

Abstract
Electrophysiological recordings and pharmacological manipulations were used to investigate the mechanisms underlying the generation of action potential burst firing and its postsynaptic consequences in visually identified rat layer 5 pyramidal neurons in vitro.
Based upon repetitive firing properties and subthreshold membrane characteristics, layer 5 pyramidal neurons were separated into three classes: regular firing and weak and strong intrinsically burst firing.
High frequency (330 ± 10 Hz) action potential burst firing was abolished or greatly weakened by the removal of Ca2+ (n = 5) from, or by the addition of the Ca2+ channel antagonist Ni2+ (250–500 μm; n = 8) to, the perfusion medium.
The blockade of apical dendritic sodium channels by the local dendritic application of TTX (100 nm; n = 5) abolished or greatly weakened action potential burst firing, as did the local apical dendritic application of Ni2+ (1 mm; n = 5).
Apical dendritic depolarisation resulted in low frequency (157 ± 26 Hz; n = 6) action potential burst firing in regular firing neurons, as classified by somatic current injection. The intensity of action potential burst discharges in intrinsically burst firing neurons was facilitated by dendritic depolarisation (n = 11).
Action potential amplitude decreased throughout a burst when recorded somatically, suggesting that later action potentials may fail to propagate axonally. Axonal recordings demonstrated that each action potential in a burst is axonally initiated and that no decrement in action potential amplitude is apparent in the axon > 30 μm from the soma.
Paired recordings (n = 16) from synaptically coupled neurons indicated that each action potential in a burst could cause transmitter release. EPSPs or EPSCs evoked by a presynaptic burst of action potentials showed use-dependent synaptic depression.
A postsynaptic, TTX-sensitive voltage-dependent amplification process ensured that later EPSPs in a burst were amplified when generated from membrane potentials positive to -60 mV, providing a postsynaptic mechanism that counteracts use-dependent depression at synapses between layer 5 pyramidal neurons.
Neurons integrate synaptic inputs to form an output signal, an action potential, that is generated in the axon and backpropagates into the dendritic tree of several types of central neurons (Stuart et al. 1997). The output of many central neurons, however, is not a single action potential, but a burst of action potentials (Llinas, 1988). In the neocortex, intrinsically burst firing neurons have a specific laminar location in many species (Connors et al. 1982; McCormick et al. 1985; Connors & Gutnick, 1990; Larkman & Mason, 1990; Silva et al. 1991). These neurons form an anatomically distinct group of layer 5 pyramidal neurons that have axons that principally project to subcortical structures, but extensively collateralise in layers 5 and 6 projecting horizontally for several millimetres (Chagnac-Amitai et al. 1990; Larkman & Mason, 1990; Kasper et al. 1994). This provides an anatomical basis for the widespread generation of low frequency synchronised activity, such as sleep rhythms and epileptogenic activity, that is known to be initiated by layer 5 pyramidal neurons (Evarts, 1964; Ball et al. 1977; Connors, 1984; Jones & Heinemann, 1988; Silva et al. 1991; Flint et al. 1997).
The cellular mechanisms underlying burst firing in neocortical layer 5 pyramidal neurons are contentious. Modelling studies have suggested that burst firing can be generated as a consequence of activation of dendritic calcium currents, subsequent to action potential backpropagation into the dendritic tree (Rhodes & Gray, 1994; Mainen & Sejnowski, 1996), but this idea has not been tested experimentally. In contrast, recent experimental studies have concluded that burst firing in these neurons is generated primarily by the activation of the persistent sodium current (Franceschetti et al. 1995; Mantegazza et al. 1998) as is also thought to be the case in CA1 pyramidal neurons (Azouz et al. 1996). Consistent with a role of dendritic calcium conductances in the generation of burst firing, bursts of action potentials have been shown to be associated with dendritic calcium electrogenesis in cerebellar Purkinje neurons (Llinas & Sugimori, 1980). Similarly, in hippocampal CA3 pyramidal and thalamic neurons the activation of voltage-activated calcium channels are thought to underlie burst firing (Wong & Prince, 1978; Llinas, 1988; Huguenard, 1996).
Critical to the importance of burst firing is the fidelity of information transfer during a burst of action potentials (Lisman, 1997). For example, do all action potentials propagate faithfully into the axonal arbour and lead to release of neurotransmitter? If all action potentials during a burst lead to neurotransmitter release, what are the postsynaptic consequences? A number of recent studies have examined synaptic transmission between layer 5 pyramidal neurons at low frequencies, typically during action potential trains (Markram et al. 1997; Thomson, 1997). At these frequencies (typically << 100 Hz) EPSPs show frequency-dependent synaptic depression, indicating that high frequency bursts of action potentials may produce unreliable postsynaptic responses questioning the importance of burst firing in cortical function.
Here we directly address the cellular mechanisms underlying burst firing in rat layer 5 neocortical pyramidal neurons in brain slices using dendritic application of voltage-activated channel antagonists and simultaneous dendritic and somatic recording. The postsynaptic consequences of burst firing were investigated using paired recordings from synaptically connected layer 5 pyramidal neurons. Our findings reveal for the first time that distinct pre- and postsynaptic amplification mechanisms act in synergy during burst firing to enhance synaptic coupling between layer 5 pyramidal neurons. Some of these data have previously appeared in abstract form (Williams et al. 1998)
METHODS
Slice preparation and electrical recording
Rats were anaesthetised with pentobarbitone (20-25 mg; i.p.) and decapitated according to methods approved by the Animal Experimentation Ethics Committee of the Australian National University. Experiments were performed on 300 μm thick coronal neocortical brain slices from 3- to 5-week-old Wistar rats. Slices containing motor and somatosensory cortices were cut starting at the level of the temporal limb of the anterior commissure. Slices were perfused with oxygenated Ringer solution of composition (mm): 125 NaCl, 25 NaHCO3, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, 1 MgCl2 and 10 glucose at 34-36°C. In some experiments NaH2PO4 was omitted and the ratio of divalent cations altered. Somatic, and simultaneous somatic and dendritic or axonal and somatic patch-clamp recordings were made from visually identified layer 5 pyramidal neurons using voltage- and current-clamp amplifiers (Dagan, USA) as described previously (Stuart et al. 1997). For all cell-attached recordings Vpipette was set at -65 mV, thereby making the transmembrane potential 0 mV. Patch electrodes were filled with (mm): 120 or 130 potassium gluconate, 7 NaCl, 10 Hepes, 10 or 0.5 EGTA, 2 Na2-ATP and 2 MgCl (pH 7.3 adjusted with KOH, osmolarity 260-280 mosmol l−1). No correction was made for liquid junction potentials. For paired recordings from neighbouring layer 5 pyramidal neurons (maximal separation 150 μm) the low EGTA recording solution was used to preserve transmitter release characteristics (Ohana & Sakmann, 1998). All action potential evoked EPSP/EPSCs were measured from the mean of six data points around peak amplitude. During dual somatic whole-cell recordings simulated EPSPs were generated by the injection of a current waveform. This waveform was made by inverting the mean (n = 30 trials) EPSC waveform that was evoked during a paired recording, by a two action potential presynaptic burst. Electrically evoked excitatory postsynaptic responses were elicited via extracellular stimulation using a patch pipette filled with extracellular solution placed within cortical layers 2-3, approximately 100 μm lateral to the recorded neuron, and driven with a constant-voltage stimulator (0.2 ms, 5-100 μA). In cell-attached recordings the stimulation current was adjusted so as to produce threshold responses, defined as the minimum current required to evoke action potentials on most trials. Voltage and current signals were filtered at 10 kHz and were acquired at 20-80 kHz, using an ITC-16/18 interface (Instrutech Corporation, Long Island, NY, USA) controlled by an Apple Power PC implementing Axograph acquisition and analysis software (kindly supplied by Dr J Clements; Division of Neuroscience, Australian National University).
Pressure application
Pipettes of similar characteristics to those used for recording were filled with either 100 nm tetrodotoxin (TTX) or 1 mm nickel dissolved in oxygenated extracellular solution and connected to a pressure line, with slight negative pressure (10-20 mmHg). The pipette was positioned close to (< 20 μm) the neuronal membrane at the desired somatodendritic location and positive pressure (120-180 mmHg) applied for 10-30 s, resulting in a clear ejection stream. The resultant ejection area has previously been shown to be approximately 100 μm in diameter (Stuart & Sakmann, 1995).
