

Summary
We have analyzed peptides associated with six human MHC class I allomorphs expressed by the U937 cell line. Peptides were isolated by mild acid elution or by MHC class I immunoprecipitation using W6/32 monoclonal antibody. A total of 85 peptides were sequenced by mass spectrometry and their putative binding alleles were assigned using bioinformatic tools. Only three peptides isolated by the two approaches were identical, suggesting that the approaches may yield distinct, partially overlapping peptide populations. Mild acid treatment-derived peptides had overall characteristics suggestive of relatively lower affinity of binding for MHC class I. Interestingly, a large proportion of putative HLA-B*5101- binding peptides was found among the mild-acid treatment eluted peptides, and to a lesser degree in the affinity-purified peptide pool. These results suggest that HLA-B*5101 may bind a potentially large pool of peptides with relatively lower affinity. We suggest that lower affinity of peptide binding may be the basis for inefficient tolerance to HLA-B*5101-binding self-peptides, a predisposing factor for the development of Behçet disease.
Keywords: HLA, antigen processing/presentation, self-peptides, autoimmunity
Introduction
The MHC class I molecules are expressed on the surface of most nucleated cells and display short peptide fragments generated by proteolytic degradation of intracellular proteins. Peptide fragments are generated in the cytoplasm by the action of the proteasome and are then transported into the endoplasmic reticulum by the transporter associated with antigen processing, where they bind to the nascent MHC class I molecules [1]. At any given time, single cells display between 1000 and 10000 different peptides [2], collectively termed peptidome [3]. The MHC class I-associated peptidome is continuously scanned by CD8+ T cells in peripheral lymphoid tissues for the presence of foreign antigens that indicate presence of intracellular pathogens. Recognition of a foreign antigen triggers the destruction of the infected cells by antigen-specific effector CD8+ T cells. In the absence of infections all peptide displayed by MHC class I are derived from self-proteins. Qualitative and/or quantitative alterations of component(s) of the self-peptide pool by transformation can also induce cytotoxic CD8+ T cell responses that form the basis of antigen-specific anti-tumor responses [4]. MHC class I bound self-peptides may also serve as targets for tissue destruction in autoimmune diseases [5] and organ rejection following transplantation [6].
Besides these roles in eliciting antigen-specific effector CD8+ T cell responses (protective or destructive), peptide/MHC class I complexes have several important roles in the physiology of T cells. Engagement of the T-cell receptors with self peptide/MHC complex is the basis for TCR repertoire formation in the thymus and thymocyte maturation [7]. Further, homeostasis of naïve T cells and maintenance of functional competence of memory T cells in the periphery depends on the constant engagement with self-peptide/MHC complexes [8]. In addition, self-peptide/MHC complexes may modulate the strength of immune responses to foreign antigens [9]. In addition to T cells, self-peptidome can impact other systems as well. For example, the function of NK cells is inhibited by self-peptide/MHC class I complexes through the engagement of killer inhibitory receptors [10]. Finally, MHC class I-bound peptides influence mating preferences in mice [11].
Given these diverse roles of peptide/MHC class I complexes, it is important to determine the rules of formation of the peptidome, as well as the individual components that may exhibit important biologic functions. This task is complicated by the complexity of the peptidome, with individual peptides being presented in low copy numbers, ranging from 10-1000 per cell [12]. In this study we isolated and characterized 85 different endogenous MHC class I bound peptides from undifferentiated U937 cells, associated with anyone of the six MHC Class I alleles expressed by this cell line. Peptides isolated by mild acid-elution from the cell surface displayed distinct characteristics, different from peptides isolated by immunoprecipitation of MHC class I. The former peptide set contained peptides with relatively looser association with MHC class I. The differential distribution of MHC class I alleles as putative binders of peptides in the two sets suggests fundamental differences between MHC class I alleles in self peptidome binding. This may explain the association of certain MHC class I alleles with autoimmune diseases.
Materials and Methods
Cell lines and reagents
The human monocytic leukemia cell U937 was cultured in stirrer flasks (Wheaton Science Products, Millville, NJ, USA) on Cellgro™ stirrers (Barnstead International, Dubuque, IA, USA) in complete RPMI-1640 medium containing 25 mM HEPES and supplemented with 10 % fetal calf serum, 2mM L-glutamine, 1mM sodium pyruvate, 4.5 g/l glucose and 10mM Penicillin/Streptomycin at an initial cell density of 1x 106cells/ml in a 5% carbon dioxide atmosphere. The murine hybridoma cells (HB-95) producing monoclonal antibody W6/32 (mouse anti human MHC Class I) reactive with HLA-A, B and C were obtained from ATCC (Manassas, Virginia, IL, USA). The hybridoma cells were grown in serum free SFM4MAB cell culture media (Hyclone Laboratories Inc. Logan, Utah, USA) supplemented with 10mM Penicillin/Streptomycin, 0.1 mM hypoxanthine, 0.016 mM thymidine, 1.0 mM sodium pyruvate at an initial cell concentration of 2x105 cells/ml.
Protease cocktail was obtained from Roche Molecular Biochemicals, Indianapolis, IN, USA. HPLC solvents, Tri Fluoro Acetic Acid (TFA) and acetonitril were obtained from Sigma, St. Louis, MO, USA. EZ-Link-sulfo-NHS biotin was from Pierce, Rockford, IL, USA. RPMI 1640, Penicillin/Steptomicin from Biosource, Camarillo, CA . Hypoxanthine and thymidine were from ATCC (Manassas, Virginia, USA). L-Glutamine and NuPAGE gel from Invitrogen, San Diego, CA, USA. N-octyl-beta-D-glucopyranoside CALBIOCHEM, La Jolla, CA, USA. Dynabeads Protein G from Dynal Biotech Inc. Lake Success, NY, USA. Nitrocellulose membrane was from Bio-Rad, Hercules, CA, USA. ECL-Plus purchased from Amersham Biosciences, Piscataway, NJ, USA.
Mild acid elution of peptides
MHC class I bound peptides were acid extracted from the surface of U937 cell line as described [13]. Briefly, up to 4 x 109 cells were washed three times with PBS and incubated with 0.1M citrate-phosphate buffer pH 3.0 for one minute. The cells were then centrifuged for 10 min at 10,000xg and the supernatant passed through the 5kDa cutoff filter and lyophilized. Peptides were reconstituted in 0.1% TFA and subjected to reverse-phase HPLC purification.
Affinity purification of MHC class I and peptide elution
A one-liter culture of hybridoma cells HB-95/W6/32 were grown in serum free media until the cell concentration reached 2x109. Cells were harvested and supernatant was precipitated with 50% (NH4)2SO4 . Protein precipitate was resuspended and anti MHC class I antibodies (IgG) were purified using a Protein G column. The purified protein was analyzed using a 4–20% NuPAGE gel stained with Coomassie Blue.
A total of 4x109 U937 cells, grown in log phase up to a concentration of 1x108/L, were harvested and washed twice in PBS, pH 8.0. Cells were resuspended in lysis buffer (50 mM Tris-HCl pH 8.0, 150 mM NaCl, 30 mM n-octyl-beta-D-glucopyranoside and protease inhibitor cocktail). The lysate was incubated on ice for 1h and centrifuged for 10 min at 10,000xg. After sample centrifugation at 100,000xg for 1 hour, supernatants were immunoprecipitated with the W6/32 monoclonal antibody and Protein G-coated magnetic beads. The optimal ratio between IgG to MHC molecules was previously determined by immunoprecipitation of biotynilated U937 cells, and plotting the immunoblot intensity of biotinylated β2m molecules versus concentrations of IgG (2-5μg of IgG /107 U937 cell lysate- data not shown). The magnetic beads were then washed 4 times with 300 μl of buffer containing 50 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.3 mM n-octyl-beta-D-glucopyranoside. Subsequently, the beads were rinsed 3 times with 300 μl of low salt buffer (20 mM Tris-HCl, pH 8.0) to remove residual salts and detergents. The immunoprecipitates were treated with 10% acetic acid, boiled for 5 minutes, centrifuged through an Ultrafree-Cl filter with 5kDa cutoff, and lyophilized by vacuum centrifugation.
