (Gm) and membrane conductance (Cm) were derived by computer-aided reconstruction following an analog-to-digital conversion (CED 1401, Cambridge, UK) using an IBM compatible PC. The software was written by Dr J. Dempster (University of Strathclyde, Glasgow, UK). Recordings were made at room temperature with pipette resistances between 1 and 4 MΩ. The basic solution in the recording pipette contained (mm): MgCl2, 2; Hepes/KOH, 10; KCl, 150; Na2ATP, 2; pH at 7.2. To this EGTA (0.65 mm) and Ca-EGTA (4.35 mm) were added in order to obtain Ca2+ activity of approximately 1000 nm, prepared as described (Neher, 1988). Intracellular [Ca2+]i was calculated assuming an apparent dissociation constant (Kd) for the Ca-EGTA complex of 150 nm (Grynkiewicz et al. 1985). If only EGTA (5 mm) was added to the pipette solution, intracellular [Ca2+]i was taken to be 0 nm. Total EGTA concentration was 5 mm, which exceeds the buffering capacity of melanotrophs (Thomas et al. 1990).
Secretory responses were measured every 100 s after the start of recordings as a change in Cm (%) relative to resting Cm, determined immediately after the establishment of a whole-cell recording. The rate of secretion was determined as the slope of the Cm signal between the start of whole-cell recording and a point 100 s after the start of dialysis.
Unless stated otherwise, statistics are given as means ±s.e.m. and differences between samples were tested by Student's t test, considering P < 0.01 to be statistically significant. Error bars on diagrams indicate s.e.m.
Fluorescence microscopy
Cells were washed with phosphate-buffered saline (PBS) at pH 7.4. After that, cells were fixed in 4% formaldehyde solution in PBS for 10 min at room temperature. Cells were again washed with PBS. We prepared a 6.6 μm phalloidin-rhodamine solution in methanol (staining solution) and placed it on coverslips for 20 min at room temperature. Cells were then washed with 1,4-diazabicyclo[2.2.2] octane (DABCO, Molecular Probes, SlowFadeTM, Oregon, USA). Mounted coverslips were viewed with an epifluorescence microscope (Zeiss IM 35). The histological effect of the toxin treatment on the actin cytoskeleton was examined by studying coverslips with a population of more than 10 000 seeded cells, controls or CST-treated cells. Two sets of cultures were examined. We counted over 100 cells per coverslip and determined whether in each cell the phalloidin stained the whole cytoplasm or only its periphery. More than 90% of the cells were of the latter morphology in the toxin treated cells.
Binary C. spiroforme toxin purification and cell treatment
The toxin was purified and prepared according to the procedure described (Popoff et al. 1989). C. spiroforme toxin is a binary toxin made of two components, Sa and Sb. Cells were treated with CST toxin by adding a bolus of the toxin stock solution (275 μg ml−1 Sa (MW 47 000) and 520 μg ml−1 Sb (MW 93 700)) into the cell culture medium or into the recording bath solution. For immunostaining cells were incubated: (i) overnight (16 h) with 3 nm Sa and 3 nm Sb components of the CST toxin; (ii) for 3 h with 1.5-3 nm Sa and 1.5-3 nm Sb components of toxin, and (iii) for 1 h with 15 nm Sa and 15 nm Sb components of toxin. In electrophysiological experiments, cell-covered coverslips were soaked for 1 h in bathing solution containing 15 nm Sa and 15 nm Sb components of toxin or denatured toxin, respectively, then thoroughly washed with bathing solution before electrophysiological recording. Toxin was denatured by immersion of the Eppendorf tube containing both toxin components in boiling water for 5 h. The toxin treatment affected the morphology of cells as viewed with phase contrast microscopy (Fig. 1). However, patch clamping these cells revealed that the steady-state membrane conductance was not affected significantly by this treatment (not shown), indicating that the membrane was not permeabilized or rendered leaky.
Figure 1. Photomicrographs of phalloidin stained rat melanotrophs.

