

Abstract
We have investigated the effects of inflammatory mediators on visceral afferent discharge and afferent responses to bradykinin (BK) in rat jejunum using a novel in vitro technique.
Prostaglandin E2 (1 μm) augmented responses to BK without affecting basal firing, while histamine (100 μm) and adenosine (100 μm) activated basal discharge and enhanced BK responses. In contrast, 5-HT (100 μm) increased basal discharge without influencing responses to BK.
Afferent discharge induced by histamine was inhibited by both H1 (pyrilamine) and H3 (thioperamide) but not H2 (ranitidine) receptor antagonists at 10 μm. In contrast, sensitization to BK induced by histamine was inhibited by ranitidine (10 μm).
Afferent discharge induced by adenosine was blocked by the A1 receptor antagonist DPCPX (10 μm) but remained unaffected by A2A receptor blockade with ZM241385 (10 μm). In contrast, sensitization of BK responses by adenosine was unaffected by both antagonists. Basal discharge and BK-induced responses were unaffected by the A3 receptor agonist IB-MECA (1 μm). While involvement of A2B receptors is not excluded, adenosine may activate afferent discharge through A1 receptors, while sensitization to BK could involve a receptor other than A1, A2A or A3, possibly the A2B receptor.
Inhibition of cyclo-oxygenase with naproxen (10 μm) prevented sensitization after histamine but not adenosine.
Sensitization was mimicked by dibutyryl cAMP. This occurred without changes in basal firing and was unaffected by naproxen.
In conclusion, afferent discharge induced by BK is augmented by histamine, adenosine and PGE2, but not by 5-HT. Evidence suggests that sensitization involves separate mechanisms from afferent activation. Sensitization may be mediated by increases in cAMP following direct activation by mediators at the nerve terminal or through indirect pathways such as the release of prostaglandins.
The gastrointestinal tract has an extensive intrinsic and extrinsic sensory innervation. Despite this, stimuli in the healthy gastrointestinal tract rarely reach the level of conscious perception. In contrast, sensations of abdominal discomfort and pain are common symptoms in patients with gastrointestinal disease. Symptoms include heartburn, chest pain, dyspepsia, bloating, abdominal cramps and feelings of incomplete rectal evacuation, all of which can arise in both organic (e.g. inflammatory) and functional disorders such as irritable bowel syndrome. Visceral afferent hypersensitivity is now a widely accepted mechanism which could explain many of these clinical symptoms associated with functional bowel disease and inflammatory diseases of the gut (Mayer & Raybould, 1990; Bueno et al. 1997). However, the mechanisms underlying peripheral sensitization of gastrointestinal afferents is poorly understood.
Much of our understanding of the mechanisms of afferent sensitization and the modulation of painful stimuli has stemmed from studies of cutaneous pain. Sensitization of cutaneous nociceptors is thought to underlie conditions of hyperalgesia (enhanced perception of pain) and allodynia where previously non-noxious stimuli can produce pain (Heller et al. 1993). Despite the lack of detailed studies on gastrointestinal afferent sensitivity, it is generally assumed that intestinal afferents behave in a similar way, being activated and/or sensitized by chemical mediators present within an ‘inflammatory soup’. Gastrointestinal afferents terminate at different levels within the gut wall (namely mucosal, muscle and serosal afferents), and reach the CNS via either vagal or spinal pathways. Evidence suggests that the spinal afferent endings in the serosa and mesentery, which have high thresholds for mechanical stimulation, could also serve a nociceptive function (Jaenig & Morrison, 1986; Ness & Gebhart, 1990). Sensitization of these spinal fibres could therefore lead to an altered perception of visceral stimuli.
Bradykinin (BK) is a pain-producing peptide generated in tissues and plasma following tissue damage or inflammation (for review see Regoli & Barabe, 1980) and can be both an algesic and a hyperalgesic agent, stimulating and sensitizing C and Aδ fibres that encode noxious stimuli (Szolscanyi, 1987). In addition, BK stimulates afferents within the gastrointestinal tract (Longhurst et al. 1984; Pan et al. 1994), is implicated in activation of abdominal visceral afferents during ischaemia (Longhurst & Dittman, 1987) and may play an important role in inflammatory bowel disease where plasma levels appear to correlate well with the onset of gastrointestinal symptoms (Cuschieri & Onabanjo, 1971). Other inflammatory mediators, particularly prostaglandins, have been shown to enhance afferent responses to noxious stimuli such as BK (Handwerker & Reeh, 1991; Nicol & Cui, 1994). We have recently characterized the action of BK on serosal afferents from rat jejunum using a novel in vitro model (Maubach & Grundy, 1999) and demonstrated the involvement of prostaglandins in this response. This simple preparation was designed to examine the sensitivity of serosal afferents more directly in the absence of the main body of the jejunum (mucosa, submucosa and muscle layers), thus minimizing the potential for secondary activation of the afferents. The aim of this current study was to investigate the sensitivity of visceral afferents to a variety of potential mediators of inflammation and to examine whether these afferents show sensitization. We have therefore examined the effects of chemical mediators on afferent discharge and explored any possible interaction between these agents and the response to BK.
METHODS
Tissue preparation
Male hooded Lister rats (350-400 g) were overdosed with urethane (1.5 g kg−1) and a mid-line laparotomy performed. A 3 cm-long piece of jejunum complete with mesenteric attachment was then carefully excised. This segment was placed in a Sylgard-lined perfusion chamber and the mesenteric arcade pulled through a small aperture into a separate recording chamber. The mesentery was then dissected free from the main body of the jejunum (which was discarded) and this mesenteric attachment pinned out. Once the aperture had been sealed with vaseline the recording chamber was filled with colourless heavy liquid paraffin (pre-warmed to 34°C) and the perfusion chamber superfused at 10 ml min−1 with bicarbonate buffer (composition (mm): Na+ 143.5, K+ 5.9, Cl− 126, Ca2+ 2.5, Mg2+ 1.2, H2PO4 1.2, SO4 1.2, HCO−3 25, glucose 10 and sodium butyrate 1, pH maintained at 7.4 with 95 % O2-5 % CO2), pre-warmed to yield a chamber temperature of 34°C. A separate preparation was used for each experiment.
Nerve recording
Under a stereomicroscope, a paravascular nerve bundle was teased out from the mesenteric arcade and wrapped around one arm of a bipolar platinum recording electrode with a length of connective tissue attached to the second indifferent electrode. The electrodes were connected to a Neurolog headstage (NL 100), and the signal amplified (NL 104, × 20 000) and filtered (NL 125; band width, 100-1000 Hz) then relayed to a spike processor (Digitimer D130) to allow discrimination of action potentials from noise using a manually set amplitude and polarity window. The whole-nerve recording was displayed on a storage oscilloscope (Tektronix 5111A) and digitized (PCM-2 A/D VCR adapter, Medical Systems Corp.) to allow recording on VHS videotape for future off-line analysis. Whole-nerve activity was continuously monitored as spike discharge (impulses (1.25 s)−1) and stored on a PC using Spike2 software (CED).