HPLC and Mass Spectrometry Analysis
The lyophilized peptides were resuspended in (A) H20/0.1% TFA and analyzed by reverse-phase HPLC using a C18 column (Vydac, Hesperia, CA; 4.6 x 250 mm) and Beckman 126 System Gold, equipped with programmable solvent module and variable wavelength detector 165 (Beckman Coulter, Inc., Fullerton, CA, USA). The analog signal was converted to digital signal using a single channel A/D converter and data acquisition software using PeakSimple Chromatography software (SRI Instruments, Torrance, CA, USA). The gradient was made by combining solution (A) with solution (B) of acetonitril/0.1% TFA at a flow rate of 1 ml/min. The reconstituted samples in solution (A) were then applied to the column and 0.31 min fractions were collected from a linear 1h gradient running from 0–60% of solution (B). The eluted peptide peaks were detected at 214 nm. Microsequencing for all fractions was done by use of MALDI-TOF-TOF (Applied Biosystems, Foster city, CA). HPLC fractions eluted between 10 and 40 min were submitted each time for Matrix-assisted laser desorption time-of-flight mass spectrometry (MALDI-TOF-TOF) for sequencing. Individual masses were assigned to peptides with strong MS/MS signals and ion scores equal or greater than 40. Peptide sequencing revealed the presence of multiple peptides in each fraction. Peptide identification was performed using the GPS system followed by BLAST.
HLA-Genotyping of U937 cells
HLA-genotyping was carried out using the PEL-FREEZ ABC SSP unitary kit (Dynal Biotech, LLC, Brown Deer, WI) according to the manufacturer’s instructions.
HLA allele binding assignment
The binding scores of each peptide to one of the six alleles expressed by U937 cells was obtained by using a profile-based prediction algorithm [14], accessible at http://mif.dfci.harvard.edu/Tools/rankpep.html. Each peptide received a numerical score for binding to any of the six alleles. For some alleles it is possible to query peptides of different lengths (e.g. nine, ten and eleven amino acid long for HLA-A*03), while in some cases there is a general allele query and a more specific one as well (e.g. HLA-A*03 and HLA-A*0301). In these cases, all possible scores were examined and the best was retained. The numerical scores were expressed as the percent of the best possible score for binding of a peptide to a given allele, supplied by the provider. Shown in Tables 1 and 2 are the two best scores for each peptide. The allele with the best relative score is considered the source of presentation of particular peptide.
Table 1.
Characteristics of peptides obtained by mild acid-elution. Bolded peptides were also found by affinity purification. Underlined are the sequences that scored best for HLA-binding. For peptides shorter than nine amino acids neighboring residues from either sides were added to search for the HLA-binding, and the ones that contributed to the binding score are indicated in parenthesis and separated from the actual sequence obtained by mass spectrometry. Double-underlined are residues conforming to previously identified binding motifs (see text for explanation).
| Peptide Number | Protein name | Sequence | Peptide length (aa) | Ion score | Calculated mass | Observed mass | HLA-binding assignment (score) |
|---|---|---|---|---|---|---|---|
| 1 | Actin ( Gama) | RVAPEEHPVL | 10 | 58 | 1146.626465 | 1146.609253 | Cw*0102(61.43); B*5101(38.16) |
| 2 | ADP-ribosylation factor 1 | DWLSNQLRNQK | 11 | 73 | 1401.723267 | 1401.721924 | A*0301(55.97); A*31(43.59) |
| 3 | Calcyclin | (AL)….IYNEALKG | 8 | 63 | 907.4882813 | 907.4818726 | A*0301(62.41); A*31(53.85) |
| 4 | Calcyclin | PLDQAIGLL | 9 | 63 | 939.5509033 | 939.541626 | Cw*0102(11.43) |
| 5 | Calcyclin | DRNKDQEVNF | 10 | 56 | 1264.591553 | 1264.580078 | Cw*0702(37.84) |
| 6 | Calcyclin | LDRNKDQEVN | 10 | 45 | 1230.6073 | 1230.605835 | B*5101(30.26) |
| 7 | Calpactin I light chain | VVHMKQKGKK | 10 | 49 | 1182.713867 | 1182.71936 | A*0301(55.64); A*31(46.15) |
| 8 | Calpactin I light chain | FVVHMKQKGKK | 11 | 51 | 1329.782349 | 1329.772461 | A*0301(58.96); A*31(46.15) |
| 9 | Calgizzarin | IGGLAMACHDSF | 12 | 60 | 1221.539063 | 1221.52417 | B*5101(24.34) |
| 10 | Cofilin, non-muscle isoform | MLPDKDCRYA | 10 | 48 | 1211.554688 | 1211.61853 | B*51(54.42); Cw*0102(24.29) |
| 11 | Deoxyuridine 5′-triphosph. nucleotidohydrolase | RIFYPEIEE | 9 | 53 | 1195.599243 | 1195.592407 | A*3101(63.01); A*0301(41.35) |
| 12 | Elongation factor 1-alpha 1 | SDYPPLGRF | 9 | 47 | 1051.52063 | 1051.515503 | Cw*0701(20.27); B*18(17.98) |
| 13 | Elongation factor 1-alpha 1 | FSDYPPLGRF | 10 | 67 | 1198.589111 | 1198.573853 | A*31(26.50); Cw*0701(20.27) |
| 14 | Fructose-bisphosphate aldolase A | (SL)….FVSNHAY | 7 | 48 | 837.388916 | 837.3813477 | A*0301(47.37); A*3101(38.15) |
| 15 | Fructose-bisphosphate aldolase A | TADDRVNPCIGG | 12 | 40 | 1217.557861 | 1217.557983 | Cw*0102(25.00); B*5101(21.71) |
| 16 | Galectin-1 | VKLPDGYEFKFPNRLNL | 17 | 48 | 2050.111816 | 2050.207031 | NA |
| 17 | Galectin-1 | TVKLPDGYEFKFPNRLNL | 18 | 40 | 2151.159668 | 2151.258057 | NA |
| 18 | Glucose-6-phosphate isomerase | IKQQREARVQ | 10 | 50 | 1255.7229 | 1255.738403 | B*5101(31.58); A*03(22.55) |
