

Abstract
ATP-sensitive potassium (KATP) channels are composed of pore-forming (Kir6.x) and regulatory sulphonylurea receptor (SURx) subunits. We have isolated a novel SUR variant (SUR1bΔ33) from a hypothalamic cDNA library. This variant lacked exon 33 and introduced a frameshift that produced a truncated protein lacking the second nucleotide binding domain (NBD2). It was expressed at low levels in hypothalamus, midbrain, heart and the insulin-secreting β-cell line MIN6.
We examined the properties of KATP channels composed of Kir6.2 and SUR1bΔ33 by recording macroscopic currents in membrane patches excised from Xenopus oocytes expressing these subunits. We also investigated the effect of truncating SUR1 at either the start (SUR1bT1) or end (SUR1bT2) of exon 33 on KATP channel properties.
Kir6.2/SUR1bΔ33 showed an enhanced open probability (Po = 0.6 at -60 mV) and a reduced ATP sensitivity (Ki, 86 μm), when compared with wild-type channels (Po = 0.3; Ki, 22 μm). However, Kir6.2/SUR1bT1 and Kir6.2/SUR1bT2 resembled the wild-type channel in their Po and ATP sensitivity.
Neither MgADP, nor the K+ channel opener diazoxide, enhanced Kir6.2/SUR1bΔ33, Kir6.2/SUR1bT1 or Kir6.2/SUR1bT2 currents, consistent with the idea that these agents require an intact NBD2 for their action. Sulphonylureas blocked KATP channels containing any of the three SUR variants, but in excised patches the extent of block was less than that for the wild-type channel. In intact cells, the extent of sulphonylurea block of Kir6.2/SUR1bΔ33 was greater than that in excised patches and was comparable to that found for wild-type channels.
Our results demonstrate that NBD2 is not essential for functional expression or sulphonylurea block, but is required for KATP channel activation by K+ channel openers and nucleotides. Some of the unusual properties of Kir6.2/SUR1bΔ33 resemble those reported for the KATP channel of ventromedial hypothalamic (VMH) neurones, but the fact that this mRNA is expressed at low levels in many other tissues makes it less likely that SUR1bΔ33 serves as the SUR subunit for the VMH KATP channel.
ATP-sensitive potassium (KATP) channels are found in a wide variety of tissues, including pancreatic β-cells, heart, smooth and skeletal muscles, neurones, peripheral axons and epithelial cells (for review see Ashcroft & Ashcroft, 1990). They play important physiological roles in all these tissues. The archetypal KATP channel, such as those found in pancreatic β-cells and cardiac muscle, is highly ATP sensitive, being half-blocked by 10-30 μm ATP. It has a single-channel conductance of 60-80 pS, is inhibited by sulphonylurea drugs and is activated by K+ channel openers and by magnesium nucleotides such as MgADP. KATP channels found in ventromedial hypothalamic (VMH) neurones appear less sensitive to ATP, with millimolar concentrations being needed to produce significant block. They also differ in their single-channel conductance and pharmacological properties (Ashford et al. 1990a,b; Sellers et al. 1992; Routh et al. 1997).
Cloning of KATP channel genes from β-cells, heart and smooth muscle has revealed that these channels are composed of two types of subunit, Kir6.x and SURx, with predicted molecular masses of 43 and 140 kDa, respectively. These subunits coassemble in an octameric (4:4) complex (Clement et al. 1997; Inagaki et al. 1997; Shyng & Nichols, 1997). Kir6.x is a member of the family of inwardly rectifying K+ channels, while SURx (the sulphonylurea receptor) belongs to the ATP-binding cassette (ABC) transporter superfamily (Inagaki et al. 1995a; Sakura et al. 1995; Aguilar-Bryan et al. 1995). Like other ABC transporters, SURx has multiple transmembrane domains and two large cytosolic loops (NBD1 and NBD2) that contain consensus sequences for nucleotide binding and hydrolysis. Kir6.x has no obvious consensus sequence for nucleotide binding; nevertheless, it is now clear that Kir6.x forms the KATP channel pore and contains the site at which ATP mediates channel inhibition (Tucker et al. 1997, 1998). SUR serves as a regulatory subunit that endows the channel with sensitivity to the stimulatory effects of MgADP and K+ channel openers, and to the inhibitory effects of sulphonylureas; in addition, it enhances the blocking action of ATP (the Ki shifts from 100 to 10 μm) and increases the channel open probability (Po) (Nichols et al. 1996; Trapp & Ashcroft, 1997; Tucker et al. 1997; Proks & Ashcroft, 1997; Gribble et al. 1997c; Shyng et al. 1997b; Ashcroft & Gribble, 1998).
To date, two Kir6.x genes have been described, Kir6.1 and Kir6.2 (Inagaki et al. 1995a,b; Sakura et al. 1995). Two closely related SURx genes, SUR1 and SUR2, have also been cloned (Aguilar-Bryan et al. 1995; Inagaki et al. 1996; Isomoto et al. 1996). A number of different splice variants of SUR2 have been described (for review, see Ashcroft & Gribble, 1998). Kir6.2 serves as a common pore-forming subunit for KATP channels in a number of tissues, while the different SURs account for the variable sensitivity to K+ channel openers and sulphonylureas. Thus, the β-cell KATP channel is composed of Kir6.2 and SUR1, that of cardiac and skeletal muscle consists of Kir6.2 and SUR2A (Inagaki et al. 1996), and in the brain single-cell PCR studies have identified Kir6.2/SUR1 and Kir6.2/SUR2B combinations in substantia nigra neurones (Liss et al. 1999), Kir6.2/SUR1 in vagal neurones (Karschin et al. 1998) and Kir6.1/SUR1 in striatal cholinergic interneurones and VMH neurones (Lee et al. 1998, 1999). In this paper we investigate whether additional types of Kir6.x and SURx genes, or splice variants of known SURs, exist in brain. We focus our studies on the ventromedial hypothalamus, as the KATP channels of this region are known to exhibit novel properties.
METHODS
Molecular biology
Isolation of mRNA
The region containing the VMH was isolated from the brains of 11 Wistar rats (∼5 mg per rat) or from 4 brains of C57bl/6J mice. These fragments inevitably also contained the arcuate nucleus, but we attempted to exclude the lateral nucleus of the hypothalamus. Animals were stunned by concussion of the brain. They were then decapitated, the brain was removed and the hypothalamus was quickly isolated. PolyA+ RNA was purified using an mRNA purification kit (Stratagene, Amsterdam, The Netherlands) and was used for making a cDNA library (600 ng, rat) or for RT-PCR (100 ng, mouse). A similar method was used to prepare polyA+ RNA from mouse heart and midbrain and from the mouse insulin-secreting β-cell line MIN6.
Screening of cDNA library
A hypothalamic cDNA library was constructed using a ZAP Express cDNA Synthesis Kit (Stratagene). This library (0.5 × 105 independent clones) was screened under low-stringency conditions (hybridised with 30 % formamide, 5 × Denhardt's, 5 × SSPE, 0.5 % SDS, 100 μg ml−1 denatured salmon sperm DNA at 37°C and washed with 2 × SSC, 0.2 % SDS at room temperature) with probes for Kir6.x and SURx. A mixed probe containing full length Kir6.1 and Kir6.2 was used for isolating Kir6.x genes. To detect SURx, the probe consisted of NBD1 and NBD2 of SUR1 and SUR2. PBK-CMV phagemids containing cDNA inserts of positive clones were excised using ExAssist helper phage (Stratagene) and sequenced.
RT-PCR
Reverse transcription was carried out with 100 ng polyA+ RNA. First strand cDNA was synthesised for 1 h at 37°C in reverse transcription buffer (Gibco BRL, Paisley, UK) containing random hexamer primers (final concentration, 5 μm; Boehringer Mannheim, Lewes, UK), dithiothreitol (final concentration, 20 mm), the four deoxyribonucleotide triphosphates (dNTPs; final concentration, 0.5 mm each; Pharmacia, St Albans, UK), 20 U ribonuclease inhibitor (Promega, Southampton, UK) and 100 U reverse transcriptase (Superscript II, Gibco-BRL). The total RT-reaction volume was 10 μl. The cDNA was kept at -70°C until PCR amplification.
PCR amplification of SUR1, SUR2A/B, Kir6.1 and Kir6.2 was carried out using the specific primers reported previously (Liss et al. 1999). In addition, we also performed PCR amplification with primers flanking exon 33 of SUR1: AAG CTC CTG GAG TAC- ACG GA (sense); CTC TGG GTC CAG GTT GAA TC (antisense). PCR amplification was carried out using a hot start protocol in a final volume of 100 μl containing 1 μl of the RT-reaction solution, 100 pmol of each primer, 0.2 mm dNTPs (Pharmacia), 1.5 mm MgCl2, 50 mm KCl, 20 mm Tris-HCl (pH 8.4) and 2.5 U Taq polymerase (Gibco BRL) in a Perkin Elmer Thermal Cycler 480C, with the following protocol: 3 min at 94°C followed by 35 cycles (94°C for 30 s, 58°C for 60 s, 72°C for 3 min) and a final elongation period of 7 min at 72°C.
