

Abstract
The pdx1 gene is essential for pancreatic organogenesis in humans and mice; pdx1 mutations have been identified in human diabetic patients. Specific inactivation of pdx1 in adult β cells revealed that this gene is required for maintenance of mature β cell function. In the following study, a Cre-lox strategy was used to remove pdx1 function specifically from embryonic β cells beginning at late-gestation, prior to islet formation. Animals in which pdx1 is lost in insulin-producing cells during embryogenesis had elevated blood glucose levels at birth and were overtly diabetic by weaning. Neonatal and adult mutant islets showed a dramatic reduction in the number of insulin+ cells and an increase in both glucagon+ and somatostatin+ cells. Lineage tracing revealed that excess glucagon+ and somatostatin+ cells did not arise by interconversion of endocrine cell types. Examination of mutant islets revealed a decrease in proliferation of insulin-producing cells just before birth and a concomitant increase in proliferation of glucagon-producing cells. We propose that pdx1 is required for proliferation and function of the β cells generated at late gestation, and that one function of normal β cells is to inhibit the proliferation of other islet cell types, resulting in the appropriate numbers of the different endocrine cell types.
Keywords: pancreas, islet, diabetes, Cre-lox, lineage tracing
Introduction
Mature pancreatic islets are composed of five different endocrine cell types (α: glucagon, β: insulin, δ: somatostatin, ε: ghrelin, and PP: pancreatic polypeptide) arranged in a typical architecture where the β cells, which make up the large majority, are found at the core and the other cell types are located at the periphery or mantle. Pancreatic endocrine cells appear in two waves during mouse pancreatic organogenesis (Pictet, et al. 1972; Pang, et al. 1994). During the first wave, which begins at embryonic day 9.5 (e9.5), the glucagon- and insulin-expressing cells that arise differ in their gene expression pattern from the mature cells found in adult islets. First wave glucagon-producing cells express pro-hormone convertase 1/3 (PC1/3) rather than PC2, which is expressed by mature α and β cells (Lee, et al. 1999; Wilson, et al. 2002), and first wave insulin-producing cells lack the glucose transporter, GLUT2, a marker of mature β cells (Pang, et al. 1994). Insulin/glucagon double-positive cells have been detected at early developmental stages and have been postulated to represent an intermediate stage of endocrine differentiation (Alpert, et al. 1988; Teitelman, et al. 1993). Lineage tracing studies, however, suggest that these cells do not in fact give rise to α or β cells of mature islets (Herrera, et al. 1994; Herrera 2000). Thus, the function of these first wave single and double hormone-positive cells remains unclear.
The second wave of endocrine differentiation occurs between e13.5–16.5, and is believed to produce the cells of the mature islets (Pictet, et al. 1972; Pang, et al. 1994). The mechanism driving the relatively sudden increase in endocrine cell formation at this particular time in development is unknown, although signals including the Notch/Ngn3 pathway that affect precursor proliferation and differentiation are likely to be involved (Apelqvist, et al. 1999; Murtaugh, et al. 2003; Jensen 2004). At late gestation and early postnatal stages, the islets begin to adopt their characteristic core/mantle architecture. Little is known about how the different endocrine cell types or their precursors interact with one another to generate the correct proportions of the various cell types. Several studies suggest that early glucagon-producing cells are required specifically for the generation of first wave insulin-producing cells (Dohrmann, et al. 2000; Prasadan, et al. 2002; Vincent, et al. 2003), but it is uncertain whether similar cell-cell interactions are involved in generating endocrine cells at the secondary transition.
Reverse genetic studies in mice are helping to elucidate the complex pathways and gene interactions involved in pancreas development (Wilson, et al. 2003). Genetic or cell deletion studies in the absence of lineage tracing techniques can, however, result in difficulties in deciphering cell differentiation pathways. Loss of one cell population in a particular mutant condition may have a profound non-cell autonomous effect on another cell population. For example, selective deletion of PP-producing cells, which results in the loss of both insulin- and somatostatin-producing cells, could be explained either by a PP-expressing precursor cell population giving rise to these two cell types, or by PP-expressing cells producing an inductive signal required for the production of β and δ cells (Herrera, et al. 1994). Lineage tracing analyses have helped distinguish between these possibilities, and suggest that mature β cells do indeed arise from cells that previously expressed PP (Herrera 2000).
The homeobox gene pdx1 is expressed within the developing pancreatic endoderm in all vertebrates so far examined (Gannon and Wright 1999). In the mouse, pdx1 expression begins at e8.0 (Guz et al., 1995; Li, et al. 1999), prior to the onset of pancreatic bud formation and islet hormone gene expression, and is initially detected throughout the pancreatic epithelium. By late gestation, pdx1 expression is selectively maintained at high levels in β cells, with low levels of expression in acinar cells (Guz, et al. 1995; Wu, et al. 1997). Loss of pdx1 function results in an early block in pancreatic outgrowth and differentiation in both mice and humans (Jonsson, et al. 1994; Offield, et al. 1996; Stoffers, et al. 1997). The pancreatic rudiment of pdx1 null mouse embryos does contain transient, first wave insulin+ cells (Ahlgren, et al. 1996), and longer-lived glucagon+ cells (Offield, et al. 1996), indicating that pdx1 is not required to generate first wave endocrine cells. In addition to an early role in pancreatic bud outgrowth, studies using tetracycline-inducible pdx1 inactivation demonstrated that pdx1 is also specifically required between e11.5 and e13.5 in order for subsequent differentiation of endocrine and exocrine cells (Holland, et al. 2002).
Mice heterozygous for a pdx1 deficiency are glucose-intolerant (Ahlgren, et al. 1998; Dutta, et al. 1998; Brissova, et al. 2002), consistent with the finding that humans carrying dominant pdx1 mutations are predisposed to a form of Type 2 diabetes called maturity onset diabetes of the young type 4 (MODY4) (Stoffers, et al. 1997; Stoffers, et al. 1997; Stoffers, et al. 1998; Macfarlane, et al. 2000). The continued essential role for pdx1 in mature β cells (Ahlgren, et al. 1998; Holland, et al. 2002) fits well with its identification as a direct activator of several β cell-specific genes that control glucose utilization and metabolism, including insulin, IAPP, glucokinase, Pax4, and pdx1 itself (Chakrabarti, et al. 2002; Cissell, et al. 2002).
Direct evidence that pdx1 is essential for maintaining mature β cell function comes from studies using tetracycline-inducible pdx1 inactivation in adult mice (Holland, et al. 2002) as well as conditional inactivation studies using an insulin promoter-driven Cre transgene that resulted in a loss of Pdx1 protein between 3–5 weeks after birth (Ahlgren, et al. 1998). This mature β cell-specific loss of pdx1 caused a dramatic decrease in insulin, Nkx6.1, and GLUT2 expression, a concomitant increase in the number of glucagon+ cells, and overt diabetes in 3–5 month old mice. The excess glucagon+ cells and large number of insulin/glucagon co-producing cells that were detected in the islets of these mice led to the suggestion that insulin+ cells acquire glucagon expression after removal of the repressive influences of Pdx1. In the absence of lineage tracing, it is impossible to determine the origin of the excess glucagon+ cells. One can envisage several ways in which such cells could arise from β cells or an insulin-expressing precursor cell type: (1) mature β cells de-differentiate to a more immature glucagon/insulin co-expressing cell type; (2) β cells slowly trans-differentiate towards a mature α cell type; or, (3) β cells loss promotes generation of new endocrine cells from an unidentified progenitor cell, which then gives rise to insulin/glucagon double-positive cells.
In summary, therefore, while pdx1 is critical early in pancreas development for global organ formation and differentiation, as well as later in mature β cells, it is unclear what role it plays at the secondary transition in generating the β cells that will contribute to mature islets. We report here the results of a Cre-lox conditional inactivation study that provides details on pdx1 function early in the definitive β cell lineage (during the second wave of endocrine differentiation), in which we assessed the consequences of pdx1 inactivation by including lineage-tracing analysis. In this study, we used an optimized rat insulin promoter-Cre transgenic line (RIP-Cre; Postic, et al. 1999; Gannon, et al. 2000) which shows functional recombination as early as e11.5 in developing pancreas and efficient (>85% of β cells) excision of loxP-flanked DNA (Postic, et al. 1999; Gannon, et al. 2000). We demonstrate that loss of pdx1 from early β cells leads to a severe reduction in the number of insulin+ cells beginning at late gestation stages. β cell-specific inactivation of pdx1 during embryogenesis results in early-onset diabetes, disrupted islet architecture, and increased numbers of glucagon+ and somatostatin+ cells at the expense of β cells. The lineage-tracing analysis allowed tracking of the fate of cells that activated the RIP-Cre transgene. We provide evidence for a linkage between the overall β cell number and the mechanisms that control the proliferation/differentiation of glucagon+ cells.
Materials and Methods
Construction of floxed pdx1 allele and generation of mice
The pdx1tm4(E2)Cvw-TKneo targeting construct (see Figure 1) was generated using the pLox-TKneo vector (Orban, et al. 1992). Standard subcloning methods were used to introduce loxP sites into the intron and just after the poly-A addition site, thus flanking the DNA binding domain-containing exon 2 (Figure 1; details available upon request). The integrity of pdx1 sequences and loxP sites was checked by diagnostic restriction enzyme digestion and sequencing. Plasmid DNA was CsCl-purified, the entire insert isolated by low-melt agarose gel electrophoresis after NotI digestion, and purified using Gelase (Epicentre Technologies).
Figure 1.