Data analysis
All data were analysed in Axograph. Numerical values are given in the text as means ±s.e.m.. Statistical analysis was performed in Microsoft Excel and Graphpad Prism. Data recorded from cells with different firing behaviour were tested by non-parametric ANOVA, while paired conditions in the same cell, or independent conditions between two groups, were analysed with Student's t test. Statistical significance was determined with P = 0.05.
RESULTS
Electrophysiological properties of regular and burst firing neurons
Previous studies of the repetitive firing properties of neocortical neurons have indicated that layer 5 pyramidal neurons can be divided into regular and burst firing based on their action potential discharge in response to threshold somatic current injection (Connors et al. 1982; Chagnac-Amitai et al. 1990; Larkman & Mason, 1990; Wang & McCormick, 1993). Consistent with these studies we found that during somatic cell-attached patch-clamp recordings the action potential response to synaptic excitation was either a high frequency burst of 2-6 action potentials (1st inter-spike interval (ISI) 299 ± 15 Hz; n = 10) or a single action potential (n = 10) (Fig. 1). Subsequent formation of somatic whole-cell recording did not change the pattern of action potential output (Fig. 1), indicating that intracellular dialysis during whole-cell recording does not alter the action potential firing pattern. The repetitive firing properties of neurons were examined by the injection of an incremental series of positive current steps from a holding potential of -65.9 ± 0.4 mV (n = 61). Regular firing neurons (26 of 61) responded to just threshold current injection with a single or more action potentials repeated at low frequency (< 10 Hz; Fig. 2). Burst firing neurons, however, responded with the generation of an initial burst of action potentials (2-6) riding upon an underlying depolarising envelope. The amount of current required to initiate action potentials (rheobase), from these holding potentials was significantly greater (P < 0.05, ANOVA) for burst firing neurons (burst firing: 0.45 ± 0.04 nA; regular firing: 0.28 ± 0.03 nA). We further categorised burst firing neurons into two groups: weak burst firing (9 of 61 neurons) and strong burst firing (26 of 61 neurons). Weak burst firing neurons generated a single burst of action potentials (2-3) followed by a train of single action potentials repeated at a lower frequency (Fig. 2), while strong burst firing neurons responded by the generation of repeated bursts of action potentials that were maintained throughout the duration of threshold current pulses (Fig. 2). Subsequent to the division of weak and strong burst firing neurons based on their firing pattern, we observed that rheobase values (weak burst firing: 0.29 ± 0.04 nA; strong burst firing: 0.50 ± 0.05 nA) and the instantaneous frequency of the first inter-spike interval of a burst were significantly greater (P < 0.05, ANOVA) for strong burst firing neurons (weak burst firing: 255.9 ± 18.1 Hz; strong burst firing: 355.1 ± 6.8 Hz). In burst firing neurons the threshold response to a step (200-500 ms) of somatic current was always a stereotyped burst discharge (1st ISI 330 ± 10 Hz; n = 35) which was independent of the prior holding potential (Fig. 3A). The inter-burst frequency increased linearly with the amplitude of injected current (500 ms steps: mean slope 3.88 ± 0.02 Hz (100 pA)−1, and burst discharges were maintained when the neuron was tonically depolarised above firing threshold for long periods (30 s; Fig. 3B).
Figure 1. Identification of burst firing neurons.
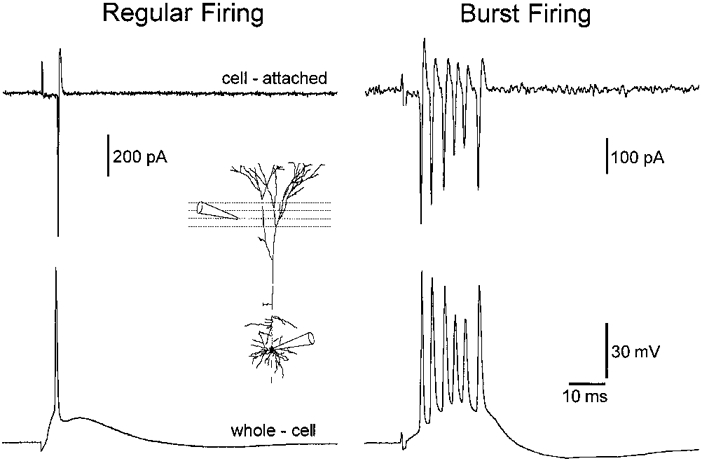
Cell-attached somatic voltage-clamp recording (Vpipette = -65 mV) of neuronal firing in response to extracellular synaptic excitation (upper traces) demonstrates two groups of layer 5 pyramidal neurons: regular and burst firing. The lower traces demonstrate that the subsequent formation of whole-cell current-clamp recording did not modify action potential firing properties when evoked from the resting membrane potential (-64 and -63 mV, regular and burst firing neuron, respectively). The experimental arrangement is shown in the inset.
Figure 2. Action potential firing patterns of layer 5 pyramidal neurons in response to somatic current injection.

Discharge patterns of three neurons in response to steps of positive current (lower superposed traces). The holding potential is shown below the lower voltage traces. Note that in the regular firing neuron a single action potential is evoked at threshold current intensity. In weak and strong burst firing neurons, however, a high frequency (> 250 Hz) burst of action potentials is the threshold response. Bursts of action potentials were repeated throughout the duration of current pulses in strong, but not weak, burst firing neurons. The number of action potentials composing a burst, however, decreased with time in strong burst firing neurons.
Figure 3. Voltage dependence of burst discharges in response to somatic current injection.

A, incremental series of somatic current steps, delivered from the indicated values of holding potential, produce families of voltage responses. Note that the threshold discharge pattern is a stereotyped burst of action potentials irrespective of the prior holding potential. Also note the highly non-liner slope resistance of this neuron. B, repeated bursts of action potentials are evoked during sustained suprathreshold depolarisation produced by the tonic passage of current through the recording electrode (values beneath traces). Lower traces show segments of the data at a faster time base: note the periodic nature of burst discharges.
We found no difference in the membrane potential (Table 1) and properties of the first action potential generated by threshold current steps in burst and regular firing neurons. The properties of each action potential composing a burst in strong burst firing neurons evoked in response to long (500 ms) threshold current steps was examined. The first action potential in the burst was found to be invariably of the largest peak amplitude, with subsequent action potentials showing a progressive decrease in peak amplitude accompanied by a decrease in the 10-90 % rise time and an increase in action potential half-width (see Figs 5 and 8). The instantaneous firing frequency was also found to decrease throughout the duration of the burst.
Table 1. Subthreshold electrophysiological properties of layer 5 pyramidal neurons.
| All cells | Regular firing | Burst firing | Weak burst firing | Strong burst firing | |
|---|---|---|---|---|---|
| Number of cells | 61 | 26 (43 %) | 35 (57 %) | 9 (14 %) | 26 (43 %) |
| Membrane potential (mV) | −63.4 ± 0.4 | −63.2 ± 0.6 | −63.5 ± 0.52 | −65.2 ± 1.1 | −62.9 ± 0.5 |
| −63 | −63 | −64 | −66 | −63 | |
| Minimum 3rd quadrant | 21.1 ± 1.2 | 24.4 ± 2.1 | 18.5 ± 1.3* | 24.6 ± 3.2 | 16.3 ± 0.9†‡ |
| peak slope resistance (MΩ) | 17.6 | 22.2 | 16.6 | 24.1 | 15.8 |
| Maximum 3rd quadrant | 28.2 ± 1.7 | 34.6 ± 3.1 | 23.2 ± 1.5* | 29.8 ± 2.45 | 21.0 ± 1.6 ‡ |
| peak slope resistance (MΩ) | 25.5 | 31.1 | 20.6 | 30.0 | 18.8 |
| Minimum 3rd quadrant steady-state slope | 17.0 ± 1.1 | 19.7 ± 1.8 | 15.0 ± 1.2 | 20.8 ± 3.6 | 12.8 ± 0.8†‡ |
| resistance (MΩ) | 15.3 | 18.3 | 3.9 | 19.3 | 12.6 |
| Maximum 3rd quadrant steady-state slope | 21.2 ± 1.4 | 26.1 ± 2.6 | 17.5 ± 1.2* | 22.82 ± 2.0 | 15.8 ± 1.3‡ |
| resistance (MΩ) | 18.98 | 24.5 | 16.1 | 22.4 | 13.6 |
| Minimum 1st quadrant | 46.2 ± 4.4 | 61.1 ± 9.2 | 36.3 ± 3.1* | 49.7 ± 5.2 | 31.3 ± 3.3†‡ |
| peak slope resistance (MΩ) | 38.9 | 54.0 | 31.8 | 47.1 | 27.6 |
| 1st quadrant steady-state | 32.2 ± 2.4 | 41.2 ± 3.9 | 26.4 ± 2.5* | 38.1 ± 4.1 | 22.1 ± 2.6†‡ |
| slope resistance (MΩ) | 28.58 | 39.8 | 22.5 | 37.3 | 17.9 |
| Subthreshold transient | 4.3 ± 0.2 | 3.6 ± 0.3 | 4.75 ± 0.3 | 2.9 ± 0.3 | 5.3 ± 0.3†‡ |
| depolarising potential (mV) | 4.3 | 3.3 | 4.8 | 2.7 | 5.5 |
Numerical values are given as means ±s.e.m. (top line) and median (lower line).