To investigate the presence and size of the amplified fragments, 15 μl aliquots of PCR products were separated by electrophoresis and visualised in ethidium bromide-stained agarose gels (2 %). The predicted sizes for SUR1 and SUR1Δ33 were 655 and 524 bp, respectively. The PCR products were verified by direct sequencing.
Expression studies
Mouse Kir6.2(GenBank D50581; Inagaki et al. 1995a; Sakura et al. 1995) was coexpressed with rat SUR1, SUR1b, SUR1bΔ33, SUR1bT1 or SUR1bT2. Synthesis of capped mRNA was carried out using the mmessage mmachine large scale in vitro transcription kit (Ambion, Austin, TX, USA).
Oocyte collection
Female Xenopus laevis were anaesthetised with MS-222 (2 g l−1 added to the water). One ovary was removed via a mini-laparotomy, the incision sutured and the animal allowed to recover. Xenopus were monitored during the recovery period from anaesthesia and checked post-operatively for complications at regular intervals. Once the wound had completely healed, the second ovary was removed in a similar operation and the animal was then killed by decapitation whilst under anaesthesia. Immature stage V-VI Xenopus oocytes were incubated for 60 min with 1.0 mg ml−1 collagenase (Type V, Sigma) and manually defolliculated. Oocytes were coinjected with ∼0.1 ng of mRNA encoding Kir6.2 and ∼2 ng of mRNA encoding SUR (wild-type or variant), giving a 1:20 ratio. The final injection volume was ∼50 nl oocyte−1. Control oocytes were injected with water. Isolated oocytes were maintained in Barth's solution and studied 1-4 days after injection (Gribble et al. 1997a).
Electrophysiology: two electrode voltage clamp
Whole-cell currents were measured using a two electrode voltage clamp (Geneclamp 500; Axon Instruments). Voltages were applied and currents recorded using a microcomputer with an Axolab interface and pCLAMP software (Axon Instruments). Currents were filtered at 1 kHz and digitised at 4 kHz. Current and voltage electrodes were filled with 3 M KCl and had resistances of 0.5-2 MΩ. Recordings were carried out at a holding potential of -10 mV in a high-potassium extracellular solution containing (mm): 90 KCl, 1 MgCl2, 1.8 CaCl2, 5 Hepes (pH 7.4 with KOH).
Electrophysiology: excised patches
Patch pipettes were pulled from thick-walled glass and had resistances of 250-500 kΩ when filled with pipette solution. Macroscopic currents were recorded from giant excised inside-out patches at a holding potential of 0 mV and at 20-24°C (Gribble et al. 1997a). Currents were evoked by repetitive 3 s voltage ramps from -110 to +100 mV and recorded using an EPC7 patch-clamp amplifier (HEKA Electronik, Lambrecht, Germany). They were filtered at 5 kHz and stored on digital audiotape. Currents were subsequently filtered at 0.5 kHz, digitised at 1 kHz using a Digidata 1200 Interface and analysed using pCLAMP software. Single-channel currents were recorded from small inside-out patches, filtered at 1 kHz and sampled at 3 kHz.
The pipette (extracellular) solution contained (mm): 140 KCl, 1.2 MgCl2, 2.6 CaCl2, 10 Hepes (pH 7.4 with KOH). The standard intracellular (bath) solution contained (mm): 107 KCl, 2 MgCl2, 1 CaCl2, 10 EGTA, 10 Hepes (pH 7.2 with KOH; final [K+]∼140 mm) and nucleotides as indicated. Mg2+-free solution contained (mm): 107 KCl, 2.6 CaCl2, 10 EDTA, 10 Hepes (pH 7.2 with KOH; final [K+]∼140 mm). Tolbutamide was made up as a 0.05 M stock solution in 0.1 M KOH and diazoxide as a 0.02 M stock solution in 0.1 M KOH. Solutions containing nucleotides were made up fresh each day. The pH of all solutions was checked and readjusted, if required, after drug and nucleotide addition. Rapid exchange of solutions was achieved by positioning the patch in the mouth of one of a series of adjacent inflow pipes placed in the bath.
The slope conductance was measured by fitting a straight line to the current-voltage relationship between -20 and -100 mV: the average of five consecutive ramps was calculated in each solution.
ATP dose-response relationships were obtained by alternating the control solution with a test ATP concentration. The conductance (G) was then expressed as a fraction of the mean of the value obtained in the control solution before and after application of the nucleotide (Gc). Dose-response curves were fitted to the Hill equation:
| (1) |
where [ATP] is the ATP concentration, Ki is the concentration at which inhibition is half-maximal and h is the slope factor (Hill coefficient).
Tolbutamide dose-response curves were fitted to the following equation (Gribble et al. 1997b):
| (2) |
where x is a term describing the high-affinity site and y a term describing the low-affinity site.
| (3) |
and
| (4) |
where [Tolb] is the tolbutamide concentration, Ki1 and Ki2 are the tolbutamide concentrations at which inhibition is half-maximal at the high- and low-affinity sites, respectively, h1 and h2 are the Hill icoefficients (slope factors) for the high- and low-affinity sites, respectively, and L is the fractional conductance remaining when all of the high-affinity inhibitory sites are occupied.
Data are given as means ± 1 s.e.m., and the symbols in the figures indicate the mean and the vertical bars indicate 1 s.e.m. (where this is larger than the symbol). Statistical significance was tested using Student's unpaired t test or ANOVA, as appropriate. P values of < 0.05 were taken to indicate that the data were significantly different.
RESULTS
Molecular biology
We isolated mRNA from the rat hypothalamus and constructed a rat hypothalamic cDNA library (5 × 105 clones). This library was screened with probes for either the Kir6.x or SURx family. Only two Kir6.x clones were isolated, both of which were identified as Kir6.2. For SUR, however, more than 40 positive clones were obtained. Analysis of 24 of these clones revealed that 15 were similar to SUR1 while 9 were identical to SUR2B. All the clones homologous to SUR1 differed from the rat SUR1 sequence originally reported by five amino acids: these included four substitutions (T487S, P835Q, G836R and G1313R) and the insertion of an additional serine residue after S741. In this paper we refer to this variant as SUR1b for clarity. However, as we show that the functional properties of SUR1b are identical to those of SUR1, it thus does not constitute a true variant and should be referred to as SUR1 elsewhere. The sequence of SUR1b is identical to that deposited in GenBank as accession no. X97279.
One clone of the SUR1b type had a 131 bp deletion before the second nucleotide-binding domain (NBD2), but was otherwise identical to SUR1b; because the missing 131 bp correspond to exon 33 of human SUR1, we call this clone SUR1bΔ33. As shown in Fig. 1, this deletion introduced a frameshift at residue 1330, and the open reading frame stopped after a further 25 amino acids, truncating the protein at residue 1355. This resulted in the presence of 25 different residues and the deletion of the whole of NBD2.
Figure 1. SUR1bΔ33 lacks exon 33.
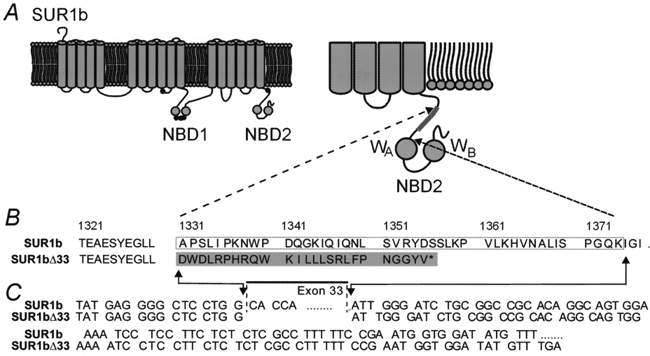
A, putative membrane topology of SUR1b (after Tusnady et al. 1997) with the position of the amino acids that are altered in SUR1b indicated (•). NBD, nucleotide binding domain. The truncation that occurs in SUR1bΔ33 is indicated in the expanded view of NBD2 (inset, right). B and C, differences in the amino acid sequence (B) and nucleotide sequence (C) of SUR1b and SUR1bΔ33. The boxed area shows exon 33 and the shaded area shows the amino acids that are different in SUR1bΔ33. WA, Walker A motif; WB, Walker B motif. The asterisk indicates the termination of translation.
We generated SUR1-specific PCR primers spanning exon 33 and used these for RT-PCR of mouse midbrain, hypothalamus and heart tissue and of the insulin-secreting β-cell line MIN6. In all cases, the major PCR product was wild-type SUR1 (655 bp) but a smaller PCR fragment of 525 bp was also identified (Fig. 2), indicating the presence of SUR1Δ33. The splice variant, however, was amplified in considerably lower abundance. The identity of the PCR products was confirmed by direct sequencing.
Figure 2. RT-PCR amplification of SUR1 and SUR1Δ33 variants from mouse tissue.