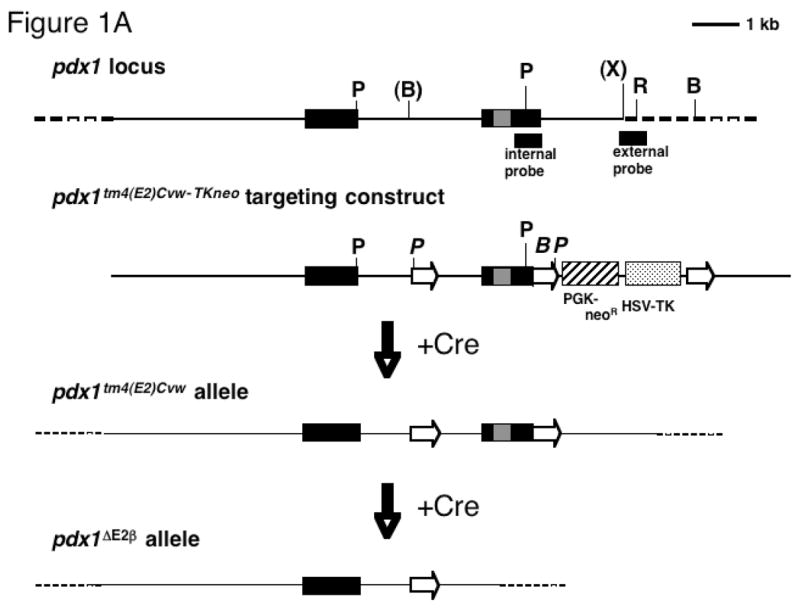
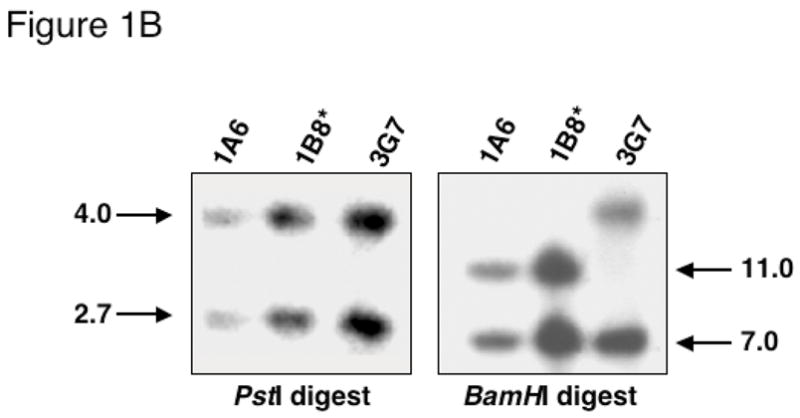
(A) Diagram of pdx1 locus, loxP-containing targeting construct, and expected products following Cre-mediated recombination. Black boxes: exons, gray box: homeodomain, solid line: genomic regions used in targeting construct, open arrows: loxP sites, cross-hatched box: neomycin resistance cassette driven by the PGK promoter, stippled box: thymidine kinase cassette driven by the HSV promoter. Location of internal and external probes for Southern blotting is indicated below the locus. Restriction enzyme sites: B, BamHI; P, PstI; X, XbaI. Sites in parentheses are lost in the generation of the targeting construct. Sites in italics are introduced in the targeting construct. (B) Southern blot of DNA from three representative neomycin-resistant ES cell clones using internal probe. PstI digest (left panel): 4.0 kb band, endogenous allele; 2.7 kb band, floxed allele. BamHI digest (right panel): 7.0 kb band, endogenous allele; 11.0 kb band, floxed allele. Asterisk: clone used for Cre electroporation.
200 μg of linearized pdx1tm4(E2)Cvw-TKneo DNA was electroporated into TL1 129 SvEv ES cells by the Vanderbilt University Transgenic/ES Cell Core, and ES cell clones were grown as described (Hogan 1994). Correctly targeted ES cell clones were identified by Southern blot analysis of genomic DNA [PstI or BamHI digest, 500 bp 3′ pdx1 cDNA internal probe (Figure 1A,B); XbaI digest, 550 bp XbaI-EcoRI external probe (data not shown)]. On PstI digestion, the 3′ cDNA probe recognizes a 4.0 kb endogenous locus band and a 2.7 kb band for the loxP-marked allele. BamHI digestion generates an endogenous allele band of 7.0 kb or an 11.0 kb band from the loxP-marked allele. A total of 543 ES cell clones were screened with a targeting efficiency of 6.8%. Thus, 37 correctly targeted clones were identified. The 1B8 ES cell clone containing the correctly targeted pdx1tm4(E2)Cvw-TKneo allele was electroporated with CMV-Cre plasmid to remove the PGK-neoR/HSV-TK cassettes. Cre-mediated excision of the selection cassette was determined using an 800 bp fragment of the neoR cassette, which recognizes a 1.0 kb PstI band. The presence of exon two was confirmed by retention of the 2.7 kb PstI band using the 3′ cDNA probe. One clone undergoing the desired recombination event (1B8/10) was injected into C57BL/6 blastocysts to generate chimeric mice. Male chimeras were bred to Black Swiss females and agouti offspring were genotyped by Southern analysis for transmission of the pdx1tm4(E2)Cvw allele (PstI digestion; 3′ pdx1 cDNA probe) from here on termed pdx1flE2. pdx1flE2 heterozygotes were bred to produce pdx1flE2/flE2 mice, which are phenotypically normal by several criteria (see Results).
The RIP-Cre line used in these studies has been described (Postic, et al. 1999; Gannon, et al. 2000). RIP-Cre transgenics were identified by Southern analysis (EcoRI digestion; 500 bp BamHI-ClaI Cre probe). R26R mice, originally from Philippe Soriano (Soriano 1999), were a gift of Richard Behringer (M.D. Anderson Cancer Center, Houston, TX) and were genotyped by Southern analysis (EcoRI digestion; 726 bp PvuI lacZ probe). RIP-Cre;R26R bigenic mice were mated with mice heterozygous for the pdx1XBKo null allele generated previously by our laboratory (Offield, et al. 1996). We will refer to these mice as pdx1+/−. pdx1+/−;RIP-Cre;R26R mice were generated at the expected frequency of 1:8. The pdx1 null allele was identified by Southern blotting (Offield, et al. 1996). pdx1flE2/flE2 mice were bred to pdx1+/−;RIP-Cre;R26R mice to generate pdx1flE2/−;RIP-Cre;R26R mice at a frequency of 1:8. pdx1flE2/−;RIP-Cre mice will be referred to as pdx1ΔE2β All animals were given water and Lab Diet #5015 mouse pellets ad lib on a 12 hour light/dark cycle.
Tissue preparation and histology
The morning of the vaginal plug was considered e0.5. Embryonic digestive organs, including pancreata, were dissected in PBS and fixed immediately in ice-cold 4% paraformaldehyde (4°C; 45 minutes to 1 hour). Tissues were dehydrated in an increasing ethanol series and embedded in paraffin for sectioning. Serial 7 μm sections mounted on glass slides with Sta-on (Surgipath) were used for histology, immunohistochemistry, and immunofluorescence analyses. Analyses were performed on at least five sections from each of at least three separate animals. Frozen tissues were fixed as above, incubated in 30% sucrose overnight at 4°C, embedded in optimum cutting temperature compound (VWR Scientific, West Chester, PA), and 5 μm sections were cut on a Leica CM 3050 S cryostat.
β-galactosidase detection
Following fixation, tissues were washed twice for 30 minutes in permeabilization solution (2 mM MgCl2, 0.01% sodium deoxycholate, 0.02% Nonidet P-40 in PBS). β-galactosidase (β-gal) activity was detected using X-gal as described (Wu, et al. 1997; Gannon, et al. 2000). Tissues were post-fixed and dehydrated for embedding as above, with isopropanol replacing xylene to minimize leaching of the blue precipitate. Images were collected on an Olympus BX41 microscope with an Olympus digital camera and Magnafire program (Optronics, Inc.). Images were all equivalently processed in Adobe Photoshop 6.0.
Immunohistochemistry and immunofluorescence
Sections were deparaffinized in xylene and rehydrated in a descending ethanol series to water. Immunoperoxidase staining for insulin, glucagon, and somatostatin was performed as previously described (Gannon, et al. 2000). Primary antibodies were used at the following dilutions: guinea pig anti-bovine insulin (Linco), 1:1000; rabbit anti-glucagon (Linco), 1:1000; guinea pig anti-glucagon (Linco), 1:500; rabbit anti-human somatostatin (Dako), 1:1000; rabbit anti-Pdx1, 1:25 (Peshavaria, et al. 1994); rabbit anti-GLUT2 (a gift of Bernard Thorens, University of Lausanne), 1:500; rabbit anti-phospho-histone H3, 1:50 (Upstate Biotechnology). Two different antibodies against β-galactosidase were utilized: a rabbit anti-β-galactosidase generated against an XlHbox4-β-gal fusion protein that specifically recognizes β-galactosidase on western blot (Gannon, et al. 2001), and rabbit anti-β-gal 1:5000 (MP Biomedicals). Antigen retrieval [10 mM citrate buffer, microwave oven 3 minutes then 1 minute at 1000W, 20 minutes at room temperature] was performed for immunodetection of Pdx1, GLUT2, phospho-histone H3, and β-gal. Samples were viewed under bright field illumination and photographed with Kodak Ektachrome 64T film, or using an Olympus BX41 microscope and digital camera (Magnafire program). For immunofluorescence, donkey anti-guinea pig CY2 and donkey anti-rabbit CY3 or CY5 were used as secondary antibodies. CY2 was excited at 543nm, CY3 at 488nm, and CY5 at 647nm using an LSM 410 confocal microscope (Zeiss). Optical sections were taken at 1 μm. TIFF images from each experiment were processed equivalently in Adobe Photoshop 6.0.
Glucose tolerance tests (GTT)
Intraperitoneal glucose tolerance tests were performed as described (Gannon, et al. 2000). Following a 16 hour fast, baseline blood glucose levels (mg/dl) were measured in 10 μl tail vein blood from mildly anesthetized mice using the Accu-Chek Advantage glucose meter and Accu-Chek test strips (Roche). Glucose (2 mg dextrose/g body weight) in sterile PBS was injected intraperitoneally and blood glucose measured 15, 30, 60, 90 and 120 minutes after injection. GTT were performed every 2 weeks for 2–4 months beginning at weaning. Insulin radioimmunoassay was performed as previously described (Gannon, et al. 2000).