Experimental protocols
Experiments were performed on preparations in which baseline afferent discharge was maintained and in which robust responses to a priming exposure to a submaximal concentration of BK (1 μm, 2 min; see Maubach & Grundy, 1999, for concentration-reponse data) could be evoked. Once discharge had returned to basal level, the preparation was allowed to stabilize for a further 15 min. Subsequent responses to BK (1 μm, 2 min) were then quantified before and 30 min after a period during which the perfusion fluid was recirculated (reperfusion period) using 60 ml of either buffer alone or buffer containing one of several chemical mediators (see Fig. 1). The first response is referred to as the control response while the second response in the presence of mediator could be compared directly to this control. Reperfusion with buffer alone served as a time control for statistical comparison. This protocol of priming challenge followed after 15 min by a control response and a second challenge after 30 min reperfusion proved reproducible, such that the second response to BK (after reperfusion) was very similar to the control response. Thus, having established this time control, mediators could be tested within the 30 min reperfusion period. The effects of the following mediators were examined: prostaglandin E2 (1 μm), histamine (100 μm), adenosine (100 μm), 5-HT (100 μm) and dibutyryl cAMP (100 μm). These concentrations were chosen following results from pilot studies and from consideration of sensitization studies in somatic nociceptors in vitro, where similar agents are often used as components of an ‘inflammatory soup’. In receptor antagonist studies and studies with naproxen, these agents were added after the priming challenge to BK and were present throughout the experiment thereafter. The response in the presence of antagonist alone (first response) could therefore be directly compared to the response after a 30 min reperfusion with antagonist together with the chemical mediator. In addition, this allowed direct comparison between BK responses without the need for extra vehicle controls. In studies with naproxen, naproxen was added after the priming challenge and left for 30 min before the control BK response was obtained. BK was then re-applied after the 30 min reperfusion period during which naproxen was still present together with the chemical mediator concerned (Fig. 4). Thus naproxen was present throughout the experiment following the priming challenge. This acted as a new time control to examine whether mediators act indirectly through the generation of prostanoids. The effects of naproxen on BK-induced discharge per se were not under investigation.
Figure 1. A representative trace of afferent discharge induced by 1 μm BK before and after a 30 min reperfusion period.
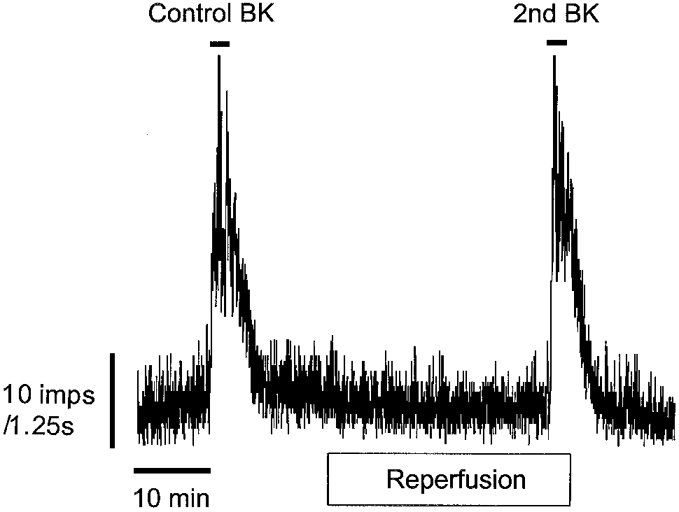
A ‘priming’ challenge with 1 μm BK was first added (not shown) and followed after 15 min of stable discharge by a second application (Control BK). BK was added again 30 min later within a fixed volume of the reperfusing solution (2nd BK).
Figure 4. Illustration of the protocol used to investigate the effects of naproxen.
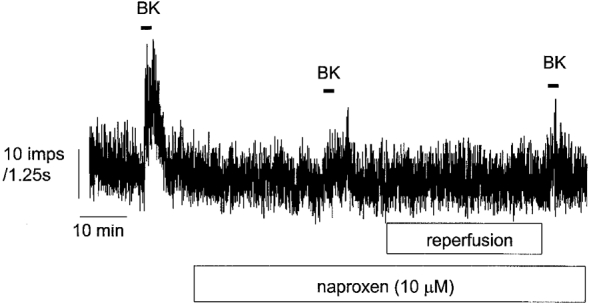
Naproxen (10 μm) was added after an initial challenge with BK and was present throughout the experiment. After 30 min, BK was reapplied followed by another addition of BK after a 30 min reperfusion period where mediators could be tested. Note that the two responses to BK in the presence of naproxen before and after reperfusion were similar following this protocol.
Drugs
The following compounds were purchased from Sigma: bradykinin (BK), prostaglandin E2 (PGE2), histamine, adenosine, 5-HT (5-hydroxytryptamine), dibutyryl cAMP (N6,2′-O-dibutyryladenosine 3′,5′-cyclic monophosphate), pyrilamine and ranitidine. DPCPX (1,3-dipropyl-8-cyclopentylxanthine) was purchased from Research Biochemicals International. ZM241385 (4-(2-[7-amino-2-(2-furyl)[1,2,4]triazolo[2,3-a][1,3,5]triazin-5-yl amino]ethyl)phenol), IB-MECA (1-deoxy-1-[6-[[(3-iodophenyl)methyl]amino]-9H-purin-9-yl]-N-methyl-β-D-ribofuranuronamide) and thioperamide were purchased from Tocris Cookson Ltd. All drug stock solutions were made in distilled water unless otherwise stated. PGE2 was dissolved in 0.1 ml absolute ethanol + 0.9 ml sodium bicarbonate (2 % w/w) giving a stock solution of 1 mg ml−1. ZM241385 was dissolved in DMSO and DPCPX was dissolved in ethanol, giving final concentrations of 0.1 and 0.01 % v/v of vehicle, respectively, in perfusion fluids.
Data analysis
BK responses are expressed as total number of impulses calculated as the number of impulses for the entire response (area under response profile) minus the number during an equal period of basal activity. These raw data are given in the text. Peak discharge frequency induced by BK was calculated as the maximum level of discharge after subtraction of the baseline discharge (impulses s−1). Mediator-induced changes in basal discharge were calculated as the mean number of impulses in 3 min following application minus mean basal activity in 3 min prior to application of the mediator, and expressed in impulses per second. Due to differences in the number of fibres in each afferent bundle and the consequent variability in baseline firing and response magnitude, afferent responses to BK were also expressed as a percentage of the control response (i.e. the second response to BK expressed as a percentage of the control response). Some data are also presented as percentage change. The change in afferent discharge upon addition of mediator or vehicle was expressed as a percentage of baseline. The increase in activity induced by BK was also expressed as a percentage of basal discharge prior to addition. Unless otherwise stated data are presented as means ± standard error of the mean (s.e.m.); n, number of experiments. Paired t tests were used to compare control and second BK responses where data were normally distributed, otherwise paired comparisons were made using the Wilcoxon signed rank test. Data involving multiple comparisons to control were analysed using Dunn's multiple comparison method following a Kruskall-Wallis one-way analysis of variance on ranks or Bonferroni's multiple comparison method on data normalized by a simple square root transformation. Values of P < 0.05 were considered significant.