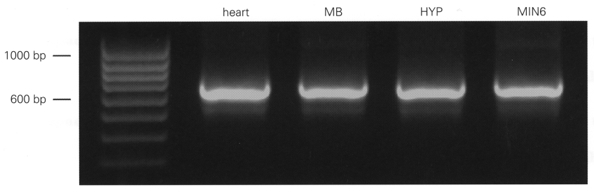
Photograph of ethidium bromide-stained gel, with RT-PCR products separated by electrophoresis in parallel with a 100 bp ladder as molecular weight marker. PCR fragments were generated with mouse SUR1-specific primers spanning exon 33. The prominent band at 655 bp corresponds to SUR1 and that at 525 bp to SUR1Δ33. MB, midbrain. HYP, hypothalamus. MIN6, mouse β-cell line MIN6.
We also carried out RT-PCR with mRNA from mouse hypothalamus using primers specific for Kir6.2, Kir6.1, SUR1 and SUR2A/B. In these experiments (n = 4), we routinely detected Kir6.1, Kir6.2, SUR1 and SUR2B, but we were unable to amplify SUR2A (data not shown).
Electrophysiological analysis
To examine whether the functional properties of KATP channels containing SUR1b or SUR1bΔ33 differ from Xenopus oocytes. In all cases, no significant currethose containing SUR1, we coexpressed the different sulphonylurea receptor variants with Kir6.2 innt was detected in the cell-attached configuration but large currents developed following patch excision into nucleotide-free solution. The mean current amplitude at -100 mV was 10.5 ± 1.8 nA (n = 19) for Kir6.2/SUR1, 7.1 ± 1.3 nA (n = 21) for Kir6.2/SUR1b and 0.9 ± 0.2 nA (n = 25) for Kir6.2/ SUR1bΔ33. Therefore, both SUR1b and SUR1bΔ33 were able to support the expression of full-length Kir6.2, although Kir6.2/SUR1bΔ33 was less effective than SUR1b. Thus NBD2 does not appear to be essential for the interaction of SUR1 with Kir6.2 that enables its functional expression.
ATP sensitivity
We next examined the effect of the different SUR1 variants on the ATP sensitivity of the channel (Fig. 3). Kir6.2/SUR1b currents were blocked by ATP with a Ki of 22 ± 3 μm and a Hill coefficient (h) of 1.30 ± 0.16 (n = 6), values which are similar to those reported for Kir6.2/SUR1 channels (Gribble et al. 1997a). However, Kir6.2/SUR1bΔ33 currents were significantly less ATP sensitive, with a Ki of 86 ± 4 μm (h = 1.20± 0.07; n = 5).
Figure 3. Effects of ATP on wild-type and mutant KATP channels.
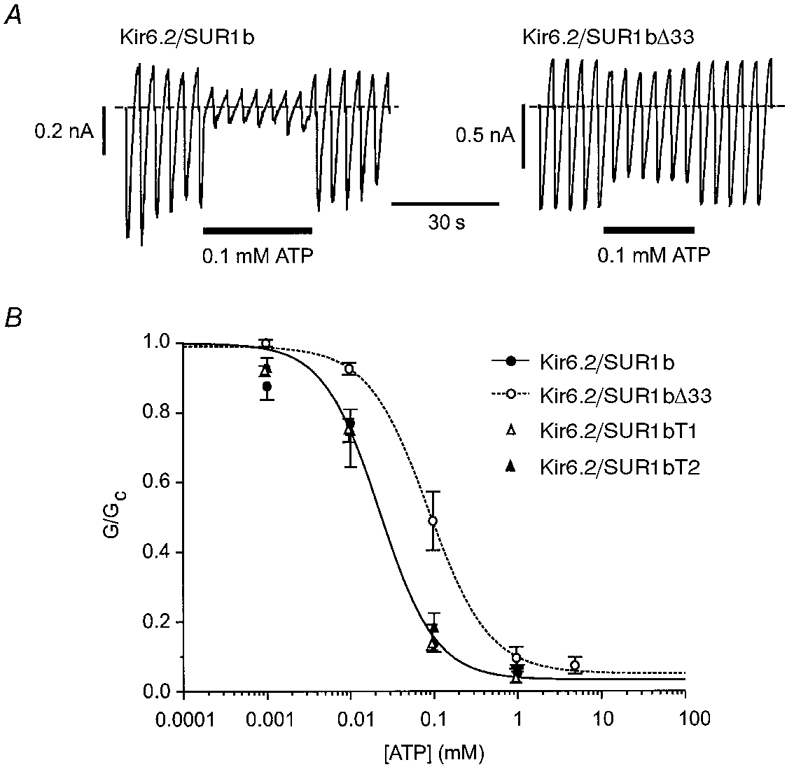
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes injected with mRNAs encoding Kir6.2 and either SUR1b (left) or SUR1bΔ33 (right). ATP was added to the intracellular solution as indicated by the horizontal bars. B, mean ATP dose-response relationships for Kir6.2/SUR1b (•, n = 6), Kir6.2/SUR1bΔ33 (○, n = 5), Kir6.2/SUR1bT1 (▵, n = 7) and Kir6.2/SUR1bT2 (▴, n = 8) currents. Test solutions were alternated with control solutions and the slope conductance (G) is expressed as a fraction of the mean (Gc) of that obtained in control solution before and after exposure to ATP. The lines are the best fits of the data to the Hill equation (eqn (1)) for SUR1b (continuous line, Ki = 22 ± 3 μm, h = 1.30± 0.16) and for SUR1bΔ33 (dotted line, Ki = 86 ± 4 μm, h = 1.20± 0.07).
In addition to lacking NBD2, SUR1bΔ33 possesses an additional 25 amino acids at its C-terminus. We explored the contribution of these amino acids to the difference in the functional properties of SUR1bΔ33 and SUR1b, by truncating SUR1b at the start (residue 1330, SUR1bT1) or at the end (residue 1374, SUR1bT2) of exon 33. Channels comprising Kir6.2 and either SUR1bT1 or SUR1bT2 expressed large currents in excised patches: 10 ± 2 nA (n = 16) and 1.8 ± 0.3 nA (n = 6), respectively. To our surprise, however, these channels had an ATP sensitivity similar to that of Kir6.2/SUR1b (Ki = 24 ± 4 μm, h = 1.1± 0.2, n = 7, for SUR1bT1; and Ki = 27 ± 3 μm, h = 1.3± 0.1, n = 8, for SUR1bT2). This suggests that the novel 25 amino acids produced by the frameshift, rather than the deletion of NBD2, are responsible for modifying the ATP sensitivity of Kir6.2/SUR1bΔ33.
One mechanism by which a mutation may indirectly alter the ATP sensitivity of the channel is by impairing the ability of the channel to close, because ATP stabilises the closed state of the channel (Shyng et al. 1997a; Trapp et al. 1998). We therefore examined the properties of the single-channel currents recorded from KATP channels containing different SUR1 variants, in the absence of nucleotide. It is evident from Fig. 4 that the open probability (Po) of Kir6.2/SUR1bΔ33 channels was greater than that of either Kir6.2/SUR1 or Kir6.2/SUR1b channels. The mean Po at -60 mV was 0.27 ± 0.05 (n = 5) for Kir6.2/SUR1, 0.29 ± 0.05 (n = 4) for Kir6.2/SUR1b and 0.63 ± 0.05 (n = 5) for Kir6.2/SUR1bΔ33. This result suggests that the reduced ATP sensitivity of SUR1bΔ33 is a secondary consequence of the change in the channel kinetics. Further support for this idea is provided by the fact that the Po of Kir6.2/SUR1bT1 and Kir6.2/SUR1bT2 channels was not markedly affected, being 0.35 ± 0.08 (n = 4) and 0.29 ± 0.06 (n = 4), respectively. The fact that truncation of SUR1b at the end of exon 32 or 33 has only a small effect on Po further suggests that it is the presence of the additional 25 amino acids in SUR1bΔ33 that is responsible for the higher Po of KATP channels containing this variant.
Figure 4. Single-channel currents.
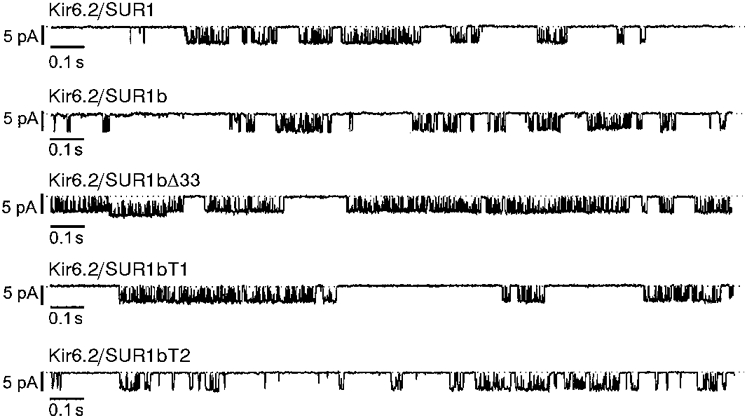
Single-channel currents recorded at -60 mV from inside-out patches excised from oocytes injected with mRNAs encoding Kir6.2 plus SUR1, SUR1b, SUR1bΔ33, SUR1bT1 or SUR1bT2. The dotted lines indicate the closed level.