Results
The floxed pdx1 allele functions as a wild type allele
In order to produce a conditional pdx1 null allele, loxP sites were placed on either side of exon two, which encodes the DNA binding homeodomain. The introduction of the 34 bp loxP sequences at these two locations did not interfere with pdx1 gene function of the floxed allele. pdx1flE2/+ and pdx1flE2/flE2 animals were born at the expected frequency, survived through adulthood, and were outwardly indistinguishable from control littermates. In neonates and one month-old animals, the gross morphology (size and shape) of the pancreata of pdx1flE2/+ and pdx1flE2/flE2 mice was identical to control littermates, as was islet number, size, and architecture (data not shown). On intraperitoneal glucose tolerance test (IP-GTT), animals of all three genotypes (+/+, flE2/+, flE2/flE2) had a fasting blood glucose level in the normal range (70–150 mg/dl) and efficiently cleared a glucose bolus from the bloodstream, returning to baseline glucose levels within a two-hour period (data not shown). We conclude that the floxed pdx1 allele is a true conditional null allele with wild type function in the non-recombined configuration.
RIP-Cre mediated pdx1 inactivation occurs during embryonic stages
To inactivate pdx1 specifically in embryonic pancreatic β cells, mice carrying the floxed pdx1 allele were bred to mice carrying a RIP-Cre transgene. Inclusion of the R26R reporter allele, in which expression of a lacZ reporter cassette depends upon Cre-mediated removal of a loxP-flanked “stop” sequence (Soriano 1999), allowed the detection of cells that have undergone recombination in our experiments. Using R26R mice, we previously characterized the RIP-Cre transgene as being active in isolated pancreatic cells at e11.5 and restricted to insulin-producing cells of the pancreas at all times examined (Gannon, et al. 2000). At P1 and in adults, recombination-based activation of β-gal expression was detected in >85% of insulin-producing cells in the islet core. Expression was never observed in glucagon-expressing cells, acinar cells, or other cells of the foregut.
We reasoned that placing the pdx1flE2 allele in trans with a pdx1 null allele (Offield, et al. 1996) would require Cre-mediated inactivation of only a single floxed pdx1 allele, facilitating the generation of pdx1 null β cells (pdx1ΔE2β). To this end, pdx1+/− mice were bred to RIP-Cre;R26R mice (see Methods). Matings between pdx1+/−;RIP-Cre;R26R mice and pdx1flE2/flE2 mice generated pdx1flE2/−;RIP-Cre and pdx1flE2/−;RIP-Cre;R26R experimental mice, both at the expected frequency of 1:8. On average, therefore, one quarter of the pups would be expected to undergo β cell-specific inactivation of pdx1 (from here called pdx1ΔE2β), and half of these pups would also carry the activatable β-gal lineage marker. The phenotypes of both pdx1ΔE2β and pdx1ΔE2β;R26R mice were identical, indicating that the presence of the R26R allele had no effect on the function of the Cre transgene, or on the consequences of pdx1 inactivation. In the subsequent studies, mice of both genotypes were used interchangeably, except for the lineage studies, which required pdx1ΔE2β and R26R in combination.
Based on the detection of RIP-Cre activity by R26R activation, we anticipated that pdx1 inactivation would occur early in the great majority of insulin-producing cells. The previous analysis of pdx1 null mutant animals revealed the presence of significant numbers of pdx1-independent first wave insulin- and glucagon-positive cells (Ahlgren, et al. 1996; Offield, et al. 1996). In the current study, pancreata from pdx1ΔE2β, pdx1ΔE2β;R26R embryos, and control littermates (pdx1+/−;RIP-Cre, pdx1flE2/+;RIP-Cre, and pdx1flE2/−) were examined during the second wave of endocrine differentiation, beginning at e14.5, to assess the effects of Cre-mediated pdx1 inactivation on the formation of endocrine cells that would go on to contribute to the mature islets.
Initially, we examined embryos at e14.5 to determine whether RIP-Cre mediated recombination resulted in the loss of Pdx1 protein expression from insulin-producing cells. At this time point, the majority of insulin-positive cells should have been generated from the second wave of endocrine differentiation. In sections from control pancreata (n=7) at this stage, nuclear Pdx1 protein was detected in 78% of insulin+ cells (n=132/170; Figure 2A), while only 46% of insulin+ cells in the e14.5 conditional mutant (n=3) pancreas co-expressed Pdx1 (n=26/57; Figure 2B) indicating that some insulin-producing cells have already undergone recombination and lost Pdx1 expression at this stage. The average number of insulin+ cells per section was identical in both mutant and control pancreata at this stage. At e17.5–e18.5, Pdx1 was detected in 75% of insulin+ cells (n=280/374) of control embryos (n=3). In contrast, only 19% of insulin+ cells (n=87/453) in mutant embryos (n=4) had detectable levels of Pdx1 at these stages (data not shown) indicating that recombination of the pdx1flE2 allele has occurred in the majority of insulin-producing cells by late gestation. At postnatal day 2, 15% of insulin+ cells found in mutant pancreata still contain detectable levels of Pdx1 protein (data not shown), in agreement with our previously published results showing that this RIP-Cre line mediates recombination in ~85% of β cells (Gannon, et al. 2000)
Figure 2.
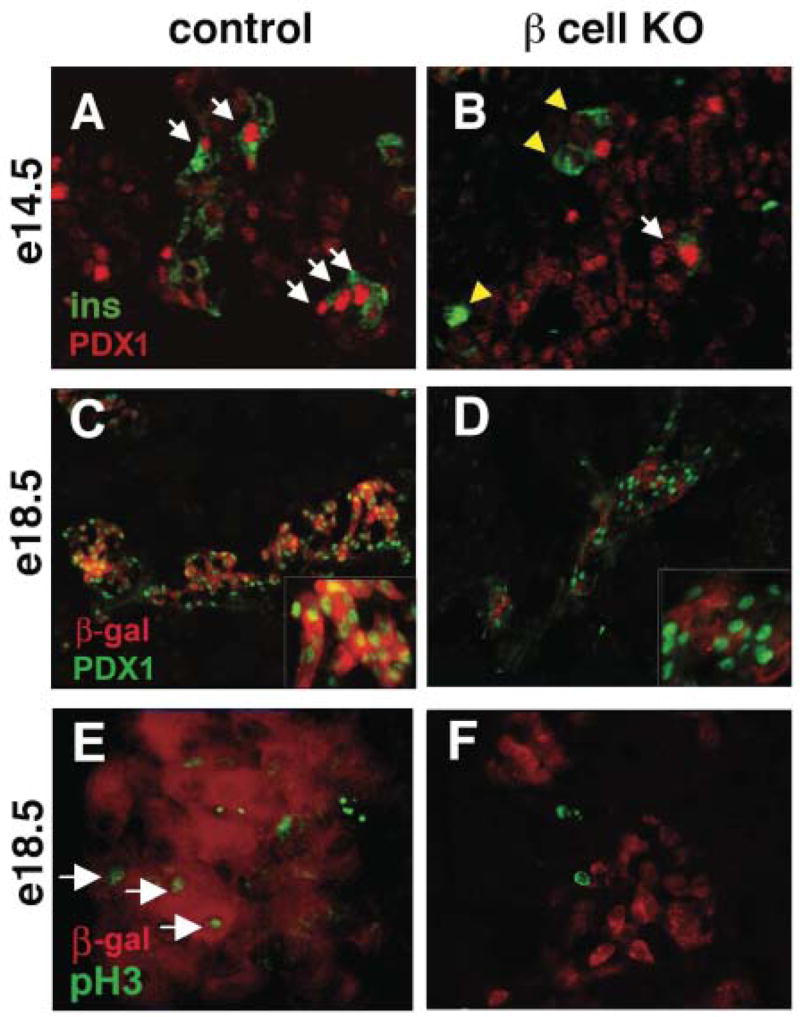
Loss of PDX1 in insulin-producing cells in pdx1ΔE2β;R26R embryos. (A) Sections from control pancreata at e14.5 show intense PDX1 protein expression (red) in the nuclei of most insulin+ cells (green; arrows). (B) In pdx1ΔE2β, most insulin+ cells showed no detectable PDX1 protein (yellow arrowheads), although PDX1+/insulin+ cells are present (arrow). Weaker PDX1 expression is observed in ducts and developing acinar cells in all embryos. (C) PDX1 protein is apparent in most nuclei of β-gal expressing (i.e., recombined/red) β cells of e18.5 RIP-Cre;R26R embryos. (D) In pdx1ΔE2β animals, recombined (β-gal+/red) cells no longer express PDX1. (E) In control embryos, proliferation, identified using a phospho-histone H3 antibody (green), is observed in some recombined β cells (red) at e18.5 (arrows). (F) β cells that have lost PDX1 (red cells) are not proliferating, although other cells within the same field are (green nuclei).
Since lineage tracing of pdx1 null mutant cells relies on equivalent efficiencies of recombination for both the pdx1flE2 and the R26R alleles, we next wanted to determine whether the dramatic Cre-mediated loss of PDX1 expression seen at e17.5–e18.5 was always associated with activation of the R26R allele. In RIP-Cre;R26R control pancreata (n=4 animals) at e18.5, Pdx1 was detected in 90% of recombined (i.e. β-gal+) nuclei (Figure 2C), consistent with its high level of expression in normal insulin-producing cells (Gamer and Wright 1995; Guz, et al. 1995). In contrast, Pdx1 was not observed in β-gal+ cells in pancreata from pdx1ΔE2β;R26R embryos at e18.5 (n=3 animals), indicating that recombination of both the R26R and pdx1flE2 allele occurred in the same cells and that any residual Pdx1 protein in these cells is below the level of detection in tissue from late gestation stage embryos (Figure 2D). Since recombination of both the pdx1flE2 and the R262R alleles appears to be coincident, the insulin+ cells present in e17.5–e18.5 mutant pancreata that still contain Pdx1 protein most likely represent newly generated β cells or β cells that are refractory to recombination (Gannon, et al. 2000). Pdx1+/β-gal− cells were present within pancreata from both control and mutant animals (Figure 2C,D). These cells likely represent a combination of un-recombined insulin+ cells, other endocrine cell types known to express Pdx1, acinar clusters, and ductal epithelial cells, although acini and ducts usually expressed lower levels of the protein.