Rat melanotrophs observed under phase contrast (A, × 40) and under fluorescence microscopy (B, × 40; C, × 100). Top panels show cells in control conditions, whereas lower panels show cells treated with CST (15 nm Sa and 15 nm Sb) for 1 h. Cell diameter is around 15 μm. Note that panels B and C display different cells.
RESULTS
Fluorescence microscopy revealed that in control cells phalloidin staining was distributed evenly throughout the cytosol with a ring of higher intensity below the plasmalemma (Fig. 1B and C, top panels). This indicates the presence of a subcortical actin network in anterior pituitary cells as reported previously (Senda et al. 1994). In contrast, in cells treated for 1 h with a high concentration of CST (15 nm) or overnight with a low concentration of CST (3 nm), phalloidin staining was negligible or faint throughout the cytoplasm, and the subplasmalemmal ring of staining was broken at several places (Fig. 1B and C, bottom panels). Treating cells with a low concentration of CST toxin (3 nm) for 3 h did not affect the appearance of phalloidin staining (not shown). Thus toxin treatment for 1 h (15 nm) or overnight (3 nm) resulted in a significant depolymerisation of actin cytoskeleton in rat melanotrophs, consistent with previous reports (Considine & Simpson, 1991; Li et al. 1994; Wex et al. 1997).
To investigate the role of actin cytoskeleton in regulated secretion of rat melanotrophs we measured changes in membrane capacitance (Cm) with the patch-clamp technique (Neher & Marty, 1982; Zorec et al. 1991). This technique allows the dialysis of cytosol with compounds soluble in the pipette solution (Marty & Neher, 1983). Therefore, the secretory activity of rat melanotrophs was stimulated by introducing a high Ca2+-containing (1000 nm) pipette solution into the cytosol. Representative results are shown on Fig. 2. In control cells Cm increased by 23 ± 4% (n = 11; mean ±s.e.m.) relative to the resting Cm, measured 400 s after the establishment of the whole-cell configuration (Fig. 2B). Similar results were reported previously (Rupnik & Zorec, 1992). In CST toxin-treated cells the relative increase in Cm was slightly higher (32 ± 5%, n = 15), but not significantly different from the changes in control cells. In cells treated with denatured CST toxin the relative increase in Cm was similar to control (25 ± 6%, n = 10).
Figure 2. Time-dependent changes in membrane capacitance with high [Ca2+]i.
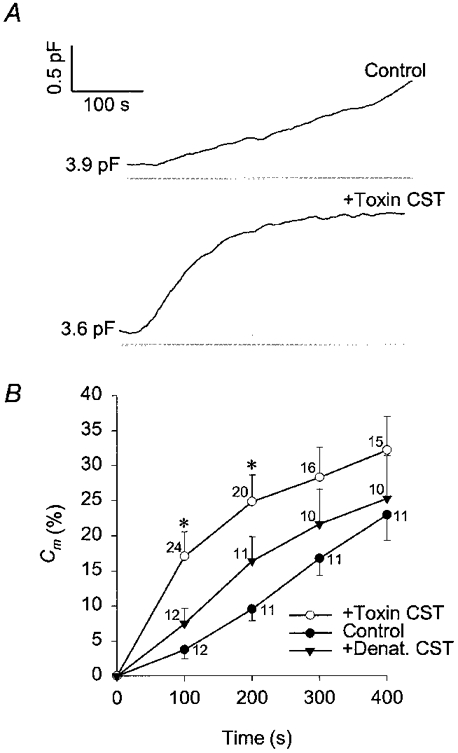
A, changes in membrane capacitance in rat melanotrophs elicited with cytosol dialysis with 1 μm . The top trace is the control and the trace below is from a CST-treated cell (15 nm Sa and 15 nm Sb, treated for 1 h). Values adjacent to traces indicate resting capacitance. B, mean changes in membrane capacitance (%Cm) relative to resting Cm with high [Ca2+]i. Changes in membrane capacitance are determined every 100 s after the start of cytosol dialysis with 1 μm . *Significant differences between the control and CST-toxin treated cells (Student's t test, P < 0.01). ○, CST toxin treated cells (+Toxin CST); ▾, cells treated with the temperature-denatured CST toxin (15 nm Sa and 15 nm Sb, treated for 1 h (+Denat. CST); •, control cells. Numbers adjacent to points indicate numbers of cells tested, bars indicate s.e.m.