The single-channel current-voltage relationship of Kir6.2/ SUR1bΔ33 currents showed slight inward rectification at potentials positive to +20 mV (Fig. 5), as is observed for Kir6.2/SUR1. The single-channel conductance, measured between -20 and -80 mV, was 63 ± 1 pS (n = 3) for Kir6.2/SUR1bΔ33 channels, a value which is not significantly different from that observed for Kir6.2/SUR1 or for Kir6.2ΔC26, a truncated Kir6.2 expressed in the absence of SURx (67-76 pS; Sakura et al. 1995; Tucker et al. 1997).
Figure 5. Single-channel currents.
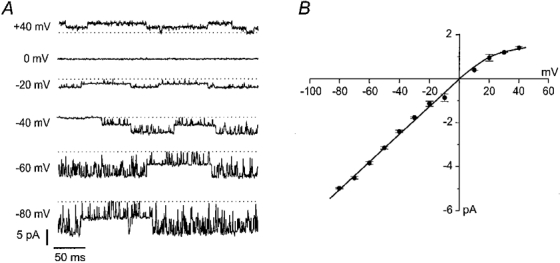
A, single-channel currents recorded at the indicated membrane potentials from an inside-out patch excised from an oocyte injected with mRNAs encoding Kir6.2 and SUR1bΔ33. The dotted lines indicate the closed level. B, mean single-channel current-voltage relationship recorded for Kir6.2/SUR1bΔ33 currents (n = 3).
Modulation by SUR
As is the case for Kir6.2/SUR1 channels, both MgADP and diazoxide were capable of enhancing the activity of Kir6.2/SUR1b currents (Fig. 6A and C). By contrast, diazoxide did not activate Kir6.2/SUR1bΔ33 currents and 100 μm MgADP was actually slightly inhibitory (Fig. 6B and C). Diazoxide was tested in the presence of MgATP because its stimulatory action is dependent upon the presence of intracellular hydrolysable nucleotides (Kozlowski et al. 1989). Similar results were obtained for Kir6.2/SUR1bT1 and Kir6.2/SUR1bT2 currents (Fig. 6C). As discussed in more detail below, the inability of MgADP and diazoxide to stimulate KATP channels containing SUR1bΔ33, SUR1bT1 or SUR1bT2 is consistent with the fact that these SUR variants lack NBD2. A reduced sensitivity to the inhibitory effect of ADP (as observed for ATP) may explain the fact that MgADP caused less inhibition of SUR1bΔ33- than of SUR1bT1- or SUR1bT2-containing channels. Indeed, Kir6.2/SUR1bΔ33 currents were blocked to the same extent in the presence and absence of Mg2+ (see Fig. 8C).
Figure 6. Effects of ADP and diazoxide on wild-type and mutant KATP channels.
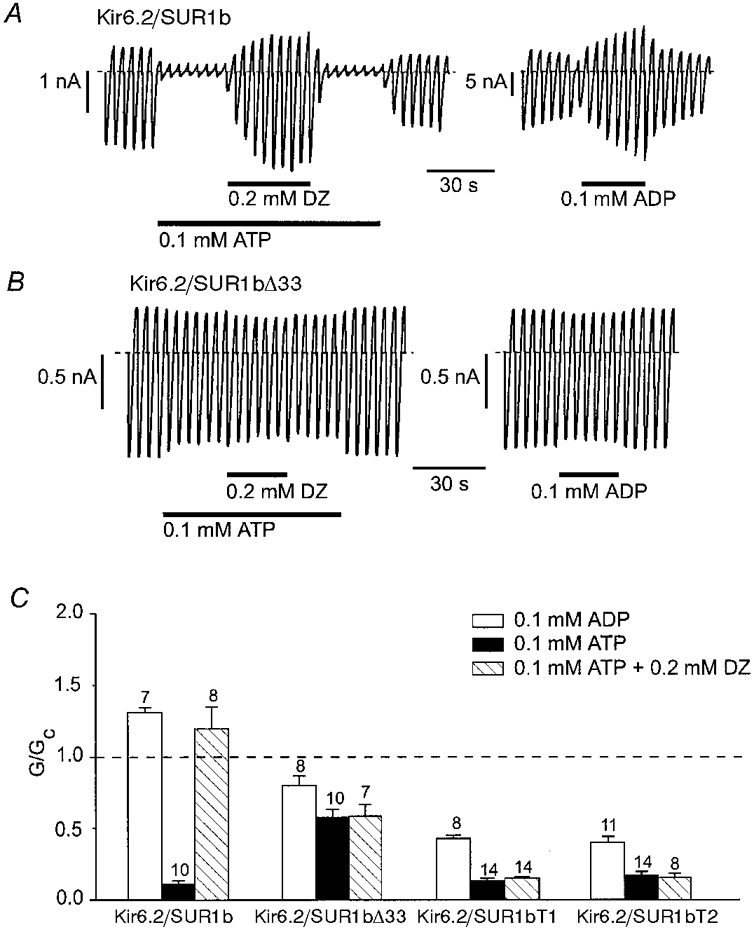
A and B, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes injected with mRNAs encoding Kir6.2 and either SUR1b (A) or SUR1bΔ33 (B). ATP, diazoxide (DZ) or MgADP were added to the intracellular solution as indicated by the horizontal bars. C, mean macroscopic slope conductance recorded in the presence of MgADP, ATP or ATP plus diazoxide, expressed as a fraction of the slope conductance in control solution (no additions), for Kir6.2/SUR1b, Kir6.2/SUR1bΔ33, Kir6.2/SUR1bT1 and Kir6.2/SUR1bT2. The dashed line indicates the current level in control solution. The number of oocytes is given above the bars.
Figure 8. Interactions between MgADP and tolbutamide.
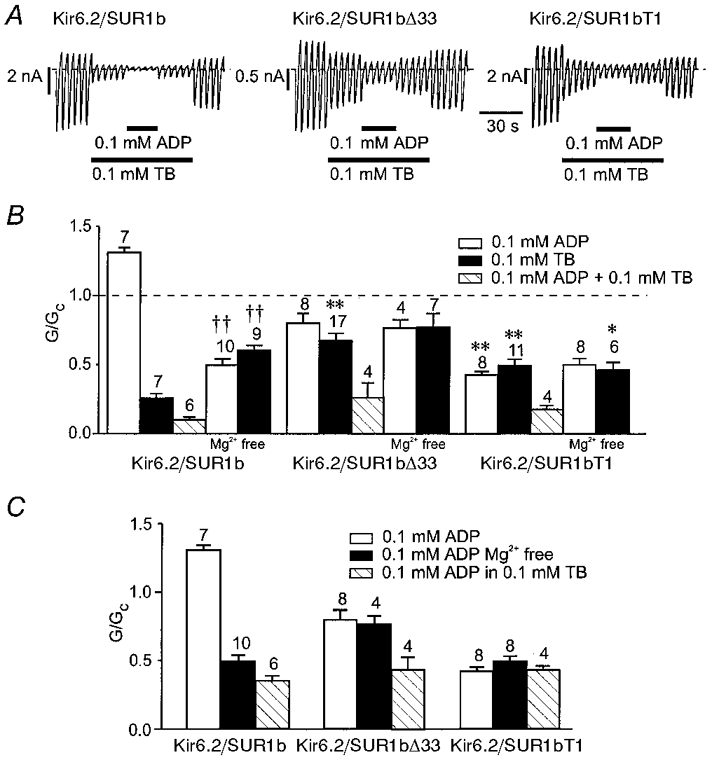
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes injected with mRNAs encoding Kir6.2 and either SUR1b, SUR1bΔ33 or SUR1bT1. Tolbutamide (TB) and MgADP were added to the intracellular solution as indicated by the horizontal bars. B, mean macroscopic slope conductance recorded in the presence of ADP, tolbutamide or ADP plus tolbutamide, expressed as a fraction of the slope conductance in control solution (no additions), for Kir6.2/SUR1b, Kir6.2/SUR1bΔ33 and Kir6.2/SUR1bT1. Experiments were carried out in the presence of Mg2+ except where indicated. The dashed line indicates the current level in control solution. The number of oocytes is given above the bars. *P < 0.05, **P < 0.01, compared with Kir6.2/SUR1b. ††P < 0.01, compared with data obtained for the same type of channel in the presence of Mg2+. C, mean macroscopic slope conductance recorded in the presence of ADP, ADP plus tolbutamide, and ADP in Mg2+-free solution for Kir6.2/SUR1b, Kir6.2/SUR1bΔ33 and Kir6.2/SUR1bT1. Currents are expressed as a fraction of the slope conductance in the absence of ADP. Experiments were carried out in the presence of Mg2+ except where indicated. The number of oocytes is given above the bars.