Significant difference between burst and regular firing cells (ANOVA).
Significant difference between weak burst and strong burst firing cells (ANOVA).
Significant difference between regular and strong burst firing cells (ANOVA).
Figure 5. Ionic basis of burst firing.
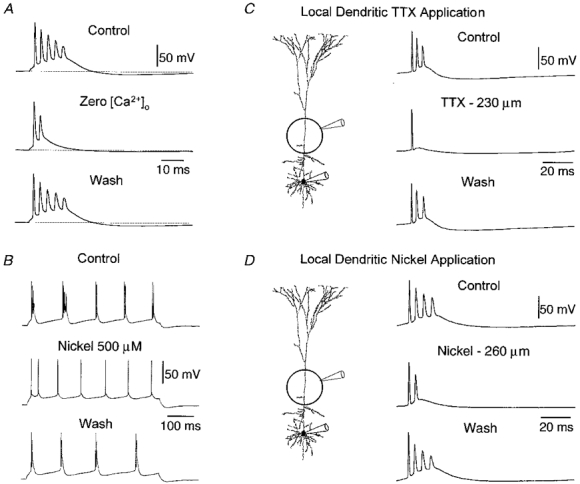
A, burst discharges evoked by a brief somatic current pulse (2 ms, 1.6 nA) recorded at the soma are significantly weakened by removal of calcium from the extracellular solution. Voltage traces obtained from control (2 mm calcium:1 mm magnesium), nominally zero calcium (3 mm magnesium) and following wash (holding potential -66 mV). B, burst discharges evoked by a long somatic current pulse (500 ms, 0.9 nA) recorded at the soma are blocked by the calcium-channel antagonist nickel (500 μm). Voltage traces obtained from control, nickel and wash conditions document the reversible blockade of burst firing (holding potential -65 mV). C, local apical dendritic application of tetrodotoxin blocks burst discharges. The experimental arrangement is shown in the inset. Traces recorded at the soma from control, following the transient local application of TTX (100 nm in pipette) to the apical dendrite 230 μm from the soma and following recovery. Note that TTX did not change the amplitude, threshold or waveform of the first somatic action potential. D, local apical dendritic application of nickel reduces the intensity of burst discharges. Traces from control, following the transient local application of nickel (1 mm in pipette) to the apical dendrite 260 μm from the soma and following recovery. Note the reduction of burst firing is not accompanied by a change in the amplitude, waveform or threshold of first action potential. Holding potential in C and D was -65 mV.
Figure 8. Action potentials are not decremental in the distal axon during a burst discharge.
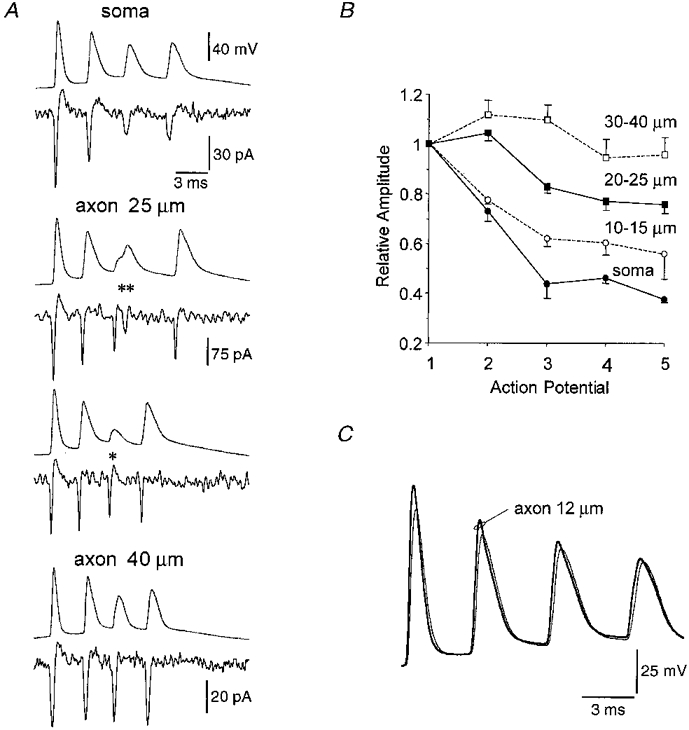
A, pairs of simultaneous recorded somatic whole-cell (top traces) and axonal cell-attached (Vpipette = -65 mV) (bottom traces) recordings at different distances from the soma during burst firing. During an action potential burst the amplitude of somatically recorded action potentials decreases, in some trials later somatically recorded action potentials show inflections in their rising phase (**) and can apparently fail (*). Simultaneous axonal recordings show that when action potential invasion of the soma almost fails (**) two axonal events are recorded, and that when somatic failure occurs the axonal action potential is largely unchanged (axonal recording 25 μm from the base of the soma, two consecutive records). The decrement in axonal action potentials during a burst is dependent upon the distance of the axonal recording from the soma. Note that each action potential of a burst is of a similar amplitude at the most distal axonal recording site (40 μm from the base of the soma). B. the relative amplitude of the second, third, fourth and fifth action potentials in a burst during cell-attached axonal recordings at different distances from the soma. Action potential amplitude was normalised to that of the first action potential in the burst. Total of 14 cells represented. C, simultaneous somatic and axonal whole-cell recording during burst firing. Note all action potentials in the burst are generated first in the axon.
Subthreshold membrane properties were found to be correlated with the type of discharge response as previously reported (Larkman & Mason, 1990; Kasper et al. 1994). The voltage-current (V-I) relationship of all neurons was highly non-linear (Fig. 4). From similar holding potentials (-65.9 ± 0.4 mV), these V-I relationships demonstrated rectification in both the depolarised (first quadrant) and hyperpolarised (third quadrant) direction. First quadrant rectification was detected as the supra-linear relationship between the amplitude of positive voltage deviations and injected current, when measured at the peak and steady state (20 ms prior to the offset of current command steps), whilst third quadrant rectification was observed as a sub-linear relationship. In 61 neurons we measured the slope of V-I relationships at three locations: (i) minimum first quadrant peak and steady-state slope resistance, which represented the linear portion of the relationship in the depolarised direction; (ii) maximum third quadrant peak and steady-state slope resistance, the maximal slope of the relationship in the hyperpolarised direction; and (iii) minimum third quadrant peak and steady-state slope resistance, the minimum slope of the relationship in the hyperpolarised direction. In all cases regression lines were fitted over at least three points. As detailed in Table 1, we observed that quantification of V-I relationships in this way revealed significant differences between neurons of different firing types, with the main difference being that the apparent peak and steady-state input resistance of strong burst firing neurons in both the first and third quadrant were significantly lower than those of either weak burst or regular firing neurons.
Figure 4. Characteristics of the voltage-current relationship in neurons exhibiting different firing behaviour.