| 19 | Glyceraldehyde 3-phosphate dehydrogenase | (DL)….MAHMASKE* | 8 | 45 | 904.4014893 | 904.3967285 | A*03(62.75); A*31(53.85) |
| 20 | Glyceraldehyde 3-phosphate dehydrogenase | YDNEFGYSNRVVDL | 14 | 78 | 1690.77063 | 1690.73877 | NA |
| 21 | Glyceraldehyde 3-phosphate dehydrogenase | ISWYDNEFGYSNRVVDL | 17 | 92 | 2076.966064 | 2076.969482 | NA |
| 22 | Glyceraldehyde 3-phosphate dehydrogenase | QERDPSKIKWGDAGAEY | 17 | 122 | 1949.935059 | 1949.892944 | NA |
| 23 | 10 kDa heat shock protein (Hsp10) | FRDGDILGKYVD | 12 | 80 | 1397.705933 | 1397.699097 | Cw*0702(32.43); B*5101(21.71) |
| 24 | Macrophage capping protein | LGERPVQHREVQGNESD | 17 | 52 | 1949.942261 | 1950.013672 | NA |
| 25 | Macrophage capping protein | LGERPVQHREVQGNESDLF | 19 | 99 | 2210.094727 | 2210.056885 | NA |
| 26 | Macrophage migration inhibitory factor | (ERL)….RISPDRVY | 8 | 41 | 1005.547546 | 1005.540588 | A*31(54.70); B*51(48.30) |
| 27 | Macrophage migration inhibitory factor | RLRISPDRVY | 10 | 39 | 1274.732666 | 1274.719238 | A*0301(27.61); B*51(22.22) |
| 28 | Macrophage migration inhibitory factor | LAERLRISPDRVY | 13 | 51 | 1587.896484 | 1587.881836 | NA |
| 29 | Macrophage migration inhibitory factor | IVNTNVPRASVPDGFLSEL | 19 | 78 | 2028.075928 | 2028.056641 | NA |
| 30 | Macrophage migration inhibitory factor | TQQLAQATGKPPQYIAVHVVPDQL | 24 | 50 | 2602.398682 | 2602.343506 | NA |
| 31 | Mannose-6-phosphate receptor binding protein | (S)….VQQQRQEQ | 8 | 42 | 1043.522827 | 1043.552612 | A*03(54.89); B*18(46.49) |
| 32 | Mannose-6-phosphate receptor binding protein | DVASVQQQRQEQSY | 14 | 59 | 1665.782593 | 1665.781494 | NA |
| 33 | NSFL1 cofactor p47 (p97 cofactor) | FIVDARPAMAATSF | 14 | 45 | 1496.756592 | 1496.731689 | NA |
| 34 | Nuclear protein ZAP3 (ZAP113) | (WDR)…DYGRPLDE | 8 | 50 | 964.4370117 | 964.5213623 | A*0301(27.30); Cw*0102(21.79) |
| 35 | Peptidyl-prolyl cis-trans isomerase A | VDGEPLGRVSFEL | 13 | 50 | 1417.732056 | 1417.721313 | NA |
| 36 | Peptidyl-prolyl cis-trans isomerase A | DIAVDGEPLGRVSF | 14 | 59 | 1474.75354 | 1474.729492 | NA |
| 37 | Peptidyl-prolyl cis-trans isomerase A | IAVDGEPLGRVSFEL | 15 | 53 | 1601.853271 | 1601.857422 | NA |
| 38 | Peptidyl-prolyl cis-trans isomerase A | ILKHTGPGILSMANAGPNTNGSQF | 24 | 110 | 2425.229248 | 2425.177979 | NA |
| 39 | Peptidyl-prolyl cis-trans isomerase A | FILKHTGPGILSMANAGPNTNGSQF | 25 | 44 | 2572.297607 | 2572.261719 | NA |
| 40 | Phosphatidylethanolamine-binding protein | PVDLSKWSGPLSL | 13 | 97 | 1398.762695 | 1398.767212 | NA |
| 41 | Placental calcium-binding protein | FEGFPDKQPRKK | 12 | 73 | 1476.795654 | 1476.801514 | A*31(43.59); B*51(38.78) |
| 42 | Placental calcium-binding protein | FFEGFPDKQPRKK | 13 | 57 | 1623.864136 | 1623.835815 | NA |
| 43 | Placental calcium-binding protein | MSNLDSNRDNEVD | 13 | 49 | 1508.628174 | 1508.631958 | NA |
| 44 | Placental calcium-binding protein | MSNLDSNRDNEVDF | 14 | 66 | 1655.696533 | 1655.693848 | NA |
| 45 | Profilin I | VNITPAEVGVLVGKDRSSF | 19 | 94 | 1988.081055 | 1988.04541 | NA |
| 46 | Profilin I | AAVPGKTFVNITPAEVGVLVGKDRSSF | 27 | 130 | 2759.508789 | 2759.43042 | NA |
| 47 | Profilin I | KEGVHGGLINKKCYEMASHLRRSQY | 25 | 41 | 2904.471924 | 2904.578125 | NA |
| 48 | Profilin I | MGKEGVHGGLINKKCYEMASHLRRSQY | 27 | 60 | 3092.533691 | 3092.452148 | NA |
| 49 | Proteasome act. complex subunit 2 (PA28beta) | YRFLPQKIIYL | 11 | 46 | 1453.856567 | 1453.854248 | Cw*0102(51.79); Cw*0702(45.95) |
| 50 | Proteasome act. complex subunit 2 (PA28beta) | LYRFLPQKIIYL | 12 | 65 | 1566.940552 | 1566.933838 | Cw*0102(51.79); Cw*0702(45.95) |
| 51 | Pyruvate kinase, M1 isozyme | (TF)….LEHMCRL | 7 | 46 | 901.4382324 | 901.432373 | Cw*0702(34.68) |
| 52 | 40S ribosomal protein S28 | LESEREARRLR | 11 | 42 | 1414.787231 | 1414.779785 | A*31(52.14); B*5101(35.53) |
| 53 | 60S acidic ribosomal protein P2 | MRYVASYLL | 9 | 55 | 1115.591675 | 1115.591187 | Cw*0702(32.43); B*5101(28.95) |
| 54 | 60S ribosomal protein L15 | ATYGKPVHH | 9 | 50 | 1009.521362 | 1009.526062 | A*31(29.91); A*03(29.41) |
| 55 | Small nuclear ribonucleoprotein Sm D1 | FILPDSLPLDTL | 12 | 45 | 1343.745605 | 1343.746582 | B*51(40.58); A*3101(32.56) |
| 56 | Synaptic vesicle membrane pr. VAT-1 homolog | LARTWWNQF | 9 | 40 | 1221.616333 | 1221.69519 | B*51(11.56) |
| 57 | Synaptic vesicle membrane pr. VAT-1 homolog | VSGVVARLLAL | 11 | 40 | 1097.703979 | 1097.750122 | B*51(45.41); Cw*0102(30.00) |
| 58 | Synaptic vesicle membrane pr. VAT-1 homolog | LVSGVVARLLAL | 12 | 40 | 1210.788086 | 1210.833374 | B*51(45.41); Cw*0102(30.00) |
| 59 | Transaldolase | ISPFVGRILDW | 11 | 40 | 1302.720459 | 1302.778198 | A*31(28.19); Cw*0702(26.58) |
| 60 | Transgelin 2 | WEGKNMACVQR | 11 | 51 | 1321.613892 | 1321.685547 | A*31(40.17); B*18(27.63) |
| 61 | Transgelin 2 | WEGKNMACVQRTL | 13 | 66 | 1535.745728 | 1535.807861 | NA |
| 62 | Transketolase | FGIDRDAIAQAVRGLITKA* | 19 | 69 | 2015.139526 | 2015.222778 | NA |
| 63 | Vacuolar ATP synthase subunit G 1 | DIRPEIHENYRING | 14 | 55 | 1725.866577 | 1725.852661 | NA |
| 64 | Zinc finger protein 236 | EEEEEHS….(DR) | 7 | 50 | 888.3217163 | 888.2773438 | A*31(40.17); B*18(31.58) |
Table 2.
