

Abstract
Modulation of intracellular free Ca2+ concentration ([Ca2+]i) by extracellular ATP was investigated in cultured adult rat brown adipocytes using the fluorescent Ca2+ indicator fura-2.
Bath application of ATP in micromolar concentrations caused a large increase in [Ca2+]i in cells previously stimulated with noradrenaline. This ATP-induced [Ca2+]i increase exhibited a monotonic decline to near the resting levels within approximately 2 min, even in the continued presence of the agonist.
The magnitude and time course of the [Ca2+]i increase in response to ATP were not significantly affected by removal of extracellular Ca2+, suggesting that a mobilization of intracellular Ca2+ primarily contributes to the increase.
The [Ca2+]i increase in response to ATP was sensitive to inhibition by suramin, suggesting the involvement of P2 purinoceptors in the response.
Thapsigargin (100 nm) evoked a gradual and irreversible increase in [Ca2+]i which was entirely dependent upon extracellular Ca2+, providing functional evidence for the expression of store-operated Ca2+ entry in these brown adipocytes.
Extracellular ATP at a concentration of 10 μm depressed this thapsigargin (100 nm)-induced [Ca2+]i increase by 92 ± 3 % (n= 8 cells), strongly suggesting that ATP inhibits an influx of Ca2+ across the plasma membrane through the store-operated pathway. Bath application of phorbol 12-myristate 13-acetate (PMA, 5 μm) did not affect the thapsigargin-induced [Ca2+]i increase, indicating that the inhibitory action of ATP is not mediated by activation of protein kinase C (PKC).
These results indicate that extracellular ATP not only mobilizes Ca2+ from the intracellular stores but also exerts a potent inhibitory effect on the store-operated Ca2+ entry process in adult rat brown adipocytes.
There is increasing evidence indicating that ATP is released as a cotransmitter with noradrenaline from sympathetic nerve terminals and elicits potent, diverse and relevant physiological effects by interacting with ATP-specific receptors on the cell surface (for reviews see Harden et al. 1995; Burnstock, 1996). Previous workers have investigated the effects of ATP as well as noradrenaline on the activities of brown adipocytes in order to elucidate the mechanisms underlying the sympathetic control of cell functions. In neonatal rats, micromolar concentrations of extracellular ATP stimulate multiple activities of brown adipocytes, namely, an elevation in cytosolic free Ca2+ (Lee & Pappone, 1997), an increase in cell membrane capacitance (Pappone & Lee, 1996), a modulation of voltage-gated K+ currents (Wilson & Pappone, 1999) and a modest facilitation of heat production (Lee & Pappone, 1997). These actions of ATP were suggested to be mediated mainly through P2Y purinoceptors. On the other hand, in brown adipocytes α-adrenoceptor activation with noradrenaline has been shown to elevate cytosolic levels of inositol 1,4,5-trisphosphate (InsP3; Nånberg & Putney, 1986; Schimmel et al. 1986) and free Ca2+ (Wilcke & Nedergaard, 1989; Lee et al. 1993), while the stimulation of β-adrenoceptors by noradrenaline is known to primarily produce an acute thermogenic effect by activating cAMP-protein kinase A signalling cascade (Nicholls & Locke, 1984). Thus, intracellular free Ca2+ levels, which have been demonstrated to modify diverse cellular processes such as gene expression and cell proliferation in brown adipocytes (Lee et al. 1993), appear to be regulated not only by noradrenaline through an α-adrenoceptor but also by ATP via a P2Y purinoceptor. However, interactions between noradrenaline and ATP in the control of intracellular free Ca2+ levels have yet to be fully clarified.
In a variety of electrically non-excitable cells, stimulation of diverse plasma membrane receptors leading to an InsP3-dependent Ca2+ release from endoplasmic reticulum Ca2+ stores has been shown to be followed by activation of Ca2+ influx across the plasma membrane through a store-operated pathway (Putney, 1986; for reviews see Putney, 1990; Berridge, 1995; Parekh & Penner, 1997). The depletion of intracellular Ca2+ stores has been proposed to activate this Ca2+ entry process, although the precise signalling cascade responsible for Ca2+ entry remains to be fully identified. A store-operated pathway has been suggested to be a major Ca2+ entry mechanism in non-excitable cells, and most Ca2+ entering the cell through this pathway is likely to refill the intracellular Ca2+ stores and thus contribute to subsequent release from the stores. The stimulation of a P2 purinoceptor has been reported to be coupled to activation of the store-operated Ca2+ entry mechanism in various cell types (C6-2B glioma cells: Munshi et al. 1993, Chiono et al. 1995; rat megakaryocytes: Somasundaram & Mahaut-Smith, 1994; HT29 colonic epithelial cells, Kerst et al. 1995; human thyrocytes, Schöfl et al. 1995a; human gastric mucous cells: Schöfl et al. 1995b; calf pulmonary artery endothelial cells: Madge et al. 1997; human glioblastoma cells: Hartmann & Verkhratsky, 1998; for review see Parekh & Penner, 1997). It still remains unknown, however, whether this Ca2+ entry mechanism also operates in brown adipocytes.
The aim of the present study was to examine (i) the presence of the store-operated Ca2+ entry mechanism and (ii) the interactions of ATP and noradrenaline with this Ca2+ entry process, using fura-2-loaded rat brown adipocytes. Our results indicate that store-operated Ca2+ entry indeed exists and also exhibits a high sensitivity to inhibition by extracellular ATP in these brown adipocytes.
METHODS
Materials
Male 3-week-old Sprague-Dawley rats were purchased from Charles River Japan Inc. (Yokohama, Japan) and fed ad libitum for at least 1 week before use. Fraction V bovine serum albumin (BSA) was purchased from Intergen (Purchase, NY, USA), class II crude collagenase from Worthington Biochemical (Freehold, NJ, USA), and DNase I from Boehringer Mannheim Co. (Tokyo, Japan). Fura-2 acetoxymethyl ester (fura-2 AM) was obtained from Dojin Chemicals (Kumamoto, Japan), and suramin sodium salt, thapsigargin and phorbol 12-myristate 13-acetate (PMA) were from Wako Pure Chemicals Industries Ltd (Osaka, Japan). Adenosine 5′-triphosphate (ATP, disodium salt), adenosine 5′-diphosphate (ADP, sodium salt), uridine 5′-triphosphate (UTP, sodium salt), (−)noradrenaline hydrochloride, adenosine 5′-O-(3-thiotriphosphate) (ATPγS, tetralithium salt), α,β-methylene adenosine 5′-triphosphate (α,β-methylene ATP, lithium salt), ionomycin, valinomycin and poly-L-lysine were purchased from Sigma Chemical Co. (St Louis, MO, USA). Rabbit anti-uncoupling protein (UCP) serum was kindly provided by Dr T. Kawada (Kyoto University, Kyoto, Japan). All other reagents were obtained from Nacalai Tesque (Kyoto, Japan).
Cell isolation and culture
The method used for isolating brown adipocytes was a modification of that described previously (Omatsu-Kanbe et al. 1996). Rats aged 4–7 weeks were kept at 5°C for 5–8 h with free access to food and water in order to deplete stored lipid in brown adipose tissues. Rats were then deeply anaesthetized by an overdose of sodium pentobarbital (50 mg kg−1, intraperitoneal injection) and were killed by decapitation. Brown adipose tissue, carefully dissected out from interscapular regions, was placed in Krebs-Ringer bicarbonate Hepes (KRBH) buffer supplemented with BSA at a concentration of 1 % (w/v) under sterile conditions. KRBH buffer contained (mm): NaCl, 120; KH2PO4, 4; CaCl2, 2; MgSO4, 1; NaHCO3, 10; Hepes, 30 (pH adjusted to 7.4 with NaOH). Brown adipose tissue obtained from these cold-exposed rats was found to contain less lipid and therefore sank easily, as has been previously reported for cold-stressed neonatal rat brown adipose tissue (Lucero & Pappone, 1989). Both muscle and other tissues were then carefully trimmed away from the brown adipose tissue mass with round-tipped forceps to avoid any damage of the surface. The tissue was then minced and digested with a combination of collagenase and DNase I (7.5 mg ml−1 and 0.5 mg ml−1, respectively) at 37°C for 20 min. During the incubation, the mixture of cells and tissue fragments was dispersed by pipetting several times with a 3 ml plastic pipette. After digestion, the reaction mixture was filtered through a 100 μm nylon mesh. The filtrate was transferred to a 15 ml centrifuge tube and centrifuged at 100 g for 5 min, and then the floating tissue debris, white adipocytes and the buffer were discarded. The pelleted cells were resuspended in KRBH-BSA buffer and washed twice by centrifugation in this buffer. The cells were then suspended in the culture medium and washed three more times by centrifugation. The culture medium consisted of Dulbecco's modified Eagle's medium (DMEM) with 10 % fetal bovine serum, 100 μg ml−1 penicillin and 0.1 mg ml−1 streptomycin. After a final wash, the cells were resuspended in the culture medium and seeded on glass cover slips pre-coated with 0.1 % poly-L-lysine in 35 mm plastic culture dishes containing 3 ml culture medium. The brown adipocytes isolated by this procedure were found to contain very little fat, and most of the cells sank to the bottom of dishes and adhered to the cover slips within 30 min. All procedures were performed in plastic containers.