Because increased Pdx1 expression has been associated with proliferation of ductal and islet cell populations (Sharma, et al. 1999; Song, et al. 1999), we examined the effect of β cell pdx1 inactivation on proliferation of recombined cells by immunohistochemical detection of phosphorylated histone H3 (pH3), an M-phase-specific marker of actively dividing cells (Gurley, et al. 1978). In e18.5 pdx1ΔE2β pancreata, β-gal+ cells (assumed to be Pdx1−) were always observed to be pH3-negative (Figure 2F), whereas 8% of β-gal+ cells in control pancreata of the same age were observed to be actively proliferating (Figure 2E). Mutant pancreata did not show a global defect in proliferation, since β-gal− cells were observed to be actively proliferating (Figure 2F). We conclude that the defect in proliferation was restricted to cells that had undergone RIP-Cre-mediated pdx1 inactivation and no longer expressed Pdx1 protein.
Loss of pdx1 from β cells causes functional and morphological abnormalities in islets
β cell-specific inactivation of pdx1 during embryogenesis resulted in very early onset diabetes. As early as postnatal day one (P1), plasma glucose levels during ad lib feeding were between 225 and 350 mg/dl in mutant pdx1ΔE2β mice (n=3), while control littermates had plasma glucose levels of 100–165 mg/dl with a mean of 145 mg/dl (n=12). Despite the elevated blood glucose, pdx1ΔE2β mice survived past weaning and into adulthood (at least to 8 months, the longest we have maintained the mutant animals).
As adults (1–5 months of age), pdx1ΔE2β male mice showed β cell dysfunction when assessed by intra-peritoneal glucose tolerance test (IP-GTT). pdx1ΔE2β male mice (n=5) had an average fasting blood glucose level of 335 mg/dl (range: 180–475 mg/dl). Upon IP administration of glucose, circulating glucose levels in pdx1ΔE2β mice became further elevated and remained high over the test period (Figure 3A). The average two-hour postprandial glucose measurement was 490 mg/dl. In contrast, control mice (n=11) had an average fasting blood glucose level of 98 mg/dl (range: 60–117 mg/dl) and an average two-hour postprandial glucose measurement of 209 mg/dl. Importantly, animals carrying the RIP-Cre transgene alone showed no defects in glucose clearance (data not shown) as has been reported for this transgene on some genetic backgrounds (Lee, Ristow et al. 2006). pdx1ΔE2β male and female mice had dramatically decreased plasma insulin following glucose administration (Figure 3B). In control mice, plasma insulin levels ranged from 225–495 pg/ml 15 minutes after glucose injection (n=6), while plasma insulin in pdx1ΔE2β animals ranged from 100–170 pg/ml (n=4), which is similar to the fasting plasma insulin levels of wild type mice (Figure 3B). Despite having a reduced number of β cells and reduced plasma insulin levels comparable to males, pdx1ΔE2β female animals showed only a mild glucose intolerance relative to control female animals (Figure 3A). This sexual dimorphism in susceptibility to diabetes has been demonstrated in other rodent models of diabetes (Ostenson, et al. 1989; Bell, et al. 1994; Zhang, et al. 2006).
Figure 3.
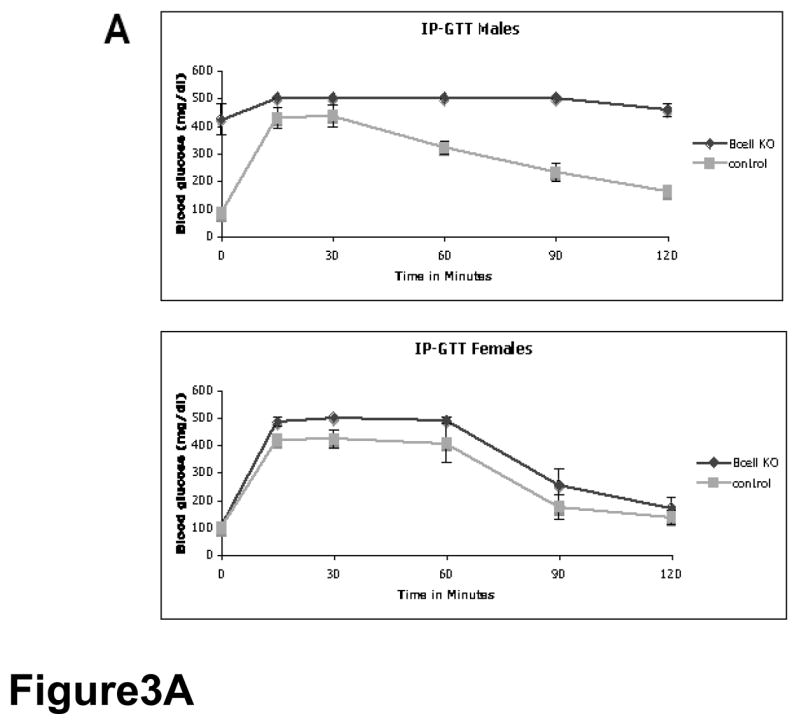

Early loss of pdx1 in β cells results in diabetes post-weaning. (A) Male animals (upper panel) in which pdx1 has been inactivated in β cells (Bcell KO: pdx1ΔE2β) are diabetic when compared with control littermates. In control animals (gray line), blood glucose levels return to baseline during the course of an IPGTT, while in the pdx1 β cell knockout animals, blood glucose levels remain elevated throughout the test (black line). Female pdx1ΔE2β mice (lower panel) show only a mild impairment in glucose tolerance compared with controls. (B) While control animals show increased plasma insulin levels 15 minutes after glucose injection (left bars), plasma insulin levels remain at fasting levels in pdx1ΔE2β animals (right bars).
Confocal immunofluorescence analysis at one month of age, when animals are already overtly diabetic, revealed several defects in mutant islets compared with controls (Figure 4). The numbers of glucagon+ and somatostatin+ cells were increased (Figure 4B, D). There was no change in the number of PP cells (data not shown). We were unsuccessful in our attempts to analyze ghrelin expression using currently available antibodies. Thus, it remains possible that pdx1-null β cells are able to convert to ghrelin-expressing cells in islets from pdx1ΔE2β animals as observed in other mouse models of embryonic β cell loss (Prado, et al. 2004). The excess α and δ cells did not co-express insulin (Figure 4F and data not shown), in contrast to the study performed by Ahlgren et. al. (1998) in which inactivation of pdx1 in adult insulin-expressing cells led to large numbers of glucagon/insulin co-expressing cells. In addition, both glucagon+ and somatostatin+ cells were scattered throughout the islets and not localized to the periphery as in control islets. Third, the level of insulin immunoreactivity in the remaining insulin+ cells was reduced, in agreement with the radioimmunoassay data (Figure 4F).
Figure 4.
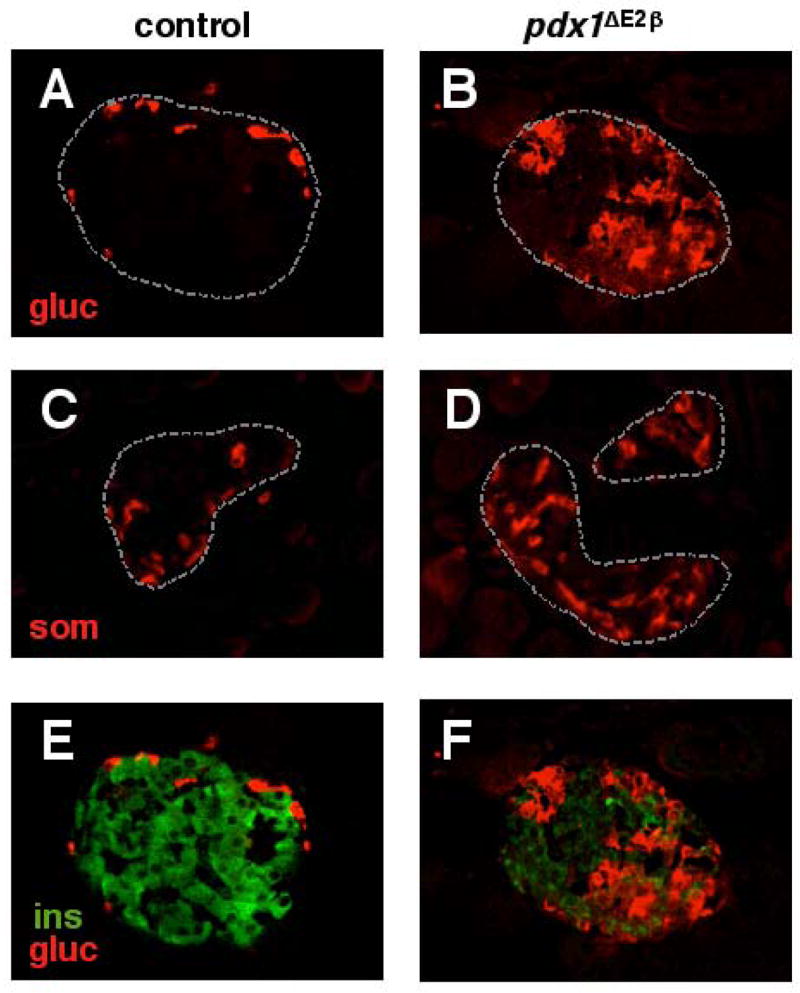
Loss of pdx1 in the β cell lineage leads to aberrant islet architecture, and increased numbers of glucagon- and somatostatin-producing cells. Confocal analysis reveals that compared to control animals at one month of age (A, C), islets of pdx1ΔE2β animals have increased numbers of glucagon+ cells (B) and somatostatin+ cells (D) that are scattered throughout the islets. pdx1ΔE2β animals also have fewer insulin+ cells (green) when compared with controls (E, F). Insulin and glucagon are not co-expressed (F).