Peptides obtained by MHC class I immunoprecipitation. Bolded peptides were also found by affinity purification. Underlined are the sequences that scored best for HLA-binding. Double-underlined are residues conforming to previously identified binding motifs (see text for explanation).
| Peptide Number | Protein name | Peptide sequence | Peptide length (aa) | Ione score | Calculated Mw(Da) | Observed Mw(Da) | HLA-binding assignment (score) |
|---|---|---|---|---|---|---|---|
| 1 | Acyl-CoA desaturase 1 | LPLRLFLII | 9 | 56 | 1097.74450 | 1097.76538 | B*51(74.15) |
| 2 | Chromatin assembly factor 1 subunit C | EERVINEEY | 9 | 50 | 1180.54797 | 1180.51867 | B*18(34.65); Cw*0702(20.27) |
| 3 | DNA-dep. protein kinase catal. subunit | DEFKIGELF | 9 | 50 | 1097.55126 | 1097.56286 | B*18(28.07) |
| 4 | Elongation factor 1-alpha 1 | FSDYPPLGRF | 10 | 41 | 1198.58911 | 1198.59143 | A*31(26.50); Cw*0702(20.27) |
| 5 | Histone H1.2 | SLAALKKALAAAGYDVEKNNSRIKLGLK | 28 | 66 | 2942.71484 | 2942.63501 | NA |
| 6 | Histone H1.3 | KVAGAATPK | 9 | 48 | 842.50933 | 842.50152 | A*03(75.49); A*31(69.23) |
| 7 | Human heat shock protein HSP 90 beta | IDIIPNPQERTLTL | 14 | 54 | 1622.91113 | 1622.91638 | NA |
| 8 | Microsomal signal peptidase | VPYIGIVTI | 9 | 61 | 974.59204 | 974.54852 | B*5101(74.34); Cw*0102(19.29) |
| 9 | Placental calcium-binding protein | FEGFPDKQPRKK | 12 | 40 | 1476.79565 | 1476.81176 | A*31(43.59); B*5101(26.32) |
| 10 | Polymyositis/scleroderma autoantigen | DEYDFYRSF | 9 | 38 | 1241.51086 | 1241.53002 | B*18(35.53); Cw*0702(24.77) |
| 11 | 26S proteasome non-ATPase regulatory subunit 7 | RIGKVGNQK | 9 | 47 | 999.60571 | 999.60681 | A*03(49.02); A*31(40.17) |
| 12 | 40S ribosomal protein S26 | DAYVLPKLYVKL | 12 | 44 | 1421.84021 | 1421.80920 | A*31(53.85); A*0301(50.38) |
| 13 | 40S ribosomal protein S30 | KVHGSLARAGKVRGQTPK | 18 | 84 | 1890.11425 | 1890.14758 | NA |
| 14 | 60S ribosomal protein L15 | ATYGKPVHH | 9 | 51 | 1009.52136 | 1009.52252 | A*31(29.91); A*03(29.41) |
| 15 | 60S ribosomal protein L18a | RIFAPNHVVAK | 11 | 62 | 1251.73193 | 1251.73999 | Cw*0102(52.14); A*3101(44.65) |
| 16 | 60S ribosomal protein L23a | RLRRQPKYPRK | 11 | 37 | 1497.92358 | 1497.94116 | A*31(65.81); A*0301(27.07) |
| 17 | 60S ribosomal protein L26 | DEVQVVRGHY | 10 | 72 | 1201.59594 | 1201.56811 | B*18(43.86) |
| 18 | 60S ribosomal protein L29 | RLAYIAHPK | 9 | 62 | 1068.63122 | 1068.61950 | A*31(70.94); A*03(55.88) |
| 19 | U5 small nuclear ribonucleoprotein | SEIELFRVF | 9 | 49 | 1139.60949 | 1139.60278 | B*18(26.32); B*51(17.69) |
| 20 | Thymidylate synthase | DAHIYLNHI | 9 | 50 | 1095.55810 | 1095.56201 | B*5101(32.89); B*18(24.56) |
| 21 | Tubulin epsilon chain | DEFPEVYRF | 9 | 54 | 1201.55236 | 1201.56945 | Cw*0702(59.01); B*18(38.60) |
Peptide binding
Peptide binding was assessed as described [15]. Peptides synthesized using FITC-labeled εNH2-lysine at position four were custom purchased from GenScript Corporation (Scotch plains, NJ). Peptides were HPLC purified and >95% pure as assessed by mass spectrometry. U937 cells (1x106/well) were incubated for 10 min on ice with various concentrations of FITC-conjugated peptides in 150μl of PBS in a 96 well polypropylene plates followed by three washes in PBS, resuspended in 300 μl PBS and immediately analyzed by Flow cytometry.
Results
General characteristics of mild acid-eluted peptides
MHC class I-binding peptides can be isolated by direct acid elution from cells or by immunoprecipitation of MHC class I followed by peptide elution. To analyze the spectrum of MHC class I-bound peptides of U937 cells we performed peptide elution by both methods. Isolated peptides were subsequently analyzed by matrix-assisted laser desorption mass spectrometry (MALDI-TOF-TOF). Typical sequencing results are shown in Fig. 1. The length distribution for the acid eluted peptides ranged from 7 to 28. A total of 64 peptides were isolated and sequenced (Table 1). 33 peptides (52%) were 8–12 aa long, 28 (44%) were larger peptides, while 3 (4%) peptides were 7aa in length. We found several nested sets of peptides (for examples see Table 1, peptides #5 and 6, 7 and 8, 12 and 13, 16 and 17, 20 and 21, 24 and 25, 26–28, 31 and 32, 35–37, 38 and 39, 41 and 42, 43 and 44, 45 and 46, 47 and 48, 49 and 50, 57 and 58, 60 and 61), which are more characteristic for MHC class II than for MHC class I molecules [16]. In contrast to MHC class II, the ends of MHC class I peptide binding groove are closed and only peptides of certain length can fit without reducing the biophysical properties of binding. Nevertheless, the nested set of peptides were recently described for MHC class I bound peptides [17], and likely result from partial proteolysis.
Figure 1.

Peptide isolation and typical mass spectrometry analysis. A-B) Reverse phase HPLC separation of MHC Class I bound peptides, isolated from U937 cells. Peptides were obtained by mild acid elution (A) or from HLA molecules purified by immunoprecipitation (B). Peptides bound to MHC Class I were released by acid treatment and passed through 5kDa cutoff filter. The peptide mixtures were separated on a C18 column (4.6x250mm) using a 0–60% acetonitril in 0.1%TFA vol/vol) gradient run over 1h at a flow rate of 1ml/min. C) A mass spectrometer spectrum for the peptide sequence of 60S Ribosomal protein L26 fractionated at t~23min and sequenced by use of matrix-assisted laser desorption mass spectrometry (MALDI-TOF-TOF).
General characteristics of affinity-purified peptides
Because of the relatively high frequency of longer peptides, as well as few peptides shorter than optimal for binding to MHC class I, we suspected that by using mild acid elution we may have isolated a subset of peptides enriched for low-affinity binding to MHC class I. To test whether the size of eluted peptides was dependent on conditions of isolation, peptides were isolated by affinity purification using an antibody specific for all human class I allomorphs (W6/32) and then analyzed by mass spectrometry in an identical manner. As expected, the frequency of peptides longer than 12aa was reduced to 3 out of a total of 21 (14%), while peptides shorter than 9aa were not detected at all (Table 2). Even though reduced in frequency, the longer peptides were still detected. One particular peptide (#13; Table 2) was isolated and identified in two separate experiments. Only three peptides identical in their sequences were common for both techniques. These were peptides derived from elongation factor 1-alpha 1, placental calcium-binding protein (calvasculin) and 60S ribosomal protein L15. The minimal overlap between the mild acid eluted and affinity purified MHC class I-eluted peptides suggests that characteristics of peptides isolated by the two approaches may differ.