The cells were then maintained at 37°C in a humidified atmosphere of 95 % O2 and 5 % CO2. The majority of the cells isolated and cultured in this way are considered to be brown adipocytes, as judged by their morphological characteristics, such as the small size of the multilocular lipid droplets and the presence of a large number of mitochondria. These brown adipocytes consisted of mature-type cells with diameters of 20–35 μm and immature-type cells, with diameters of approximately 15 μm, classified as brown pre-adipocytes (Goglia et al. 1992). The mature brown adipocytes accumulated numerous lipid droplets during culture and retained the ability to respond to both noradrenaline and ATP for at least 6 days, while the immature-type cells neither responded to these stimulants nor accumulated any lipid, even after 6 days of culture. The mature-type brown adipocytes cultured for 1–4 days were used for experiments for the measurement of [Ca2+]i. All experiments were carried out according to the guidelines laid down by the Shiga University of Medical Science Animal Care Committee.
Immunofluorescent staining
The cells were fixed and immunostained based on standard immunohistochemical methods for tissue sections. Cells cultured on cover slips were rinsed with ice-cold 0.1 M phosphate-buffered saline (PBS, pH adjusted to 7.4), and were fixed with 4 % paraformaldehyde, 0.2 % picric acid and 0.35 % glutaraldehyde in 0.1 M phosphate buffer (pH adjusted to 7.4) at 5°C for 20 min. The fixed cells were washed three times with 15 % sucrose in 0.1 M phosphate buffer (pH adjusted to 7.4) and washed three more times with 0.1 M PBS containing 0.3 % Triton X-100. The cells were then incubated with anti-UCP antiserum (1:500) at 5°C for 3 days and stained with fluorescein isothiocyanate (FITC)-labelled anti-rabbit immunoglobulin G (1:100). After washing with PBS-Triton X-100, the cells were examined using a confocal laser microscope (MRC 600, BioRad, UK; Diaphot 300, Nikon, Tokyo, Japan).
Measurement of [Ca2+]i
Changes in [Ca2+]i were measured using the fluorescent Ca2+ indicator fura-2 from cells in a recording chamber perfused with the standard bath solution consisting of KRBH buffer with 5.6 mm glucose and 0.2 % BSA. The cells cultured on cover slips were loaded with membrane-permeant fura-2 AM (5 μm) in the dark for 30 min at 37°C and were washed twice in the standard bath solution. Fura-2-loaded cells were then incubated in the standard bath solution for 30 min at 37°C and were transferred to the recording chamber (approximately 0.5 ml in volume) mounted on the stage of an inverted microscope (Diaphot, Nikon, Tokyo, Japan). The recording chamber was maintained at 36°C and was perfused continuously at a rate of 1.5 ml min−1 with the standard bath solution or test solutions. The fluorescence measurement of [Ca2+]i was performed using a microspectrofluorometer (CAM-220, Japan Spectroscopic Co., Tokyo, Japan). The fluorescence intensity increased with an increase in incubation time and became saturated after 30–60 min of incubation, as has been described for neonatal rat brown adipocytes (Lee et al. 1993).
The [Ca2+]i values were calculated from the fluorescence ratios (R) acquired at 340 and 380 nm excitation wavelengths using the following equation (Grynkiewicz et al. 1985):
where Kd is the dissociation constant of fura-2 for Ca2+ (Kd= 224 nm, Grynkiewicz et al. 1985), Sf2/Sb2 is the fluorescence ratio of the 380 nm signal in the absence of Ca2+ to that in the presence of saturating Ca2+ (Sf2/Sb2= 2.35 ± 0.11, mean ±s.e.m., n= 8 cells), and Rmin and Rmax are minimal and maximal fluorescence ratios, respectively. Rmin, measured by incubating cells with a nominally Ca2+-free bath solution supplemented with 5 mm EGTA and 5 μm ionomycin, was 0.47 ± 0.03 (mean ±s.e.m., n= 8 cells) and an Rmax value of 4.85 ± 0.22 was obtained by incubating cells with the standard bath solution supplemented with 3 mm CaCl2 (total [Ca2+]o= 5 mm) and 5 μm ionomycin. All data were stored either on videotape after being digitized by a PCM processor (PCM-501 modified, SONY, Tokyo, Japan) or on a digital audiotape using a PCM data recorder (RD-120TE, TEAC, Tokyo, Japan) and were then transferred to a magnetic optical disk through a DigiData 1200 interface (Axon Instruments Co., Foster City, CA, USA) for a later analysis with pCLAMP software (Axon Instruments Co.).
The nominally Ca2+-free bath solution used for some experiments shown in Figs 4 and 7 was prepared by simply omitting CaCl2 from the standard bath solution.
Figure 4. Effects of the removal of extracellular Ca2+ on the noradrenaline- and ATP-evoked [Ca2+]i responses.
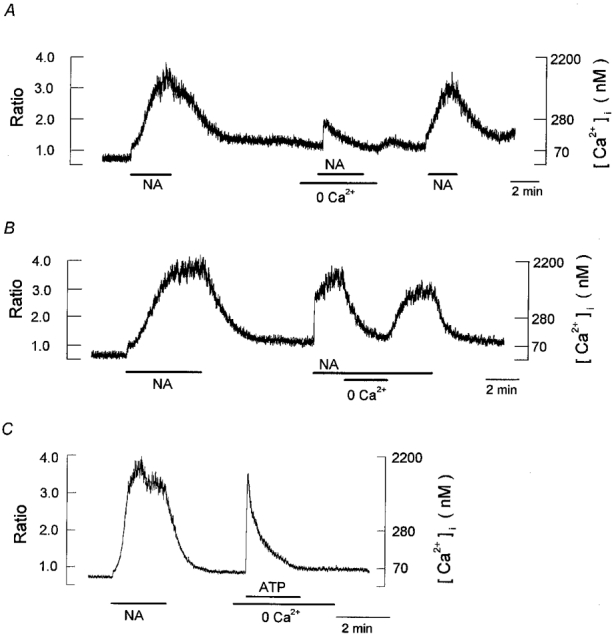
Periods of exposure to 1 μm noradrenaline (NA), 10 μm ATP and the nominally Ca2+-free bath solution (0 Ca2+) were indicated by the horizontal bars below the records. A, a single cell was exposed to noradrenaline in the standard bath solution ([Ca2+]o= 2.0 mm) and in nominally Ca2+-free bath solution, as indicated. Note that noradrenaline evoked only a transient and small increase in [Ca2+]i in the nominally Ca2+-free solution. B, the bath solution was changed to the nominally Ca2+-free solution with noradrenaline present. The noradrenaline-induced increase in [Ca2+]i was nearly completely abolished in the absence of extracellular Ca2+. C, a single cell was stimulated with noradrenaline in standard bath solution and then with ATP in nominally Ca2+-free bath solution. Each trace is obtained from different cells and is representative of 6–8 cells.
Figure 7. Presence of store-operated Ca2+ entry in brown adipocytes.