The lack of insulin co-expression in the excess α and δ cells in our model suggests two possibilities: 1. β → α or δ conversion initiates earlier in development and is complete by the time we examined them at one month of age, or, 2. the excess glucagon+ and somatostatin+ cells we observe in our model do not arise from a cell that previously expressed insulin. The subsequent studies address each of these possibilities.
Expansion of the α cell population in mutant pancreata is observed at e18.5
We used confocal immunofluorescence analysis of pancreata from several stages (gestation, birth, and up to one month of age) to determine when the increase in glucagon-expressing cells could be detected, and whether significant numbers of insulin+/glucagon+ cells exist at any stage in our mutant pancreata. In contrast to the finding in islets at one month of age, there was no significant difference in the number of glucagon+ cells between control and pdx1 β cell knockout pancreata at e14.5 or e17.5 (Figure 5A and E, and data not shown). The earliest time at which increased numbers of glucagon+ cells could be distinguished in mutant pancreata was e18.5 (Figure 5B and F). By P1, the increased proportion of glucagon+ cells was dramatic (compare Figure 5C, D with G, H). The timing of this increase in glucagon+ cells correlated with a severe reduction in the number of insulin-producing cells per islet. In both control and pdx1ΔE2β mutant pancreata, clusters of insulin+ cells also expressed GLUT2, a marker of second wave β cells, at the membrane at sites of β cell-β cell contacts (data not shown). GLUT2 expression was not detected in insulin− cells.
Figure 5.
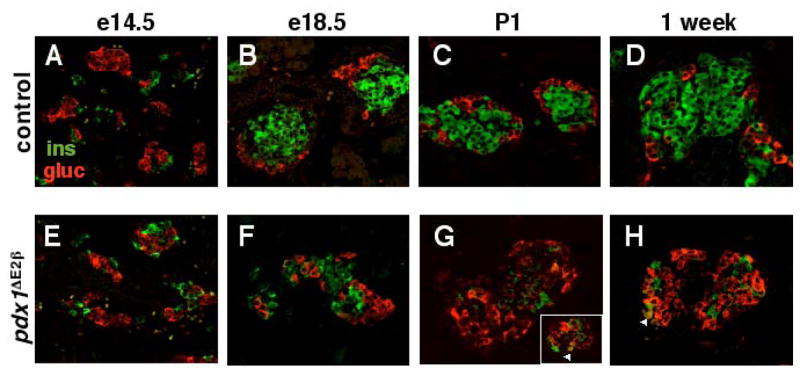
Alterations in the proportion of α and β cells in pdx1ΔE2β animals are first apparent at e18.5. Confocal analysis of insulin (green) and glucagon (red) expression indicates that the proportion of α and β cells is identical in control and pdx1ΔE2β embryos at e14.5 (compare A with E) and e17.5 (not shown). Expansion of the α cell population and decline in the β cell population begins at e18.5 (compare B with F) and continues into postnatal development (G, H). The vast majority of cells express either one hormone or the other. Cells co-expressing both hormones are occasionally detected (arrowheads in G and H).
No glucagon/insulin co-expressing cells were detected at any embryonic time point examined (e14.5, e17.5, e18.5) in either control or mutant pancreata. At P1, very few cells (0.1% of all glucagon+ cells; arrowheads in Figure 5G and H) co-expressing insulin and glucagon were detected within mutant islets, a frequency that was consistent across many tissue sections sampled from several different animals. Thus, we find no evidence for significant numbers of glucagon/insulin co-expressing cells prior to the expansion of the α cell population, nor at times when this expansion first becomes apparent. These results are inconsistent with the idea that the excess glucagon+ cells arose via a transitional cell type in which insulin-producing cells convert to glucagon-producing cells. This type of analysis, however, cannot exclude the possibility that the β → α transition occurs too rapidly to be detected at the chosen static time points. To determine more directly whether the excess glucagon+ and somatostatin+ cells present within the β cell knockout islets arose from cells that expressed insulin at an earlier time point, we performed lineage-tracing analyses using the R26R reporter mouse.
Excess glucagon+ and somatostatin+ cells do not arise from insulin-producing cells
In RIP-Cre;R26R mice, Cre-mediated β-gal expression is detected specifically in cells that activated the rat insulin promoter transgene. The cell type-independence of the ROSA26 promoter means that progeny of the original cell undergoing recombination stably inherit β-gal expression. Therefore, because the ROSA26 promoter is active in all pancreatic endocrine cell types (Gannon, et al. 2000; Kawaguchi, et al. 2002), cells derived from insulin-expressing cells should be detectable regardless of their subsequent insulin expression status.
In RIP-Cre;R26R control pancreata from P1 animals, β-gal expression was restricted, as expected, to insulin-producing cells in the islet core as expected (Figure 6A, C), and absent from glucagon- and somatostatin-producing cells. At P1, >90% of somatostatin+ cells were β-gal− (Figure 6C), we did detect co-expression of somatostatin and β-gal in ~8% of δ cells (n=12/156) in control pancreata (data not shown). The small percentage of somatostatin+ cells expressing β-gal could reflect leakiness of the RIP-Cre transgene in this lineage, or alternatively, that a proportion of δ cells may arise from an insulin/somatostatin co-expressing intermediate, as suggested previously (Fernandes, et al. 1997; Sosa-Pineda, et al. 1997). In P1 pdx1ΔE2β pancreata, very few β-gal+ cells were detected within the islets (Figure 6B, D), consistent with the vast decrease in insulin+ cells noted above (e.g. Figure 5G). Significantly, the β-gal+ cells present at P1 do not co-express either glucagon (0.2%; n=2/849) or somatostatin (0.7%; n=1/142), indicating that the excess α and δ cells did not arise from cells that were previously insulin-producing. Taken together, these data suggest that the increase in glucagon+ and somatostatin+ cells in pdx1ΔE2β pancreata is likely caused by increased proliferation of these cell types or their precursors, and not by conversion of pdx1 null “former-β cells” to other endocrine cell types.
Figure 6.
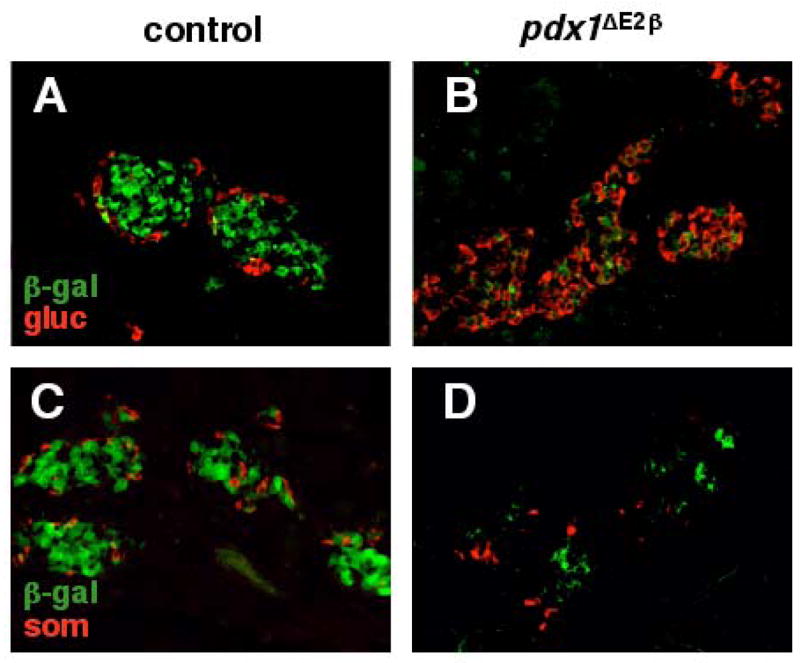
Lineage tracing analysis at P1 reveals that excess glucagon+ and somatostatin+ cells in pdx1 β cell knockout animals do not arise from insulin-producing cells. In RIP-Cre;R26R control animals (A,C), insulin-producing cells located at the core of the islet express β-gal (green) as expected. Glucagon+ (A) and somatostatin+ (C) cells (red) are located at the periphery and do not co-express β-gal. In pdx1ΔE2β animals, the number of β-gal expressing cells is dramatically reduced (B, C). β-gal expression does not co-localize with glucagon (B) or somatostatin (C) indicating that these cells derive from cells that did not express RIP-Cre.
We next analyzed the proliferation status of differentiated endocrine cells during the second wave of endocrine differentiation (e14.5, e17.5, and e18.5). This developmental window represents the time during which the proportions of the endocrine cell types become abnormal in the conditional mutant animals. pH3-immunopositive cells were scored on at least ten tissue sections containing endocrine cells from two litters at each developmental time point. We failed to detect any difference in proliferation between control (n=4) and mutant (n=2) pancreata at e14.5 (data not shown). Compared with wild type pancreata (n=2), a three-fold increase in pH3-positive glucagon+ cells [2% (n=2/113): wild-type; 6% (n=47/755): mutant] was first detected at e17.5 in β cell knockout pancreata (n=3) and was maintained at e18.5 (Figure 7A–C and data not shown). The proportion of proliferating insulin+ cells at e17.5 (~1–2%; data not shown) was indistinguishable between control (n=2/196) and mutant (n=7/410) pancreata but, at e18.5, was dramatically reduced in conditional mutant tissue (10% in wild-type n=43/438, 2% in mutants n=12/538; Figure 7D,E). Proliferation rates in the acinar tissue were not different between control and mutant pancreata. We also examined whether insulin+ cells in mutant pancreata were undergoing apoptosis. TUNEL labeling was performed on control and mutant pancreata from e14.5 and e18.5 embryos, P1 and adult. At no time point did we observe apoptotic insulin+ cells in either control or mutant animals (data not shown).