MHC class I allele binding assignment
To predict MHC class I allele binding to peptides we first identified all alleles present in U937 cells by HLA genotyping. This analysis revealed presence of two alleles for each HLA-A (A*0301 and A*3101), HLA-B (B*1801 and B*5101) and HLA-C (Cw*0102 and Cw*0702) locus (data not shown). To assign particular peptides to given alleles we used a profile based prediction algorithm [14]. Binding of long peptides to MHC class I has been reported only sporadically [18–23], hence the rules for their binding to class I molecules are not well defined. Therefore the assignment of the MHC class I allele binding was done only for peptides of 12aa or less. For peptides 7aa and 8aa in length, the optimal binding allele was searched with N-terminal and C-terminal 3aa extensions (or 4aa N-terminal extension for peptides located at the C-terminus) obtained from the original proteins by blast search. Two top scores for each peptide are shown in Tables 1 and 2. The allele with the highest binding score is hereafter considered as the presenting MHC molecule. We believe that these HLA assignment results are accurate for two reasons. First, four out of five putative HLA-B*1801-binding peptides were confirmed in a recent study that identified peptides binding to soluble HLA-B*1801 [17]. Second, all affinity purified peptides and 72.4% of mild acid-eluted peptides with assigned HLA allele contained at least one or both anchor residues (double underligned in Tables 1 and 2) defining the motifs for binding to the particular allele [24, 25]. These motifs are: L, V or M at position 2 and K, Y, or F at position 9 (HLA-A*03); R at position 9 and auxilliary anchors l, V, Y, or F at position 2 and/or F, L, Y, or W at position 3 (HLA-A*31); E at position 2 and F or Y at position 9 (HLA-B*18); A, P, or G at position 2 and V or I at position 9 (HLA-B*51); A, or L at position 2 and L at position 9 (HLA-Cw*01); and Y, F, or L at position 9 and auxilliary anchors Y or P at position 2, l, V, Y, L, M or F at position 5 and/or V, I, L, or M at position 6 (HLA-Cw*07).
Overall, peptides with scores indicative of binding to all MHC class I alleles were isolated by both mild acid elution and affinity purification. However, the alleles were not equally represented (Table 3). The most mild acid-eluted peptides appeared to bind to HLA-B*5101 (eight), followed by HLA-A*3101 (seven). On the other end of the spectrum, no peptides appeared to have been derived from HLA-B*1801. In the affinity purified peptide set, however, the distribution of putative binding alleles was more balanced between the HLA-A and HLA-B molecules, with the exception of HLA-A*0301. In addition, peptides binding to either of the HLA-C molecules were not abundant. The average binding scores for each individual allele were higher in the affinity-purification-derived peptides than in the mild acid elution-derived group (Table 3). In addition, the proportion of peptides with scores obtained for a fraction of the identified peptide (“mismatched peptides”) is much higher in the mild acid-eluted-group (Table 3). The implication of the “mismatch” is that the binding score for the entire peptide would be lower than the most optimal one. The lower average binding scores and higher proportion of the “mismatched peptides” suggest that peptides eluted by mild-acid treatment bind to the HLA alleles with generally lower affinity. In turn, the relative abundance of peptides binding to the particular allele derived by mild acid elution or affinity purification suggests the overall relative affinity of that allele for binding U937-derived peptides. For example, HLA-B*1801 appears to bind peptides with relatively high affinity since five HLA-B*1801-binders were identified in the affinity purification-derived peptides, while none were assigned from the mild acid elution-derived group. Conversely, HLA-B*5101 appears to be a poor peptide binder since the proportion of peptides assigned to this allele is significantly reduced in the group of affinity purification-derived peptides.
Table 3.
Comparison of predictive binding of peptides isolated by mild acid elution or MHC class I precipitation to six HLA alleles.
| Mild acid elution | Affinity purification | |||||||
|---|---|---|---|---|---|---|---|---|
| HLA allele | Total # of peptides | # of peptides with score/length missmatch* | # of peptides with no anchor residues | Average binding score | Total # of peptides | # of peptides with score/length missmatch* | # of peptides with no anchor residues | Average binding score |
| A*03 | 4 | 3 | 1 | 49.55 | 2 | 0 | 0 | 62.26 |
| A*31 | 7 | 5 | 2 | 40.50 | 6 | 4 | 0 | 48.43 |
| B*18 | 0 | 0 | 0 | - | 5 | 1 | 0 | 33.69 |
| B*51 | 8 | 6 | 4 | 35.45 | 3 | 0 | 0 | 60.46 |
| Cw01 | 5 | 4 | 1 | 40.28 | 1 | 1 | 0 | 52.14 |
| Cw07 | 4 | 2 | 0 | 30.74 | 1 | 0 | 0 | 59.01 |
| Total | 28 | 20 (71.4%) | 8 (28.6%) | 37.24 | 18 | 6 (33.3%) | 0 | 48.67 |
Score/length missmatch is defined by HLA-binding scores assigned to peptides either shorter or longer than the actual sequence obtained by mass spectrometry (see tables I and II for examples).
Protein source of the peptides
The sequenced peptides were found to originate from a variety of proteins. Two questions seem to be the most relevant in this context. First, is abundance of peptide presentation by MHC class I in the function of the level of protein expression? The U937 cell line is very convenient model to address this question because protein expression 2D gel data has been made available on the web (www.expasy.org). Of the 22 proteins, which were shown to be highly expressed, only three proteins were the source of MHC class I-bound peptides: actin, glyceraldehyde-dehydrogenase and peptidyl-prolyl cis-trans isomerase A. Interestingly, all peptides from these proteins were identified by the mild acid elution method. No peptide sequence homology for any of the other highly expressed proteins of U937 was found.
Second, are MHC class I-binding peptides preferentially derived from ribonucleoproteins, as suggested recently by the study on HLA-B*1801 peptidome [17]. Seven of the 21 affinity purified peptides (33.3%) were derived from nucleoproteins (Fig. 2). These results suggest that ribonucleoproteins indeed preferentially serve as a source of MHC class I-binding peptides. Interestingly, among the mild acid-eluted peptide group, the frequency of ribonucleoproteins as a source is significantly lower (3 out of 37 <13aa long peptides- 8.1%) suggesting that lower affinity binding peptides are rarely produced from ribonucleoproteins. The converse is true for membrane associated proteins. Only one of the seven ribonucleoprotein-derived peptides binds to HLA-B*1801. The remaining peptides bind to HLA-A*3101 (4 of 7), HLA-Cw*0102 (1 of 7), while the binding HLA for one peptide 18aa long has not been assigned. Therefore, the distribution of allele binding of ribonucleoprotein-derived peptides is relatively unbiased (with most peptides binding neither to the “well binding allele HLA-B*1801, nor the poor binding HLA-B*5101).
Figure 2.

Subcellular distribution of protein donors of MHC class I-associated peptides. A) Peptides isolated by mild acid elution. B) Peptides isolated by affinity purification.
HLA-B*1801- binding synthetic peptide with low binding score binds better than HLA-B*5101- binding peptide with high binding score
An implication of the above findings is that peptide binding to certain HLA class I alleles (i.e. HLA-B*1801) may be relatively strong, while relatively weak to the others (HLA-B*5101). Furthermore, relatively high binding scores can be obtained with peptides binding to both alleles, and yet the numbers of peptides binding to HLA-B*1801 or HLA-B*5101 is severely reduced in the peptide set obtained by mild acid elution or affinity purification, respectively. An assumption can therefore be made that binding to HLA-B*1801 of peptides with relatively poor binding scores may be of higher affinity than binding to HLA-B*5101 of peptides with relatively high binding scores. To test this hypothesis we identified from the literature a highly-scoring (score: 77.1) HLA-B*5101-binding peptide [26], and a poorly scoring HLA-B*1801-binding peptide from the present study ( peptide number 3 from Table 2; score: 28.1). These two peptides were chosen because both have lysine at position 4, whose side chain can be labeled with FITC, and the binding of labeled peptides to U937 cells can be comparatively assessed. Indeed, U937 cells stained brighter in the presence of equimolar concentrations of HLA-B*1801-binding peptide (Fig. 3).