Families of voltage (upper, superposed traces) and current (lower, superposed traces) records demonstrate the typical rectification properties of regular, weak burst and strong burst firing neurons. Recordings were made from similar holding potentials (-65 mV). Note, in the strong burst firing neuron a clear transient depolarising potential was generated at the onset of positive voltage responses. The lower graphs show voltage-current relationship when measured at the peak (○) and steady-state, 20 ms prior to the offset of current command steps (•). The subtraction of these measurements yielded the curves shown by ▵
In all neurons first quadrant rectification was found to be time dependent, as reflected by the generation of a transient depolarising potential (TDP) at the onset of positive voltage deviations (Fig. 4). The magnitude of the TDP was quantified by the subtraction of the value of steady-state voltage deviations from peak (subtraction curves are shown for the different neuronal types in Fig. 4). In both regular firing and weak burst firing neurons the amplitude of this TDP tended to increase linearly or sub-linearly with injected current, whilst in strong burst firing neurons the amplitude of the TDP increased supra-linearly and was most prominent at potentials just subthreshold for the generation of action potentials. Accordingly the amplitude of the TDP, when evoked by just subthreshold current steps, was found to be significantly greater in strong burst firing neurons (Table 1). In four strong burst firing neurons we observed that TTX (0.5-1 μm) greatly reduced the amplitude of TDPs (not illustrated), indicating that this potential is, in part, mediated by the activation of voltage-activated sodium channels
Role of voltage-activated conductances in burst firing
To examine the role of calcium currents in the generation of burst firing we replaced extracellular calcium with magnesium. This resulted in a statistically significant (P < 0.05, t test) reduction of the number of action potentials in a burst from, on average, 3.5 ± 1 to 1.5 ± 0.3 (n = 5; Fig. 5A). This action was accompanied by a significant increase in the half-width of the first action potential (0.65 ± 0.04 to 0.83 ± 0.07 ms, P < 0.05, t test), while the peak amplitude and rate of rise of action potentials were unaffected. In common with non-cortical burst firing neurons, we observed that the application of nickel (250 or 500 μm), a concentration range that is required to reduce burst firing in non-cortical neurons (Llinas, 1988; Huguenard, 1996), significantly reduced (P < 0.05, t test) the number of action potentials in a burst or abolished burst generation (3.3 ± 0.3 to 2.0 (250 μm); 3.0 ± 0.7 to 1.0 (500 μm); n = 8; Fig. 5B), without altering the properties of the first action potential of a burst (peak amplitude: 91.1 ± 2.6 to 87.3 ± 0.9 mV; half-width: 0.58 ± 0.03 to 0.6 ± 0.04 ms; P > 0.05, t test). The effects of nominally zero [Ca2+]o and nickel on action potential burst firing were similar for all values of current used to construct current-discharge relationships (not shown).
These results indicate that activation of nickel-sensitive calcium channels is required for the generation of burst firing. Furthermore, they suggest a distribution of labour amongst calcium channels, as the removal of calcium from the extracellular medium both decreased the intensity of burst firing and led to an increase in the duration of somatic action potentials, indicating an involvement of calcium-dependent ion channels in somatic action potential repolarisation. The application of nickel, however, failed to alter somatic action potential duration suggesting that nickel-sensitive calcium channels are not coupled with the currents that repolarise somatic action potentials.
Next we examined the role of dendritic mechanisms in the generation of burst firing as suggested by modelling studies (Rhodes & Gray, 1994; Mainen & Sejnowski, 1996). To address the role of backpropagating action potentials we investigated the effect of the local application of the sodium channel blocker tetrodotoxin (TTX) to the apical dendrite. Pressure ejection of TTX at sites greater than 150 μm from the soma (mean distance 208 ± 8 μm; n = 5) significantly and reversibly reduced (P < 0.05, t test) the number of action potentials composing a burst from 4.0 ± 1.1 to 1.3 ± 0.3 without effecting the threshold or amplitude of the first action potential at the soma (Fig. 5C). Application of TTX to sites more proximal to the soma (mean distance 90 ± 17 μm; n = 5) resulted in blockade of action potential initiation or a change in action potential amplitude and threshold. These data suggest that action potential backpropagation is necessary for the initiation of burst firing. One possibility is that backpropagating action potentials have this effect via activation of dendritic calcium channels (Amitai et al. 1993; Yuste et al. 1994; Markram et al. 1995; Schiller et al. 1997; Stuart et al. 1997). Consistent with this idea, the local application of nickel (1 mm in pipette) to the apical dendrite (> 250 μm from soma) significantly and reversibly reduced (P < 0.05, t test) the number of action potentials composing a burst (3.5 ± 0.4 to 1.6 ± 0.7; n = 5) (Fig. 5D), without influencing the properties of the first action potential of a burst. Application of nickel to the region of the basal dendrites also reduced, although not significantly, the number of action potentials composing a burst (3.5 ± 0.5 to 2.0; n = 3) (not shown). These actions of nickel were not a product of a screening charge effect as the bath application of 1 mm nickel was found to block burst firing without changing the amplitude, time course, rheobase or threshold of the first action potential (n = 5). Similarly in regular firing neurons the properties of action potentials were not altered by 1 mm nickel (n = 3). These results indicate that activation of dendritic nickel-sensitive calcium channels by backpropagation of action potentials into the apical dendritic tree is necessary for the generation of burst firing.
Effect of dendritic depolarisation on burst firing
Simultaneous dendritic and somatic whole-cell recordings (n = 22) were used to examine further the role of dendritic voltage-dependent conductances in the generation of burst firing. In regular firing neurons we observed that current steps injected through the dendritic pipette could evoke at threshold a burst of 2-3 action potentials at low frequency (1st ISI 157 ± 26 Hz; n = 6; Fig. 6A). In some neurons, dendritically evoked burst discharges only occurred following sustained suprathreshold dendritic depolarisation (Fig. 6B). When this was the case a progressive increase in the duration of dendritically recorded action potentials was observed, until the falling shoulder of the dendritic action potential led to the recruitment of a further action potential, thereby generating a low-frequency burst (Fig. 6B, right). Previous studies have shown that the shoulder on the falling phase of backpropagating action potentials is mediated by activation of voltage-activated calcium channels (Kim & Connors, 1993; Stuart et al. 1997). These results, therefore, provide further evidence that dendritic calcium conductances are important for the generation of burst firing. This behaviour was dependent upon the site of dendritic current injection; burst discharges were only evoked in regular firing neurons when dendritic recordings were made more than 150 μm from the soma (247 ± 27 μm; n = 6: no difference in firing pattern was observed for more proximal sites, 132 ± 6 μm; n = 5). Simultaneous dendritic and somatic recordings from intrinsically burst firing neurons (n = 11) revealed that the strength of burst discharges was greater when current was injected through the dendritic pipette (Fig. 7A). In both weak and strong intrinsically bursting neurons the characteristic fading of the burst intensity with time during somatic current injection (Fig. 7A andB, left), was replaced by a time-dependent increase in the strength of burst discharges when evoked by dendritic current injection (Fig. 7A and B, right). This effect was stronger the more distant the dendritic current injection was made from the soma (mean dendritic recording distance 270 ± 35 μm from soma); in a manner similar to regular firing neurons the discharge pattern evoked by somatic or dendritic current injection was similar if the dendritic recording site was < 150 μm from the soma. These results suggest that the slow activation or inactivation of dendritic conductances play a role in the control of burst discharge intensity. In intrinsically bursting neurons where a brief (2 ms) somatic current step, that more closely resembles a synaptic potential, failed to evoke a burst discharges (9 of 35 neurons), a brief dendritic current pulse could generate burst firing (n = 4), further supporting the notion that dendritic depolarisation increases the propensity for the generation of burst firing.
Figure 6. Burst firing is promoted in regular firing neurons by dendritic depolarisation.

A, all traces during simultaneous dendritic and somatic recording, as symbolised in the inset. The threshold discharge response to a somatic current pulse (Iinj) is a single action potential (left), while a burst of action potentials is evoked by threshold dendritic current injection 250 μm from the soma (right). B, somatic current injection results in a train of action potentials typical of a regular firing neuron (left), whereas dendritic current injection 200 μm from the soma evokes a single action potential at threshold; more intense dendritic depolarisation, however, leads to the generation of bursts of two action potentials later in the train (right). Note the progressive increase in the duration of dendritic action potentials during the dendritic current injection (right) is absent during somatic current injection (left). The values of injected current are shown at the left of voltage records.
Figure 7. Burst firing is strengthened by dendritic depolarisation.