Figure 7.

Loss of pdx1 in the β cell lineage results in increased proliferation of glucagon+ cells and decreased proliferation of insulin+ cells. Co-labelling of e17.5 and e18.5 pancreatic sections for either glucagon (green, A–C) or insulin (green, D–F) and phospho-histone H3 (red, A–F) reveals that in pdx1ΔE2β animals, the fraction of proliferating α cells is increased at e17.5 (compare arrowheads in A with B,C), while the proportion of insulin-expressing β cells actively proliferating is dramatically decreased at e18.5 (compare arrowheads in D with E,F). No difference was observed at e14.5 (not shown).
Discussion
The findings we report here add to our understanding of the continual requirement for pdx1 in the life history of β cells. The accumulating evidence demonstrates that Pdx1 plays essential regulatory roles both during global pancreas formation and specifically in β cells during embryonic and adult stages. Homozygous null pdx1 mice have a very early block in pancreas development, but do undergo limited bud outgrowth (Offield, et al. 1996). These animals form neither mature islets nor acinar tissue (Jonsson, et al. 1994; Offield, et al. 1996). Thus, pdx1 is not required to specify the pancreatic field or to initiate outgrowth from the endodermal epithelium. Loss of pdx1 function after initial pancreatic bud outgrowth, using a doxycycline-inducible system, results in a block in further pancreas development (Holland, et al. 2002), while pdx1 inactivation at later embryonic stages results in a specific defect in development of the exocrine pancreas (Hale, et al. 2005). These studies did not evaluate the effects of pdx1 removal on second wave endocrine differentiation and expansion. Inactivation of pdx1 specifically in mature, adult β cells results in diabetes, loss of β cell markers such as Nkx6.1 and GLUT2, and a great decrease in the number of insulin-producing cells (Ahlgren, et al. 1998; Holland, et al. 2002). Thus, neither the whole animal pdx1 knockout studies, nor the published conditional inactivation studies address the requirement for pdx1 specifically during late gestation, the time during which the majority of the insulin-producing cells that will give rise to the mature islet are generated.
pdx1 function in late gestation β cells differs from its function in adult β cells
The current study is the first to define a role for pdx1 in the late gestation embryonic β cells that are assumed to incorporate into mature islets. In these studies, we used a Cre transgenic line defined via R26R activation for its early and efficient expression in insulin-producing cells within the pancreas. In addition, we included the first lineage-tracing of pdx1-deficient cells, and studies of the effect of pdx1 inactivation on cell proliferation. Mice in which pdx1 was specifically inactivated in β cells during embryogenesis showed a decrease in proliferation of insulin+ cells at late gestation that translated into a decreased number of β cells. pdx1ΔE2β animals showed a concomitant increase in proliferation of glucagon+ cells at late gestation, elevated postprandial blood glucose levels in neonates, and overt diabetes at weaning.
Although both the current study and the one performed by Ahlgren et. al. (1998) used a similar strategy to inactivate pdx1 in the β cell lineage, Cre-mediated recombination in our study occurred much earlier in the life history of the β cell. Thus, in our experimental system, loss of Pdx1 was observed in a substantial number of insulin-producing cells at e14.5, while a significant decrease in Pdx1 expression was not detected until 3–5 weeks postnatally in the Ahlgren study. This difference in the timing of pdx1 inactivation is perhaps due to a higher efficiency of recombination driven by our RIP-Cre transgene, or because our experimental design required inactivation of only one floxed pdx1 allele to reach the null condition. Although it is difficult to rule out, we believe it is less likely that the large difference in timing of pdx1 inactivation between the two studies is related to differences in the genetic background of the various mice involved.
The fortuitous production of two models of β cell-specific pdx1 inactivation has therefore revealed specific requirements for pdx1 in embryonic and adult β cells. In the study by Ahlgren et. al. (1998), pdx1 inactivation specifically in mature β cells resulted in a reduced number of insulin-producing cells and an associated increase in the number of glucagon-expressing cells, a large fraction of which co-expressed both hormones, leading to the hypothesis that insulin-producing cells were converting to glucagon-producing cells following loss of Pdx1 protein. We also observed a large increase in glucagon+ cells, as well as somatostatin+ cells, following the selective inactivation of pdx1 in embryonic β cells. This increase in α and δ cell number was apparent as early as e18.5. Lineage tracing of pdx1 null cells revealed that these excess α and δ cells did not arise from insulin-producing cells. Thus, loss of pdx1 in late gestation β cells affects these cells differently from loss of pdx1 in a mature β cell.
The increase in α cell number is associated with increased proliferation of glucagon-producing cells and a decrease in proliferation of insulin-producing cells. Other studies have shown an association between pdx1 and proliferation of pancreatic cells (Ahlgren, et al. 1996; Sharma, et al. 1999; Hart, et al. 2000), consistent with a loss of pdx1 leading to decreased proliferation in the β cell lineage. Intriguingly, the reduction in insulin-producing cells in mutant animals was most dramatic at P1 and one week. By one month of age, we consistently observed an increase in the number of insulin+ cells (compare Figures 4 and 5). Because the RIP-Cre transgene used here reproducibly showed activity in approximately 85–90% of β cells at P1 (Gannon, et al. 2000; Sund, et al. 2001), it is possible that a population of β cells is refractory to recombination and thus maintains PDX1 expression. These insulin+/Pdx1+ cells likely remain capable of proliferating since β cell mass continues to increase even after weaning via replication of pre-existing β cells (Kaung 1994; Finegood, et al. 1995). Thus, the clusters of insulin+ cells observed in adult mutant islets may represent clonal expansion of unrecombined β cells.
Lineage tracing provides information about the fate of mutant cells
Organogenesis involves the gradual restriction of potential cell fate from multi-potency towards expression of specific differentiated cellular phenotypes. A general regulatory principle for such programs is the sophisticated interplay between cell-autonomous processes and intercellular communication. The current study suggests a previously unappreciated interaction between β cells and other endocrine cells (α and δ) or their progenitors that establishes the appropriate proportions of endocrine cells during islet development. The R26R lineage tracer revealed that the excess glucagon+ or somatostatin+ cells we observe in pdx1ΔE2β mutants did not arise from β cells. This finding leads to the idea that the decrease in β cell number results from an increased flux from multipotential or lineage-restricted endocrine progenitors towards the α/δ cell fates, and/or an increased rate of proliferation of differentiated non-β cells in the absence of appropriate numbers of β cells.
All five of the islet endocrine cell types are currently believed to derive from a common multipotential precursor, although confirmation of this hypothesis awaits rigorous testing by lineage-tracing techniques. Regardless, endocrine cells arise from progenitors that at some time in their history expressed pdx1 and ngn3 (Gu, et al. 2002). The production of the five major endocrine cell types must be regulated in order to consistently produce the proportions seen in normal mature islets. Presumably, environmental cues and lineage-specific combinations of regulatory factors modulate the direction of differentiation of multipotent or lineage-restricted amplifying populations towards one endocrine cell type or another. Very little is known about these instructive processes. Our findings are consistent with a type of feedback effect whereby β cells regulate α cell proliferation in a paracrine fashion (Figure 8), and we propose a similar mechanism to be responsible for the increased numbers of somatostatin+ cells in β cell-specific pdx1 knockout animals. A previous study in which insulin-producing cells were specifically ablated using a toxic β cell-specific transgene did not show an effect on the development of α and δ cells (Herrera, et al. 1994). In that study, however, endocrine differentiation was assessed only at e16.5, and it is possible, based on our findings, that this is prior to the time at which an effect of β cell loss on α and δ cell development would begin to be apparent. While the nature of the β cell-derived paracrine inhibitory signal is unknown, it is unlikely to be insulin itself since mice lacking both insulin genes show normal numbers of α cells (Duvillie, et al. 1997; Duvillie, et al. 2002).
Figure 8.
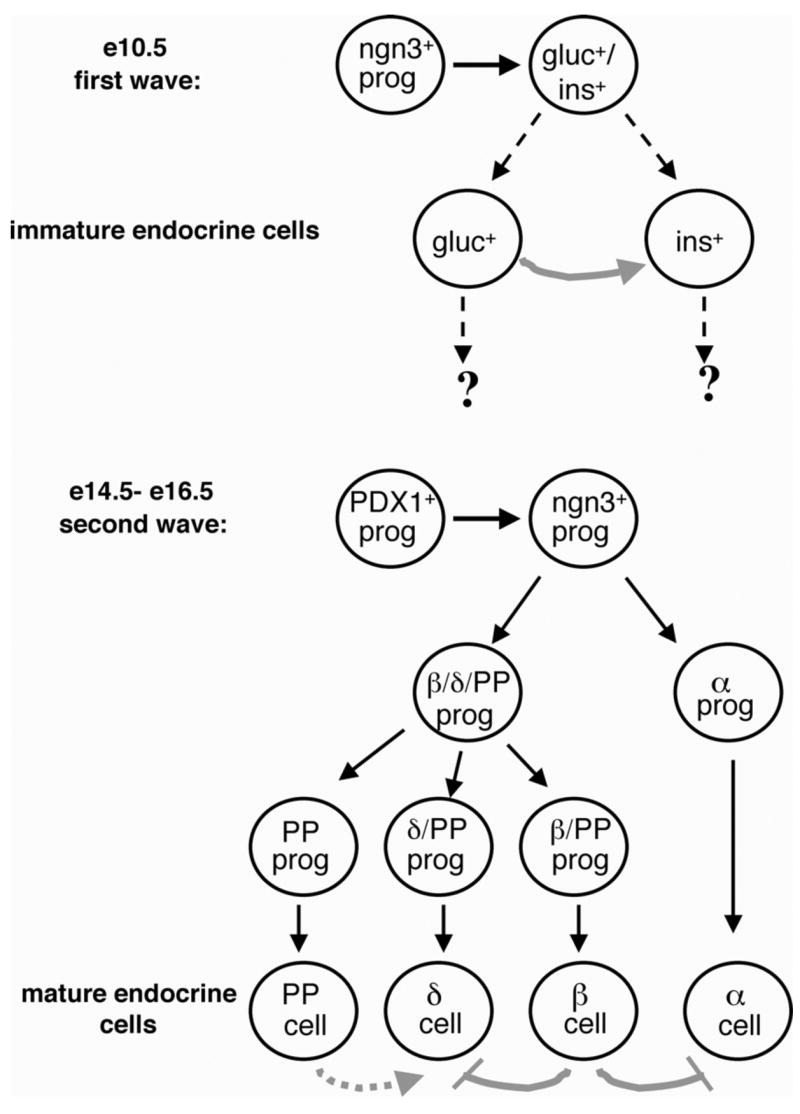
Model of pancreatic endocrine differentiation and endocrine lineage relationships (solid black lines indicate direct linage relationships, while dashed black lines indicate presumed relationships). Endocrine cells arise from ngn3-expressing progenitors regardless of whether they are generated in the first or second wave of endocrine differentiation. In the first wave, which begins at around e10.5 in the mouse, immature endocrine cells are generated, some of which co-express insulin and glucagon. The fate of these cells is currently unknown, however some studies suggest that the early glucagon+ cells function in the generation of early insulin+ cells (gray arrow). In the second wave of endocrine differentiation, which begins at around e14.5, mature endocrine cells are generated. These cells arise from a PDX1+ cell. The α cell lineage is thought to diverge early, while the existence of a multi-hormone positive cell that gives rise to β, δ, and PP cells has been observed. Mature β cells (and presumably δ cells) arise from a PP-expressing precursor. A paracrine effect of PP cells on δ cell development has not been ruled out (dashed gray arrow). In the current study, we have evidence for an inhibitory effect of β cells on the differentiation/proliferation of α and δ cells (gray lines).