Figure 3.
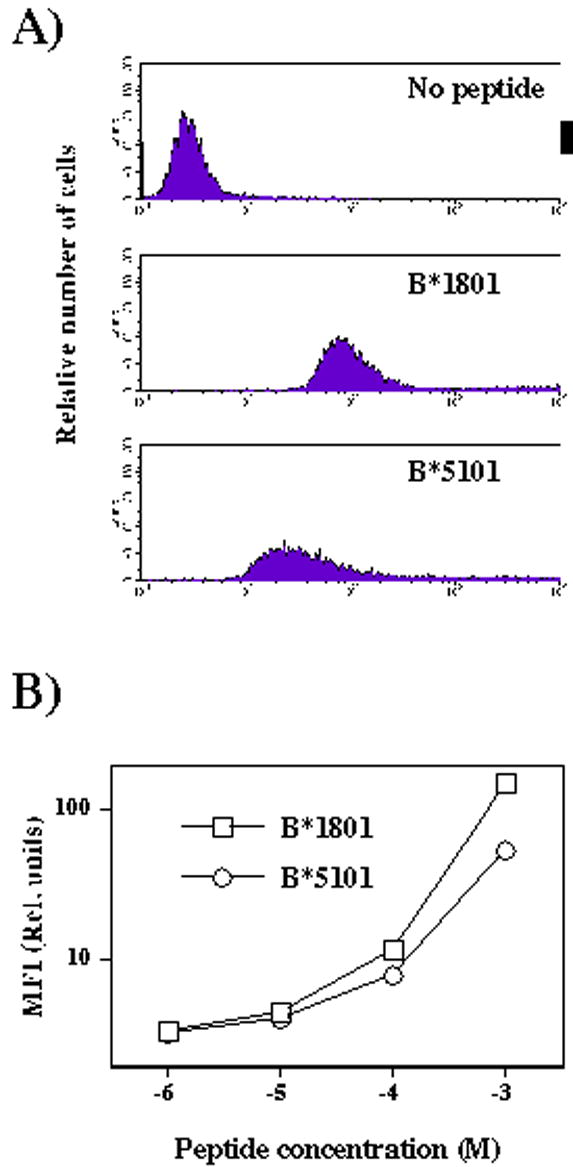
Binding of fluoresceinated peptides to U937 cells. A) Representative histograms of U937 cells incubated in the absence or presence of HLA-B*1801- (sequence: DEFKIGELF) or HLA-B*5101- (LPEKDSWTV) binding peptides (1x10−3M). B) Graph displaying mean fluorescence intensity of U937 cells incubated with indicated concentrations of HLA-B*1801- or HLA-B*5101-binding peptides.
Discussion
We have analyzed characteristics of peptides eluted from six MHC class I molecules expressed by the U937 cell line. This cell line was chosen because it is frequently used model for epitope identification from infectious agents and because of existing considerable data on intracellular protein composition. Peptides were simultaneously isolated either by mild acid elution from living cells, or by affinity purification using antibody with specificity for a common epitope of MHC class I. Very little overlap in obtained peptide sequences indicated that the two peptide subsets have broadly different characteristics. Several lines of evidence, discussed in more detail below, indicate that mild acid elution produces a peptide subset that binds to MHC class I with lower affinity. Assignment of binding MHC class I molecules to individual peptides revealed preferential representation: HLA-B*1801 was represented mostly in the peptides isolated by affinity purification, whereas HLA-B*5101, and to a lesser degree HLA-Cw*0102 and HLA-Cw*0702 were preferentially represented among the peptides obtained by mild acid elution. These findings suggest that the latter group of MHC class I molecules binds peptides with reduced affinity, relative to other MHC class I molecules.
There are several differences between the self-peptide sets obtained by mild acid elution or MHC class I immunoprecipitation. First, the former peptide set was more heterogeneous with respect to the peptide size and contained more peptides longer and shorter than optimal for binding to MHC class I. Although there is no direct proof that these peptides actually bind MHC class I, prior evidence for binding of unusually long peptides to MHC class I [18–23], together with heavy bias of the HLA-binding scores for alleles expressed by the U937 cells (data not shown), suggest that these peptides are associated with MHC class I. Second, the same peptide population contained more examples of nested sets of peptides. Both of these characteristics are suggestive of lower binding affinity of the former peptide set. Further support for this notion is provided by predictive scores of binding, that were generally lower in the former set. In addition, the scores in the former set were more frequently assigned to peptides that were one or more amino acid shorter than the actual sequence obtained by mass-spectrometry analysis, indicating that these scores for the actual (longer) peptides should in effect be even lower. Collectively, these data make a strong case in favor of less tight association with MHC class I of peptides which were obtained by mild acid elution.
Interestingly, significant portion of mild acid-eluted peptides represent C-terminal sequences of proteins. Fourteen of the total 64 sequenced peptides from eleven proteins were derived from the C-terminal portion, in contrast to none of the peptides in the affinity-purified group. Eight of the fourteen C-terminal peptides were either shorter or longer than optimal length. These data suggests that this form of processing for MHC class I presentation may not be best suited for generation of optimal MHC class I binding peptides. The C-terminal epitope generation, as originally described, involves the action of ER-resident amino terminal peptidase cleaving the C-terminus of the ER-resident proteins [27]. Subsequently, it was suggested that C-terminal peptides can be liberated from proteins with virtually any subcellular localization [28]. Indeed, the eleven donor proteins in our study were membrane, cytoplasmic, mitochondrial, ribosomal and vacuolar proteins. Furthermore, the higher abundance of C-terminal peptides in the mild acid-eluted group suggests that processing C-termini of proteins yields in general peptides with suboptimal capacity for binding to MHC class I. Most likely, this is the reason why C-terminal processing was originally discovered in transporter associated with antigen processing-deficient cells.
The finding that mild acid-eluted peptides have a lower binding affinity for MHC class I than the affinity-purified peptides, together with the differences in allele-binding assignments in the two groups of peptides provided an insight into the relative efficiency of distinct MHC class I alleles in binding peptides. The biggest differences were seen with HLA-B*1801 and HLA-B*5101, which were preferentially found as binding alleles in the affinity-purified- or mild acid-eluted groups of peptides, respectively. This observation indicates that HLA-B*1801 may bind peptides with higher affinity than other alleles expressed by the U937 cells, whereas HLA-B*5101 may be on the other end of the spectrum of binding affinity for U937 cell derived self-peptides. Affinity and stability of binding of peptides to MHC molecules can be determined on an individual peptide basis, and binding to HLA-B*5101 was previously found to be lower than individual peptide binding to closely related HLA-B*3501 [29]. Our results extend these findings suggesting that peptide binding to HLA-B*5101 is globally of low affinity relative to not only to closely related but also different HLA molecules. Direct binding assays suggested that even the worst scoring HLA-B*1801-binding peptide has a higher affinity than the relatively well scoring HLA-B*5101-binding peptide (Fig. 3), albeit the binding of peptides to other HLA alleles in this assay has not been excluded. The structural basis for the low affinity peptide binding may be a small size of the A pocket in the HLA-B*5101 that accommodates only very small amino acids and forces a non-standard zig-zag peptide conformation with the peptide N- and C-termini protruding out of the groove more than in other HLA molecules [30].