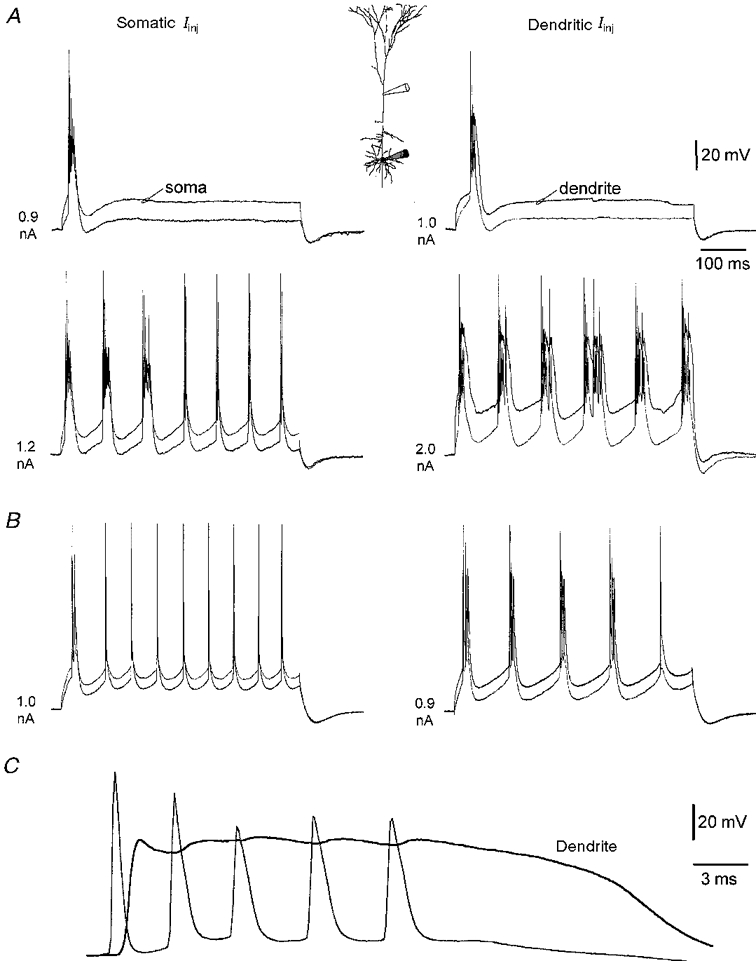
A, all traces during simultaneous dendritic and somatic recording, as represented in the inset. In a strong burst firing neuron, threshold somatic (left) or dendritic (right) current injection leads to a burst discharge. A larger somatic current step evoked a series of burst discharges that become weaker later in the pulse (left), whereas dendritic current injection 300 μm from the soma generates strong burst discharges throughout the duration of the current injection (right). B, in a weak bursting neuron, the transformation of the firing pattern from burst to single action potentials in response to somatic current injection (left) is replaced by the appearance of repeated burst discharges in response to dendritic current injection made 230 μm from the soma (right). C, a burst during dendritic current injection made 480 μm from the soma is shown on an expanded time base, note the presence of a large depolarising envelope in the dendritic recording and that each action potential in the burst is observed to occur first at the somatic recording site.
In all burst firing neurons we observed that each action potential composing a burst was generated first at the somatic recording site (n = 10; Fig. 7C). We also found no difference in the amplitude of the first dendritic action potential in regular and burst firing neurons (regular firing: 69.7 ± 4.8 mV at 195 ± 23 μm, n = 11; burst firing: 66.1 ± 3.4 mV at 213 ± 20 μm, n = 10).
Properties of axonal action potentials during burst firing
Action potentials recorded at the soma during a burst, showed a progressive decrease in amplitude and increase in both rise-time and duration (Fig. 8A). Furthermore, later action potentials in a burst often showed clear inflections on their rising phase (** in Fig. 8A) and could apparently fail (* in Fig. 8A). These findings suggest that during burst firing some action potentials may fail to propagate down the axon and evoke transmitter release. To address this possibility we made simultaneous somatic whole-cell and axonal cell-attached recordings, in order to map the degree of action potential decrement in the axon at different distances from the soma (Fig. 8A). Extracellularly recorded action potentials in these cell-attached patch recordings reflect, predominantly, the first derivative of the intracellular action potential as the vast majority of sodium channels in the patch were inactivated by holding the pipette voltage at approximately -65 mV (i.e. transmembrane potential close to zero) and so may be described as capacitive. In contrast to somatic recordings, we observed that for increasingly more distal axonal recording sites the degree of action potential decrement was reduced and finally abolished (Fig. 8B). In cases where somatic action potentials showed inflections on their rising phase, the first derivative of which had two peaks (not shown), action potentials recorded from axonal sites (< 30 μm from the soma) exhibited two peaks with the first being consistently of the largest amplitude (** in Fig. 8A; n = 3 of 14). On some trials the second component of the somatic action potential apparently failed (* in Fig. 8A), while the axonal action potential was largely unchanged. These results indicate that invasion of axonal action potentials into the soma during burst firing is decremental and can fail. The axonal initiation of all action potentials in a burst was directly confirmed during simultaneous axonal and somatic whole-cell recordings (n = 4), where each action potential arose first in the axon (Fig. 8C). These results confirm the axonal generation of each action potential in a burst discharge and demonstrate that action potentials propagate forward in the axon in a secure manner, but that backpropagation from axon to soma is insecure during high frequency burst discharges, such that action potential propagation into the soma is decremental and may fail.
Postsynaptic consequences of burst firing
In order to ascertain if each action potential in a burst faithfully propagates into axon terminals to release neurotransmitter paired recordings were made from neighbouring synaptically connected layer 5 pyramidal neurons. Figure 9 shows that each action potential in a burst can lead to transmitter release, as was true in all other paired recordings from burst firing neurons (n = 16). The average response over a number of trials was excitatory postsynaptic potentials (EPSPs) or currents (EPSCs) that showed step-wise synaptic depression in 14 of 16 pairs examined (Fig. 9, bottom).
Figure 9. Each action potential in a burst can lead to transmitter release.
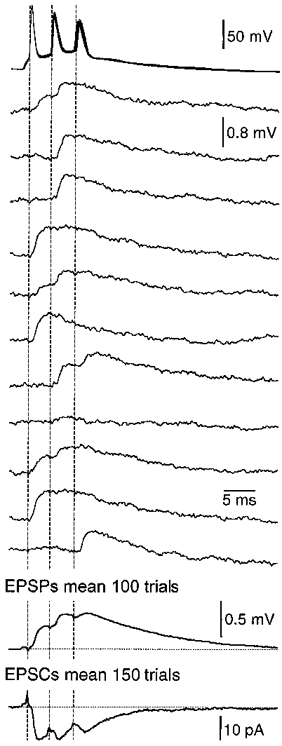
Examples of single sweeps of unitary EPSPs recorded from synaptically coupled layer 5 pyramidal neurons during burst firing. Presynaptic action potentials are shown at the top (11 superposed trials), followed by the evoked postsynaptic potentials. The mean of 100 trails is shown at the bottom together with the mean synaptic current (150 trials). Note that both the mean EPSPs and EPSCs show step-wise use-dependent synaptic depression.
Depression of synaptic transmission between neocortical pyramidal neurons during action potential trains has been shown to be frequency dependent over a range of 10-200 Hz, and dependent upon pre- and postsynaptic mechanisms (Markram et al. 1997; Thomson, 1997). During a high frequency burst of action potentials we show that this use-dependent depression is compensated for by interactions with postsynaptic voltage-gated ion channels, leading to facilitation of EPSPs at membrane potentials near firing threshold (Fig. 10). The amount of amplification at depolarised potentials depended upon the number of action potentials in the burst, with bursts containing more action potentials leading to larger postsynaptic responses due to greater amplification and temporal summation of later EPSPs (Fig. 10).
Figure 10. The postsynaptic consequence of varying the membrane potential and the number of action potentials in a burst.
Overlayed mean (n = 50 trials) traces of EPSPs recorded at the indicated holding potentials, in response to one, two and three action potentials. Arrows indicate the peak of the first and second EPSP at the most depolarised holding potential. The number of action potentials evoked in a burst was controlled by applying positive- followed by negative-current sequences to the presynaptic neuron.