Combining gene inactivation with lineage tracing allows one to determine which, if any, developmental pathways are still open to cells that have lost a particular gene product. For example, the original global inactivation of Ptf1a concluded that this gene is required for generation of the exocrine pancreas (Krapp, et al. 1998). Lineage-tracing studies using Cre-mediated R26R activation (Kawaguchi, et al. 2002), showed a previously unappreciated expression of Ptf1a in precursors of both exocrine and endocrine cells. In homozygous mutants, lineage tracing revealed a conversion of most Ptf1a-deficient pancreatic progenitors into normal duodenal cell fates. Similarly, analysis of Pax6 mutant animals using hormone expression as an indicator of endocrine differentiation led to the conclusion that Pax6 is required for differentiation of α cells and normal numbers of other endocrine cell types (Sander, et al. 1997; St-Onge, et al. 1997). Conditional inactivation of Pax6 in the pancreas combined with lineage tracing of null mutant cells revealed that normal numbers of endocrine cells actually form in the mutant animals, but that these cells lack certain critical terminal differentiation markers, resulting in impaired α and β cell function (Ashery-Padan, et al. 2004).
In addition to lineage-restricted transcriptional regulators (reviewed in (Wilson, et al. 2003), it is becoming apparent that pancreatic endocrine cell development depends on interactions and communications between the different islet endocrine lineages (Herrera, et al. 1994; Prasadan, et al. 2002; Vincent, et al. 2003). Additional work is needed to define the nature of the signals produced by the different islet endocrine lineages and how these signals promote differentiation and proliferation to yield fully functional islets.
Acknowledgments
We would like to thank Drs. Roland Stein and Anna Means for helpful comments on the manuscript. We would also like to thank Michael Ray for technical assistance, Wendell Nicholson for performing insulin measurements, Amanda Ackermann for performing the TUNEL assay, and Dr. Patricia Labosky for her expertise in ES cell culture. Experiments were performed in part through the use of the VUMC Cell Image Shared Resource and the Transgenic Mouse/ES Cell Shared Resource (both supported by NIH grants CA68485, DK20593, DK58404 and HD15052). This work was supported by NIH grants DK42502 to C.V.E.W., DK42502 and DK42612 to M.A.M., and a postdoctoral fellowship (397019) and Career Development Award (2-2002-583) from the Juvenile Diabetes Research Foundation International to M.G.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahlgren U, Jonsson J, et al. The morphogenesis of the pancreatic mesenchyme is uncoupled from that of the pancreatic epithelium in IPF1/PDX1-deficient mice. Development. 1996;122(5):1409–16. doi: 10.1242/dev.122.5.1409. [DOI] [PubMed] [Google Scholar]
- Ahlgren U, Jonsson J, et al. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998;12(12):1763–8. doi: 10.1101/gad.12.12.1763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpert S, Hanahan D, et al. Hybrid insulin genes reveal a developmental lineage for pancreatic endocrine cells and imply a relationship with neurons. Cell. 1988;53(2):295–308. doi: 10.1016/0092-8674(88)90391-1. [DOI] [PubMed] [Google Scholar]
- Apelqvist A, Li H, et al. Notch signalling controls pancreatic cell differentiation. Nature. 1999;400(6747):877–81. doi: 10.1038/23716. [DOI] [PubMed] [Google Scholar]
- Ashery-Padan R, Zhou X, et al. Conditional inactivation of Pax6 in the pancreas causes early onset of diabetes. Dev Biol. 2004;269(2):479–88. doi: 10.1016/j.ydbio.2004.01.040. [DOI] [PubMed] [Google Scholar]
- Bell RC, Khurana M, et al. Gender differences in the metabolic response to graded numbers of transplanted islets of Langerhans. Endocrinology. 1994;135(6):2681–7. doi: 10.1210/endo.135.6.7988458. [DOI] [PubMed] [Google Scholar]
- Brissova M, Shiota M, et al. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem. 2002;277(13):11225–32. doi: 10.1074/jbc.M111272200. [DOI] [PubMed] [Google Scholar]
- Chakrabarti SK, James JC, et al. Quantitative assessment of gene targeting in vitro and in vivo by the pancreatic transcription factor, Pdx1. Importance of chromatin structure in directing promoter binding. J Biol Chem. 2002;277(15):13286–93. doi: 10.1074/jbc.M111857200. Epub 2002 Feb 1. [DOI] [PubMed] [Google Scholar]
- Cissell MA, Zhao L, et al. Transcription factor occupancy of the insulin gene in vivo: evidence for direct regulation by Nkx2.2. J Biol Chem. 2002;7:7. doi: 10.1074/jbc.M205905200. [DOI] [PubMed] [Google Scholar]
- Dohrmann C, Gruss P, et al. Pax genes and the differentiation of hormone-producing endocrine cells in the pancreas. Mech Dev. 2000;92(1):47–54. doi: 10.1016/s0925-4773(99)00324-x. [DOI] [PubMed] [Google Scholar]
- Dutta S, Bonner-Weir S, et al. Regulatory factor linked to late-onset diabetes? [letter] Nature. 1998;392(6676):560. doi: 10.1038/33311. [DOI] [PubMed] [Google Scholar]
- Duvillie B, Cordonnier N, et al. Phenotypic alterations in insulin-deficient mutant mice. Proc Natl Acad Sci U S A. 1997;94(10):5137–40. doi: 10.1073/pnas.94.10.5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvillie B, Currie C, et al. Increased islet cell proliferation, decreased apoptosis, and greater vascularization leading to beta-cell hyperplasia in mutant mice lacking insulin. Endocrinology. 2002;143(4):1530–7. doi: 10.1210/endo.143.4.8753. [DOI] [PubMed] [Google Scholar]
- Fernandes A, King LC, et al. Differentiation of new insulin-producing cells is induced by injury in adult pancreatic islets. Endocrinology. 1997;138(4):1750–62. doi: 10.1210/endo.138.4.5049. [DOI] [PubMed] [Google Scholar]
- Finegood DT, Scaglia L, et al. Dynamics of beta-cell mass in the growing rat pancreas. Estimation with a simple mathematical model. Diabetes. 1995;44(3):249–56. doi: 10.2337/diab.44.3.249. [DOI] [PubMed] [Google Scholar]
- Gamer LW, Wright CV. Autonomous endodermal determination in Xenopus: regulation of expression of the pancreatic gene XlHbox 8. Dev Biol. 1995;171(1):240–51. doi: 10.1006/dbio.1995.1275. [DOI] [PubMed] [Google Scholar]
- Gannon M, Gamer LW, et al. Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev Biol. 2001;238(1):185–201. doi: 10.1006/dbio.2001.0359. [DOI] [PubMed] [Google Scholar]
- Gannon M, Herrera PL, et al. Mosaic Cre-mediated recombination in pancreas using the pdx-1 enhancer/promoter. Genesis. 2000;26(2):143–4. doi: 10.1002/(sici)1526-968x(200002)26:2<143::aid-gene13>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- Gannon M, Ray MK, et al. Persistent expression of HNF6 in islet endocrine cells causes disrupted islet architecture and loss of beta cell function. Development. 2000;127(13):2883–95. doi: 10.1242/dev.127.13.2883. [DOI] [PubMed] [Google Scholar]
- Gannon M, Shiota C, et al. Analysis of the Cre-mediated recombination driven by rat insulin promoter in embryonic and adult mouse pancreas. Genesis. 2000;26(2):139–42. doi: 10.1002/(sici)1526-968x(200002)26:2<139::aid-gene12>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- Gannon M, Wright CVE. Endodermal Patterning and Organogenesis. In: Moody SA, editor. Cell Lineage and Fate Determination. San Diego: Academic Press; 1999. pp. 583–615. [Google Scholar]
- Gu G, Dubauskaite J, et al. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129(10):2447–57. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Gurley LR, D’Anna JA, et al. Histone phosphorylation and chromatin structure during mitosis in Chinese hamster cells. Eur J Biochem. 1978;84(1):1–15. doi: 10.1111/j.1432-1033.1978.tb12135.x. [DOI] [PubMed] [Google Scholar]
- Guz Y, Montminy MR, et al. Expression of murine STF-1, a putative insulin gene transcription factor, in beta cells of pancreas, duodenal epithelium and pancreatic exocrine and endocrine progenitors during ontogeny. Development. 1995;121(1):11–8. doi: 10.1242/dev.121.1.11. [DOI] [PubMed] [Google Scholar]
- Hale MA, Kagami H, et al. The homeodomain protein PDX1 is required at mid-pancreatic development for the formation of the exocrine pancreas. Dev Biol. 2005;286(1):225–37. doi: 10.1016/j.ydbio.2005.07.026. [DOI] [PubMed] [Google Scholar]