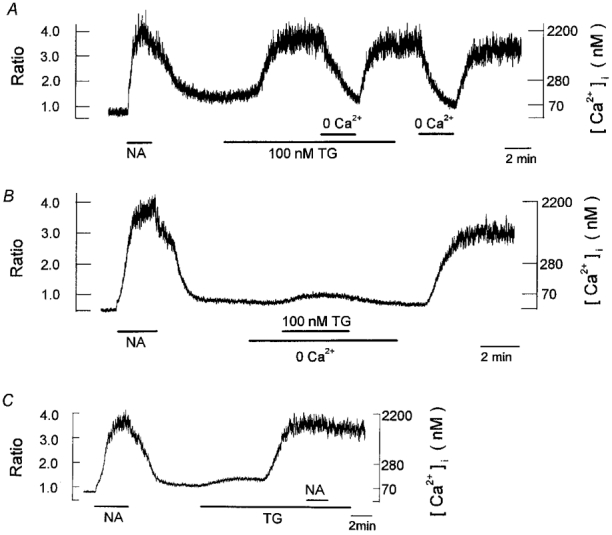
Periods of exposure to 1 μm noradrenaline (NA), 100 nm thapsigargin (TG), and the nominally Ca2+-free bath solution (0 Ca2+) were indicated by the horizontal bars below the records. A, a single cell was successively exposed to noradrenaline and thapsigargin. Extracellular Ca2+ was transiently removed during the presence of and after a washout of thapsigargin. B, a cell, previously stimulated with noradrenaline, was exposed to thapsigargin in the absence of extracellular Ca2+. Note that even after the withdrawal of thapsigargin a large and irreversible increase in [Ca2+]i was evoked by restoring extracellular Ca2+. C, a cell, previously stimulated with noradrenaline, was exposed to thapsigargin, which was followed by the addition of noradrenaline in the presence of thapsigargin. Each trace is obtained from different cells and is representative of 6–8 cells.
Data analysis and statistics
The results are expressed as means ±s.e.m. and n is the number of cells studied. Statistical comparisons were made using Student's t test for paired or unpaired data where appropriate, and differences were considered to be significant at P < 0.05.
RESULTS
Expression of uncoupling protein in cultured brown adipocytes
Brown adipocytes have been demonstrated to express the uncoupling protein (UCP) in their mitochondria, which uncouples substrate oxidation from ATP production, thus resulting in non-shivering thermogenesis. Using immunofluorescence techniques we first examined whether the mitochondrial UCP is expressed in cultured adult rat brown adipocytes prepared using the procedures described above. Figure 1 demonstrates representative results obtained using the well-characterized immune serum (Kawada et al. 1991; Hikichi et al. 1993). The protein expression of UCP can be clearly detected as bright signals in cells which had been cultured for 1 (left panel) and 4 days (right panel). Single brown adipocytes were thus found to maintain the characteristic expression of mitochondrial UCP for a period of 1–4 days of culture.
Figure 1. Immunofluorescent labelling of the mitochondrial UCP in cultured brown adipocytes.
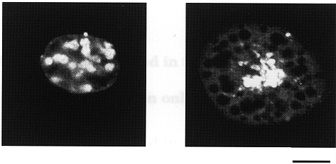
Cells cultured for 1 day (left) and 4 days (right) were fixed and incubated with anti-UCP antiserum. Immunoreactive proteins were stained with FITC-labelled anti-rabbit immunoglobulin G and were then examined using a confocal laser microscope. Immunolabelled proteins are detected as bright signals. Numerous lipid droplets accumulating in the cells are seen as dark circles. Calibration bar in the right panel represents 10 μm and also refers to the left panel.
Properties of noradrenaline- and ATP-evoked [Ca2+]i responses
We then examined [Ca2+]i responses of brown adipocytes to extracellular noradrenaline and ATP using the Ca2+-sensitive fluorescent dye fura-2. In a standard bath solution the resting [Ca2+]i averaged 57 ± 4 nm (n= 70 cells) and bath application of 1 μm noradrenaline evoked a sustained increase in [Ca2+]i with a mean amplitude of 1533 ± 154 nm (n= 10; Fig. 2A). This [Ca2+]i response to noradrenaline was concentration dependent and was saturated at 0.5 μm noradrenaline (data not shown), which is in good agreement with the concentration dependence of noradrenaline-induced [Ca2+]i responses in neonatal rat brown adipocytes (Lee et al. 1993). Successive applications of 1 μm noradrenaline at 5–6 min intervals caused increases in [Ca2+]i of a similar magnitude. However, the [Ca2+]i response during the first exposure to noradrenaline was found to develop much more slowly than that during subsequent exposure to noradrenaline. The times required to reach a peak level of [Ca2+]i after the first and second applications of 1 μm noradrenaline were 155.1 ± 12.8 and 30.3 ± 4.0 s (P < 0.0001, n= 10), respectively. The [Ca2+]i response to noradrenaline was completely abolished by 1 μm phentolamine (Fig. 2B), thus indicating the involvement of an α-adrenoceptor in [Ca2+]i responses.
Figure 2. Comparison of [Ca2+]i increases in response to noradrenaline and ATP.
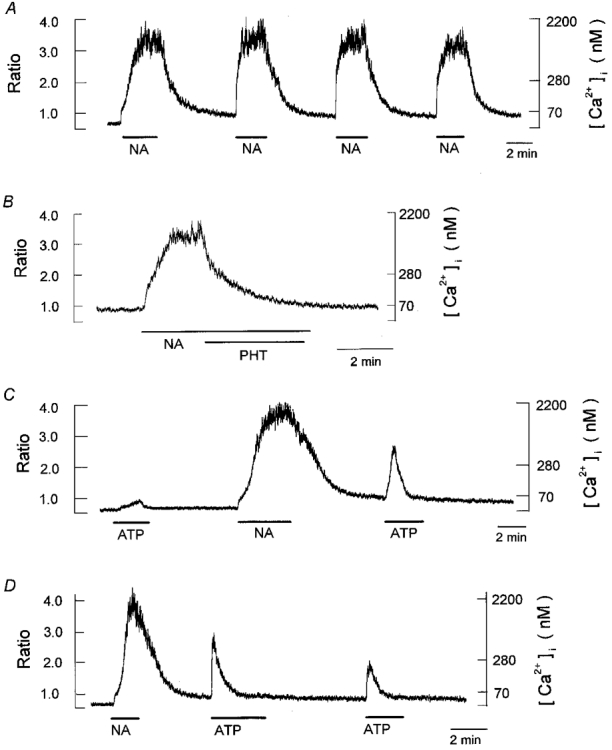
Chart record of [Ca2+]i responses during exposure to 1 μm noradrenaline (NA) and 10 μm ATP, indicated by the horizontal bars below the records. Here and in subsequent figures except Fig. 3, left scales give the fluorescence ratio and right scales the corresponding [Ca2+]i. A, [Ca2+]i responses during four successive applications of noradrenaline. B, inhibition of noradrenaline-induced [Ca2+]i increase by the α-adrenoceptor blocker phentolamine (1 μm, PHT). C, [Ca2+]i response in a cell initially exposed to ATP and then to noradrenaline, followed by a second application of ATP. D, [Ca2+]i responses during two successive applications of ATP at 5 min intervals in a cell previously exposed to noradrenaline. Each trace is obtained from different cells and is representative of 6–8 cells. Note the different time scale in panel B.
Bath application of 10 μm ATP induced only a small increase in [Ca2+]i with a mean amplitude of 53 ± 2 nm (n= 8), while application of ATP at the same concentration to cells previously stimulated with noradrenaline evoked a rapid increase in [Ca2+]i of 1057 ± 114 nm (n= 10; Fig. 2C). The time-to-peak [Ca2+]i in response to 10 μm ATP averaged 9.7 ± 1.9 s (n= 10). It should be noted that the ATP-evoked increase in [Ca2+]i subsided monotonically towards the resting levels within approximately 2 min, even in the continued presence of the agonist. Extracellular ATP at concentrations of 10 μm caused such a transient [Ca2+]i response in almost all (209/212) cells previously stimulated with 1 μm noradrenaline. Repeated applications of equimolar ATP with short wash-out periods were consistently accompanied by the diminished [Ca2+]i responses. As demonstrated in Fig. 2D, a second application of 10 μm ATP after a 5 min interval evoked only about 60 % of the [Ca2+]i response observed during the first application of ATP. When the interval between ATP applications was less than 2 min, the [Ca2+]i response during the second application of 10 μm ATP became negligibly small (data not shown). The ATP-evoked [Ca2+]i responses thus appear to be subject to some desensitization process, as previously reported for P2 purinoceptor-mediated [Ca2+]i increases in various cell types (Nobles et al. 1995; Bouyer et al. 1998).
The concentration-response relationship for the ATP-induced [Ca2+]i increase is illustrated in Fig. 3A. The [Ca2+]i response was evoked by extracellular ATP at concentrations of more than 0.1 μm and the maximal response was obtained with 10 μm ATP. The [Ca2+]i response was somewhat reduced when the concentration of ATP was further increased to 100 μm.
Figure 3. Properties of the ATP-induced [Ca2+]i response.