In control cells the rate of increase in Cm appeared constant, whereas the time course of Cm in CST-treated cells was characterized by a fast increase following the establishment of the whole-cell configuration (see Fig. 2). Therefore, the rate of secretory activity was measured as the rate of increase in Cm (Fig. 3). In control cells the maximal rate of secretory activity was 4.27 ± 0.85 fF s−1 (n = 12; measured 200 s after the start of patch rupture), whereas it was 8.0 ± 1.35 fF s−1 (n = 23; measured 75 s after the start of patch rupture) in CST toxin-treated cells and significantly different (P < 0.01; see Fig. 3B). A rise in Cm is indicative of net exocytosis (Neher & Marty, 1982; Zupancic et al. 1994), therefore one can conclude that treatment with the actin depolymerising CST toxin augments the initial rate of exocytosis.
Figure 3. Time-dependent changes in the rate of the secretory response (dCm/dt) with high [Ca2+]i.
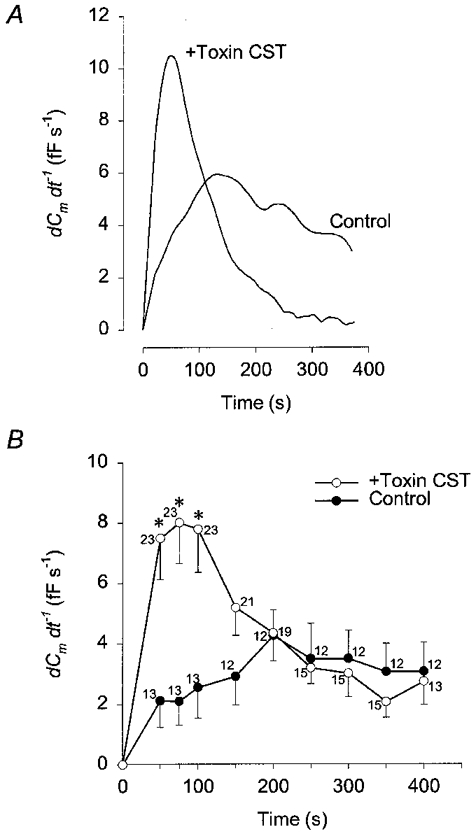
A, representative time-dependent changes in the rate of the secretory response (dCm/dt) in control and in a CST-treated cell after cytosol dialysis with 1 μm . B, mean time-dependent changes in the rate of Cm change (dCm/dt) determined at various time points after the start of cytosol dialysis with 1 μm in control (•) and CST-treated cells (○). *Significant differences between the control and CST- treated cells (Student's t test, P < 0.01). Numbers adjacent to points indicate numbers of cells tested.
On the other hand, changes in Cm may also be influenced by endocytosis (Zupancic et al. 1994), therefore the CST toxin-induced increased rate of Cm rise shown on Fig. 3 may be due to a transient block of endocytosis. It was found previously that endocytosis determines the time course of Cm in rat melanotrophs, if cytosol is dialysed with a solution containing low Ca2+ (Rupnik & Zorec, 1992, 1995). Therefore, we studied the effect of CST toxin treatment on the time course of Cm in melanotrophs dialysed with a low Ca2+-containing pipette solution. Representative results are shown in Fig. 4. In control cells and in cells treated with denatured CST toxin, Cm was decreasing with a slow steady rate to a value of -9 ± 3% (n = 7) and -9 ± 5% (n = 4), respectively. Whereas in CST toxin-treated cells Cm was increasing steadily to a value of 11 ± 3% (n = 7), measured 400 s after the start of cytosol dialysis (Fig. 4B). This effect may account in part for the increases in Cm recorded in CST toxin-treated cells at high cytosolic Ca2+ (Fig. 2).