- Hart AW, Baeza N, et al. Attenuation of FGF signalling in mouse beta-cells leads to diabetes. Nature. 2000;408(6814):864–8. doi: 10.1038/35048589. [DOI] [PubMed] [Google Scholar]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127(11):2317–22. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Herrera PL, Huarte J, et al. Ablation of islet endocrine cells by targeted expression of hormone-promoter-driven toxigenes. Proc Natl Acad Sci U S A. 1994;91(26):12999–3003. doi: 10.1073/pnas.91.26.12999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan B, Beddington R, Costantini F, Lacy E. Manipulating the Mouse Embryo. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory; 1994. [Google Scholar]
- Holland AM, Hale MA, et al. Experimental control of pancreatic development and maintenance. Proc Natl Acad Sci U S A. 2002;99(19):12236–41. doi: 10.1073/pnas.192255099. Epub 2002 Sep 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen J. Gene regulatory factors in pancreatic development. Dev Dyn. 2004;229(1):176–200. doi: 10.1002/dvdy.10460. [DOI] [PubMed] [Google Scholar]
- Jonsson J, Carlsson L, et al. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature. 1994;371(6498):606–9. doi: 10.1038/371606a0. [DOI] [PubMed] [Google Scholar]
- Kaung HL. Growth dynamics of pancreatic islet cell populations during fetal and neonatal development of the rat. Dev Dyn. 1994;200(2):163–75. doi: 10.1002/aja.1002000208. [DOI] [PubMed] [Google Scholar]
- Kawaguchi Y, Cooper B, et al. The role of the transcriptional regulator Ptf1a in converting intestinal to pancreatic progenitors. Nat Genet. 2002;32(1):128–34. doi: 10.1038/ng959. [DOI] [PubMed] [Google Scholar]
- Krapp A, Knofler M, et al. The bHLH protein PTF1-p48 is essential for the formation of the exocrine and the correct spatial organization of the endocrine pancreas. Genes Dev. 1998;12(23):3752–63. doi: 10.1101/gad.12.23.3752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Ristow M, et al. RIP-Cre revisited, evidence for impairments of pancreatic beta-cell function. J Biol Chem. 2006;281(5):2649–53. doi: 10.1074/jbc.M512373200. [DOI] [PubMed] [Google Scholar]
- Lee YC, Damholt AB, et al. Developmental expression of proprotein convertase 1/3 in the rat. Mol Cell Endocrinol. 1999;155(1–2):27–35. doi: 10.1016/s0303-7207(99)00119-7. [DOI] [PubMed] [Google Scholar]
- Li H, Arber S, et al. Selective agenesis of the dorsal pancreas in mice lacking homeobox gene Hlxb9. Nat Genet. 1999;23(1):67–70. doi: 10.1038/12669. [DOI] [PubMed] [Google Scholar]
- Macfarlane WM, Frayling TM, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes [In Process Citation] J Clin Invest. 2000;106(5):717. [PubMed] [Google Scholar]
- Murtaugh LC, Stanger BZ, et al. Notch signaling controls multiple steps of pancreatic differentiation. Proc Natl Acad Sci U S A. 2003;100(25):14920–5. doi: 10.1073/pnas.2436557100. Epub 2003 Dec 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Offield MF, Jetton TL, et al. PDX-1 is required for pancreatic outgrowth and differentiation of the rostral duodenum. Development. 1996;122(3):983–95. doi: 10.1242/dev.122.3.983. [DOI] [PubMed] [Google Scholar]
- Orban PC, Chui D, et al. Tissue- and site-specific DNA recombination in transgenic mice. Proc Natl Acad Sci U S A. 1992;89(15):6861–5. doi: 10.1073/pnas.89.15.6861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ostenson CG, Grill V, et al. Studies on sex dependency of B-cell susceptibility to streptozotocin in a rat model of type II diabetes mellitus. Exp Clin Endocrinol. 1989;93(2–3):241–7. doi: 10.1055/s-0029-1210863. [DOI] [PubMed] [Google Scholar]
- Pang K, Mukonoweshuro C, et al. Beta cells arise from glucose transporter type 2 (Glut2)-expressing epithelial cells of the developing rat pancreas. Proc Natl Acad Sci U S A. 1994;91(20):9559–63. doi: 10.1073/pnas.91.20.9559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peshavaria M, Gamer L, et al. XIHbox 8, an endoderm-specific Xenopus homeodomain protein, is closely related to a mammalian insulin gene transcription factor. Mol Endocrinol. 1994;8(6):806–16. doi: 10.1210/mend.8.6.7935494. [DOI] [PubMed] [Google Scholar]
- Pictet RL, Clark WR, et al. An ultrastructural analysis of the developing embryonic pancreas. Dev Biol. 1972;29(4):436–67. doi: 10.1016/0012-1606(72)90083-8. [DOI] [PubMed] [Google Scholar]
- Postic C, Shiota M, et al. Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J Biol Chem. 1999;274(1):305–15. doi: 10.1074/jbc.274.1.305. [DOI] [PubMed] [Google Scholar]
- Prado CL, Pugh-Bernard AE, et al. Ghrelin cells replace insulin-producing {beta} cells in two mouse models of pancreas development. Proc Natl Acad Sci U S A. 2004;101(9):2924–2929. doi: 10.1073/pnas.0308604100. Epub 2004 Feb 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasadan K, Daume E, et al. Glucagon is required for early insulin-positive differentiation in the developing mouse pancreas. Diabetes. 2002;51(11):3229–36. doi: 10.2337/diabetes.51.11.3229. [DOI] [PubMed] [Google Scholar]
- Sander M, Neubuser A, et al. Genetic analysis reveals that PAX6 is required for normal transcription of pancreatic hormone genes and islet development. Genes Dev. 1997;11(13):1662–73. doi: 10.1101/gad.11.13.1662. [DOI] [PubMed] [Google Scholar]
- Sharma A, Zangen DH, et al. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes. 1999;48(3):507–13. doi: 10.2337/diabetes.48.3.507. [DOI] [PubMed] [Google Scholar]
- Song SY, Gannon M, et al. Expansion of Pdx1-expressing pancreatic epithelium and islet neogenesis in transgenic mice overexpressing transforming growth factor alpha. Gastroenterology. 1999;117(6):1416–26. doi: 10.1016/s0016-5085(99)70292-1. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21(1):70–1. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Sosa-Pineda B, Chowdhury K, et al. The Pax4 gene is essential for differentiation of insulin-producing beta cells in the mammalian pancreas. Nature. 1997;386(6623):399–402. doi: 10.1038/386399a0. [DOI] [PubMed] [Google Scholar]
- St-Onge L, Sosa-Pineda B, et al. Pax6 is required for differentiation of glucagon-producing alpha-cells in mouse pancreas. Nature. 1997;387(6631):406–9. doi: 10.1038/387406a0. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Ferrer J, et al. Early-onset type-II diabetes mellitus (MODY4) linked to IPF1 [letter] Nat Genet. 1997;17(2):138–9. doi: 10.1038/ng1097-138. [DOI] [PubMed] [Google Scholar]
- Stoffers DA, Stanojevic V, et al. Insulin promoter factor-1 gene mutation linked to early-onset type 2 diabetes mellitus directs expression of a dominant negative isoprotein. J Clin Invest. 1998;102(1):232–41. doi: 10.1172/JCI2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoffers DA, Zinkin NT, et al. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet. 1997;15(1):106–10. doi: 10.1038/ng0197-106. [DOI] [PubMed] [Google Scholar]
- Sund NJ, Vatamaniuk MZ, et al. Tissue-specific deletion of Foxa2 in pancreatic beta cells results in hyperinsulinemic hypoglycemia. Genes Dev. 2001;15(13):1706–15. doi: 10.1101/gad.901601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teitelman G, Alpert S, et al. Precursor cells of mouse endocrine pancreas coexpress insulin, glucagon and the neuronal proteins tyrosine hydroxylase and neuropeptide Y, but not pancreatic polypeptide. Development. 1993;118(4):1031–9. doi: 10.1242/dev.118.4.1031. [DOI] [PubMed] [Google Scholar]
- Vincent M, Guz Y, et al. Abrogation of protein convertase 2 activity results in delayed islet cell differentiation and maturation, increased alpha-cell proliferation, and islet neogenesis. Endocrinology. 2003;144(9):4061–9. doi: 10.1210/en.2003-0088. [DOI] [PubMed] [Google Scholar]
- Wilson ME, Kalamaras JA, et al. Expression pattern of IAPP and prohormone convertase 1/3 reveals a distinctive set of endocrine cells in the embryonic pancreas. Mech Dev. 2002;115(1–2):171–6. doi: 10.1016/s0925-4773(02)00118-1. [DOI] [PubMed] [Google Scholar]
- Wilson ME, Scheel D, et al. Gene expression cascades in pancreatic development. Mech Dev. 2003;120(1):65–80. doi: 10.1016/s0925-4773(02)00333-7. [DOI] [PubMed] [Google Scholar]
- Wu KL, Gannon M, et al. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol Cell Biol. 1997;17(10):6002–13. doi: 10.1128/mcb.17.10.6002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Ackermann AM, et al. The Foxm1 Transcription Factor is Required to Maintain Pancreatic Beta Cell Mass. Mol Endocrinol. 2006 doi: 10.1210/me.2006-0056. [DOI] [PubMed] [Google Scholar]