We next compared the ability of sulphonylureas to block KATP channels which contain different SUR1 variants. Figure 7A shows that both tolbutamide and glibenclamide were significantly less effective at blocking Kir6.2/ SUR1bΔ33 than Kir6.2/SUR1b currents. Indeed, the effect of glibenclamide was so slow that it was difficult to distinguish from rundown. The mean dose-response relationships for tolbutamide are given in Fig. 7B and, like that for Kir6.2/SUR1 (Gribble et al. 1997b), were best fitted by assuming that the drug interacts with both a high-affinity and a low-affinity site (eqn (2)). The low-affinity site lies on Kir6.2 (Gribble et al. 1997b) and it is therefore not surprising that the Ki for this site was independent of the SUR1 variant, being 1.2 ± 0.6 mm (n = 4) and 1.4 ± 0.1 mm (n = 7) for Kir6.2/SUR1b and Kir6.2/ SUR1bΔ33, respectively. The Ki values for the high-affinity site - which lies on SUR1 - were also comparable: 2.1 ± 0.8 μm for Kir6.2/SUR1b and 1.2 ± 0.4 μm for Kir6.2/SUR1bΔ33. Similar values of Ki were also found for both the high- (2.0 μm) and low- (1.8 mm) affinity sites of Kir6.2/SUR1 channels (Gribble et al. 1997b). Although the affinity for tolbutamide was unaffected by deletion of NBD2, there was a marked difference in the ratio of high-to low-affinity inhibition. The percentage of tolbutamide block attributable to the high-affinity site was 57 % for Kir6.2/SUR1 (Gribble et al. 1997b) and 72 ± 4 % (n = 4) for Kir6.2/SUR1b, but only 26 ± 6 % (n = 7) for Kir6.2/ SUR1bΔ33.
Figure 7. Effect of sulphonylureas on Kir6.2/SUR1b and Kir6.2/SUR1bΔ33 currents.
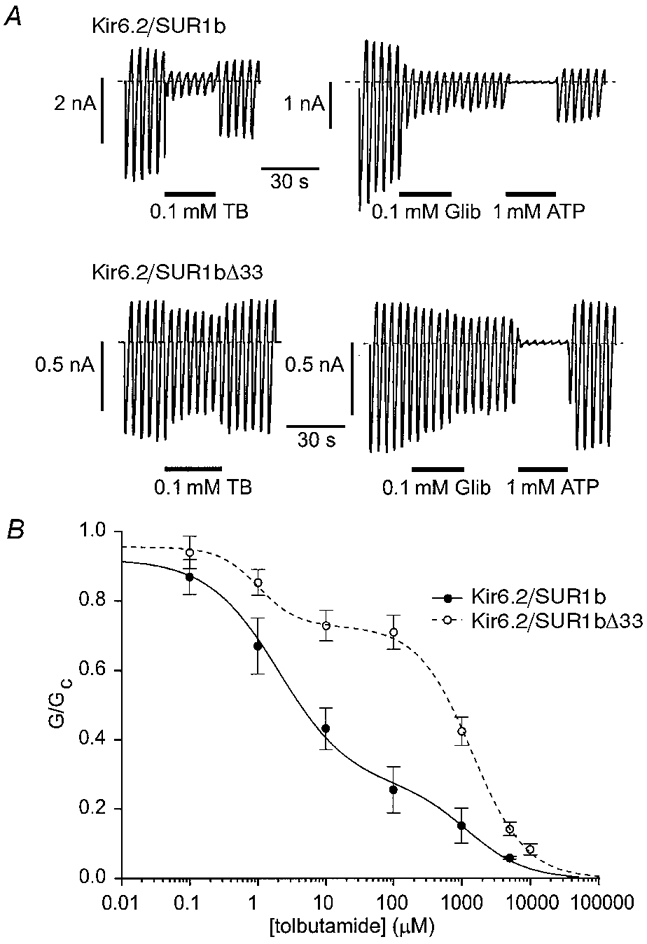
A, macroscopic currents recorded from inside-out patches in response to a series of voltage ramps from -110 to +100 mV from oocytes injected with mRNAs encoding Kir6.2 and either SUR1b (top) or SUR1bΔ33 (bottom). Tolbutamide (TB), glibenclamide (Glib) or ATP were added to the intracellular solution as indicated by the horizontal bars. B, mean relationships between tolbutamide concentration and the macroscopic KATP conductance, expressed as a fraction of its amplitude in the absence of the drug. •, continuous line: Kir6.2/SUR1b currents (n = 4). ○, dashed line: Kir6.2/SUR1bΔ33 currents (n = 7). The lines are the best fit of the data to eqn (2). Kir6.2/SUR1b currents: Ki1 = 2.1 μm, h1 = 0.83, Ki2 = 1.2 mm, h2 = 1.0, L = 26%. Kir6.2/SUR1bΔ33 currents: Ki1 = 1.2 μm, h1 = 1.5, Ki2 = 1.4 mm, h2 = 1.1, L = 72%.
The apparent sulphonylurea sensitivity of both cloned and native β-cell KATP channels is enhanced by MgADP. It has been hypothesised that tolbutamide prevents the stimulatory effect of MgADP (mediated by SUR1), thus unmasking its inhibitory effect on Kir6.2 (Gribble et al. 1997c). The inhibitory effect of MgADP sums with that of tolbutamide to enhance the apparent block by the drug. We investigated the effect of MgADP on the inhibitory effect of 100 μm tolbutamide, a concentration that produces saturation of high-affinity inhibition in both wild-type and mutant channels (Fig. 7). Figure 8 shows that MgADP blocked, rather than activated, Kir6.2/SUR1b channels in the presence of tolbutamide. The amount of inhibition produced by MgADP in the presence of tolbutamide was comparable to that observed in the absence of Mg2+ (when ADP acts solely as a blocker), thus supporting the idea that tolbutamide prevents the stimulatory action of the magnesium nucleotide. We next explored the effect of MgADP in the presence of tolbutamide on Kir6.2/ SUR1bΔ33 and Kir6.2/SUR1bT1 channels, which are not activated by MgADP. As Fig. 8C shows, MgADP was an equally effective blocker of Kir6.2/SUR1bT1 in the presence and absence of tolbutamide. In contrast, inhibition of Kir6.2/SUR1bΔ33 currents by MgADP was greater in the presence of the drug (P < 0.01).
The Mg2+ ion itself has been reported to enhance the efficacy of tolbutamide block of native β-cell KATP channels (Lee et al. 1994). This was also the case for Kir6.2/SUR1b, because removal of Mg2+ caused a significant reduction in the inhibitory effect of the drug. It also abolished the stimulatory effect of ADP. In contrast, tolbutamide block of both Kir6.2/SUR1bΔ33 and Kir6.2/SUR1bT1 channels was unaffected by Mg2+. Furthermore, in the absence of Mg2+, the extent of tolbutamide block of Kir6.2/SUR1bT1 and Kir6.2/SUR1b channels was barely significantly different. This raises the possibility that the ability of Mg2+ to enhance tolbutamide inhibition involves NBD2 of SUR1, or requires the functional integrity of both NBDs (as is the case for MgADP; Gribble et al. 1997c) or of the whole C-terminus.
Whole-cell studies
To explore the effect of metabolic inhibition on Kir6.2/SUR1, Kir6.2/SUR1b, Kir6.2/ SUR1bΔ33 and Kir6.2/SUR1bT1 channels, we recorded whole-cell KATP currents from intact oocytes and used 3 mm azide as a metabolic poison. We also examined the effect of metabolic inhibition on Kir6.2ΔC26, expressed in the absence of a sulphonylurea receptor: this truncated form of Kir6.2 is able to express independently of SUR (Tucker et al. 1997). In control solution, oocytes expressing all five types of channel exhibited very small current amplitudes, similar to those of water-injected oocytes. As shown in Fig. 9, metabolic poisoning produced a large increase in Kir6.2/SUR1 and Kir6.2/SUR1b currents but a very much smaller increase in Kir6.2/SUR1bΔ33, Kir6.2/SUR1bT1 and Kir6.2ΔC26 currents. Nevertheless, it is clear from the inset to Fig. 9 that Kir6.2/SUR1bΔ33 currents were activated about 5-fold and that the activated current could be inhibited by tolbutamide and glibenclamide, indicating that it flows through KATP channels. Likewise, azide induced ∼4-fold activation of Kir6.2ΔC26 currents and 3-fold activation of Kir6.2/SUR1bT1 currents. It is also noteworthy that, in contrast to what was observed for excised patches, both tolbutamide and glibenclamide blocked Kir6.2/SUR1bΔ33 currents in intact oocytes almost completely (Fig. 9).
Figure 9. Effects of metabolic inhibition.