RESULTS
Extracellular recordings were made from a total of 28 serosal afferent preparations. All multiunit recordings showed a continuous pattern of on-going discharge, the extent of which was highly variable between preparations because of differences in the number of viable units in individual paravascular nerve bundles.
Response to BK
During a 2 min exposure to 1 μm BK there was a marked increase in whole-nerve discharge after a latency of 28.8 ± 1.9 s reaching a peak of 41.7 ± 3.7 impulses s−1 from a baseline level of 12.6 ± 1.4 impulses s−1 (n = 28, P < 0.001). Discharge returned to baseline levels after the BK challenge and was well maintained for the 30 min reperfusion period (Fig. 1). In time control experiments the baseline discharge before and after 30 min reperfusion was 9.16 ± 3 vs. 8.5 ± 3 impulses s−1, respectively (n = 5, P > 0.05). Moreover, the response to BK was also unaffected by the 30 min reperfusion period. The overall effect of BK, expressed as the area under the response curve, was 1962 ± 334 impulses on first exposure compared to 1731 ± 183 impulses during the second challenge (P > 0.05), such that the second response to BK represented 96 ± 16 % of control (n = 5). During subsequent experimental protocols the effect of various mediators on baseline discharge and the magnitude of the response to BK were examined.
Baseline discharge
The effects of PGE2 (1 μm), histamine (100 μm), adenosine (100 μm) and 5-HT (100 μm) on baseline discharge were examined in separate experiments and representative responses are illustrated in Fig. 2. Discharge was unaffected by PGE2 (10.6 ± 2.4 vs. 10.5 ± 2.5 impulses s−1, n = 6, P > 0.05). In contrast, histamine (17.7 ± 2.7 vs. 23.2 ± 3.9 impulses s−1, n = 4, P < 0.05) and adenosine (7.1 ± 1.9 vs. 10 ± 2.7 impulses s−1, n = 4, P < 0.05) caused increases within 3 min, which were sustained throughout the reperfusion. A transient but significant increase in basal discharge was also observed with 5-HT (10.8 ± 3.2 vs. 17.4 ± 1.9 impulses s−1, n = 4, P < 0.05). A summary of the effects of these agents on baseline discharge is shown in Table 1.
Figure 2. Chemical mediators have differential effects on afferent discharge and discharge induced by BK.
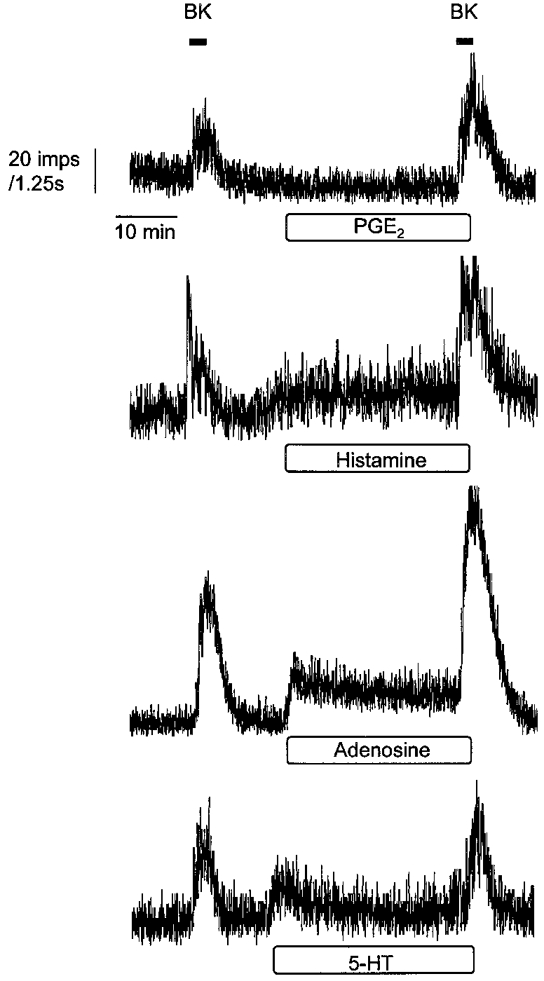
Examples of the differential effects of 1 μm PGE2, 100 μm histamine, 100 μm adenosine and 100 μm 5-HT on basal afferent discharge and afferent activity induced by BK. Mediators were present within the 30 min reperfusion period indicated by the horizontal box and were still present during the second application of BK.
Table 1. Summary of the effects of chemical mediators on activation of afferent discharge and sensitization of responses to BK.
| Basal discharge(%) | BK-induced discharge(%) | |
|---|---|---|
| Time control (n = 6) | −9.8 ± 3.9 | −7.7 ± 12 |
| PGE2 (n = 6) | −2.4 ± 4.0 | +117 ± 48 |
| Histamine (n = 4) | +29.9 ± 6.1 | +76.4 ± 25 |
| Adenosine (n = 4) | +39.3 ± 4.4 | +88.7 ± 27 |
| 5-HT(n = 4) | +56.2 ± 18.3 | −13.8 ± 5.3 |
| Dibutyryl cAMP (n = 4) | −5.1 ± 3.7 | +123.5 ± 28 |
Data are expressed as the percentage change (means ± s.e.m.s) for comparison, while statistical analysis was performed on the raw data presented in the text.
Sensitization of BK responses
The response to BK was significantly enhanced by PGE2 (4179 ± 944 vs. 7882 ± 1356 impulses, n = 6, P < 0.05). Similar augmentation of the BK response was observed with histamine (5051 ± 1357 vs. 8732 ± 2397 impulses, n = 4, P < 0.05) and adenosine (2604 ± 704 vs. 4672 ± 949 impulses, n = 4, P < 0.05). In contrast, 5-HT had no significant effect on BK-induced afferent discharge (5186 ± 933 vs. 4315 ± 514 impulses, n = 4, P > 0.05). These data are expressed as percentage changes in Table 1 to emphasize the differential effects of these mediators on baseline discharge and response to BK. Mediators had no effect on the latency of onset of the response to BK (data not shown).
Since the effect of 5-HT on baseline discharge was only transient, it is possible that the lack of any sensitization of the response to BK could reflect receptor desensitization or reuptake/breakdown of the 5-HT within the 30 min reperfusion period. In a second set of experiments with 5-HT, BK was applied during an on-going response to 5-HT (added during the last 10 min of the reperfusion period). The response to BK was 83.5 ± 14 % of the control response (n = 4, P > 0.05) and still comparable to that seen in the time control group.