The [Ca2+]i response to extracellular ATP was measured in cells previously stimulated with noradrenaline (1 μm). A, concentration-response relationship for the increase in [Ca2+]i induced by extracellular ATP. The amplitude of the [Ca2+]i increase was calculated by subtracting the resting [Ca2+]i from the peak [Ca2+]i during exposure to ATP in each cell. Only one concentration of ATP was tested in a given cell to exclude the diminished response to multiple applications of ATP (cf. Fig. 2D). B, concentration-response relationship for the inhibition of the ATP-induced increase in [Ca2+]i by suramin. Cells were pretreated with suramin for 2 min and were then exposed to 10 μm ATP in the presence of suramin. Peak amplitudes of the ATP (10 μm)-induced [Ca2+]i increase are plotted against concentrations of suramin. Data in panels A and B represent the means ±s.e.m. of 8 cells.
A P2 purinoceptor antagonist suramin (for reviews see Burnstock, 1996; North & Barnard, 1997) was found to suppress the ATP-induced [Ca2+]i increase in a concentration-dependent manner (Fig. 3B). The pretreatment of cells with suramin at concentrations of 500 μm or more nearly completely inhibited the peak amplitudes of the [Ca2+]i response evoked by 10 μm ATP. This result suggests that extracellular ATP evokes a [Ca2+]i response in these brown adipocytes by binding to a P2 purinoceptor, consistent with a previous report on neonatal brown adipocytes (Lee & Pappone, 1997).
In order to elucidate whether the flux of extracellular Ca2+ into the cells is involved in agonist-induced [Ca2+]i increases, the effect of extracellular Ca2+ removal was examined in the experiments shown in Fig. 4. In the absence of extracellular Ca2+, bath application of 1 μm noradrenaline evoked a small transient increase in [Ca2+]i (224 ± 25 nm, n= 8; Fig. 4A), thus suggesting that the release of Ca2+ from the intracellular stores contributes at least partly to the initial rising phase of the [Ca2+]i response. The stimulation of α-adrenoceptor in brown adipocytes was demonstrated to induce an increase in InsP3 formation through the activation of phospholipase C (Nånberg & Putney, 1986; Schimmel et al. 1986). The release of Ca2+ from InsP3-sensitive stores such as the endoplasmic reticulum is therefore most likely to underlie noradrenaline-induced increases in [Ca2+]i recorded in the absence of extracellular Ca2+ (Fig. 4A), as has been previously suggested by Lee et al. (1993).
When extracellular Ca2+ was removed during a sustained elevation in [Ca2+]i in response to 1 μm noradrenaline, the [Ca2+]i declined to near the resting levels and this effect was reversible upon returning to the standard bath solution (Fig. 4B), thus indicating that the sustained elevation in [Ca2+]i during exposure to noradrenaline is produced primarily by an influx of Ca2+ from the extracellular solution. However, this noradrenaline-evoked sustained increase in [Ca2+]i was not appreciably affected by the L-type Ca2+ channel blocker nifedipine (1 μm), the T-type Ca2+ channel blocker flunarizine (1 μm), the P/Q-type Ca2+ channel blocker ω-conotoxin MVIIC (1 μm) and the N-type Ca2+ channel blocker ω-conotoxin MVIIA (1 μm) (data not shown), thus suggesting that the noradrenaline-induced Ca2+ entry which underlies the sustained increase in [Ca2+]i is not mediated through the activation of these voltage-dependent Ca2+ channels.
In contrast, in the absence of extracellular Ca2+, bath application of 10 μm ATP consistently evoked a rapid (time to peak, 9.2 ± 3.3 s; n= 8) increase in [Ca2+]i with a mean amplitude of 1004 ± 92 nm (n= 8) in cells previously stimulated with noradrenaline in the standard bath solution (Fig. 4C). The time course and amplitude of the ATP-induced [Ca2+]i increase in the absence of extracellular Ca2+ are similar to those (time to peak, 9.7 ± 1.9 s; amplitude, 1057 ± 114 nm; n= 10) of the ATP-induced [Ca2+]i increase recorded in the presence of standard bath solution ([Ca2+]o= 2 mm; cf. Fig. 1C and D), indicating that the [Ca2+]i increase evoked by ATP is primarily due to a mobilization of Ca2+ from the intracellular stores.
Interactions of P2 purinergic agonists and noradrenaline in the regulation of [Ca2+]i
To further characterize the effects of ATP on changes in [Ca2+]i, the interactions of ATP and noradrenaline in the regulation of [Ca2+]i were examined. In the experiments shown in Fig. 5A, the cell was previously stimulated with 1 μm noradrenaline, and 10 μm ATP was subsequently added to the bath solution about 2 min after starting a second application of noradrenaline. Extracellular ATP was found to greatly diminish the sustained levels of noradrenaline-induced [Ca2+]i increase without producing any additional increase in [Ca2+]i. In a total of eight cells, 10 μm ATP reduced the sustained levels of noradrenaline (1 μm)-induced [Ca2+]i increases by 92 ± 2 %. This ATP-induced depression consistently persisted after withdrawal of the ATP, and a subsequent application of ATP after a washout period of about 5 min did not have any appreciable effect on [Ca2+]i.
Figure 5. Inhibitory effect of extracellular ATP on the noradrenaline-evoked [Ca2+]i increase.

Periods of exposure to 1 μm noradrenaline (NA) and 10 μm ATP are indicated by horizontal bars below chart records of [Ca2+]i. A, ATP was added to the bath twice at an interval of about 5 min in the presence of noradrenaline, as indicated. The initial application of ATP greatly depressed the noradrenaline-induced increase in [Ca2+]i, the depression persisting after the withdrawal of ATP, and the second application was associated with a negligibly small change in [Ca2+]i. B, effects of pretreatment by ATP on the noradrenaline-induced [Ca2+]i increase. Application of noradrenaline in the presence of ATP was associated with a transient increase in [Ca2+]i. Each trace is obtained from different cells and is representative of 6–8 cells.
In the presence of 10 μm ATP, bath application of 1 μm noradrenaline only evoked a transient increase in [Ca2+]i of 254 ± 33 nm, (n= 8; Fig. 5B), which resembles in both magnitude and time course the [Ca2+]i response to noradrenaline recorded in the absence of extracellular Ca2+ (cf. Fig. 4A). Assuming that the sustained increase in [Ca2+]i during exposure to noradrenaline is largely due to noradrenaline-stimulated Ca2+ influx across the plasma membrane, it is reasonable to propose that extracellular ATP blocks noradrenaline-stimulated Ca2+ influx. Extracellular ATP was thus found not only to mobilize Ca2+ from intracellular stores (Fig. 4C) but also to exert an inhibitory effect on the process of Ca2+ influx mediating the sustained increase in [Ca2+]i in response to noradrenaline (Fig. 5).
We next examined the effect of other P2 purinergic agonists on both resting [Ca2+]i and the noradrenaline-evoked [Ca2+]i elevation. Bath application of 10 μm ADP induced a sustained increase in [Ca2+]i of 734 ± 74 nm (n= 6) in cells previously stimulated with 1 μm noradrenaline (Fig. 6A). In contrast to the effects of ATP, extracellular ADP did not exert any inhibitory effect on the noradrenaline-evoked sustained increase in [Ca2+]i (Fig. 6B, cf. Fig. 5A). As demonstrated in Fig. 6C, the external application of 10 μm UTP produced a sustained [Ca2+]i increase of 485 ± 44 nm (n= 6). Figure 6D illustrates the effects of 10 μm UTP on the noradrenaline-evoked [Ca2+]i increase. We could not detect any depression by UTP at concentrations of up to 100 μm of the noradrenaline-evoked [Ca2+]i increase. Bath application of AMP at concentrations of up to 100 μm did not elicit any effect on resting [Ca2+]i or the noradrenaline-stimulated [Ca2+]i elevation (data not shown).
Figure 6. Effects of ADP and UTP on resting [Ca2+]i and the noradrenaline-evoked [Ca2+]i increase.