Figure 4. Time-dependent changes in membrane capacitance with low [Ca2+]i.

A, representative time-dependent changes in Cm in control (bottom trace) and in a CST-treated cell. Both cells were dialysed with a pipette-filling solution containing 5 mm EGTA with no added Ca2+. Values adjacent to traces indicate resting capacitance. B, mean changes in membrane capacitance (%Cm) relative to resting Cm with low [Ca2+]i determined every 100 s after the start of cytosol dialysis. *Significant differences between the control (•) and CST-treated cells (○, Student's t test, P < 0.01). Differences between control cells and those treated with denatured CST (▾) were found not to be significantly different. Numbers adjacent to points indicate numbers of cells tested.
To see whether the time course in Cm elicited with high Ca2+ in CST toxin-treated cells was due to a transient inhibition of endocytosis, we also measured the rate of change in Cm at several times after the start of cytosol dialysis. At low cytosolic [Ca2+]i the rate of increase of Cm in CST toxin-treated cells was fairly constant over time (see slope of line in Fig. 4B), and was not transient. Hence the mechanism of CST action on the Cm time course differs, depending on whether the cytosol is dialysed with low or high cytosolic [Ca2+]i.
DISCUSSION
The aim of this work was to study the relationship between the actin cytoskeleton and the dynamics of the plasmalemmal area in single rat melanotrophs, to learn about the role of cytoskeleton in regulated secretion at cellular level. There are at least three approaches to study the role of cytoskeleton in regulated secretion. Firstly, perhaps the most commonly used approach is to stimulate secretory activity while the secretory response and the reorganisation of the cytoskeletal network is monitored (Vitale et al. 1995 and references therein). Secondly, manipulation of proteins directly affecting the organisation of the cytoskeleton may also affect exocytosis (Zhang et al. 1996; Norman et al. 1996). Thirdly, another possibility is to measure secretory activity when the actin network is depolymerised with specific toxins (see Considine & Simpson, 1991; Li et al. 1994; Wex et al. 1997). This approach was used in this study.
Actin cytoskeleton was disaggregated with the Clostridium spiroforme toxin, a specific actin-depolymerising toxin (Considine & Simpson, 1991). This resulted in a significant reduction of phalloidin staining of rat melanotrophs (Fig. 1), which is consistent with previous studies (Considine & Simpson, 1991; Li et al. 1994; Wex et al. 1997). The secretory activity of CST-treated cells was compared with the non-treated cells, or with cells treated by the denatured toxin. We used the patch-clamp capacitance measurements to monitor the dynamics of membrane capacitance (Cm), reporting changes in the area of the plasmalemma (Neher & Marty, 1982; Zupancic et al. 1994). An increase in Cm is indicative of net exocytosis, whereas a decrease in Cm is indicative of net endocytosis. As reported previously (Rupnik & Zorec, 1992), we found that introducing high Ca2+ into the cytosol results in a rise in Cm (Fig. 2), whereas with a low Ca2+-containing pipette solution a steady decline in Cm was recorded (Fig. 4).
When cells were treated with the CST toxin, the relative Ca2+-induced increase in Cm measured 400 s after patch rupture was slightly higher, but not significantly different from controls (Fig. 2B). However, the maximal rate of rise of Ca2+-induced changes in Cm increased by a factor of two after CST treatment (Fig. 3A). In these cells the maximal rate of Cm increase was recorded at 75 s after the start of cytosol dialysis and returned to control values 200 s after the start of cytosol dialysis (Fig. 3B). This transient increase in the rate of Cm increase is compatible with a transient stimulation of the rate of exocytosis (Burgoyne, 1990), suggesting that cortical actin cytoskeleton may act as a barrier to exocytosis (see Linsted & Kelly, 1987). On the other hand, one has to consider that changes in membrane capacitance report a net change in the surface area and therefore a transient increase in the rate of Cm increase may be due to a transient inhibition of endocytosis.