Comparison of the amplitude of the second EPSP in an action potential burst with that of the first shows that at hyperpolarised membrane potentials the second EPSP is depressed, while at near firing threshold the second EPSP is preferentially amplified (Fig. 11A and B). This voltage-dependent relationship was quantified in 14 paired recordings where the amplitude of the second EPSP of a burst was found to exceed the first when generated from membrane potentials positive to -60 mV (Fig. 11A and B). Previous studies have shown that EPSPs can be amplified at depolarised membrane potentials by voltage-activated sodium channels (Stuart & Sakmann, 1995; Markram et al. 1997). To investigate whether the same conductance is involved in the amplification of later EPSPs during a burst, the EPSC generated during a burst was injected into the soma of layer 5 pyramidal neurons during simultaneous somatic recordings (Fig. 11C), where one pipette acted as a current source and the other was used for voltage measurements, obviating bridge balancing artifacts (Stuart & Sakmann, 1995). Amplification of these simulated EPSPs at depolarised membrane potentials was similar to that observed for synaptic EPSPs (compare Fig. 11A and B with 11C and D), and was blocked by bath application of TTX (Fig. 11C and D). Comparison of the amplitude of the second simulated EPSP with the first indicates that at depolarised membrane potentials the second EPSP was preferentially amplified by a TTX-sensitive mechanism (Fig. 11D). These results show that despite significant use-dependent depression of the synaptic current between layer 5 neurons during a burst, the postsynaptic response is facilitated near action potential threshold due to the activation of voltage-activated sodium channels.
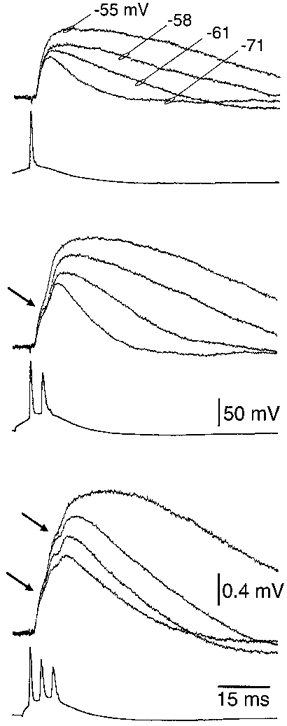
Figure 11. Postsynaptic amplification of burst discharges.

A, superposed consecutive traces (n = 10) demonstrating the amplification of the second EPSP in a burst when evoked at near threshold membrane potentials (-55 mV), and its depression at more negative holding potentials (-75 mV). The lower action potential waveform is the mean of 20 trials. B, scatter-plot demonstrating the voltage dependence of the amplification of the second EPSP in a burst. Each point represents mean (n = 30-50) EPSP amplitude at a single holding potential, where EPSP1 is the amplitude of the first EPSP in the burst and EPSP2 is the peak amplitude of the response to a burst of two action potentials. Data pooled from 12 paired recordings. C, amplification of later EPSPs in a burst is tetrodotoxin sensitive. Simulated EPSPs produced by somatic current injection of the mean synaptic current generated during a burst (derived from the cell illustrated in A). The voltage dependence of simulated EPSP amplification is shown in the upper superposed traces at the indicated holding potentials. The addition of TTX (1 μm) abolished amplification (lower superposed traces). D, scatter plot demonstrating the pooled voltage dependence of amplification of the second simulated EPSP in a burst in control (n = 7) and in TTX (n = 5).
DISCUSSION
We show that the recruitment of dendritic nickel-sensitive calcium channels by backpropagating action potentials underlies burst firing in layer 5 pyramidal neurons. Each action potential of a burst could lead to transmitter release, giving rise to a series of excitatory postsynaptic currents that showed marked depression. A postsynaptic amplification process, however, produced facilitation of EPSPs near firing threshold leading to significant temporal summation. These findings reveal unique cellular mechanisms that will enhance synchronisation between cortical neurons during burst firing.
Cellular mechanism of burst firing
Our results provide direct experimental evidence for the following scheme for the generation and termination of burst discharges in layer 5 cortical pyramidal neurons. The initiation of an action potential in the axon backpropagates into the dendrites of layer 5 neurons leading to a regenerative sequence that involves the recruitment of dendritic nickel-sensitive calcium channels. The ability of backpropagating action potentials to evoke dendritic calcium electrogenesis can be facilitated in regular and burst-firing neurons by direct dendritic current injection. This facilitated interaction between backpropagating action potentials and dendritic calcium channel activation may be simply a consequence of dendritic depolarisation increasing the likelihood of backpropagating action potential achieving the activation threshold for dendritic calcium channels. In addition, in the distal apical dendrite depolarisation may increase the amplitude of the backpropagating action potential (Magee & Johnston, 1997), as a consequence of increased dendritic sodium channel activation, that in turn will increase the likelihood of calcium channel activation. Feedback of dendritic depolarisation to the soma will be supplemented by capacitive discharge of the dendritic membrane (Nelson & Burke, 1967; Mainen & Sejnowski, 1996), and possibly the activation of the persistent sodium current (Franceschetti et al. 1995; Mantegazza et al. 1998), but is checked by activation of voltage- and calcium-dependent potassium currents (Schwindt et al. 1988; Silva et al. 1991; Friedman & Gutnick, 1992). Only if the net depolarisation at the soma and axon is sufficient, will a further axonal action potential be initiated forming a burst discharge.
Previous studies have indicated a primary role of the persistent sodium current in the genesis of burst firing in layer 5 pyramidal neurons (Franceschetti et al. 1995; Azouz et al. 1996; Mantegazza et al. 1998). This conclusion was based on a difference in the time course for blockade of somatic action potentials and burst discharges during bath application of TTX. This observation can be explained, by our scheme, if the amplitude of backpropagating dendritic action potentials is more sensitive to this form of TTX application than are somatic action potentials. Given the presumed high density of sodium channels in the axon relative to that in the soma and dendrites (Stuart et al. 1997), such a location-specific difference in the sensitivity of somatic and dendritic action potential amplitude to bath application of TTX would be expected (S. R. Williams & G. J. Stuart, unpublished observations) and has been directly demonstrated in cultured cortical neurons (Mackenzie & Murphy, 1998).
Our data directly show a critical role for dendritic nickel-sensitive calcium channels in the generation of burst firing. Both calcium imaging and electrophysiological evidence suggests the presence of nickel-sensitive calcium channels in the apical dendrites of both layer 5 and hippocampal pyramidal neurons (Markram & Sakmann, 1994; Johnston et al. 1996). Furthermore, [Ca2+]i measurements in vivo indicate that burst firing in layer 5 neurons is associated with a large calcium transient in the apical dendrites, but not in the apical tuft (Helmchen et al. 1998).
The above scheme of burst firing stresses the importance of activation of dendritic voltage-activated channels in the initiation and control of burst firing. Previous observations from layer 5 neocortical and CA1 pyramidal neurons have demonstrated that direct or indirect depolarisation of apical dendrites can transform the firing pattern of a neuron from single action potentials to a burst discharge firing pattern (Wong & Stewart, 1992; Schwindt & Crill, 1997). We show here, that if action potential firing is triggered by direct apical dendritic current injection made at a sufficient distance from the soma, then the generation of burst discharges is the inevitable consequence. In regular firing neurons, dendritically evoked burst discharges were found to evolve over time, as a consequence of the broadening of backpropagating action potentials. Similarly in burst firing neurons, the characteristic weakening of burst discharges with time when evoked with long somatic current steps was replaced by a progressive augmentation of burst firing intensity during dendritic current injection. We suggest that this results from the progressive inactivation of a dendritically located potassium current. In support of this idea the apical dendrites of layer 5 pyramidal neurons are known to contain a high density of slowly inactivating, but not fast inactivating, potassium channels (Bekkers & Stuart, 1998). These results indicate that under physiological conditions synaptically mediated depolarisation of apical dendrites of layer 5 pyramidal neurons will facilitate the generation of burst discharges, as has recently been observed (Larkum et al. 1999).