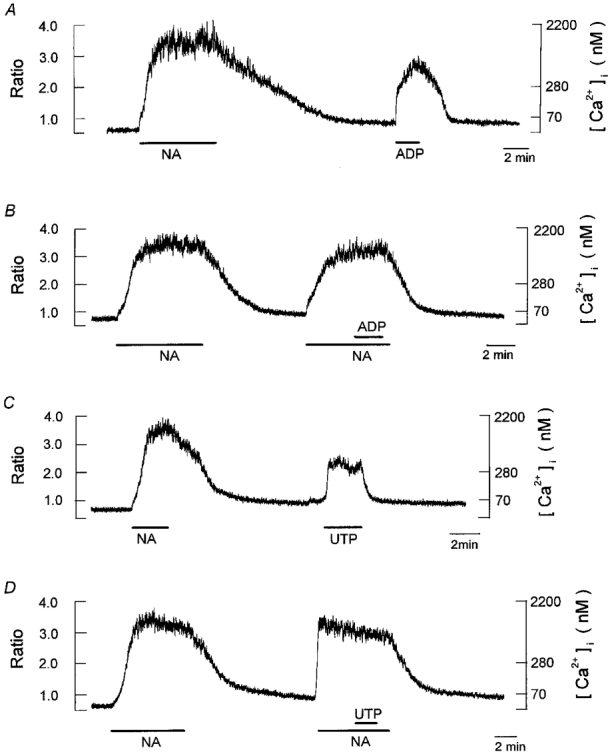
Periods of exposure to 1 μm noradrenaline (NA), 10 μm ADP and 10 μm UTP are indicated by horizontal bars below chart records of [Ca2+]i. A, a single cell was successively exposed to noradrenaline and ADP, as indicated. B, effect of addition of ADP on noradrenaline-induced increase in [Ca2+]i. C, a cell was initially exposed to noradrenaline and then to UTP. D, effects of application of UTP on the noradrenaline-induced increase in [Ca2+]i. Increases in [Ca2+]i evoked by ADP and UTP were not accompanied by a monotonic decline during the continued presence of the agonists. Neither ADP nor UTP exerted an inhibitory effect on the noradrenaline-induced increases in [Ca2+]i. Each trace is obtained from different cells and is representative of 6–8 cells.
Presence of store-operated Ca2+ entry in rat brown adipocytes
It has been reported in a number of electrically non-excitable cells that the depletion of intracellular Ca2+ stores triggers Ca2+ entry across the plasma membrane (Putney, 1986; for reviews see Putney, 1990; Berridge, 1995; Parekh & Penner, 1997). In order to determine whether the store-operated Ca2+ entry mechanism operates in rat brown adipocytes, we examined the effect on [Ca2+]i of thapsigargin, an agent which gradually depletes endoplasmic reticulum Ca2+ stores by specifically and irreversibly inhibiting Ca2+ uptake through endoplasmic reticulum Ca2+ pumps (Thastrup et al. 1990; Premack et al. 1994). In the presence of extracellular Ca2+, bath application of 100 nm thapsigargin consistently caused a gradual increase in [Ca2+]i of 1552 ± 141 nm (n= 8) in cells previously stimulated with 1 μm noradrenaline (Fig. 7A). This [Ca2+]i response to thapsigargin was reversibly abolished by removing extracellular Ca2+ and persisted even after withdrawal of the drug in the presence of extracellular Ca2+. When 100 nm thapsigargin was applied to the cell in the absence of extracellular Ca2+, the cell responded with a small elevation in [Ca2+]i of 84 ± 2 nm (n= 4), which presumably reflects the small amount of releasable Ca2+ stored in the endoplasmic reticulum in these brown adipocytes. However, once extracellular Ca2+ was restored, a large and irreversible elevation in [Ca2+]i was evoked, even after withdrawal of the thapsigargin (Fig. 7B). A similar [Ca2+]i response to thapsigargin was observed in cells which were not previously exposed to noradrenaline (data not shown). These rat brown adipocytes were thus found to possess store-operated Ca2+ entry mechanisms.
Noradrenaline has been suggested to activate InsP3-dependent mobilization of intracellular Ca2+ through stimulation of α-adrenoceptors in brown adipocytes (Nånberg & Putney, 1986; Schimmel et al. 1986; Lee et al. 1993). The question then arises as to whether the resultant emptying of the endoplasmic reticulum Ca2+ stores is followed by the activation of store-operated Ca2+ entry in association with a noradrenaline-induced sustained increase in [Ca2+]i which is entirely dependent upon extracellular Ca2+ (cf. Fig. 4B). In order to address this issue, we tested whether the stimulatory effect of noradrenaline on [Ca2+]i was additive to that of thapsigargin. In the experiments shown in Fig. 7C, the cell was initially exposed to 100 nm thapsigargin, which resulted in a sustained elevation in [Ca2+]i through the activation of store-operated Ca2+ entry. After the [Ca2+]i response to thapsigargin reached a steady state, noradrenaline at a maximally effective concentration (1 μm) was then added. In all eight cells examined subsequent addition of 1 μm noradrenaline in the presence of thapsigargin failed to cause any additional increases in [Ca2+]i, thus suggesting that the Ca2+ influx pathway activated in response to noradrenaline is the same as that activated by thapsigargin, i.e. store-operated Ca2+ entry.
Inhibition of the thapsigargin-induced [Ca2+]i increase by ATP and ATPγS
We then investigated the effects of extracellular ATP on [Ca2+]i elevation evoked by activation of store-operated Ca2+ entry (Fig. 8). When the gradual [Ca2+]i elevation in response to 100 nm thapsigargin reached a steady state, 10 μm ATP was added to the bath solution. In a total of eight cells, 10 μm ATP reduced the thapsigargin (100 nm)-induced [Ca2+]i elevation by 92 ± 3 % (upper panel), thus strongly suggesting that an influx of Ca2+ through the store-operated pathway is potently inhibited by extracellular ATP. It should be noted that the inhibition persisted for a relatively long period (≥ 10 min) after withdrawal of the ATP. Addition of the Ca2+ ionophore ionomycin (10 μm) to the bath solution after ATP application consistently induced an abrupt increase in [Ca2+]i (n= 4 cells), confirming that the dye was functioning normally and that the cells were viable. In addition, fluorescence signals following excitation at 340 and 380 nm (F340 and F380, respectively) were found to shift qualitatively as if they were mirror images during the course of the experiment (middle and lower panels), confirming again that [Ca2+]i was successfully recorded from intact cells. Therefore, the irreversible nature of the ATP action can be regarded as a genuine cellular response.
Figure 8. Inhibition of the thapsigargin-induced [Ca2+]i elevation by extracellular ATP.
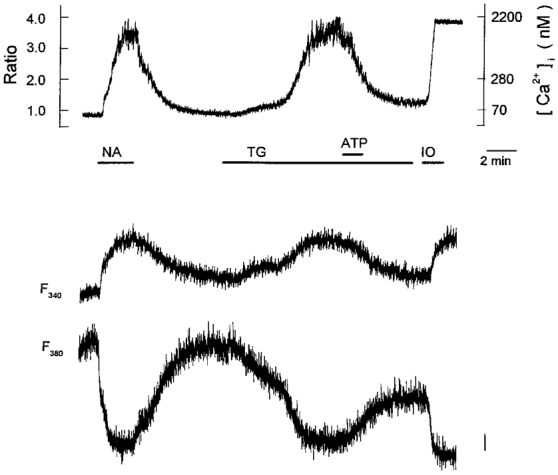
Periods of exposure to 1 μm noradrenaline (NA), 100 nm thapsigargin (TG), 10 μm ATP and 10 μm ionomycin (IO) are indicated by horizontal bars below the chart record of [Ca2+]i (upper panel). Fluorescence intensities following excitation at 340 and 380 nm are shown in the middle and lower panels, respectively. The vertical bar in the lower panel represents 50 arbitrary fluorescence intensity units and refers also to the middle panel. The trace is representative of 6 cells.
We tested whether the synthetic ATP analogues ATPγS and α,β-methylene ATP can mimic the inhibitory action of ATP on thapsigargin-induced [Ca2+]i elevation. Bath application of 10 μm ATPγS not only induced a transient increase in [Ca2+]i of 632 ± 79 nm (n= 6 cells) in cells previously stimulated with noradrenaline, but also reduced the thapsigargin-induced [Ca2+]i increase by 85 ± 2 % (Fig. 9A). This inhibitory effect of ATPγS also persisted for a period of at least 10 min after withdrawal of the ATPγS. In contrast, 10 μmα,β-methylene ATP neither elevated [Ca2+]i nor decreased thapsigargin-induced [Ca2+]i elevation (Fig. 9B).
Figure 9. Effects of ATPγS and α,β-methylene ATP on resting [Ca2+]i and the thapsigargin-evoked [Ca2+]i increase.

Periods of exposure to 1 μm noradrenaline (NA), 100 nm thapsigargin (TG), 10 μm ATPγS, and 10 μmα,β-methylene ATP (αβ-meATP) are indicated by horizontal bars below chart records of [Ca2+]i. The effects of ATPγS (A) and α,β-methylene ATP (B) on resting [Ca2+]i and the thapsigargin-induced [Ca2+]i increase were examined, as indicated. Each trace was obtained from different cells and is representative of 6 cells.