In a different set of experiments, in which cytosol was dialysed with 5 mm EGTA and with no added Ca2+, changes in Cm were dominated by endocytosis (Fig. 4). The CST treatment resulted in an increase in Cm, which was not transient but sustained. The CST-mediated reversal of a sustained decrease in Cm to a sustained increase in Cm (Fig. 4B) may indicate that: (i) the disassembly of cortical actin cytoskeleton stimulates constitutive exocytosis and basal release, and/or (ii) that endocytosis requires an intact actin cytoskeleton. The first possibility is unlikely. Böttinger et al. (1987) examined the effects of binary toxins on histamine release from rat peritoneal mast cells and reported that the toxin did not affect basal release. Studying 3H-noradrenaline release from PC12 cells, Matter et al. (1989) also report that binary toxin treatment did not change the basal rate of catecholamine secretion. Hence, the second possibility is that the disassembly of cortical actin cytoskeleton inhibits endocytosis in rat melanotrophs. In support of this, the actin cytoskeleton is required for endocytosis in yeast (Kubler & Riezman, 1993) and in mammalian cells (Lamaze et al. 1997). The action of CST on changes in Cm is different at low and high cytosolic [Ca2+], which suggests that actin cytoskeleton plays a distinct role in the plasma membrane area dynamics at low and high [Ca2+]. While we do not know the mechanism of such specificity, specific actin functions in membrane turnover may be mediated by various actin-binding proteins (Molitoris, 1997) such as cofilin, a member of the cofilin/ADF family of small actin-binding proteins (Moon & Drubin, 1995). Cofilin yeast mutants provided an opportunity to distinguish those actin functions that depend specifically on endocytosis from those that do not (Lappalainen & Drubin, 1997).
In cells dialysed with 0 μm CST treatment resulted in a small but steady rate of Cm increase, which is consistent with a Ca2+-insensitive exocytosis. This component of exocytosis may be due to constitutive membrane turnover. The rate of around 1 fF s−1 compares well to the rate of putative constitutive exocytosis determined by cell-attached measurements of discrete steps in membrane capacitance in rat melanotrophs (Kreft & Zorec, 1997).
In summary, we studied whether the depolymerisation of actin cytoskeleton by CST affects the time course of plasma membrane area changes in rat melanotrophs. We found that the early Ca2+-induced rise in Cm is increased in CST-treated cells, which is consistent with a barrier role of cortical actin cytoskeleton in triggered exocytosis. Moreover, under low [Ca2+]i conditions we also found evidence for a CST-mediated inhibition of endocytosis, suggesting that the actin cytoskeleton may be required for a steady uptake of the membrane in melanotrophs.
Acknowledgments
We thank Sonja Grilc for cell cultures and Marjan Rupnik for the help with immunocytochemistry. This work was supported by the Ministry of Sciences and Technology of the Republic of Slovenia with grants J3 6027 and J 8722.