HLA-B*5101 allele expression is associated with Behçet disease [31, 32], a multisystemic inflammatory disorder possibly of autoimmune nature [33, 34]. The linkage is very suggestive of involvement of peptide presentation function, since Behçet disease is not associated with HLA-B*5201, which differs by only two amino acids located in the pocket B of the peptide-binding groove [34]. However, although the general characteristics of HLA-B*5101 and other related allomorphs have been studied [26, 35], the identity of peptide(s) responsible for development of the Behçet disease is not known. Responses to Herpes Simplex Virus, streptococcal and other bacterial antigens have been proposed as a triggering event for subsequent cross-reactive responses to self-antigens [33, 34]. Peptides derived from thymidilate synthase, guanine-nucleotide binding protein b subunit-like protein 12.3, or yeast protein UBC5 have been proposed as possible autoantigens involved in the pathogenesis of Behçet Disease [33]. The sequence of the first of these three peptides has been identified in this study (Table 2, peptide #20). T cell activation and oligoclonal expansions of T cells are frequently found in Behçet Disease patients, but high individual variability of dominant Vβ subsets makes a single underlying antigen or auto-antigen involvement unlikely [36]. A unifying hypothesis linking potentially multiple (auto)antigens to the selective involvement of HLA-B*5101 in the pathogenesis of the disease is missing.
Our findings of generally decreased peptide binding to HLA-B*5101 may provide the missing link. Tolerance to self-peptide/MHC complexes is most efficiently established for self-peptides with high affinity binding for MHC [37]. Peptides that bind with low affinity are unable to establish tolerance, but are also unable to induce immune responses. However, peptides with intermediate affinity are poor at inducing tolerance, but are capable of inducing the immune responses. In fact, this group of peptides is considered for immunotherapy approaches against cancer. The same group, of course, carries the potential of initiating autoimmune responses. Thus, lower affinity binding of peptides by HLA-B*5101 may predispose development of distinct autoimmune responses, which could explain presumed multiple specificities in Behçet Disease patients. The question remains, how T cell responses to different antigen-specificities can produce the single clinical entity of Behçet disease? Presumably, different autoantigens may have different tissue distribution and would thus favor different individual pathology. This is to a degree indeed the case, as Behçet disease may present with relatively heterogeneous symptomatology [33, 34]. In addition, animal models of autoimmunity have clearly demonstrated that an autoimmune response to ubiquitous autoantigen can lead to localized pathology [38], further complicating the relationship between the localization of causative autoantigen and the clinical outcome of the disease.
Assessing the binding of a population of different peptides to MHC class I is technically difficult because of the individual variability in peptide binding. MHC-binding prediction algorithms provide a tool for initial estimates of the binding abilities of distinct MHC molecules. The present study provides an example of how prediction algorithms together with different methodologies of isolating MHC class I-binding peptides, followed by mass spectrometry sequencing, can provide this needed information on binding of peptidomes to distinct MHC class I molecules. Because MHC alleles are associated with a number of different types of diseases [39], this information may be important for understanding the pathogenesis of the MHC-associated diseases, as here postulated for the association of HLA-B*5101 with Behçet disease.
Acknowledgments
The authors would like to thank Eric Ecclestone for the help with mass spectrometry analysis and Daniela Papini for the help with flow cytometry. Footnotes
Footnotes
This work was supported by the CRI Discovery Fund awarded to HS and NIH grants AI48837 and AI41573 to SV.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Pamer E, Cresswell P. Mechanisms of MHC class I--restricted antigen processing. AnnuRevImmunol. 1998;16:323. doi: 10.1146/annurev.immunol.16.1.323. [DOI] [PubMed] [Google Scholar]
- 2.Engelhard VH. Structure of peptides associated with Class I and Class II MHC molecules. Ann Rev Immunol. 1994;12:181. doi: 10.1146/annurev.iy.12.040194.001145. [DOI] [PubMed] [Google Scholar]
- 3.Yewdell JW. Immunology. Hide and seek in the peptidome. Science. 2003;301:1334. doi: 10.1126/science.1089553. [DOI] [PubMed] [Google Scholar]
- 4.Boon T, Cerottini J-C, Van den Eynde B, van der Bruggen P, Van Pel A. Tumor antigens recognized by T lymphocytes. AnnRevImmunol. 1994;12:337. doi: 10.1146/annurev.iy.12.040194.002005. [DOI] [PubMed] [Google Scholar]
- 5.Vizler C, Bercovici N, Cornet A, Cambouris C, Liblau R. Role of autoreactive CD8+ T cells in organ-specific autoimmune diseases: insight from transgenic mouse models. Immunol Rev. 1999;169:81. doi: 10.1111/j.1600-065x.1999.tb01308.x. [DOI] [PubMed] [Google Scholar]
- 6.Rocha PN, Plumb TJ, Crowley SD, Coffman TM. Effector mechanisms in transplant rejection. Immunol Rev. 2003;196:51. doi: 10.1046/j.1600-065x.2003.00090.x. [DOI] [PubMed] [Google Scholar]
- 7.Goldrath AW, Bevan MJ. Selecting and maintaining a diverse T-cell repertoire. Nature. 1999;402:255. doi: 10.1038/46218. [DOI] [PubMed] [Google Scholar]
- 8.Marleau AM, Sarvetnick N. T cell homeostasis in tolerance and immunity. J Leukoc Biol. 2005;78:575. doi: 10.1189/jlb.0105050. [DOI] [PubMed] [Google Scholar]
- 9.Vukmanovic S, Santori FR. Self-peptide/MHC and TCR antagonism: physiological role and therapeutic potential. Cell Immunol. 2005;233:75. doi: 10.1016/j.cellimm.2005.04.011. [DOI] [PubMed] [Google Scholar]
- 10.Vilches C, Parham P. KIR: diverse, rapidly evolving receptors of innate and adaptive immunity. Annu Rev Immunol. 2002;20:217. doi: 10.1146/annurev.immunol.20.092501.134942. [DOI] [PubMed] [Google Scholar]
- 11.Ziegler A, Kentenich H, Uchanska-Ziegler B. Female choice and the MHC. Trends Immunol. 2005;26:496. doi: 10.1016/j.it.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 12.Perreault C, Roy DC, Fortin C. Immunodominant minor histocompatibility antigens: the major ones. ImmunolToday. 1998;19:69. doi: 10.1016/s0167-5699(97)01185-7. [DOI] [PubMed] [Google Scholar]
- 13.Sugawara S, Abo T, Kumagai K. A simple method to eliminate the antigenicity of surface Class I MHC molecules from the membrane of viable cells by acid treatment at pH 3. J Immunol Methods. 1987;100:83. doi: 10.1016/0022-1759(87)90175-x. [DOI] [PubMed] [Google Scholar]
- 14.Reche PA, Glutting JP, Reinherz EL. Prediction of MHC Class I Binding Peptides Using Profile Motifs. Hum Immunol. 2002;63:701. doi: 10.1016/s0198-8859(02)00432-9. [DOI] [PubMed] [Google Scholar]
