

Abstract
Multiple system atrophy (MSA) is by nature a ‘sporadic’ disease with no evidence of familial aggregation observed. However the SNCA multiplication families have clinically displayed parkinsonism and autonomic dysfunction. The present study did not find any SNCA multiplications in a series of 58 pathologically confirmed MSA cases excluding this event as a common cause of MSA. The question of a genetic component in MSA remains to be answered.
Keywords: Semi-quantitative PCR, alpha-synuclein, genomic multiplication, multiple system atropy
Introduction
Genomic multiplication of the α-synuclein locus (SNCA) has been observed to cause parkinsonism, dementia and autonomic dysfunction [1,2]. The α-synuclein protein has also been demonstrated to be a major constituent of Lewy bodies in Parkinson’s disease (PD) and the glial cytoplasmic inclusions (GCI) which are the pathognomonic hallmark of multiple system atrophy (MSA) [3–6]. MSA is a neurodegenerative disorder with a mixed clinical presentation combining autonomic dysfunction, parkinsonism and cerebellar or pyramidal symptoms [7,8].
MSA is classified based on the predominant clinical features – parkinsonism is the major feature of eighty percent (MSA-P) while cerebellar features (MSA-C) are the most prominent findings in twenty percent of patients [7]. The initial clinical signs of MSA-P can make it difficult to differentially diagnose it from early PD. In addition up to thirty percent of MSA-P patients may have a transient response to levodopa therapy. Both MSA and PD are classified as synucleinopathies along with dementia with Lewy bodies (DLB) based on predominate α-synuclein pathology. The clinical spectrum and overlap of these disorders are reflected in the SNCA multiplication families [1,2].
Although no pathogenic mutation has been found to cause MSA, the identification of SNCA multiplication families suggest that over-expression of α-synuclein can result in parkinsonism with autonomic dysfunction. Although this hypothesis is supported by increased levels of the α-synuclein protein in MSA blood plasma, two other studies show no differential SNCA mRNA expression pattern in brain samples of patients with MSA in comparison to controls [9–11]. These contrary findings highlight tissue-specific expression and it is noteworthy that in contrast to brain tissue phosphorylation of α-synuclein serine 129 does not occur in platelets of MSA patients [12].
Herein we investigate a series of pathologically confirmed definite MSA cases for SNCA multiplication using a novel semi-quantitative PCR methodology we have developed.
Methods
A total of 58 cases with neuropathological report of definite MSA, based upon the presence of neuronal loss and gliosis in vulnerable basal ganglia and brainstem nuclei, as well as pathognomonic α-synuclein immunoreactive GCIs [13]. Cases were obtained from the Mayo Clinic Jacksonville brain bank and University of Miami/NPF Brain Endowment Bank™ and the University of Saskatoon. Institutional Ethical review boards approved the study and informed written consent was provided by all donors. All cases were examined for evidence of SNCA multiplication.
DNA was extracted from frozen brain tissue for each case by standard protocol with Wizard®Genomic DNA Purification (Promega). To determine whole gene multiplication primer pairs and probes were designed to exon 5 of the SNCA gene, (Forward primer 5′GGAGTCCAAGCTGAATCTTTCTAACA3′ & Reverse primer 5′GAGCATGTAGAGAGCAAATGATTCTCA3′). A fluorescent-labeled (5′ FAM) DNA probe to measure expression was designed to SNCA exon 5 sequence (5′CTGTCATTGTCACATTTC3′). An endogenous control assay designed against exon 5 of the Presenilin2 gene (Forward 5′CCTTCTCCCTCAGCATCTACAC3′, Reverse 5′GTGTTCAGCACGGAGTTGAG3′, & 5′FAM-labeled probe 5′ATTCACTGAGGACACACCC3′). All primers and probes were designed using File Builder® software and purchased from Applied Biosystems. Quantitative PCR was carried out using TaqMan® expression chemistry protocol, 25ng genomic DNA was amplified with 0.25μl primer probe, 2.5 μl TaqMan 2X Universal PCR Master Mix (Applied Biosystems) The thermal cycle conditions were performed at 50°C for 2min, 95 °C for 10 min, followed by 40 cycles at 95 °C for 15 s for denaturation and 60 °C for 1 min for annealing and extension. Each sample was run in triplicate and control DNA from known SNCA multiplication cases were included as controls on each run. All assays were performed on the ABI 7900HT Fast Real-Time PCR System. The fluorescent signal was analyzed and genomic copy number determined using ABI SDS 2.2.2 Software (Applied Biosystems).