Involvement of cAMP in afferent sensitivity
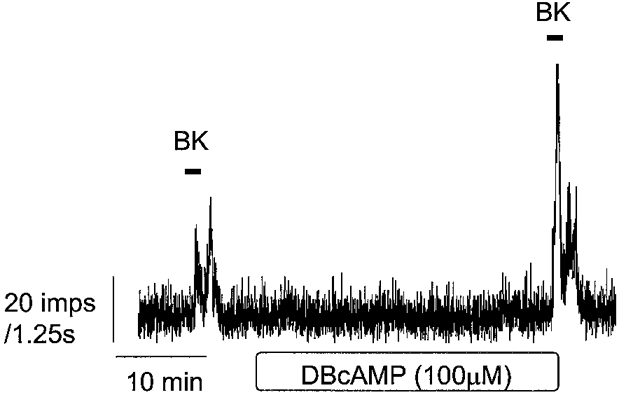
The cell-permeant form of cAMP (dibutyryl cAMP, 100 μm) was used to examine the effects of elevating this intracellular second messenger on the afferent responses to BK. Despite no effect on basal discharge (15.8 ± 4.2 vs. 15.1 ± 4.2 impulses s−1, n = 4, P > 0.05), dibutyryl cAMP significantly augmented responses to BK (2650 ± 755 vs. 5784 ± 1508 impulses, n = 4, P < 0.05) without influencing the response latency (27.5 ± 2.9 vs. 27.5 ± 7 s, n = 4, P > 0.05). The effects of dibutyryl cAMP are also summarized in Table 1 and a representative response is shown in Fig. 3.
Figure 3. Example of the effects of reperfusion with dibutyryl cAMP (DBcAMP) on basal afferent discharge and afferent activity induced by BK.
Effects of naproxen
The involvement of prostanoid synthesis in mediator-induced sensitization of the BK response was investigated using the cyclo-oxygenase (Cox 1 and 2) inhibitor naproxen (10 μm). These studies were confined to the effects of histamine and adenosine since 5-HT did not cause sensitization and PGE2 has already been shown to restore BK responses after naproxen (Maubach & Grundy, 1999). Naproxen (10 μm) had a negligible effect on baseline discharge (16.9 ± 1.6 vs. 15.1 ± 1.3 impulses s−1) but caused a 49 ± 7 % reduction in the BK-induced discharge (4716 ± 661 vs. 2189 ± 346 impulses, n = 12, P < 0.01) (Fig. 4). During the subsequent reperfusion period there was no change in basal discharge in time control experiments (10.9 ± 1.2 vs. 10.5 ± 0.9 impulses s−1, n = 4, P > 0.05). The increases in baseline firing by histamine and adenosine were absent after treatment with naproxen (16.9 ± 3.8 vs. 17.6 ± 3.9 impulses s−1, n = 5, P > 0.05 and 13 ± 2.7 vs. 14.2 ± 2.6 impulses s−1, n = 5, P > 0.05, before and after histamine and adenosine, respectively). Similarly there was no change in basal discharge following dibutyryl cAMP after treatment with naproxen (9.5 ± 1.6 vs. 8.2 ± 1.0 impulses s−1, n = 4, P > 0.05).
The histamine-induced augmentation of the BK response was also absent in the presence of naproxen (81 ± 17 %, n = 5, P > 0.05) with a response comparable to that observed in time control experiments (76 ± 14 %, n = 4). In contrast, adenosine still caused a significant increase (156 ± 25 %, n = 5, P < 0.05) in the presence of naproxen. In addition, the sensitizing effect of dibutyryl cAMP still occurred in the presence of naproxen (167 ± 13 % of control, n = 4, P < 0.05). Sensitization induced by adenosine and dibutyryl cAMP, unlike histamine, is therefore independent of prostanoid formation (Fig. 5).
Figure 5. Afferent discharge induced by BK after histamine (100 μm), adenosine (100 μm) and dibutyryl cAMP (100 μm) in the presence of naproxen.
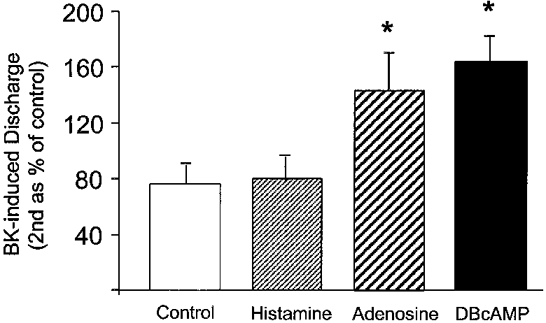
Responses to BK are expressed as percentage of control response (means and s.e.m., n = 4-5) and were compared against the time control using Bonferroni's multiple comparison test on data normalized following a simple square root transformation (* P < 0.05).
Effects of histamine antagonists
The effects of histamine on baseline discharge and augmentation of the BK response were compared in the presence of selective histamine H1 (pyrilamine), H2 (ranitidine) and H3 (thioperamide) receptor antagonists, all at 10 μm. This concentration was chosen on the basis of effects of histamine antagonists (10-100 μm) on submucosal nerves in guinea-pig colon (Frieling et al. 1993). The increase in basal discharge induced by histamine was prevented by pretreatment with either pyrilamine or thioperamide but not ranitidine (Fig. 6A). In contrast, the augmentation of the BK response by histamine was unaffected by thioperamide but reduced by pyrilamine and ranitidine although only the latter reached significance (Fig. 6B).
Figure 6. Effects of histamine antagonists.
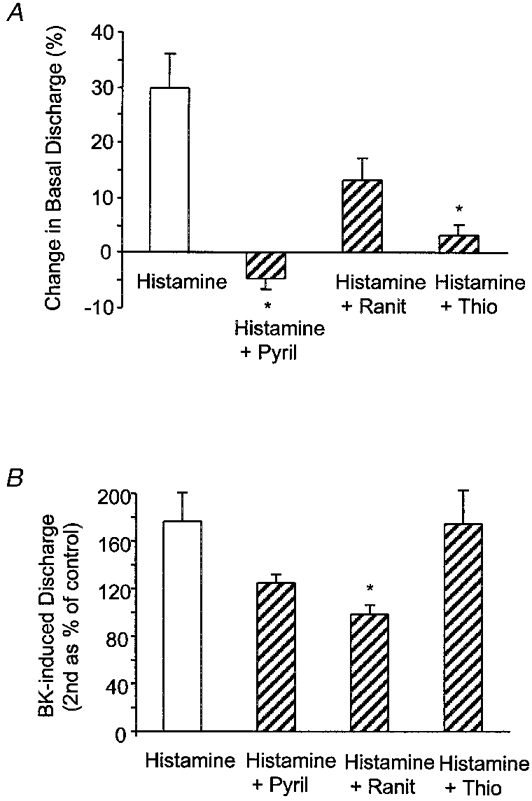
Effects of the histamine antagonists pyrilamine (Pyril), ranitidine (Ranit) and thioperamide (Thio) at 10 μm on the afferent responses to histamine (A) and BK-induced discharge following reperfusion with histamine (100 μm; B). Data are presented as means and s.e.m. (n = 4-6) and were compared statistically against control (Histamine) using Dunn's method for multiple comparison (* P < 0.05).
Effects of adenosine antagonists
The effect of adenosine on baseline discharge and the augmented BK response was examined in the presence of antagonists of adenosine A1 and A2A receptors, namely DPCPX and ZM241385, respectively, at 10 μm and the A3 receptor agonist IB-MECA at 1 μm. The increase in basal discharge induced by adenosine was clearly abolished by DPCPX but not by ZM241385 (Fig. 7A). In contrast, neither antagonist influenced the adenosine-induced augmentation of afferent responses to BK (Fig. 7B). The A3 receptor agonist IB-MECA had no effect on basal discharge (11.8 ± 3.4 vs. 12.1 ± 3.3 impulses s−1, n = 3, P > 0.05, paired t test) and while BK-induced discharge was augmented (174 ± 4 %), this was similar to vehicle controls (DMSO 0.1 % v/v, 162 ± 24 %).