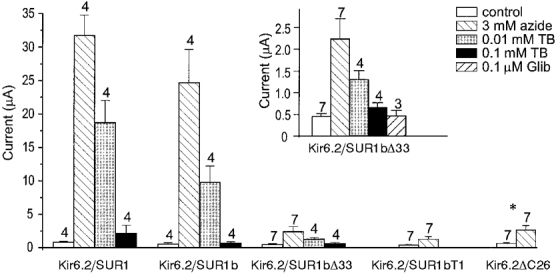
Mean whole-cell current amplitudes recorded in response to a 300 ms voltage step to -100 mV in control solution (□), 15 min after exposure to 3 mm azide ( ), and in the continued presence of azide plus either 0.01 mm tolbutamide (TB;
), and in the continued presence of azide plus either 0.01 mm tolbutamide (TB;  ) or 0.1 mm tolbutamide (▪). Oocytes were coinjected with mRNAs encoding Kir6.2 and SUR1, SUR1b, SUR1bΔ33 or SUR1bT1, as indicated, or were injected with mRNA encoding Kir6.2ΔC26. The number of oocytes is given above the bars. *Data taken from Tucker et al. (1997). Inset, mean whole-cell current amplitudes recorded at -100 mV in control solution, in the presence of 3 mm azide, in the presence of 3 mm azide plus 0.01 mm tolbutamide, 0.1 mm tolbutamide or 0.1 μm glibenclamide (Glib), as indicated. Oocytes were coinjected with mRNAs encoding Kir6.2 and SUR1bΔ33. The number of oocytes is given above the bars.
) or 0.1 mm tolbutamide (▪). Oocytes were coinjected with mRNAs encoding Kir6.2 and SUR1, SUR1b, SUR1bΔ33 or SUR1bT1, as indicated, or were injected with mRNA encoding Kir6.2ΔC26. The number of oocytes is given above the bars. *Data taken from Tucker et al. (1997). Inset, mean whole-cell current amplitudes recorded at -100 mV in control solution, in the presence of 3 mm azide, in the presence of 3 mm azide plus 0.01 mm tolbutamide, 0.1 mm tolbutamide or 0.1 μm glibenclamide (Glib), as indicated. Oocytes were coinjected with mRNAs encoding Kir6.2 and SUR1bΔ33. The number of oocytes is given above the bars.
There are two possible reasons for the smaller currents evoked by metabolic inhibition in oocytes expressing Kir6.2/SUR1bΔ33, Kir6.2/SUR1bT1 and Kir6.2ΔC26: either the number of KATP channels present in the plasma membrane is less than that found for the wild-type channel, or a similar number of KATP channels are present but they are less sensitive to metabolic inhibition. An indication of the level of functional channel expression can be obtained from the amplitude of the macroscopic current recorded in excised patches. Figure 10 therefore shows a comparison of the amplitude of the current at -100 mV activated by metabolic inhibition in the intact oocyte with that observed in the inside-out patch. In each case, we have expressed the current as a fraction of that recorded for Kir6.2/SUR1. It is evident that Kir6.2/SUR1bΔ33 and Kir6.2ΔC26 expressed much smaller currents than Kir6.2/SUR1, in both whole-cell and excised patch configurations. Nevertheless, the relative amount of current in the two configurations was similar, suggesting that the smaller currents evoked by metabolic inhibition are at least partly due to a reduced level of functional channel expression. The lower activation of Kir6.2/SUR1bT1 currents on metabolic poisoning, however, cannot be attributed to a reduced level of expression, because this channel exhibited large currents in excised patches, with amplitudes comparable to those observed for Kir6.2/SUR1b. Moreover, there was a profound difference in the relative amount of current observed in the whole-cell and excised patch configurations for this variant. This variant, therefore, appears to exhibit a marked reduction in the sensitivity of the channel to metabolic inhibition.
Figure 10.
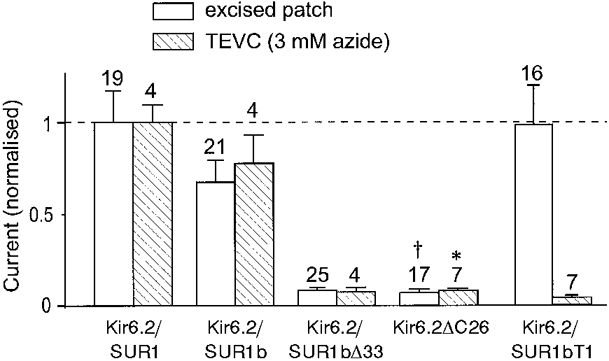
Mean current amplitudes recorded at -100 mV in the excised patch or whole-cell (TEVC) configuration, expressed as a fraction of the mean current recorded for Kir6.2/SUR1. Whole-cell currents were recorded in the presence of 3 mm azide. Oocytes were coinjected with mRNAs encoding Kir6.2 and SUR1, SUR1b, SUR1bΔ33 or SUR1bT1, as indicated, or were injected with mRNA encoding Kir6.2ΔC26. *Data taken from Tucker et al. (1997). † Data from Trapp et al. (1998). The number of oocytes is given above the bars.
DISCUSSION
The results of our cDNA library screening and RT-PCR experiments demonstrate that Kir6.2, SUR1 and SUR2B are expressed in the rodent hypothalamus. Thus it can be expected that both Kir6.2/SUR1 and Kir6.2/SUR2B channels exist in this region of the brain. Similar findings have been reported for the substantia nigra (Liss et al. 1999). In contrast, in an earlier single-cell PCR study of rat VMH neurones only SUR1 and Kir6.1 were detected (Lee et al. 1999). The sequence of SUR1 we identified differed from the original sequence by five amino acids. However, when SUR1b was coexpressed with Kir6.2, the properties of the KATP currents were not different from those of Kir6.2/SUR1 channels. Thus, these five amino acids are probably simply polymorphisms and in future SUR1b should be referred to as SUR1.
We also detected a novel variant of SUR1, SUR1bΔ33, which was expressed at low levels in hypothalamus, midbrain, heart and the insulin-secreting β-cell line MIN6. This variant would not have been distinguished by the primers used by Lee et al. (1999), because its sequence is identical to that of SUR1 in the amplified region. Several of the properties of the channels formed by coexpression of Kir6.2 and SUR1bΔ33 resemble those of Kir6.2ΔC26 expressed in the absence of the sulphonylurea receptor. This raises the question of whether SUR1bΔ33 facilitates the surface expression of full-length Kir6.2, but is unable to modify its functional properties fully. We believe this is unlikely for several reasons. Firstly, the Po of Kir6.2 is lower in the absence of SUR1 than in its presence (Proks & Ashcroft, 1997; Trapp et al. 1998; John et al. 1998; Mikhailov et al. 1998), whereas the Po of Kir6.2/ SUR1bΔ33 is substantially higher than that of Kir6.2ΔC26. Secondly, the tolbutamide dose-response curve for Kir6.2/ SUR1bΔ33 is best fitted assuming a two-site model, rather than the one-site model as for Kir6.2ΔC26 (Gribble et al. 1997b). Thirdly, a Ki of 86 μm for ATP inhibition would not be expected for Kir6.2ΔC26 if the Po were as high as that observed for Kir6.2/SUR1bΔ33 (Trapp et al. 1998). At a Po of 0.63, a Ki in the millimolar range is predicted. Thus it appears that SUR1bΔ33 is capable of functionally coupling to Kir6.2.
We did not detect SUR2A, either by RT-PCR or by cDNA library screening, which suggests that this subunit is not expressed to any marked extent in the hypothalamus. We detected Kir6.1 by RT-PCR of mouse hypothalamic mRNA, but not by screening a rat hypothalamic cDNA library. It seems unlikely that this is a species difference, but is rather a result of the greater sensitivity of the RT-PCR method.
ATP sensitivity
The ATP sensitivity of Kir6.2/SUR1bΔ33 channels is approximately 4-fold lower than that of Kir6.2/SUR1 channels. One might expect the ATP sensitivity of Kir6.2/SUR1bΔ33 channels to be higher than that of wild-type KATP channels, since mutations within the Walker A and Walker B motifs of NBD2 enhance channel inhibition by ATP (Gribble et al. 1997c, 1998). This is because MgATP (like MgADP) enhances KATP channel activity by interaction with the NBDs of SUR1, and thereby produces an apparent reduction in ATP sensitivity (Gribble et al. 1998). One possible reason for the paradoxical reduction in ATP sensitivity of Kir6.2/SUR1bΔ33 is that the loss of NBD2 might prevent the ability of SUR1 to ‘sensitise’ Kir6.2 to ATP. Tucker et al. (1997) have shown that when a truncated form of Kir6.2 is expressed by itself the Ki for ATP inhibition is ∼100 μm, but that this is shifted to 10-30 μm in the presence of SUR1. This idea seems unlikely, however, because Kir6.2/SUR1bT1 and Kir6.2/ SUR1bT2 channels lack NBD2 yet exhibit normal ATP sensitivity. An alternative explanation for the reduced ATP sensitivity of Kir6.2/SUR1bΔ33 channels is that it results from the higher channel open probability. This idea is supported by the fact that mutations within Kir6.2 which enhance Po produce a concomitant reduction in ATP sensitivity (Shyng et al. 1997a; Trapp et al. 1998). Furthermore, the open probabilities of Kir6.2/SUR1bT1 and Kir6.2/SUR1bT2, like their ATP sensitivity, are similar to that of Kir6.2/SUR1b channels.