Results and Discussion
This study investigated pathologically confirmed MSA cases for evidence of genomic multiplication of the SNCA locus based on the hypothesis that over-expression of the α-synuclein protein can lead to disease. We developed a semi-quantitative PCR-based method using ABI Taqman chemistry to identify duplication or triplication of the SNCA locus (Figure 1). Our probe was designed to identify whole gene multiplication and does not detect single exon deletion, multiplications or rearrangements that may effect differential isoform expression. Therefore, although we did not observe any SNCA gene multiplication in MSA cases, we can not rule out a role for increased expression of the α-synuclein protein in MSA. Of note, a patient with parkinsonism and early autonomic dysfunction who received a diagnosis of ‘probable MSA’ has been identified who was found to harbor a SNCA duplication [14]. This proband has a family history of autosomal dominant parkinsonism.
Figure 1.
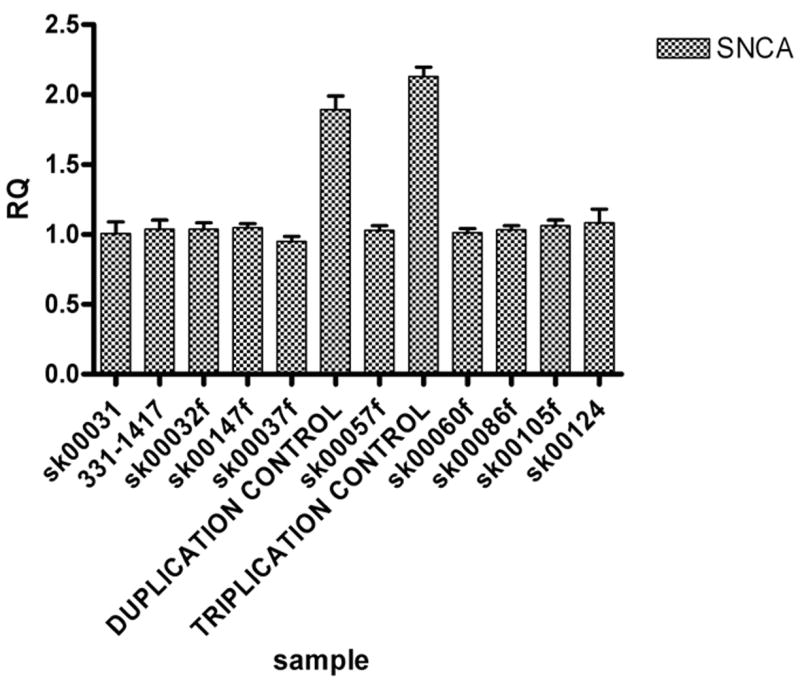
The relative quantification (RQ) results obtained from the ABI 7900HT Fast Real-Time PCR System. The DNA from subjects carrying a SNCA duplication or triplication was included on each assay as positive controls. There was no evidence of any of our 58 MSA cases harboring a SNCA multiplication event.
This case would suggest that over-expression of the SNCA gene resulting in increased levels of α-synuclein protein can lead to autonomic dysfunction and a clinical presentation of MSA; however, direct sequencing of the exons of the SNCA gene in eleven pathologically confirmed MSA cases did not reveal any coding variants [15]. Recently, Ozawa and colleagues (2006) generated a set of twenty-one haplotype-tagging SNPs (htSNPs) that capture 95% of haplotypic diversity of the SNCA gene [16]. This study performed by the European MSA study group did not observe any frequency difference between the MSA cases and controls for any of the individual SNPs or the defined haplotypes within the SNCA gene [16].
This group also recently employed the same methodology and study design to report no association between the UCH-L1 gene and MSA [17]. In these studies, as in the present series, no distinction was made between MSA-C and MSA-P and it may be that SNCA and other parkinsonism-related genes may have a more important role in the MSA-P cases. A number of other candidate genes including MAPT, APO-E, synphilin and cytochrome P-450 have been examined in MSA although no association has been confirmed [18–21].
MSA is regarded as a sporadic disease with little evidence of familial aggregation. Recent genetic findings in PD, another ‘sporadic disorder’, have implicated pathogenic mutations in at least six genes, which calls for a reconsideration of the diagnostic criteria [22]. Similar to PD, a family history of disease is regarded as an exclusion criterion for a clinical diagnosis of MSA [7]. The lack of family history creates a scenario whereby population-based studies may offer the best hope for addressing this question. Genome-wide association studies may be one approach and through the collaborative efforts of the MSA study groups (European-MDSA and North American-MSA) the increased sample size may provide enough power to detect the genetic loci or highlight key pathways in the pathophysiology of MSA. The identification of genetic factors influencing susceptibility to and the pathogenesis of MSA will provide novel therapeutic targets.