Figure 7. Effects of adenosine antagonists.
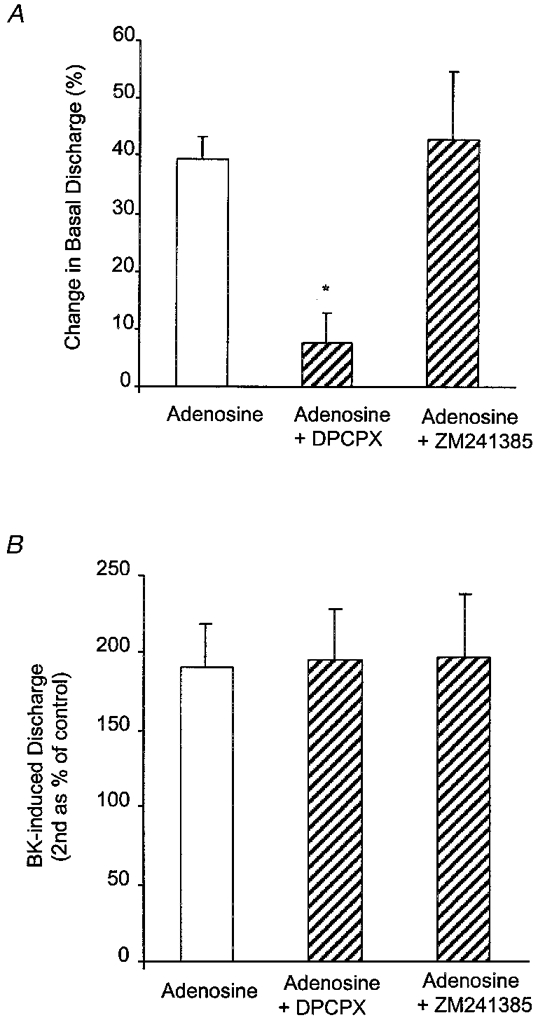
Effects of the adenosine A1 antagonist DPCPX and the A2A antagonist ZM241385 at 10 μm on the afferent discharge induced by adenosine (A) and BK-induced discharge following reperfusion with adenosine (100 μm; B). Data are presented as means and s.e.m. (n = 4-6) and were statistically compared against control (Adenosine) using Dunn's method for multiple comparison (* P < 0.05).
DISCUSSION
This study extends our earlier observations that the algesic peptide BK activates mesenteric afferents from rat jejunum in vitro via B2 receptors and interacts with prostanoids at the level of the afferent nerve terminal (Maubach & Grundy, 1999). Thus the response to BK was attenuated by cyclo-oxygenase inhibitors such as naproxen, an effect that could be reversed upon addition of PGE2 (Maubach & Grundy, 1999). The present study demonstrates that mesenteric afferents are also influenced by other inflammatory mediators, including histamine, adenosine and 5-HT. However, unlike PGE2, which augments the response to BK without affecting baseline afferent discharge, these three agents activated afferent activity in the absence of BK. Moreover, with the exception of 5-HT, they also enhanced the afferent response to BK. Thus inflammatory agents can be divided into those which stimulate afferents, those which sensitize the response to BK and those which can both stimulate and sensitize. Clearly, excitation and sensitization of sensory nerves may be mediated by separate mechanisms. In addition, mediators could enhance the BK response by increasing the sensitivity of active fibres or possibly by causing the recruitment of previously ‘silent’ afferents. Clearly, single unit analysis is required to investigate this further. Given that these mesenteric afferents could be nociceptive (spinal origin) and that these chemical mediators can be released in pathological states, the afferent responses described in the present study could provide the basis for peripheral sensitization leading to the production of pain in inflamed, damaged or ischaemic intestine.
Effects of PGE2
The effects of PGE2 were tested since this is the predominant prostanoid found in inflamed tissues. Prostaglandins are known to increase the sensitivity of cutaneous sensory nerves to chemical mediators of inflammation and pain including BK (see Handwerker & Reeh, 1991). The mechanism of sensitization involved in these present studies was independent of activation of basal discharge. This may be similar to that seen in embryonic rat sensory neurones in culture where PGE2 brought about a 3-fold increase in the number of action potentials evoked by BK without altering resting membrane potential (Nicol & Cui, 1994). In contrast, however, prostaglandins are themselves powerful stimuli of mesenteric afferents in vivo (Mense, 1981; Longhurst & Dittman, 1987), although in vivo there is considerable potential for multiple mechanisms, both direct and indirect, to influence afferent firing. Indeed, recent evidence (Haupt et al. 1999) suggests that PGE2 activates mesenteric afferents in vivo by a direct action on mucosal afferents and indirectly following an increase in intraluminal pressure (ILP). This may explain the lack of effect of PGE2 on serosal afferent discharge in vitro.
PGE2 produces many of its biological effects through the generation of cAMP (e.g. Wellman & Schwabe, 1973). Raised intracellular cAMP, following stimulation by PGE2, may be involved in sensitizing these mesenteric nerves to BK since the effect of PGE2 was mimicked by the addition of dibutyryl cAMP. cAMP has also been implicated in a mechanism for hyperalgesia produced by prostaglandins in rat paw (Ferreira & Nakamura, 1979) and enhanced responses to BK (Cui & Nicol, 1995) and capsaicin (Pitchford & Levine, 1991) in sensory nerve cultures. The lack of effect of PGE2 on baseline afferent activation suggests that baseline firing is independent of cAMP since directly raising cAMP with dibutyryl cAMP also failed to affect afferent discharge.
Effects of histamine
Histamine is an important mediator in gastrointestinal hypersensitivity reactions (Perdue et al. 1984), and can be released from mast cells which are in turn closely associated with afferent nerves in the gastrointestinal tract (Williams et al. 1995). In the present study histamine both increased basal discharge and sensitized the afferent response to BK. Inhibition of histamine-induced discharge by pyrilamine and thioperamide, but not by ranitidine, would implicate H1 and H3 receptors in the response. While we are cautious about this interpretation in the absence of concentration- response data it appears that H2 receptors are unlikely to mediate afferent activation by histamine. This contrasts with effects of histamine on intrinsic submucosal nerves in guinea-pig colon where both postsynaptic H2 and presynaptic H3 receptors are involved (Frieling et al. 1993). This may reflect the differential expression of receptors on intrinsic and extrinsic nerves. However, in the small intestine of the cat in vivo, histamine-induced afferent discharge was attenuated by the H2 antagonist cimetidine (Akoev et al. 1996), although this response may have been secondary to motor responses triggered by high concentrations of histamine. Other studies have implicated the involvement of H1 receptors in mediating the actions of endogenous histamine on ischaemically sensitive visceral afferents in the cat (Fu et al. 1997) and the activation of mesenteric afferents in the rat (Kreis et al. 1998). In contrast to the effect on baseline discharge, the H2 antagonist ranitidine significantly inhibited the ability of histamine to sensitize afferents to BK. However, the actions of histamine may involve indirect effects on afferents through the release of other mediators of sensitization. For example, histamine can generate prostaglandins in the gastrointestinal tract and these in turn mediate histamine-induced Cl− secretion in rat small intestine (Hardcastle & Hardcastle, 1987). Indeed, the sensitization of BK-induced afferent discharge by histamine seen in the present study was inhibited by naproxen. Thus, sensitization of mesenteric afferents by histamine may occur indirectly following prostaglandin release which in turn raises intracellular cAMP.