Tolbutamide sensitivity
In excised patches, Kir6.2/SUR1bΔ33 channels exhibit a lower tolbutamide sensitivity than wild-type KATP channels. Our results suggest that this effect is not a consequence of a reduced binding affinity for the drug, since the Ki for tolbutamide block is unaffected and Kir6.2/SUR1bΔ33 channels exhibit a normal tolbutamide sensitivity in intact oocytes. Rather, they suggest that the reduced drug sensitivity results because the transduction of tolbutamide binding to SUR1bΔ33 into channel closure is impaired. This is suggested by the fact that the fraction of block associated with the high-affinity site is lower for Kir6.2/SUR1bΔ33 channels than for Kir6.2/SUR1b channels. There are at least three possible explanations for this finding. Firstly, residues within the C-terminus of SUR1, not present in SUR1bΔ33, may be directly involved in coupling SUR to Kir6.2. Secondly, the reduced drug potency may be a consequence of the higher Po of Kir6.2/SUR1bΔ33 channels, if tolbutamide stabilises the long closed state (Gillis et al. 1989). Indeed, we have shown elsewhere that an increased Po, resulting from a mutation in Kir6.2, is associated with a reduced block by tolbutamide (Trapp et al. 1998). Thirdly, the fact that Mg2+ is unable to enhance tolbutamide block of Kir6.2/SUR1bΔ33 currents may contribute to the lower efficacy of the drug. The first explanation is unlikely because tolbutamide blocks wild-type, Kir6.2/SUR1bΔ33 and Kir6.2/SUR1bT1 channels to a similar extent in the absence of Mg2+. The fact that Kir6.2/SUR1bT1 and Kir6.2/SUR1bΔ33 differ in their open probabilities supports the second possibility. The third explanation may also play a contributory role.
As is the case for native VMH KATP channels, the sulphonylurea sensitivity of Kir6.2/SUR1bΔ33 channels varies markedly between the intact cell and the excised patch. Glibenclamide, for example, produces no clear inhibition in the excised patch, yet causes complete block of the whole-cell current. A possible explanation for this difference is afforded by the fact that the ability of sulphonylureas to inhibit both the wild-type and Kir6.2/SUR1bΔ33 channel is enhanced by intracellular MgADP. In intact cells, magnesium nucleotides are likely to be present and will therefore contribute to the enhanced block by sulphonylureas. Indeed, both Kir6.2/SUR1b and Kir6.2/SUR1bΔ33 channels show greater inhibition by tolbutamide in intact cells than in excised patches (as is also the case for Kir6.2/SUR1 channels; compare Sakura et al. 1995 with Gribble et al. 1997b).
It is noteworthy that the inhibitory effect of MgADP on Kir6.2/SUR1bΔ33 channels in the presence of tolbutamide is greater than that observed for ADP in either the presence or the absence of Mg2+. This would not be expected if our hypothesis that tolbutamide abolishes the stimulatory effect of MgADP on wild-type channels is correct: rather, there should be no difference in the extent of block. The most likely explanation for this result is that the reduction in Po produced by tolbutamide leads to an enhanced sensitivity of Kir6.2 to nucleotide block. The fact that Kir6.2/SUR1bT1 channels, which have a Po close to wild-type, are blocked to a similar extent by ADP in the absence and presence of tolbutamide supports this view. Because the relationship between Po and nucleotides is exponential (Shyng et al. 1997a), ADP inhibition of Kir6.2/SUR1b and Kir6.2/SUR1bT1 channels (which have a lower intrinsic Po) is unaffected by tolbutamide.
Metabolic activation
In contrast to Kir6.2/SUR1 and Kir6.2/SUR1b, Kir6.2/SUR1bΔ33 and Kir6.2/SUR1bT1 channels show only a small activation by metabolic inhibition when expressed in Xenopus oocytes. Because Kir6.2/SUR1bT1 shows substantial currents in excised patches, it appears that the metabolic sensitivity of this channel is reduced. Thus, deletion of NBD2 prevents channel activation by metabolic inhibition, consistent with previous studies in which NBD2 mutations that prevent magnesium nucleotide activation also abolished the ability of metabolic block to stimulate channel activity (Nichols et al. 1996; Shyng et al. 1997b, 1998).
Current amplitudes observed in inside-out patches were much smaller for Kir6.2/SUR1bΔ33 than for Kir6.2/SUR1b. This suggests that the inability of metabolic inhibition to activate large Kir6.2/SUR1bΔ33 currents in intact oocytes is primarily due to a reduced level of surface expression. In contrast to Kir6.2/SUR1bT1, however, azide induced an ∼5-fold activation of Kir6.2/SUR1bΔ33. A similar increase was found for Kir6.2ΔC26 expressed in the absence of SUR. This cannot reflect magnesium nucleotide stimulation (mediated by SUR) of channel activity by metabolic inhibition, as this is not observed for either of these channels. We therefore speculate that it may be a consequence of a fall in intracellular ATP induced by azide, which is sensed by the Kir6.2 subunit. This is more evident for Kir6.2/SUR1bΔ33 (and Kir6.2ΔC26), than for Kir6.2/SUR1bT1, because both the former two channels have a lower ATP sensitivity. It seems likely, however, that activation of Kir6.2/SUR1bT1 channels would also occur, were ATP levels to fall further.
Is SUR1bΔ33 part of the VMH KATP channel?
Many of the properties of the rat VMH KATP channel differ from those of the archetypal KATP channel. Compared with Kir6.2/SUR1, the VMH channel has a reduced ATP sensitivity (Ki, 1-3 mm) and a different single-channel conductance (46 or 140 pS); it is also not activated by K+ channel openers, and is blocked by tolbutamide in intact cells but not in excised patches. Because the single-channel conductance and ATP inhibition of archetypal KATP channels are intrinsic to Kir6.2, while the sulphonylurea and K+ channel opener sensitivities are conferred by SUR, these results suggest that both the pore-forming and regulatory subunits of the VMH KATP channel may differ from those of archetypal KATP channels.
Channels formed by coexpression of SUR1bΔ33 with Kir6.2 share many properties with those of the native KATP channel of VMH neurones. In particular, the cloned channels are not activated by diazoxide or MgADP, and are blocked less by tolbutamide in excised patches than in intact oocytes. It is therefore possible that the clone we have isolated may encode the SUR subunit of the VMH KATP channel. One argument against this idea is the fact that only a single SUR1bΔ33 clone was detected in the hypothalamic cDNA library, compared with 14 SUR1b clones. This suggests that the expression level of SUR1bΔ33 in the hypothalamus is low. Furthermore, SUR1bΔ33 is also expressed at similarly low levels in midbrain, heart and the β-cell line MIN6, which possess classical KATP channels. In addition, SUR1 mRNA has been identified in individual VMH neurones by single-cell PCR (Lee et al. 1999). Nevertheless, it remains possible that SUR1bΔ33, but not SUR1b, is present in a specific neuronal population within the hypothalamus. A careful analysis of the mRNAs present in the VMH glucose-sensing neurones by single-cell PCR is needed, therefore, to confirm whether or not SUR1bΔ33 does indeed constitute the SUR subunit of VMH neurones.
Identification of the pore-forming subunit of the KATP channel from VMH neurones is complicated by the fact that single-channel conductances of 46 and ∼150 pS have been reported for rat (Ashford et al. 1990a; Routh et al. 1997)and 70 pS for mouse (Rowe et al. 1998). The latter value is close to that found for Kir6.2 (Sakura et al. 1995; Tucker et al. 1997), but the former values suggest that a different protein is involved. The reduced ATP sensitivity of the VMH KATP channel also supports this idea.
Functional consequences of C-terminal deletions of SUR1
The results presented in this paper indicate that the C-terminus of SUR1 influences KATP channel gating, that it plays an important role in the regulation of KATP channel activity by nucleotides and drugs, and that it is not needed for functional channel expression.
Previous studies have shown that truncation of up to 49 amino acids from the C-terminus of SUR1 prevents the cell surface expression of SUR1, as measured by luminometry using c-myc-labelled protein (Sharma et al. 1999). These authors also suggest that truncation of 221 amino acids prevents cell surface expression of SUR1, based on the fact that the protein is not glycosylated. We observed, however, that truncation of 253 amino acids (SUR1bT1) did not affect the magnitude of the macroscopic currents observed in excised patches, and that truncation of 210 amino acids (SUR1bT2) produced macroscopic current with a magnitude about 20 % of that observed for wild-type SUR1b, when coexpressed with Kir6.2. This indicates that C-terminal truncations of SUR1 do not necessarily prevent cell surface expression of the KATP channel.