In neonatal rat brown adipocytes extracellular ATP was shown to activate a non-selective cation conductance which would depolarize the cell membrane to some extent (Lee & Pappone, 1997). Membrane depolarization attenuates the magnitude of Ca2+ entry through the store-operated pathway due to a reduced driving force for Ca2+ (Hoth & Penner, 1992). It is therefore probable that the inhibitory effect of ATP occurred secondarily to membrane depolarization. To test this possibility, we investigated the effect of ATP on thapsigargin-induced [Ca2+]i elevation in the presence of the K+ ionophore valinomycin (1 μm) which is expected to clamp the cell membrane near the reversal potential of K+. In the presence of valinomycin, 10 μm ATP still decreased thapsigargin-induced [Ca2+]i elevation by 93 ± 2 % (n= 6 cells; Fig. 10), which is similar to the decrease observed in the absence of valinomycin (92 ± 3 %, n= 8 cells; cf. Fig. 8). Thus, the inhibitory effect of ATP was not attenuated by stabilization of cell membrane potential with valinomycin, suggesting that the possible depolarization of cell membrane is not involved.
Figure 10. Effect of valinomycin on the inhibitory action of ATP on the thapsigargin-induced [Ca2+]i increase.
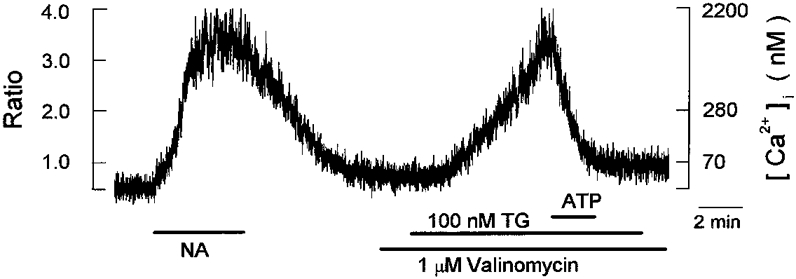
Periods of exposure to 1 μm noradrenaline (NA), 100 nm thapsigargin (TG), 10 μm ATP, 1 μm valinomycin are indicated by horizontal bars below the chart records of [Ca2+]i. The effect of ATP on the thapsigargin-induced [Ca2+]i increase was examined in the presence of valinomycin, as indicated. The trace is representative of 6 cells.
In a number of cell types the activation of protein kinase C (PKC) has been shown to inhibit Ca2+ influx through the store-operated pathway (e.g. HL-60 promyelocytes: Montero et al. 1993; Song et al. 1998; rat basophilic leukaemia cells: Parekh & Penner, 1995; rat mesangial cells: Mene et al. 1997; guinea-pig enteric glial cells: Sarosi et al. 1998). In order to address the question of whether PKC was involved in the inhibitory effect of ATP on the thapsigargin-induced [Ca2+]i increase, we examined whether PMA, a direct activator of PKC, could mimic the action of ATP. Bath application of 5 μm PMA did not produce any appreciable effects on the thapsigargin-induced [Ca2+]i increase in all six cells examined (Fig. 11), thus suggesting that an inhibitory action of ATP is not mediated by activation of PKC in rat brown adipocytes.
Figure 11. Effect of PMA on thapsigargin-evoked [Ca2+]i increase.
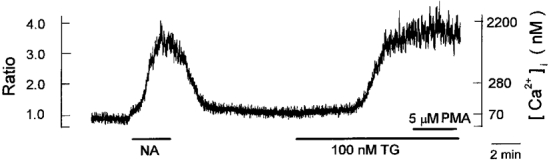
Periods of exposure to 1 μm noradrenaline (NA), 100 nm thapsigargin (TG) and 5 μm PMA are indicated by horizontal bars below chart records of [Ca2+]i. The trace is representative of 6 cells.
DISCUSSION
In the present study we characterized the [Ca2+]i responses to extracellular ATP in adult rat brown adipocytes. External application of ATP in micromolar concentrations evokes a large and transient increase in [Ca2+]i in cells previously stimulated with noradrenaline (Fig. 2). In addition, extracellular ATP exerts a potent inhibitory effect on both noradrenaline- and thapsigargin-induced [Ca2+]i increases (Figs 5 and 8), thus clearly showing that extracellular ATP exerts a dual effect on [Ca2+]i in these brown adipocytes.
Properties of the stimulatory effects of ATP on [Ca2+]i response
The [Ca2+]i response of brown adipocytes to extracellular ATP was first analysed in neonatal rats by Lee & Pappone (1997). These investigators demonstrated that bath application of ATP in micromolar concentrations evokes an increase in [Ca2+]i by activating both Ca2+ influx and Ca2+ release from intracellular stores. Our results in adult rat brown adipocytes, however, show that the removal of extracellular Ca2+ did not produce any appreciable effect on the ATP-induced increase in [Ca2+]i (Fig. 4C), which indicates that ATP exerts a stimulatory effect on [Ca2+]i mainly through mobilization of Ca2+ from the intracellular Ca2+ stores. The observation that the initial application of ATP is accompanied by a small increase in [Ca2+]i (Fig. 2C) may reflect the fact that the intracellular stores contain a low amount of releasable Ca2+ in these adult rat brown adipocytes under unstimulated basal conditions.
The suramin sensitivity of the ATP-induced [Ca2+]i increase (Fig. 3B) suggests the involvement of a P2 purinoceptor. In recent years P2 purinoceptors have been classified into two main subfamilies, namely, P2X purinoceptors comprising ligand-gated cation channels (an ionotropic receptor; Valera et al. 1994; Brake et al. 1994) and P2Y purinoceptors comprising G protein-coupled receptors (a metabotropic receptor; Lustig et al. 1993; Webb et al. 1993; for reviews see Burnstock, 1996; Burnstock & King, 1996; North & Barnard, 1997). The stimulation of both P2X and P2Y purinoceptors in various cell types has been shown to lead to an increase in [Ca2+]i through distinct intracellular mechanisms. An elevation in [Ca2+]i associated with P2X purinoceptor stimulation is elicited by the influx of Ca2+ through the ATP-gated non-selective cation channel (Michel et al. 1996; Troadec et al. 1998) and/or via the voltage-gated Ca2+ channel which is secondarily activated by membrane depolarization due to the influx of Na+ through the ATP-gated channel (Hirano et al. 1991). Extracellular Ca2+ is therefore essential for the [Ca2+]i increases via P2X purinoceptors (for review see Harden et al. 1995).
On the other hand, the P2Y purinoceptor-mediated [Ca2+]i increase in various cell types has been shown to be primarily evoked by the release of Ca2+ from InsP3-sensitive internal stores, i.e. the endoplasmic reticulum; thus this response is not obligatorily dependent upon the presence of extracellular Ca2+ (Nobles et al. 1995; Bouyer et al. 1998; Hartmann & Verkhratsky, 1998). Judging from the observation that the removal of extracellular Ca2+ was without effect (Fig. 4C), extracellular ATP is most likely to evoke a transient [Ca2+]i increase through a P2Y purinoceptor rather than via a P2X purinoceptor in this preparation. The finding that micromolar levels of UTP increased [Ca2+]i with a high potency (Fig. 6C) further supports the involvement of a P2Y purinoceptor in this [Ca2+]i response, since none of P2X purinoceptor subtypes (P2X1-P2X7) was effectively activated by UTP (for review see Burnstock & King, 1996). The post-receptor process mediating the [Ca2+]i increase in response to extracellular ATP is presently unclear; however, most of metabotropic P2Y purinoceptors have been reported to be coupled to phospholipase Cβ through a GTP binding protein, resulting in activation of InsP3-mediated mobilization of intracellular Ca2+ (for review see Burnstock & King, 1996). Further experiments are called for to explore the possible involvement of the InsP3-dependent process in ATP-induced [Ca2+]i elevation in brown adipocytes.
Presence of store-operated Ca2+ entry and its inhibition by extracellular ATP
It has been demonstrated in a variety of electrically non-excitable cells that InsP3-mediated Ca2+ release and the resultant depletion of endoplasmic reticulum Ca2+ stores trigger Ca2+ entry across the plasma membrane through a store-operated pathway (Putney, 1986; for reviews see Putney, 1990; Berridge, 1995; Parekh & Penner, 1997). This store-operated Ca2+ entry mechanism can also be pharmacologically activated by thapsigargin, a highly specific and irreversible inhibitor of endoplasmic reticulum Ca2+-ATPase (Thastrup et al. 1990; Premack et al. 1994). The present experiments clearly demonstrated that the application of thapsigargin consistently evoked an irreversible increase in [Ca2+]i that was entirely dependent upon extracellular Ca2+ (Fig. 7A and B), which strongly indicates the presence of store-operated Ca2+ entry in these brown adipocytes.