References
- Böttinger H, Reuner KH, Aktories K. Inhibition of histamine release from rat mast cells by botulinum C2 toxin. International Archives of Allergy and Applied Immunology. 1987;84:380–384. doi: 10.1159/000234453. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD. Secretory vesicle-associated proteins and their role in exocytosis. Annual Review of Physiology. 1990;52:647–659. doi: 10.1146/annurev.ph.52.030190.003243. [DOI] [PubMed] [Google Scholar]
- Burgoyne RD, Morgan A. Regulated exocytosis. Biochemical Journal. 1993;293:305–316. doi: 10.1042/bj2930305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek TR, Burgoyne RD. Nicotine-evoked disassembly of cortical actin filaments in adrenal chromaffin cells. FEBS Letters. 1986;207:110–114. doi: 10.1016/0014-5793(86)80022-9. [DOI] [PubMed] [Google Scholar]
- Considine RV, Simpson LL. Cellular and molecular actions of binary toxins possessing ADP-ribosyltransferase activity. Toxicon. 1991;29:913–936. doi: 10.1016/0041-0101(91)90076-4. [DOI] [PubMed] [Google Scholar]
- Estes JE, Selden LA, Gershman LC. Mechanism of action of phalloidin on the polymerisation of muscle actin. Biochemistry. 1981;20:708–712. doi: 10.1021/bi00507a006. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Kreft M, Zorec R. Cell-attached measurements of attofarad capacitance steps in rat melanotrophs. Pflügers Archiv. 1997;434:212–214. doi: 10.1007/s004240050387. [DOI] [PubMed] [Google Scholar]
- Kubler E, Riezman H. Actin and fimbrin are required for the internalization step of endocytosis in yeast. EMBO Journal. 1993;12:1855–1862. doi: 10.1002/j.1460-2075.1993.tb05947.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamaze C, Fujimoto LM, Yin HL, Schmid SL. The actin cytoskeleton is required for receptor-mediated endocytosis in mammalian cells. Journal of Biological Chemistry. 1997;272:20332–20335. doi: 10.1074/jbc.272.33.20332. [DOI] [PubMed] [Google Scholar]
- Lappalainen P, Drubin DG. Cofilin promotes rapid actin filament turnover in vivo. Nature. 1997;388:78–82. doi: 10.1038/40418. [DOI] [PubMed] [Google Scholar]
- Li G, Rungger-Brandle E, Just I, Jonas JC, Aktories K, Wollheim CB. Effect of disruption of actin filaments by Clostridium botulinum C2 toxin on insulin secretion in HIT-T15 cells and pancreatic islets. Molecular Biology of the Cell. 1994;5:1199–1213. doi: 10.1091/mbc.5.11.1199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linstedt AD, Kelly RB. Overcoming barriers to exocytosis. Trends in Neurosciences. 1987;10:446–448. [Google Scholar]
- Mains RE, Eipper BA. Synthesis and secretion of corticotropins, melanotropins and endorphins by rat intermediate pituitary cells. Journal of Biological Chemistry. 1979;254:7885–7894. [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum Press; 1983. pp. 107–121. [Google Scholar]
- Matter K, Dreyer F, Aktories K. Actin involvement in exocytosis from PC12 cells: studies on the influence of botulinum C2 toxin on stimulated noradrenaline release. Journal of Neurochemistry. 1989;52:370–376. doi: 10.1111/j.1471-4159.1989.tb09131.x. [DOI] [PubMed] [Google Scholar]
- Molitoris BA. Putting the actin cytoskeleton into perspective: pathophysiology of ischemic alterations. American Journal of Physiology. 1997;272:F430–433. doi: 10.1152/ajprenal.1997.272.4.F430. [DOI] [PubMed] [Google Scholar]