Effects of adenosine
Adenosine can be generated in large amounts in hypoxic and ischaemic tissue (Edlund & Sollevi, 1993), activates unmyelinated afferents (Cherniak et al. 1987) and causes pain in human blister base studies (Bleehen & Keele, 1977). Adenosine is also a potential sensitizing mediator since it is implicated in mucosal inflammation (Pratt et al. 1986). Recent work has shown clear activation by adenosine of mesenteric afferents in rat intestine in vivo via an action at both A1 and A2B-like receptors with no apparent contribution from A2A and A3 receptors (Kirkup et al. 1998). In the present in vitro study adenosine was seen to both stimulate and sensitize serosal afferents. The stimulation was abolished by the A1 receptor antagonist DPCPX but not by an equal concentration of the A2A antagonist ZM241385. Since these antagonists have very similar potencies at A1 and A2A receptors (DPCPX, -log of antagonist concentration producing a 2-fold rightward shift of the dose-response curve (pA2) = 8.5; and ZM241385, pA2 = 9, respectively), and ZM241385 can maintain selectivity even at 10 μm (Poucher et al. 1995), these results suggest the involvement of A1 receptors in the stimulation of mesenteric afferents. While these studies cannot exclude the involvement of A2B receptors, A3 receptors are unlikely to mediate the actions of adenosine since the A3 receptor agonist IB-MECA had no effect on basal discharge. The concentration of IB-MECA used (1 μm) is not excessive and should have been adequate to activate any A3 receptors in our preparation since this compound relaxes mesenteric artery with an EC50 of approximately 4 μm (Prentice et al. 1997).
In contrast to the clear inhibition of adenosine-induced afferent discharge by DPCPX, this A1 receptor antagonist had no effect on the ability of adenosine to sensitize afferents to BK. Sensitization was also unaffected by the A2A antagonist (ZM241385). It seems unlikely therefore that A1 receptors mediate adenosine-induced sensitization of mesenteric afferents to BK. Again, this supports the idea that afferent sensitization and stimulation are mediated by separate mechanisms. The A3 receptor agonist IB-MECA did not sensitize afferents to BK and while we must be cautious in excluding the involvement of A3 receptors on this basis, we speculate that the A2B receptor remains a likely candidate for mediating afferent sensitization by adenosine. Furthermore, adenosine A2B receptors, unlike A1 and A3 receptors, are positively coupled to adenylate cyclase (Snyder, 1985). In addition, adenosine stimulates Cl− secretion in the T84 colonic cell line by an A2B-mediated rise in intracellular cAMP (Strohmeier et al. 1995). Thus, adenosine-induced sensitization of BK responses may be mediated by a rise in intracellular cAMP, possibly generated following activation of adenosine A2B receptors. Adenosine may therefore act ‘directly’ at the afferent terminals. However, while sensitization by adenosine is independent of prostanoids, we cannot exclude possible interactions of adenosine with non-neural cells such as mast cells or even blood vessels within the mesentery.
5-HT
Serotonin (5-HT), while involved in many gastrointestinal functions, is also implicated in the regulation of nociception. Activation of 5-HT1 (Sufka et al. 1992), 5-HT2 (Grubb et al. 1988; Abbott et al. 1996) and 5-HT3 receptors (Eschalier et al. 1989) has been associated with pain and hyperalgesia. The novel 5-HT3 receptor antagonist alosetron attenuates c-fos expression in the spinal cord following noxious levels of colonic distension in the rat (Kozlowski et al. 1999). In the present study, 5-HT caused transient increases in basal afferent discharge but did not sensitize these afferents to BK. This contrasts with sensitization of BK responses by 5-HT in cutaneous preparations in vitro (Lang et al. 1990) and the ability of 5-HT to reduce mechanical nociceptive thresholds in rat hindpaw (Taiwo & Levine, 1992). This discrepancy could be due to other secondary effects of 5-HT or simply to differences in receptor subtypes present on cutaneous and mesenteric afferents. We have yet to identify the 5-HT receptor responsible for activation of these serosal afferents. While 5-HT3 receptors mediate a direct action of 5-HT on vagal mucosal afferents in rat jejunum (Hillsley & Grundy, 1998), spinal fibres could also possess 5-HT3 receptors (Fu & Longhurst, 1998). The 5-HT3 receptor is linked to a ligand-gated cation channel (Hoyer et al. 1993) and as such is unlikely to sensitize the nerve ending to BK, which appears to rely on adenylate cyclase.
Role for cAMP in a mechanism of afferent sensitization to BK
Our present findings implicate a role for cAMP in a mechanism of afferent sensitization to BK. While there is a great deal of evidence already implicating cAMP/cAMP-dependent protein kinase in afferent sensitization and hyperalgesia in cutaneous systems (see Heller et al. 1993), our results now support a similar role for cAMP in visceral afferents from the gastrointestinal tract. In general, neuronal excitability can be influenced by changes in intracellular cAMP following the phosphorylation of membrane ion channels by cAMP-dependent protein kinase. Indeed, hyperalgesia induced by forskolin (an activator of adenylate cyclase) in rat hindpaw is antagonized by an inhibitor of cAMP-dependent protein kinase (Taiwo & Levine, 1991). However, the precise actions further downstream are not fully understood. cAMP has a variety of actions on sensory nerves depending on the preparation used. In rat embryonic dorsal root ganglion (DRG) cells, PGE2 (which acts through cAMP) increased the number of action potentials induced by BK without altering resting membrane potential (Nicol & Cui, 1994). Thus, cAMP may act on ion channels that do not normally contribute to resting potential. Indeed, cAMP has also been implicated in prolonging Ca2+-dependent action potentials in cultured mouse DRG cells (Grega & Macdonald, 1987) through inhibition of voltage-dependent K+ channels. Changes in repolarization could therefore affect sensitivity. Other studies where prostaglandins influence Ca2+ currents suggest that cAMP could alter excitability through changes in Ca2+ conductance. However, augmentation of BK-induced peptide release by PGE2 is unaffected by L-, N- or P-type Ca2+ channel blockade in rat DRG cells (Evans et al. 1996). Finally, cAMP could influence receptor expression, as demonstrated in rat mesangial cells where BK receptors are upregulated by cAMP (Castano et al. 1998).