The stimulation of α1-adrenoceptors with noradrenaline has been shown to cause a sustained increase in [Ca2+]i in brown adipocytes of hamster (Wilcke & Nedergaard, 1989) and neonatal rat (Lee et al. 1993). This α1-adrenoceptor-mediated [Ca2+]i increase was clearly demonstrated to arise from two distinct pathways, i.e. an initial mobilization of intracellular Ca2+ followed by a sustained Ca2+ influx process (Lee et al. 1993). We confirmed in adult rat brown adipocytes that a similar consecutive activation of Ca2+ release and Ca2+ influx underlies an increase in [Ca2+]i in the presence of noradrenaline (Fig. 4A and B). The present experiments also show that the stimulatory effect of noradrenaline on [Ca2+]i is not additive to that of thapsigargin (Fig. 7C), which suggests that the Ca2+ influx process associated with noradrenaline stimulation is evoked through the store-operated pathway. Taken together, in rat brown adipocytes, noradrenaline appears to increase [Ca2+]i by initially mobilizing internal Ca2+ through an InsP3-dependent pathway, followed by the activation of Ca2+ influx through the store-operated pathway triggered by the resultant depletion of endoplasmic reticulum Ca2+ stores. The slow onset of the [Ca2+]i increase during the first exposure to noradrenaline (Fig. 2A) is, again, thought to be related to the nature of intracellular Ca2+ stores containing a small amount of releasable Ca2+ under unstimulated conditions.
Micromolar levels of extracellular ATP nearly completely reduced the sustained increase in [Ca2+]i evoked by thapsigargin (Fig. 8), thus strongly suggesting that in rat brown adipocytes store-operated Ca2+ entry is potently inhibited by extracellular ATP. This inhibitory action of ATP was not affected by the K+ ionophore valinomycin (Fig. 10), ruling out the possible involvement of membrane potential changes. Stimulation of PKC with PMA did not mimic the inhibitory effect of ATP (Fig. 11), indicating that the inhibitory effect is not mediated by activation of PKC. Extracellular ATP at 10 μm reduced the sustained increases in [Ca2+]i evoked by noradrenaline and thapsigargin to a similar extent (92 ± 2 % reduction with noradrenaline, n= 8 cells; 92 ± 3 % reduction with thapsigargin, n= 8 cells). In addition, ATP irreversibly depressed both the noradrenaline- (Fig. 5A) and thapsigargin-induced [Ca2+]i increases (Fig. 8). These similarities in the sensitivity of depression of [Ca2+]i increases to extracellular ATP are also consistent with the activation of store-operated Ca2+ entry in the presence of noradrenaline. The inhibitory effect of ATP on the noradrenaline- or thapsigargin-induced increase in [Ca2+]i was mimicked by ATPγS (Fig. 9A) but not by ADP, UTP or α,β-methylene ATP (Figs 6B and D, and 9B). It still remains unclear whether extracellular ATP inhibits store-operated Ca2+ entry by binding to a P2 purinoceptor in brown adipocytes; however, none of the P2X and P2Y purinoceptors identified to date exhibit such a high agonist selectivity for ATP and ATPγS (for reviews see Burnstock & King, 1996; North & Barnard, 1997).
The ATP-induced [Ca2+]i elevation is consistently accompanied by a monotonic decline to near resting levels, even in the continued presence of ATP, which forms a striking contrast to the noradrenaline-induced [Ca2+]i increase, which remains stable near peak levels during the entire period of agonist application (Fig. 2). In several cell types, extracellular ATP has been shown to induce InsP3-dependent Ca2+ release followed by the activation of store-operated Ca2+ entry, by stimulating metabotropic purinoceptors (Munshi et al. 1993; Somasundaram & Mahaut-Smith, 1994; Chiono et al. 1995; Kerst et al. 1995; Schöfl et al. 1995a,b; Madge et al. 1997; Hartmann & Verkhratsky, 1998). Assuming that these two cellular processes are consecutively activated during exposure to ATP in brown adipocytes, the subsequently activated store-operated Ca2+ entry should be almost completely inhibited by the presence of extracellular ATP; as a result, only cytosolic Ca2+ initially released from the endoplasmic reticulum can contribute to the changes in [Ca2+]i, and this phenomenon may account for the characteristic transient form of the ATP-induced [Ca2+]i elevation.
Acknowledgments
We would like to thank Mr T. Yamamoto (Central Research Laboratory, Shiga University of Medical Science) for assisting in the use of the confocal laser microscope, Dr B. Quinn for critically reading the manuscript and Ms Y. Tanigaki for her secretarial assistance. This study was supported by a Grant-in-Aid for Scientific Research on Priority Areas (no. 07276102) from the Ministry of Education, Science, Sports and Culture of Japan.
References
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouyer P, Paulais M, Cougnon M, Hulin P, Anagnostopoulos T, Planelles G. Extracellular ATP raises cytosolic calcium and activates basolateral chloride conductance in Necturus proximal tubule. The Journal of Physiology. 1998;510:535–548. doi: 10.1111/j.1469-7793.1998.535bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brake AJ, Wagenbach MJ, Julius D. New structural motif for ligand-gated ion channels defined by an ionotropic ATP receptor. Nature. 1994;371:519–523. doi: 10.1038/371519a0. [DOI] [PubMed] [Google Scholar]
- Burnstock G. P2 purinoceptors: historical perspective and classification. Ciba Foundation Symposium. 1996;198:1–28. doi: 10.1002/9780470514900.ch1. [DOI] [PubMed] [Google Scholar]
- Burnstock G, King BF. Numbering of cloned P2 purinoceptors. Drug Development Research. 1996;38:67–71. [Google Scholar]
- Chiono M, Mahey R, Tate G, Cooper DMF. Capacitative Ca2+ entry exclusively inhibits cAMP synthesis in C6–2B glioma cells: evidence that physiologically evoked Ca2+ entry regulates Ca2+-inhibitable adenylyl cyclase in non-excitable cells. Journal of Biological Chemistry. 1995;270:1149–1155. doi: 10.1074/jbc.270.3.1149. [DOI] [PubMed] [Google Scholar]
- Goglia F, Géloën A, Lanni A, Minaire Y, Bukowiecki LJ. Morphometric-sterologic analysis of brown adipocytes differentiation in adult mice. American Journal of Physiology. 1992;262:C1018–1023. doi: 10.1152/ajpcell.1992.262.4.C1018. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Harden TK, Boyer JL, Nicholas RA. P2-purinergic receptors: subtype-associated signaling responses and structure. Annual Review of Pharmacology and Toxicology. 1995;35:541–579. doi: 10.1146/annurev.pa.35.040195.002545. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Verkhratsky A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. The Journal of Physiology. 1998;513:411–424. doi: 10.1111/j.1469-7793.1998.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hikichi Y, Sugihara H, Sugimoto E. Differentiation of brown adipose cells in three-dimensional collagen gel culture. Pathology Research and Practice. 1993;189:73–82. doi: 10.1016/S0344-0338(11)80119-6. [DOI] [PubMed] [Google Scholar]
- Hirano Y, Abe S, Sawanobori T, Hiraoka M. External ATP-induced changes in [Ca2+]i and membrane currents in mammalian atrial myocytes. American Journal of Physiology. 1991;260:C673–680. doi: 10.1152/ajpcell.1991.260.4.C673. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Kawada T, Sakabe S, Aoki N, Watanabe T, Higeta K, Iwai K, Sugimoto E. Intake of sweeteners and pungent ingredients increases the thermogenin content in brown adipose tissue of rat. Journal of Agricultural Food Chemistry. 1991;39:651–654. [Google Scholar]
- Kerst G, Fischer K-G, Normann C, Kramer A, Leipziger J, Greger R. Ca2+ influx induced by store release and cytosolic Ca2+ chelation in HT29 colonic carcinoma cells. Pflügers Archiv. 1995;430:653–665. doi: 10.1007/BF00386159. [DOI] [PubMed] [Google Scholar]