Persistent hyperinsulinaemic hypoglycaemia of infancy (PHHI) results from mutations in SUR1 (usually) or Kir6.2 (rarely) that lead to the loss of functional KATP channels in the β-cell membrane and thus to continuous insulin secretion even at very low blood glucose concentrations (Aguilar-Bryan & Bryan, 1999). PHHI mutations within SUR1 either prevent surface expression of the KATP channel, or abolish the ability of MgADP to cause channel activation. In both cases, this leads to a loss of KATP channel activity in response to metabolic inhibition. It is interesting that a splice site mutation within intron 32, which causes premature protein truncation, produces PHHI (Thomas et al. 1995). This truncation is very similar to that found when exon 33 is deleted: it contains an additional 27 novel amino acids, rather than 25 novel residues, although the sequence is different. It would be interesting to know whether the intron 32 mutation is able to produce small KATP currents in excised patches exposed to nucleotide-free solution (when coexpressed with Kir6.2), as is found for the exon 33 deletion. And, in addition, whether any such currents show a response to metabolic inhibition similar to that of SUR1bΔ33 or whether, like SUR1bT1, they are not activated by metabolic inhibition.
Acknowledgments
We thank the Wellcome Trust and the British Diabetic Association for support.
References
- Aguilar-Bryan L, Bryan A. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocrine Reviews. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β-cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Ashcroft FM, Gribble FM. Correlating structure and function in ATP-sensitive K+ channels. Trends in Neurosciences. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- Ashcroft SJ, Ashcroft FM. Properties and functions of ATP-sensitive K-channels. Cellular Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. [DOI] [PubMed] [Google Scholar]
- Ashford ML, Boden PR, Treherne JM. Glucose-induced excitation of hypothalamic neurones is mediated by ATP-sensitive K+ channels. Pflügers Archiv. 1990a;415:479–483. doi: 10.1007/BF00373626. [DOI] [PubMed] [Google Scholar]
- Ashford ML, Boden PR, Treherne JM. Tolbutamide excites rat glucoreceptive ventromedial hypothalamic neurones by indirect inhibition of ATP-K+ channels. British Journal of Pharmacology. 1990b;101:531–540. doi: 10.1111/j.1476-5381.1990.tb14116.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clement JP, IV, Kunjilwar K, Gonzalez G, Schwanstecher M, Panten U, Aguilar-Bryan L, Bryan J. Association and stoichiometry of K-ATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- Gillis KD, Gee WM, Hammoud A, McDaniel ML, Falke LC, Misler S. Effects of sulfonamides on a metabolite-regulated intracellular ATP-sensitive potassium channel in rat pancreatic B-cells. American Journal of Physiology. 1989;257:C1119–1127. doi: 10.1152/ajpcell.1989.257.6.C1119. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Ashfield R, Ämmälä C, Ashcroft FM. Properties of cloned ATP-sensitive K+ currents expressed in Xenopus oocytes. The Journal of Physiology. 1997a;498:87–98. doi: 10.1113/jphysiol.1997.sp021843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The interaction of nucleotides with the tolbutamide block of cloned ATP-sensitive K+ channel currents expressed in Xenopus oocytes: a reinterpretation. The Journal of Physiology. 1997b;504:35–45. doi: 10.1111/j.1469-7793.1997.00035.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO Journal. 1997c;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Haug T, Ashcroft FM. MgATP activates the β cell KATP channel by interaction with its SUR1 subunit. Proceedings of the National Academy of Sciences of the USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Namba N, Inazawa J, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995a;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Seino S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Letters. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. Journal of Biological Chemistry. 1995b;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Isomoto S, Kondo C, Yamada M, Matsumoto S, Higashiguchi O, Horio Y, Matsuzawa Y, Kurachi Y. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. Journal of Biological Chemistry. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- John SA, Monck JR, Weiss JN, Ribalet B. The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293) The Journal of Physiology. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karschin A, Brockhaus J, Ballanyi K. KATP channel formation by the sulphonylurea receptors SUR1 with Kir6.2 subunits in rat dorsal vagal neurons in situ. Journal of Physiology. 1998;509:339–346. doi: 10.1111/j.1469-7793.1998.339bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlowski RZ, Hales CN, Ashford ML. Dual effects of diazoxide on ATP-K+ currents recorded from an insulin-secreting cell line. British Journal of Pharmacology. 1989;97:1039–1050. doi: 10.1111/j.1476-5381.1989.tb12560.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Dixon AK, Freeman TC, Richardson PJ. Identification of an ATP-sensitive potassium channel current in rat striatal cholinergic interneurones. The Journal of Physiology. 1998;510:441–453. doi: 10.1111/j.1469-7793.1998.441bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Dixon AK, Richardson PJ, Pinnock RD. Glucose-receptive neurones in the rat ventromedial hypothalamus express KATP channels composed of Kir6.1 and SUR1 subunits. The Journal of Physiology. 1999;515:439–452. doi: 10.1111/j.1469-7793.1999.439ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Ozanne SE, Hales CN, Ashford ML. Mg2+-dependent inhibition of KATP by sulphonylureas in CRI-G1 insulin-secreting cells. British Journal of Pharmacology. 1994;111:632–640. doi: 10.1111/j.1476-5381.1994.tb14783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liss B, Bruns R, Roeper J. Alternative sulphonylurea receptor expression defines metabolic sensitivity of K-ATP channels in dopaminergic midbrain neurons. EMBO Journal. 1999;18:833–846. doi: 10.1093/emboj/18.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikhailov MV, Proks P, Ashcroft FM, Ashcroft SJH. Expression of functionally active ATP-sensitive K-channels in insect cells using baculovirus. FEBS Letters. 1998;429:390–394. doi: 10.1016/s0014-5793(98)00640-1. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng SL, Nestorowicz A, Glaser B, Clement JP, IV, Gonzalez G, Aguilar-Bryan L, Permutt MA, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Proks P, Ashcroft FM. Phentolamine block of KATP channels is mediated by Kir6.2. Proceedings of the National Academy of Sciences of the USA. 1997;94:11716–11720. doi: 10.1073/pnas.94.21.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Routh VH, McArdle JJ, Levin BE. Phosphorylation modulates the activity of the ATP-sensitive K+ channel in the ventromedial hypothalamic nucleus. Brain Research. 1997;778:107–119. doi: 10.1016/s0006-8993(97)01043-3. [DOI] [PubMed] [Google Scholar]
- Rowe ICM, Spanswick D, Ashford MLJ. The characterization of potassium channels present in glucose- and sulphonylurea-sensitive hypothalamic neurones in isolation and in brain slices of the mf1 mouse. The Journal of Physiology. 1998;506.P:152P. [Google Scholar]
- Sakura H, Ämmälä C, Smith PA, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Letters. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Sellers AJ, Boden PR, Ashford ML. Lack of effect of potassium channel openers on ATP-modulated potassium channels recorded from rat ventromedial hypothalamic neurones. British Journal of Pharmacology. 1992;107:1068–1074. doi: 10.1111/j.1476-5381.1992.tb13408.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma N, Crane A, Clement JP, IV, Gonzalez G, Babenko AP, Bryan J, Aguilar-Bryan L. The C terminus of SUR1 is required for trafficking of KATP channels. Journal of Biological Chemistry. 1999;274:20628–20632. doi: 10.1074/jbc.274.29.20628. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Control of rectification and gating of cloned K-ATP channels by the Kir6.2 subunit. Journal of General Physiology. 1997a;110:141–153. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Nichols CG. Regulation of KATP channel activity by diazoxide and MgADP: Distinct functions of the two nucleotide binding folds of the sulfonylurea receptor. Journal of General Physiology. 1997b;110:643–654. doi: 10.1085/jgp.110.6.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Ferrigni T, Shepard JB, Nestorowicz A, Glaser B, Permutt MA, Nichols CG. Functional analyses of novel mutations in the sulfonylurea receptor 1 associated with persistent hyperinsulinemic hypoglycemia of infancy. Diabetes. 1998;47:1145–1151. doi: 10.2337/diabetes.47.7.1145. [DOI] [PubMed] [Google Scholar]
- Shyng SL, Nichols CG. Octameric stoichiometry of the KATP channel complex. Journal of General Physiology. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PM, Cote GJ, Wohllk N, Haddad B, Mathew PM, Rabl W, Aguilar-Bryan L, Gagel RF, Bryan J. Mutations in the sulphonylurea receptor gene in familial persistent hyperinsulinaemic hypoglycaemia of infancy. Science. 1995;268:425–429. doi: 10.1126/science.7716548. [DOI] [PubMed] [Google Scholar]
- Trapp S, Ashcroft FM. A metabolic sensor in action: News from the ATP-sensitive K+-channel. News in Physiological Sciences. 1997;12:255–263. [Google Scholar]
- Trapp S, Proks P, Tucker SJ, Ashcroft FM. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. Journal of General Physiology. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Proks P, Trapp S, Ryder TJ, Haug T, Reimann F, Ashcroft FM. Molecular determinants of KATP channel inhibition by ATP. EMBO Journal. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker SJ, Gribble FM, Zhao C, Trapp S, Ashcroft FM. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Tusnady GE, Bakos E, Varadi A, Sarkadi B. Membrane topology distinguishes a subfamily of the ATP-binding cassette (ABC) transporters. FEBS Letters. 1997;402:1–3. doi: 10.1016/s0014-5793(96)01478-0. [DOI] [PubMed] [Google Scholar]