Conclusions
The present investigation demonstrates that certain potential inflammatory mediators can sensitize serosal afferents to BK in rat jejunum in vitro. Sensitization was independent of activation of basal discharge and could be mediated by cAMP. Agents such as PGE2 and adenosine may act ‘directly’ while histamine acts indirectly through the generation of prostanoids which in turn can generate cAMP. Since these afferents are spinal in origin and readily activated by BK, they may be implicated in signalling nociceptive stimuli from the gastrointestinal tract. While afferent hypersensitivity is known to underlie conditions of hyperalgesia in many cutaneous models, analogous mechanisms could also operate in visceral organs. Such sensitization, which could occur in the presence of elevated levels of inflammatory mediators or products of ischaemia, may eventually lead to an altered perception of physiological stimuli and possibly the type of symptom commonly associated with both organic and functional bowel disorders.
Acknowledgments
We thank the BBSRC for financially supporting this research.
References
- Abbott FV, Hong Y, Blier P. Activation of 5-HT2A receptors potentiates pain produced by inflammatory mediators. Neuropharmacology. 1996;35:99–110. doi: 10.1016/0028-3908(95)00136-0. [DOI] [PubMed] [Google Scholar]
- Akoev GN, Filippova LV, Sherman NO. Mast cell mediators excite the afferents of cat small intestine. Neuroscience. 1996;71:1163–1166. doi: 10.1016/0306-4522(95)00479-3. [DOI] [PubMed] [Google Scholar]
- Bleehen T, Keele CA. Observations on the algogenic actions of adenosine compounds on the human blister base preparation. Pain. 1977;4:367–377. doi: 10.1016/0304-3959(77)90066-5. [DOI] [PubMed] [Google Scholar]
- Bueno L, Fioramonti J, Delvaux M, Frexinos J. Mediators and pharmacology of visceral sensitivity: from basic to clinical investigations. Gastroenterology. 1997;112:1714–1743. doi: 10.1016/s0016-5085(97)70056-8. [DOI] [PubMed] [Google Scholar]
- Castano ME, Schanstra JP, Hirtz C, Pesquero JB, Pecher C, Girolami JP, Bascands JL. B2 kinin receptor upregulation by cAMP is associated with BK-induced PGE2 production in rat mesangial cells. American Journal of Physiology. 1998;274:F532–540. doi: 10.1152/ajprenal.1998.274.3.F532. [DOI] [PubMed] [Google Scholar]
- Cherniak NS, Runold M, Prabhakar NR, Mitra J. Effect of adenosine on vagal sensory pulmonary afferents. Federation Proceedings, Federation of the American Society of Biology. 1987;46:825. [Google Scholar]
- Cui M, Nicol GD. Cyclic AMP mediates the prostaglandin E2 induced potentiation of BK excitation in rat sensory nerves. Neuroscience. 1995;66:459–466. doi: 10.1016/0306-4522(94)00567-o. [DOI] [PubMed] [Google Scholar]
- Cuschieri A, Onabanjo OA. Kinin release after gastric surgery. British Medical Journal. 1971;3:565–566. doi: 10.1136/bmj.3.5774.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edlund A, Sollevi A. Renal effects of I.V. adenosine infusion in humans. Clinical Physiology. 1993;13:361–371. doi: 10.1111/j.1475-097x.1993.tb00336.x. [DOI] [PubMed] [Google Scholar]
- Eschalier A, Kayser V, Guilbaud G. Influence of a specific 5-HT3 antagonist on carrageenan-induced hyperalgesia in rats. Pain. 1989;36:249–255. doi: 10.1016/0304-3959(89)90030-4. [DOI] [PubMed] [Google Scholar]
- Evans AR, Nicol GD, Vasko MR. Differential regulation of evoked peptide release by voltage-sensitive calcium channels in rat sensory neurons. Brain Research. 1996;712:265–273. doi: 10.1016/0006-8993(95)01447-0. [DOI] [PubMed] [Google Scholar]
- Ferreira SH, Nakamura M. Prostaglandin hyperalgesia, a cAMP/Ca2+ dependent process. Prostaglandins. 1979;18:179–190. doi: 10.1016/0090-6980(79)90103-5. [DOI] [PubMed] [Google Scholar]
- Frieling T, Cooke HJ, Wood JD. Histamine receptors on submucous neurons in guinea pig colon. American Journal of Physiology. 1993;264:G74–80. doi: 10.1152/ajpgi.1993.264.1.G74. [DOI] [PubMed] [Google Scholar]
- Fu LW, Longhurst JC. Role of 5-HT3 receptors in activation of abdominal sympathetic C fibre afferents during ischaemia in cats. The Journal of Physiology. 1998;509:3729–3740. doi: 10.1111/j.1469-7793.1998.729bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu LW, Pan HL, Longhurst JC. Endogenous histamine stimulates ischaemically sensitive abdominal visceral afferents through H1 receptors. American Journal of Physiology. 1997;273:H2726–2737. doi: 10.1152/ajpheart.1997.273.6.H2726. [DOI] [PubMed] [Google Scholar]
- Grega DS, Macdonald RL. Activators of adenylate cyclase and cyclic AMP prolong calcium-dependent action potentials of mouse sensory neurons in culture by reducing a voltage-dependent potassium conductance. Journal of Neuroscience. 1987;7:700–707. doi: 10.1523/JNEUROSCI.07-03-00700.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grubb BD, McQueen DS, Iggo A, Birrell GJ, Dutia MB. A study of 5-HT-receptors associated with afferent nerves located in normal and inflamed rat ankle joints. Agents and Actions. 1988;25:216–218. doi: 10.1007/BF01965015. [DOI] [PubMed] [Google Scholar]
- Handwerker HO, Reeh PW. Pain and inflammation. In: Bond MR, Charlton JE, Woolf CJ, editors. Proceedings of the VIth World Congress on Pain. Elsevier Science Publishers BV; 1991. pp. 59–70. chap. 7. [Google Scholar]
- Hardcastle J, Hardcastle PT. The secretory actions of histamine in rat small intestine. The Journal of Physiology. 1987;388:521–532. doi: 10.1113/jphysiol.1987.sp016629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haupt W, Kreiss ME, Becker H, Grundy D. Prostaglandin E2 stimulates mesenteric afferent nerves in rat small intestine via EP1 and EP2 receptors. Gastroenterology. 1999;116:G4356. [Google Scholar]
- Heller P, Taiwo Y, Ahlgren S, Gold M, Levine J. The irritated nociceptor. In: Mayer EA, Raybould HE, editors. Basic and Clinical Aspects of Chronic Abdominal Pain. Elsevier Science Publishers BV; 1993. pp. 189–199. chap. 15. [Google Scholar]
- Hillsley K, Kirkup AJ, Grundy D. Direct and indirect actions of 5-hydroxytryptamine on the discharge of mesenteric afferent fibres innervating the rat jejunum. The Journal of Physiology. 1998;506:551–561. doi: 10.1111/j.1469-7793.1998.551bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer D, Clarke DE, Fozard JR, Hartig PR, Martin GR, Mylecharane EJ, Saxena PR, Humphrey PPA. International Union of Pharmacology classification of receptors for 5-hydroxytryptamine (serotonin) Pharmacological Reviews. 1993;46:157–203. [PubMed] [Google Scholar]