- Lee SC, Nuccitelli R, Pappone PA. Adrenergically activated Ca2+ increases in brown fat cells: effect of Ca2+, K+, and K channel block. American Journal of Physiology. 1993;264:C217–228. doi: 10.1152/ajpcell.1993.264.1.C217. [DOI] [PubMed] [Google Scholar]
- Lee SC, Pappone PA. Effects of P2 purinergic receptor stimulation in brown adipocytes. American Journal of Physiology. 1997;273:C679–686. doi: 10.1152/ajpcell.1997.273.2.C679. [DOI] [PubMed] [Google Scholar]
- Lucero MT, Pappone PA. Voltage-gated potassium channels in brown fat cells. Journal of General Physiology. 1989;93:451–472. doi: 10.1085/jgp.93.3.451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lustig KD, Shiau AK, Brake AJ, Julius D. Expression cloning of an ATP receptor from mouse neuroblastoma cells. Proceedings of the National Academy of Sciences of the USA. 1993;90:5113–5117. doi: 10.1073/pnas.90.11.5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madge L, Marshall ICB, Taylor CW. Delayed autoregulation of the Ca2+ signals resulting from capacitative Ca2+ entry in bovine pulmonary artery endothelial cells. The Journal of Physiology. 1997;498:351–369. doi: 10.1113/jphysiol.1997.sp021863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mene P, Pugliese G, Pricci F, Di Mario U, Cinotti GA, Pugliese F. High glucose level inhibits capacitative Ca2+ influx in cultured rat mesangial cells by a protein kinase C-dependent mechanism. Diabetologia. 1997;40:521–527. doi: 10.1007/s001250050710. [DOI] [PubMed] [Google Scholar]
- Michel AD, Grahames CB, Humphrey PP. Functional characterisation of P2 purinoceptors in PC12 cells by measurement of radiolabelled calcium influx. Naunyn-Schmiedeberg's Archives of Pharmacology. 1996;354:562–571. doi: 10.1007/BF00170829. [DOI] [PubMed] [Google Scholar]
- Montero M, Garcia-Sancho J, Alvarez J. Inhibition of the calcium store-operated calcium entry pathway by chemotactic peptide and by phorbol ester develops gradually and independently along differentiation of HL60 cells. Journal of Biological Chemistry. 1993;268:26911–26919. [PubMed] [Google Scholar]
- Munshi R, Debernardi MA, Brooker G. P2U-purinergic receptors on C6–2B rat glioma cells: modulation of cytosolic Ca2+ and cAMP levels by protein kinase C. Molecular Pharmacology. 1993;44:1185–1191. [PubMed] [Google Scholar]
- Nånberg E, Connolly E, Nedergaard J. Presence of a Ca2+-dependent K+ channel in brown adipocytes. Possible role in maintenance of α1-adrenergic stimulation. Biochimica et Biophysica Acta. 1985;844:42–49. doi: 10.1016/0167-4889(85)90231-9. [DOI] [PubMed] [Google Scholar]
- Nånberg E, Putney J., Jr α1-Adrenergic activation of brown adipocytes leads to an increased formation of inositol polyphosphates. FEBS Letters. 1986;195:319–322. doi: 10.1016/0014-5793(86)80185-5. [DOI] [PubMed] [Google Scholar]
- Nicholls DG, Locke RM. Thermogenic mechanisms in brown fat. Physiological Reviews. 1984;64:1–64. doi: 10.1152/physrev.1984.64.1.1. [DOI] [PubMed] [Google Scholar]
- Nobles M, Revest PA, Couraud P-O, Abbott NJ. Characteristics of nucleotide receptors that cause elevation of cytoplasmic calcium in immortalized rat brain endothelial cells (RBE4) and in primary cultures. British Journal of Pharmacology. 1995;115:1245–1252. doi: 10.1111/j.1476-5381.1995.tb15032.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Current Opinion in Neurobiology. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Omatsu-Kanbe M, Ding W-G, Hashiramoto M, Kitasato H. Immunohistochemical localization of cellubrevin on secretory granules in pancreatic B-cells. Archives of Histology and Cytology. 1997;60:289–295. doi: 10.1679/aohc.60.289. [DOI] [PubMed] [Google Scholar]
- Omatsu-Kanbe M, Zarnowski MJ, Cushman SW. Hormonal regulation of glucose transport in a brown adipose cell preparation isolated from rats that shows a large response to insulin. Biochemical Journal. 1996;315:25–31. doi: 10.1042/bj3150025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappone PA, Lee SC. Purinergic receptor stimulation increases membrane trafficking in brown adipocytes. Journal of General Physiology. 1996;108:393–404. doi: 10.1085/jgp.108.5.393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Depletion-activated calcium current is inhibited by protein kinase in RBL-2H3 cells. Proceedings of the National Academy of Sciences of the USA. 1995;92:7907–7911. doi: 10.1073/pnas.92.17.7907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Premack BA, McDonald TV, Gardner P. Activation of Ca2+ current in Jurkat T cells following the depletion of Ca2+ stores by microsomal Ca2+-ATPase inhibitors. Journal of Immunology. 1994;152:5226–5240. [PubMed] [Google Scholar]
- Putney JW., Jr A model for receptor-regulated calcium entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW., Jr Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Sarosi GA, Barnhart DC, Turner DJ, Mulholland MW. Capacitative Ca2+ entry in enteric glia induced by thapsigargin and extracellular ATP. American Journal of Physiology. 1998;275:G550–555. doi: 10.1152/ajpgi.1998.275.3.G550. [DOI] [PubMed] [Google Scholar]
- Schimmel RJ, Dzierzanowski D, Elliott ME, Honeyman TW. Stimulation of phosphoinositide metabolism in hamster brown adipocytes exposed to α1-adrenergic agents and its inhibition with phorbol esters. Biochemical Journal. 1986;236:757–764. doi: 10.1042/bj2360757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schöfl C, Rössig L, Pötter E, Von Zur Mühlen A, Brabaut G. Extracellular ATP and UTP increase cytosolic free calcium by activating a common P2U-receptor in single human thyrocytes. Biochemical and Biophysical Research Communications. 1995a;213:928–934. doi: 10.1006/bbrc.1995.2218. [DOI] [PubMed] [Google Scholar]
- Schöfl C, Rössig L, Von Zur Mühlen A, Beil W, Jähne J, Manns MP, Wagner S. Extracellular nucleotides increase cytosolic free calcium by activating P2U-receptors in single human gastric mucous cells. Biochemical and Biophysical Research Communications. 1995b;216:636–641. doi: 10.1006/bbrc.1995.2669. [DOI] [PubMed] [Google Scholar]
- Somasundaram B, Mahaut-Smith MP. Three cation influx currents activated by purinergic receptor stimulation in rat megakaryocytes. The Journal of Physiology. 1994;480:225–231. doi: 10.1113/jphysiol.1994.sp020355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song SK, Choi SY, Kim KT. Opposing effects of protein kinase A and C on capacitative calcium entry into HL-60 promyelocytes. Biochemical Pharmacology. 1998;56:561–567. doi: 10.1016/s0006-2952(97)00660-6. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Troadec J-D, Thirion S, Nicaise G, Lemos JR, Dayanithi G. ATP-evoked increases in [Ca2+]i and peptide release from rat isolated neurohypophysial terminals via a P2X2 purinoceptor. The Journal of Physiology. 1998;511:89–103. doi: 10.1111/j.1469-7793.1998.089bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valera S, Hussy N, Evans RJ, Adami N, North RA, Surprenant A, Buell G. A new class of ligand-gated ion channel defined by P2X receptor for extracellular ATP. Nature. 1994;371:516–519. doi: 10.1038/371516a0. [DOI] [PubMed] [Google Scholar]
- Webb TE, Simon J, Krishek BJ, Bateson AN, Smart TG, King BF, Burnstock G, Barnard EA. Cloning and functional expression of a brain G-protein-coupled ATP receptor. FEBS Letters. 1993;324:219–225. doi: 10.1016/0014-5793(93)81397-i. [DOI] [PubMed] [Google Scholar]
- Wilcke M, Nedergaard J. α1- and β-adrenergic regulation of intracellular Ca2+ levels in brown adipocytes. Biochemical and Biophysical Research Communications. 1989;163:292–300. doi: 10.1016/0006-291x(89)92134-7. [DOI] [PubMed] [Google Scholar]
- Wilson SM, Pappone PA. P2 receptor modulation of voltage-gated potassium currents in brown adipocytes. Journal of General Physiology. 1999;113:125–138. doi: 10.1085/jgp.113.1.125. [DOI] [PMC free article] [PubMed] [Google Scholar]