- Moon A, Drubin DG. The ADF-cofilin proteins: stimulus responsive modulators of actin dynamics. Molecular Biology of the Cell. 1995;6:1423–1431. doi: 10.1091/mbc.6.11.1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muallem S, Kwiatkowska K, Xu X, Yin HL. Actin filament disassembly is a sufficient final trigger for exocytosis in nonexcitable cells. Journal of Cell Biology. 1995;128:589–598. doi: 10.1083/jcb.128.4.589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The influence of intracellular calcium concentration on degranulation of dialysed mast cells from rat peritoneum. The Journal of Physiology. 1988;395:193–214. doi: 10.1113/jphysiol.1988.sp016914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E, Marty A. Discrete changes of cell membrane capacitance observed under conditions of enhanced secretion in bovine adrenal chromaffin cells. Proceedings of the National Academy of Sciences of the USA. 1982;79:6712–6716. doi: 10.1073/pnas.79.21.6712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman JC, Price LS, Ridley AJ, Koffer A. The small GTP-binding proteins Rac and Rho, regulate cytoskeletal organisation and exocytosis in mast cells by parallel pathways. Molecular Biology of the Cell. 1996;7:1429–1442. doi: 10.1091/mbc.7.9.1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popoff MR, Bouquet P. Clostridium spiroforme toxin is a binary toxin which ADP-ribosylates cellular actin. Biochemical and Biophysical Research Communications. 1988;152:1361–1368. doi: 10.1016/s0006-291x(88)80435-2. [DOI] [PubMed] [Google Scholar]
- Popoff MR, Milward FW, Bancillion B, Bouquet P. Purification of the Clostridium spiroforme binary toxin and the activity of the toxin on HEp-2 Cells. Infection and Immunity. 1989;57:2462–2469. doi: 10.1128/iai.57.8.2462-2469.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rupnik M, Zorec R. Cytosolic chloride ions stimulate Ca2+-induced exocytosis in melanotrophs. FEBS Letters. 1992;303:221–223. doi: 10.1016/0014-5793(92)80524-k. [DOI] [PubMed] [Google Scholar]
- Rupnik M, Zorec R. Intracellular Cl− modulates Ca2+-induced exocytosis from rat melanotrophs through GTP-binding proteins. Pflügers Archiv. 1995;431:76–83. doi: 10.1007/BF00374379. [DOI] [PubMed] [Google Scholar]
- Senda T, Okabe T, Matsuda M, Fujita H. Quick-freeze, deep-etch visualisation of exocytosis in anterior pituitary secretory cells: localisation and possible roles of actin and annexin II. Cell and Tissue Research. 1994;277:51–60. doi: 10.1007/BF00303080. [DOI] [PubMed] [Google Scholar]
- Thomas P, Surprenant A, Almers W. Cytosolic Ca2+, exocytosis and endocytosis in single melanotrophs of the rat pituitary. Neuron. 1990;5:723–733. doi: 10.1016/0896-6273(90)90226-6. [DOI] [PubMed] [Google Scholar]
- Trifaró J-M, Vitale ML. Cytoskeleton dynamics during neurotransmitter release. Trends in Neurosciences. 1993;16:466–472. doi: 10.1016/0166-2236(93)90079-2. [DOI] [PubMed] [Google Scholar]
- Vitale ML, Seward EP, Trifaró J-M. Chromaffin cell cortical actin network dynamics control the size of the release-ready vesicle pool and the initial rate of exocytosis. Neuron. 1995;14:353–363. doi: 10.1016/0896-6273(95)90291-0. [DOI] [PubMed] [Google Scholar]
- Wex CBA, Koch G, Aktories K. Effects of Clostridium botulinum C2 toxin-induced depolymerisation of actin on degranulation of suspended and attached mast cells. Archives in Pharmacology. 1997;355:319–327. doi: 10.1007/pl00004949. [DOI] [PubMed] [Google Scholar]
- Zhang L, Marcu MG, Nau-Staudt K, Trifaró J-M. Recombinant scinderin enhances exocytosis, an effect blocked by two scinderin-derived actin-binding peptides and PIP2. Neuron. 1996;17:287–296. doi: 10.1016/s0896-6273(00)80160-9. [DOI] [PubMed] [Google Scholar]
- Zorec R. Calcium signaling and secretion in pituitary cells. Trends in Endocrinology and Metabolism. 1996;7:384–388. doi: 10.1016/s1043-2760(96)00169-5. [DOI] [PubMed] [Google Scholar]
- Zorec R, Henigman F, Mason WT, Kordas M. Electrophysiological study of hormone secretion by single adenohypophysal cells. Methods in Neurosciences. 1991;4:194–210. [Google Scholar]
- Zupancic G, Kocmur L, Veranic P, Grilc S, Kordas M, Zorec R. The separation of exocytosis from endocytosis in rat melanotroph membrane capacitance records. The Journal of Physiology. 1994;480:539–552. doi: 10.1113/jphysiol.1994.sp020382. [DOI] [PMC free article] [PubMed] [Google Scholar]