- Jaenig W, Morrison JFB. Functional properties of spinal visceral afferents supplying abdominal and pelvic organs with special emphasis on visceral nociception. In: Cervero F, Morrison JFB, editors. Progress in Brain Research. Vol. 67. New York: Elsevier; 1986. pp. 87–144. [DOI] [PubMed] [Google Scholar]
- Kirkup AJ, Eastwood C, Grundy D, Chessell IP, Humphrey PPA. Characterisation of adenosine receptors evoking excitation of mesenteric afferents in the rat. British Journal of Pharmacology. 1998;125:1352–1360. doi: 10.1038/sj.bjp.0702202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski CM, Green A, Grundy D, Boissonade FM, Bountra C. Alosetron inhibits the colorectal distension-induced depressor response and spinal c-fos expression in the anaesthetised rat. Pain. 1999 doi: 10.1136/gut.46.4.474. in the Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kreis ME, Haupt W, Kirkup AJ, Grundy D. Histamine sensitivity of mesenteric afferent nerves in the rat jejunum. American Journal of Physiology. 1998;275:G675–680. doi: 10.1152/ajpgi.1998.275.4.G675. [DOI] [PubMed] [Google Scholar]
- Lang E, Novak A, Reeh PW, Handwerker HO. Chemosensitivity of fine afferents from rat skin in vitro. Journal of Neurophysiology. 1990;63:887–901. doi: 10.1152/jn.1990.63.4.887. [DOI] [PubMed] [Google Scholar]
- Longhurst JC, Dittman LE. Hypoxia, bradykinin, and prostaglandins stimulate ischaemically sensitive visceral afferents. American Journal of Physiology. 1987;253:H556–567. doi: 10.1152/ajpheart.1987.253.3.H556. [DOI] [PubMed] [Google Scholar]
- Longhurst JC, Kaufman MP, Ordway GA, Musch TI. Effects of bradykinin and capsaicin on endings of afferent fibers from abdominal visceral organs. American Journal of Physiology. 1984;247:R552–559. doi: 10.1152/ajpregu.1984.247.3.R552. [DOI] [PubMed] [Google Scholar]
- Maubach K, Grundy D. The role of prostaglandins in the bradykinin-induced activation of serosal afferents of the rat jejunum in vitro. The Journal of Physiology. 1999;515:277–285. doi: 10.1111/j.1469-7793.1999.277ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayer EA, Raybould HE. Role of visceral afferent mechanisms in functional bowel disorders. Gastroenterology. 1990;99:1688–1704. doi: 10.1016/0016-5085(90)90475-g. [DOI] [PubMed] [Google Scholar]
- Mense S. Sensitization of group IV muscle receptors to bradykinin by 5-hydroxytryptamine and prostaglandin E2. Brain Research. 1981;225:95–105. doi: 10.1016/0006-8993(81)90320-6. [DOI] [PubMed] [Google Scholar]
- Ness TJ, Gebhart GF. Visceral pain: a review of experimental studies. Pain. 1990;41:167–234. doi: 10.1016/0304-3959(90)90021-5. [DOI] [PubMed] [Google Scholar]
- Nicol GD, Cui M. Enhancement by prostaglandin E2 of bradykinin activation of embryonic rat sensory neurones. The Journal of Physiology. 1994;480:485–492. doi: 10.1113/jphysiol.1994.sp020377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan HL, Stahl GL, Rendig SV, Carretero OA, Longhurst JC. Endogenous BK stimulates ischemically sensitive abdominal visceral C fiber afferents through kinin B2 receptors. American Journal of Physiology. 1994;267:H2398–2406. doi: 10.1152/ajpheart.1994.267.6.H2398. [DOI] [PubMed] [Google Scholar]
- Perdue MH, Chung M, Gall DG. Effect of intestinal anaphylaxis on gut function in the rat. Gastroenterology. 1984;86:391–397. [PubMed] [Google Scholar]
- Pitchford S, Levine JD. Prostaglandins sensitize nociceptors in cell culture. Neuroscience Letters. 1991;132:105–108. doi: 10.1016/0304-3940(91)90444-x. [DOI] [PubMed] [Google Scholar]
- Poucher SM, Keddie JR, Singh P, Stoggall SM, Caulkett PW, Jones G, Coll MG. The in vitro pharmacology of ZM 241385, a potent, non-xanthine A2a selective adenosine receptor antagonist. British Journal of Pharmacology. 1995;115:1096–1102. doi: 10.1111/j.1476-5381.1995.tb15923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt AD, Clancy G, Welsh MJ. Mucosal adenosine stimulates chloride secretion in canine tracheal epithelium. American Journal of Physiology. 1986;251:C167–174. doi: 10.1152/ajpcell.1986.251.2.C167. [DOI] [PubMed] [Google Scholar]
- Prentice DJ, Payne SL, Hourani SM. Activation of two sites by adenosine receptor agonists to cause relaxation in rat isolated mesenteric artery. British Journal of Pharmacology. 1997;122:1509–1515. doi: 10.1038/sj.bjp.0701524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regoli D, Barabe J. Pharmacology of bradykinin and related kinins. Pharmacological Reviews. 1980;32:1–46. [PubMed] [Google Scholar]
- Snyder SH. Adenosine as a neuromodulator. Annual Review of Neuroscience. 1985;8:103–124. doi: 10.1146/annurev.ne.08.030185.000535. [DOI] [PubMed] [Google Scholar]
- Strohmeier GR, Reppert SM, Lencer WI, Madara JL. The A2B adenosine receptor mediates cAMP responses to adenosine receptor agonists in human intestinal epithelia. Journal of Biological Chemistry. 1995;270:2387–2395. doi: 10.1074/jbc.270.5.2387. [DOI] [PubMed] [Google Scholar]
- Sufka KJ, Schomburg FM, Giordano J. Receptor mediation of 5-HT-induced inflammation and nociception in rats. Pharmacology, Biochemistry and Behavior. 1992;41:53–56. doi: 10.1016/0091-3057(92)90058-n. [DOI] [PubMed] [Google Scholar]
- Szolcsanyi J. Selective responsiveness of polymodal nociceptors of the rabbit ear to capsaicin, bradykinin and ultra-violet irradiation. The Journal of Physiology. 1987;388:9–23. doi: 10.1113/jphysiol.1987.sp016598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Further confirmation of the role of adenyl cyclase and of cAMP-dependent protein kinase in primary afferent hyperalgesia. Neuroscience. 1991;44:131–135. doi: 10.1016/0306-4522(91)90255-m. [DOI] [PubMed] [Google Scholar]
- Taiwo YO, Levine JD. Serotonin as a directly-acting hyperalgesic agent in the rat. Neuroscience. 1992;48:485–490. doi: 10.1016/0306-4522(92)90508-y. [DOI] [PubMed] [Google Scholar]
- Wellmann W, Schwabe U. Effects of prostaglandin E1 E2 and F2a on cyclic AMP levels in brain in vivo. Brain Research. 1973;59:371–378. doi: 10.1016/0006-8993(73)90275-8. [DOI] [PubMed] [Google Scholar]
- Williams RM, Bienenstock J, Stead RH. Mast cells: the neuroimmune connection. Chemical Immunology. 1995;61:208–235. [PubMed] [Google Scholar]