- 15.Day PM, Esquivel F, Lukszo J, Bennink JR, Yewdell JW. Effect of TAP on the generation and intracellular trafficking of peptide-receptive major histocompatibility complex class I molecules. Immunity. 1995;2:137. doi: 10.1016/s1074-7613(95)80014-x. [DOI] [PubMed] [Google Scholar]
- 16.Rammensee H-G. Chemistry of peptides associated with MHC class I and class II molecules. CurrOpinImmunol. 1995;7:85. doi: 10.1016/0952-7915(95)80033-6. [DOI] [PubMed] [Google Scholar]
- 17.Hickman HD, Luis AD, Buchli R, Few SR, Sathiamurthy M, VanGundy RS, Giberson CF, Hildebrand WH. Toward a definition of self: proteomic evaluation of the class I peptide repertoire. J Immunol. 2004;172:2944. doi: 10.4049/jimmunol.172.5.2944. [DOI] [PubMed] [Google Scholar]
- 18.Urban RG, Chicz RM, Lane WS, Strominger JL, Rehm A, Kenter MJ, UytdeHaag FG, Ploegh H, Uchanska-Ziegler B, Ziegler A. A subset of HLA-B27 molecules contains peptides much longer than nonamers. Proc Nat Acad Sci USA. 1994;91:1534. doi: 10.1073/pnas.91.4.1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Olsen AC, Pedersen LO, Hansen AS, Nissen MH, Olsen M, Hansen PR, Holm A, Buus S. A quantitative assay to measure the interaction between immunogenic peptides and purified class I major histocompatibility complex molecules. Eur J Immunol. 1994;24:385. doi: 10.1002/eji.1830240218. [DOI] [PubMed] [Google Scholar]
- 20.Boisgerault F, Mounier J, Tieng V, Stolzenberg MC, Khalil-Daher I, Schmid M, Sansonetti P, Charron D, Toubert A. Alteration of HLA-B27 peptide presentation after infection of transfected murine L cells by Shigella flexneri. Infect Immun. 1998;66:4484. doi: 10.1128/iai.66.9.4484-4490.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Horig H, Young AC, Papadopoulos NJ, DiLorenzo TP, Nathenson SG. Binding of longer peptides to the H-2Kb heterodimer is restricted to peptides extended at their C terminus: refinement of the inherent MHC class I peptide binding criteria. J Immunol. 1999;163:4434. [PubMed] [Google Scholar]
- 22.Stryhn A, Pedersen LO, Holm A, Buus S. Longer peptide can be accommodated in the MHC class I binding site by a protrusion mechanism. Eur J Immunol. 2000;30:3089. doi: 10.1002/1521-4141(200011)30:11<3089::AID-IMMU3089>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 23.Probst-Kepper M, Stroobant V, Kridel R, Gaugler B, Landry C, Brasseur F, Cosyns J, Weynand B, Boon T, Van Den Eynde BJ. An alternative open reading frame of the human macrophage colony-stimulating factor gene is independently translated and codes for an antigenic peptide of 14 amino acids recognized by tumor-infiltrating CD8 T lymphocytes. J Exp Med. 2001;193:1189. doi: 10.1084/jem.193.10.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Rammensee H-G, Friede T, Stevanovic S. MHC ligands and peptide motifs: first listing. Immunogenetics. 1995;41:178. doi: 10.1007/BF00172063. [DOI] [PubMed] [Google Scholar]
- 25.Rammensee H-G, Bachmann J, Emmerich NP, Bachor OA, Stevanovic S. SYFPEITHI: database for MHC ligands and peptide motifs. Immunogenetics. 1999;50:213. doi: 10.1007/s002510050595. [DOI] [PubMed] [Google Scholar]
- 26.Kikuchi A, Sakaguchi T, Miwa K, Takamiya Y, Rammensee HG, Kaneko Y, Takiguchi M. Binding of nonamer peptides to three HLA-B51 molecules which differ by a single amino acid substitution in the A-pocket. Immunogenetics. 1996;43:268. doi: 10.1007/BF02440994. [DOI] [PubMed] [Google Scholar]
- 27.Snyder HL, Bacik I, Bennink JR, Kearns G, Behrens TW, Bachi T, Orlowski M, Yewdell JW. Two novel routes of transporter associated with antigen processing (TAP)-independent major histocompatibility complex class I antigen processing. J Exp Med. 1997:1087. doi: 10.1084/jem.186.7.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Snyder HL, Bacik I, Yewdell JW, Behrens TW, Bennink JR. Promiscuous liberation of MHC-class I-binding peptides from the C termini of membrane and soluble proteins in the secretory pathway. EurJImmunol. 1998;28:1339. doi: 10.1002/(SICI)1521-4141(199804)28:04<1339::AID-IMMU1339>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- 29.Kubo H, Ikeda-Moore Y, Kikuchi A, Miwa K, Nokihara K, Schonbach C, Takiguchi M. Residue 116 determines the C-terminal anchor residue of HLA-B*3501 and -B*5101 binding peptides but does not explain the general affinity difference. Immunogenetics. 1998;47:256. doi: 10.1007/s002510050355. [DOI] [PubMed] [Google Scholar]
- 30.Maenaka K, Maenaka T, Tomiyama H, Takiguchi M, Stuart DI, Jones EY. Nonstandard peptide binding revealed by crystal structures of HLA-B*5101 complexed with HIV immunodominant epitopes. J Immunol. 2000;165:3260. doi: 10.4049/jimmunol.165.6.3260. [DOI] [PubMed] [Google Scholar]
- 31.Ohno S, Ohguchi M, Hirose S, Matsuda H, Wakisaka A, Aizawa M. Close association of HLA-Bw51 with Behcet's disease. Arch Ophthalmol. 1982;100:1455. doi: 10.1001/archopht.1982.01030040433013. [DOI] [PubMed] [Google Scholar]
- 32.Mizuki N, Ota M, Katsuyama Y, Yabuki K, Ando H, Yoshida M, Onari K, Nikbin B, Davatchi F, Chams H, Ghaderi AA, Ohno S, Inoko H. HLA class I genotyping including HLA-B*51 allele typing in the Iranian patients with Behcet's diseas. Tissue Antigens. 2001;57:457. doi: 10.1034/j.1399-0039.2001.057005457.x. [DOI] [PubMed] [Google Scholar]
- 33.Direskeneli H. Behçet disease: infectious aetiology, mew autoantigens, and HLA-B51. Ann Rheum Dis. 2001;60:996. doi: 10.1136/ard.60.11.996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Verity DH, Wallace GR, Vaughan RW, Stanford MR. Behçet's disease: from Hippocrates to the third millennium. Br J Ophtalmol. 2003;87:1175. doi: 10.1136/bjo.87.9.1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Falk K, Rotzschke O, Takiguchi M, Gnau V, Stevanovic S, Jung G, Rammensee HG. Peptide motifs of HLA-B51, -B52 and -B78 molecules, and implications for Behcet's disease. Int Immunol. 1995;7:223. doi: 10.1093/intimm/7.2.223. [DOI] [PubMed] [Google Scholar]
- 36.Direskeneli H, Eksioglu-Demiralp E, Kibaroglu A, Yavuz S, Ergun T, Akoglu T. Oligoclonal T cell expansions in patients with Behcet's disease. Clin Exp Immunol. 1999;117:166. doi: 10.1046/j.1365-2249.1999.00931.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fairchild PJ, Wraith DC. Lowering the tone: mechanisms of immunodominance among epitopes with low affinity for MHC. ImmunolToday. 1996;17:80. doi: 10.1016/0167-5699(96)80584-6. [DOI] [PubMed] [Google Scholar]
- 38.Matsumoto I, Maccioni M, Lee DM, Maurice M, Simmons B, Brenner M, Mathis D, Benoist C. How antibodies to a ubiquitous cytoplasmic enzyme may provoke joint-specific autoimmune disease. Nat Immunol. 2002;3:360. doi: 10.1038/ni772. [DOI] [PubMed] [Google Scholar]
- 39.Vukmanovic S, Neubert TA, Santori FR. Could TCR antagonism explain associations between MHC genes and disease? Trends Mol Med. 2003;9:139. doi: 10.1016/s1471-4914(03)00029-7. [DOI] [PubMed] [Google Scholar]