It has been suggested that the difference in the firing pattern between layer 5 pyramidal neurons may result from either a difference in the ability of action potentials to backpropagate into the dendrites of these neurons (Kim & Connors, 1993), and/or a difference in the degree of electrical coupling between somatic and apical dendritic compartments (Mainen & Sejnowski, 1996). The amplitude of backpropagating action potentials were, however, found to be similar in regular and burst firing neurons. These data suggest that the degree of electrical coupling between soma and dendrites may be similar between these classes of neurons. A detailed analysis of the passive coupling between soma and dendrites is, however, required to clarify this point. Furthermore, it remains to be established if an increased density of dendritic calcium and/or a decreased density of opposing potassium channels contributes to the difference in firing pattern of layer 5 pyramidal neurons.
Axonal action potential generation
Simultaneous axonal and somatic recordings demonstrated that each action potential composing a burst discharge is axonally generated, indicating that the depolarisation initiated by dendritic calcium channels propagates to the axon to evoke further action potential initiation. When recorded from the soma, a progressive decrease in action potential amplitude was observed during a burst discharge, as has been shown for other burst firing neurons (Llinas, 1988), and occasionally action potentials appeared to fail at the somatic level. Such a decrement in action potential amplitude was not observed in cell-attached recordings made at axonal recording sites > 30 μm from the soma. This result shows that action potential backpropagation from the axon to the soma is decremental and in some cases fails to evoke a somato-dendritic spike during high frequency burst firing. This phenomena is best explained by an accumulation of somato-dendritic sodium channel inactivation (Colbert et al. 1997; Jung et al. 1997). These findings may suggest that the properties of distal axonal sodium channels are distinct, and that these channels may fail to enter, or enter for briefer periods, states of inactivation and so be available for activation at higher frequencies. These data are also supportive of a high density of sodium channels at relatively distal axonal sites, with a lower sodium channel density in the proximal axon initial segment and soma (Colbert & Johnston, 1996). The large reserve of sodium channels at more distal axonal sites, therefore, not only provides the focus for action potential initiation (Stuart et al. 1997), but also allows initiation to be repeated at high frequencies.
Postsynaptic consequences of burst firing
The postsynaptic consequence of neurotransmitter release during a burst discharge was found to be dominated by a step-wise depression of EPSC amplitude. Despite this synaptic depression, a postsynaptic voltage-dependent amplification process ensured that later EPSPs in a burst were amplified at membrane potentials near firing threshold. This amplification mechanism, therefore, converts synaptic depression to facilitation, thereby producing significant temporal summation of EPSPs near firing threshold and so increasing the likelihood of action potential initiation in the postsynaptic neuron in response to a presynaptic burst discharge. The TTX sensitivity and voltage dependence of this amplification mechanism indicates the involvement of voltage-activated sodium channels of the persistent type (Crill, 1996). We suggest that later EPSPs in a burst are preferentially amplified as the temporal summation of EPSPs allows later EPSPs in a burst to attain the voltage threshold for sodium channel activation. Furthermore, the summation of EPSPs will also provide a depolarising input that lasts longer than a single EPSP and so is more likely to initiate feedforward amplification due to the progressive recruitment of sodium channels. This mechanism may operate physiologically, as the membrane potential of cortical pyramidal neurons in vivo is maintained at potentials near firing threshold (Pare et al. 1998). We suggest that the increased likelihood of action potential initiation produced by this amplification process may underlie the critical informational role of burst firing in the cortex (Lisman, 1997; Snider et al. 1998) despite significant synaptic depression at this distributed synapse. A dynamic coupling of pre- and postsynaptic amplification mechanisms therefore enhances the strength of synaptic transmission between layer 5 pyramidal neurons during action potential burst firing, providing a physiological basis for the synchronisation of large assemblies of these neurons in vivo.
Acknowledgments
This work was supported by the Wellcome Trust and the Human Frontiers Science Program.
References
- Amitai Y, Friedman A, Connors BW, Gutnick MJ. Regenerative activity in apical dendrites of pyramidal cells in neocortex. Cerebral Cortex. 1993;3:26–38. doi: 10.1093/cercor/3.1.26. [DOI] [PubMed] [Google Scholar]
- Azouz R, Jensen MS, Yaari Y. Ionic basis of spike after-depolarization and burst generation in adult rat hippocampal CA1 pyramidal cells. The Journal of Physiology. 1996;492:211–223. doi: 10.1113/jphysiol.1996.sp021302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball GJ, Gloor P, Schaul N. The cortical electromicrophysiology of pathological delta waves in the electroencephalogram of cats. Electroencephalography and Clinical Neurophysiology. 1977;43:346–361. doi: 10.1016/0013-4694(77)90258-9. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stuart G. Distribution and properties of potassium channels in the soma and apical dendrites of layer 5 cortical pyramidal neurons. Society for Neuroscience Abstracts. 1998;24:2019–807.8. [Google Scholar]
- Chagnac-Amitai Y, Luhmann HJ, Prince DA. Burst generating and regular spiking layer 5 pyramidal neurons of rat neocortex have different morphological features. Journal of Comparative Neurology. 1990;296:598–613. doi: 10.1002/cne.902960407. [DOI] [PubMed] [Google Scholar]
- Colbert CM, Johnston D. Axonal action-potential initiation and Na+ channel densities in the soma and axon initial segment of subicular pyramidal neurons. Journal of Neuroscience. 1996;16:6676–6686. doi: 10.1523/JNEUROSCI.16-21-06676.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. Journal of Neuroscience. 1997;17:6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connors BW. Initiation of synchronized neuronal bursting in neocortex. Nature. 1984;310:685–687. doi: 10.1038/310685a0. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ. Intrinsic firing patterns of diverse neocortical neurons. Trends in Neurosciences. 1990;13:99–104. doi: 10.1016/0166-2236(90)90185-d. [DOI] [PubMed] [Google Scholar]
- Connors BW, Gutnick MJ, Prince DA. Electrophysiological properties of neocortical neurons in vitro. Journal of Neurophysiology. 1982;48:1302–1320. doi: 10.1152/jn.1982.48.6.1302. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annual Review of Physiology. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Evarts EV. Temporal patterns of discharge of pyramidal tract neurons during sleep and waking in the monkey. Journal of Neurophysiology. 1964;27:152–171. doi: 10.1152/jn.1964.27.2.152. [DOI] [PubMed] [Google Scholar]
- Flint AC, Maisch US, Kriegstein AR. Postnatal development of low [Mg2+] oscillations in neocortex. Journal of Neurophysiology. 1997;78:1990–1996. doi: 10.1152/jn.1997.78.4.1990. [DOI] [PubMed] [Google Scholar]
- Franceschetti S, Guatteo E, Panzica F, Sancini G, Wanke E, Avanzini G. Ionic mechanisms underlying burst firing in pyramidal neurons: intracellular study in rat sensorimotor cortex. Brain Research. 1995;696:127–139. doi: 10.1016/0006-8993(95)00807-3. [DOI] [PubMed] [Google Scholar]
- Friedman A, Gutnick MJ. Intracellular calcium and control of burst generation in neurons in guinea-pig neocortex in vitro. European Journal of Neuroscience. 1992;1:374–381. doi: 10.1111/j.1460-9568.1989.tb00802.x. [DOI] [PubMed] [Google Scholar]
- Helmchen F, Svoboda K, Denk K, Tank DW. Dendritic calcium dynamics in deep neocortical pyramidal neurons measured in vivo using two-photon microscopy. Society for Neuroscience Abstracts. 1998;24:79–37.6. [Google Scholar]
- Huguenard JR. Low-threshold calcium currents in central nervous system neurons. Annual Review of Physiology. 1996;58:329–348. doi: 10.1146/annurev.ph.58.030196.001553. [DOI] [PubMed] [Google Scholar]
- Johnston D, Magee JC, Colbert CM, Cristie BR. Active properties of neuronal dendrites. Annual Review of Neuroscience. 1996;19:165–186. doi: 10.1146/annurev.ne.19.030196.001121. [DOI] [PubMed] [Google Scholar]
- Jones RSG, Heinemann U. Synaptic and intrinsic responses of medical entorhinal cortical cells in normal and magnesium-free medium in vitro. Journal of Neurophysiology. 1988;59:1476–1496. doi: 10.1152/jn.1988.59.5.1476. [DOI] [PubMed] [Google Scholar]
- Jung HY, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. Journal of Neuroscience. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasper EM, Larkman AU, Lubke J, Blakemore C. Pyramidal neurons in layer 5 of the rat visual cortex. I. Correlation among cell morphology, intrinsic electrophysiological properties, and axon targets. Journal of Comparative Neurology. 1994;339:459–474. doi: 10.1002/cne.903390402. [DOI] [PubMed] [Google Scholar]
- Kim HG, Connors BW. Apical dendrites of the neocortex: correlation between sodium- and calcium-dependent spiking and pyramidal cell morphology. Journal of Neuroscience. 1993;13:5301–5311. doi: 10.1523/JNEUROSCI.13-12-05301.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkman A, Mason A. Correlations between morphology and electrophysiology of pyramidal neurons in slices of rat visual cortex. II Electrophysiology. Journal of Neuroscience. 1990;10:1415–1428. doi: 10.1523/JNEUROSCI.10-05-01415.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkum ME, Zhu JJ, Sakmann B. A new cellular mechanism for coupling inputs arriving at different cortical layers. Nature. 1999;398:338–341. doi: 10.1038/18686. [DOI] [PubMed] [Google Scholar]
- Lisman JE. Bursts as a unit of neural information: making unreliable synapses reliable. Trends in Neurosciences. 1997;20:38–43. doi: 10.1016/S0166-2236(96)10070-9. [DOI] [PubMed] [Google Scholar]
- Llinas R. The intrinsic electrophysiological properties of mammalian neurons insights into central nervous system function. Science. 1988;242:1654–1664. doi: 10.1126/science.3059497. [DOI] [PubMed] [Google Scholar]
- Llinas R, Sugimori M. Electrophysiological properties of in vitro Purkinje cell dendrites in mammalian cerebellar slices. The Journal of Physiology. 1980;305:197–213. doi: 10.1113/jphysiol.1980.sp013358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick DA, Connors BW, Lighthall JW, Prince DA. Comparative electrophysiology of pyramidal and sparsely spiny stellate neurons of the neocortex. Journal of Neurophysiology. 1985;54:782–806. doi: 10.1152/jn.1985.54.4.782. [DOI] [PubMed] [Google Scholar]
- Mackenzie PJ, Murphy TH. High safety factor for action potential conduction along axons but not dendrites of cultured hippocampal and cortical neurons. Journal of Neurophysiology. 1998;80:2089–2101. doi: 10.1152/jn.1998.80.4.2089. [DOI] [PubMed] [Google Scholar]
- Magee JC, Johnston D. A synaptically controlled, associative signal for Hebbian plasticity in hippocampal neurons. Science. 1997;275:209–213. doi: 10.1126/science.275.5297.209. [DOI] [PubMed] [Google Scholar]
- Mainen ZF, Sejnowski TJ. Influence of dendritic structure on firing pattern in model neocortical neurons. Nature. 1996;382:363–366. doi: 10.1038/382363a0. [DOI] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G. Anemone toxin (ATX II)-induced increase in persistent sodium current: effects on the firing properties of rat neocortical pyramidal neurones. The Journal of Physiology. 1998;507:105–116. doi: 10.1111/j.1469-7793.1998.105bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Helm PJ, Sakmann B. Dendritic calcium transients evoked by single back-propagating action potentials in rat neocortical pyramidal neurons. The Journal of Physiology. 1995;485:1–20. doi: 10.1113/jphysiol.1995.sp020708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Lubke J, Frotscher M, Roth A, Sakmann B. Physiology and anatomy of synaptic connections between thick tufted pyramidal neurones in the developing rat neocortex. The Journal of Physiology. 1997;500:409–440. doi: 10.1113/jphysiol.1997.sp022031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markram H, Sakmann B. Calcium transients in dendrites of neocortical neurons evoked by single subthreshold excitatory postsynaptic potentials via low-voltage- activated calcium channels. Proceedings of the National Academy of Sciences of the USA. 1994;91:5207–5211. doi: 10.1073/pnas.91.11.5207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson PG, Burke RE. Delayed depolarization in cat spinal motoneurons. Experimental Neurology. 1967;17:16–26. doi: 10.1016/0014-4886(67)90118-5. [DOI] [PubMed] [Google Scholar]
- Ohana O, Sakmann B. Transmitter release modulation in nerve terminals of rat neocortical pyramidal cells by intracellular calcium buffers. The Journal of Physiology. 1998;513:135–148. doi: 10.1111/j.1469-7793.1998.135by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pare D, Shink E, Gaudreau H, Destexhe A, Lang EJ. Impact of spontaneous synaptic activity on the resting properties of cat neocortical pyramidal neurons in vivo. Journal of Neurophysiology. 1998;79:1450–1460. doi: 10.1152/jn.1998.79.3.1450. [DOI] [PubMed] [Google Scholar]
- Rhodes PA, Gray CM. Simulations of intrinsically bursting neocortical pyramidal neurones. Neural Computation. 1994;6:1086–1110. [Google Scholar]
- Schiller J, Schiller Y, Stuart G, Sakmann B. Calcium action potentials restricted to distal apical dendrites of rat neocortical pyramidal neurons. The Journal of Physiology. 1997;505:605–616. doi: 10.1111/j.1469-7793.1997.605ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Local and propagated dendritic action potentials evoked by glutamate iontophoresis on rat neocortical pyramidal neurons. Journal of Neurophysiology. 1997;77:2466–2483. doi: 10.1152/jn.1997.77.5.2466. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Spain WJ, Foehring RC, Stafstrom CE, Chubb MC, Crill WE. Multiple potassium conductances and their functions in neurons from cat sensorimotor cortex in vitro. Journal of Neurophysiology. 1988;59:424–449. doi: 10.1152/jn.1988.59.2.424. [DOI] [PubMed] [Google Scholar]
- Silva LR, Amitai Y, Connors BW. Intrinsic oscillations of neocortex generated by layer 5 pyramidal neurons. Science. 1991;251:432–435. doi: 10.1126/science.1824881. [DOI] [PubMed] [Google Scholar]
- Snider RK, Kabara JF, Roig BR, Bonds AB. Burst firing and modulation of functional connectivity in cat striate cortex. Journal of Neurophysiology. 1998;80:730–744. doi: 10.1152/jn.1998.80.2.730. [DOI] [PubMed] [Google Scholar]
- Stuart G, Sakmann B. Amplification of EPSPs by axosomatic sodium channels in neocortical pyramidal neurons. Neuron. 1995;15:1065–1076. doi: 10.1016/0896-6273(95)90095-0. [DOI] [PubMed] [Google Scholar]
- Stuart G, Schiller J, Sakmann B. Action potential initiation and propagation in rat neocortical pyramidal neurons. The Journal of Physiology. 1997;505:617–632. doi: 10.1111/j.1469-7793.1997.617ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart G, Spruston N, Sakmann B, Hausser M. Action potential initiation and backpropagation in neurons of the mammalian CNS. Trends in Neurosciences. 1997;20:125–131. doi: 10.1016/s0166-2236(96)10075-8. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Activity-dependent properties of synaptic transmission at two classes of connections made by rat neocortical pyramidal axons in vitro. The Journal of Physiology. 1997;502:131–147. doi: 10.1111/j.1469-7793.1997.131bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, McCormick DA. Control of firing mode of corticotectal and corticopontine layer V burst-generating neurons by norepinephrine, acetylcholine, and 1S,3R-ACPD. Journal of Neuroscience. 1993;13:2199–2216. doi: 10.1523/JNEUROSCI.13-05-02199.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams SR, Hendry I, Stuart G. Mechanisms of action potential burst firing in layer 5 neocortical neurons. Society for Neuroscience Abstracts. 1998;24:2018–807.3. [Google Scholar]
- Wong RK, Prince DA. Participation of calcium spikes during intrinsic burst firing in hippocampal neurons. Brain Research. 1978;159:385–390. doi: 10.1016/0006-8993(78)90544-9. [DOI] [PubMed] [Google Scholar]
- Wong RK, Stewart M. Different firing patterns generated in dendrites and somata of CA1 pyramidal neurones in guinea-pig hippocampus. The Journal of Physiology. 1992;457:675–687. doi: 10.1113/jphysiol.1992.sp019401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuste R, Gutnick MJ, Saar D, Delaney KR, Tank DW. Ca2+ accumulations in dendrites of neocortical pyramidal neurons: an apical band and evidence for two functional compartments. Neuron. 1994;13:23–43. doi: 10.1016/0896-6273(94)90457-x. [DOI] [PubMed] [Google Scholar]