Acknowledgments
Mayo Clinic Jacksonville is a Morris K. Udall Parkinson’s Disease Research Center of Excellence (NINDS P01 #NS40256). We are grateful to the Parkinson Society Canada. The Brain Endowment Bank sponsored in part by the NPF Inc. (Miami, FL 33136 USA). We thank Minnie Schreiber for technical assistance. We would like to thank all those who have contributed to our research, particularly the patients and their families.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Singleton AB, Farrer M, Johnson J, Singleton A, Hague S, Kachergus J, Hulihan M, Peuralinna T, Dutra A, Nussbaum R, Lincoln S, Crawley A, Hanson M, Maraganore D, Adler C, Cookson MR, Muenter M, Baptista M, Miller D, Blancato J, Hardy J, Gwinn-Hardy K. alpha-Synuclein locus triplication causes Parkinson’s disease. Science. 2003;302:841. doi: 10.1126/science.1090278. [DOI] [PubMed] [Google Scholar]
- 2.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann Neurol. 2004;55:174–9. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 3.Papp MI, Kahn JE, Lantos PL. Glial cytoplasmic inclusions in the CNS of patients with multiple system atrophy (striatonigral degeneration, olivopontocerebellar atrophy and Shy-Drager syndrome) J Neurol Sci. 1989;94:79–100. doi: 10.1016/0022-510x(89)90219-0. [DOI] [PubMed] [Google Scholar]
- 4.Spillantini MG, Schmidt ML, Lee VM, Trojanowski JQ, Jakes R, Goedert M. Alpha-synuclein in Lewy bodies. Nature. 1997;388:839–40. doi: 10.1038/42166. [DOI] [PubMed] [Google Scholar]
- 5.Wakabayashi K, Takahashi H. Cellular pathology in multiple system atrophy. Neuropathology. 2006;26:338–45. doi: 10.1111/j.1440-1789.2006.00713.x. [DOI] [PubMed] [Google Scholar]
- 6.Armstrong RA, Cairns NJ, Lantos PL. Multiple system atrophy (MSA): Topographic distribution of the α-synuclein-associated pathological changes. Parkinson Rel Dis. 2002;12:356–362. doi: 10.1016/j.parkreldis.2006.02.005. [DOI] [PubMed] [Google Scholar]
- 7.Wenning GK, Colosimo C, Geser F, Poewe W. Multiple system atrophy. Lancet Neurol. 2004;3:93–103. doi: 10.1016/s1474-4422(03)00662-8. [DOI] [PubMed] [Google Scholar]
- 8.Wenning GK, Jellinger KA. The role of alpha-synuclein in the pathogenesis of multiple system atrophy. Acta Neuropathol (Berl) 2005;109:129–40. doi: 10.1007/s00401-004-0935-y. [DOI] [PubMed] [Google Scholar]
- 9.Lee PH, Lee G, Park HJ, Bang OY, Joo IS, Huh K. The plasma alpha-synuclein levels in patients with Parkinson’s disease and multiple system atrophy. J Neural Transm. 2006;113:1435–9. doi: 10.1007/s00702-005-0427-9. [DOI] [PubMed] [Google Scholar]
- 10.Ozawa T, Okuizumi K, Ikeuchi T, Wakabayashi K, Takahashi H, Tsuji S. Analysis of the expression level of α-synuclein mRNA using postmortem brain samples from pathologically confirmed cases of multiple system atrophy. Acta Neuropathol (Berl) 2001;102:188–90. doi: 10.1007/s004010100367. [DOI] [PubMed] [Google Scholar]
- 11.Vogt IR, Lees AJ, Evert BO, Klockgether T, Bonin M, Wullner U. Transcriptional changes in multiple system atrophy and Parkinson’s disease putamen. Exp Neurol. 2006;199:465–78. doi: 10.1016/j.expneurol.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 12.Shults CW, Barrett JM, Fontaine D. α-synuclein from platelets is not phosphorylated at serine 129 in Parkinson’s disease and multiple system atrophy. Neurosci Lett. 2006;405:223–25. doi: 10.1016/j.neulet.2006.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Lantos PL. The definition of multiple system atrophy: a review of recent developments. J Neuropathol Exp Neurol. 1998;57:1099–11. doi: 10.1097/00005072-199812000-00001. [DOI] [PubMed] [Google Scholar]
- 14.Fuchs J, Nilsson C, Kachergus J, Munz M, Larsson EM, Schule B, Langston JW, Middleton F, Ross OA, Hulihan M, Gasser T, Farrer M. Phenotypic variation in a large Swedish pedigree due to SNCA duplication and triplication. Neurology. doi: 10.1212/01.wnl.0000254458.17630.c5. In press. [DOI] [PubMed] [Google Scholar]
- 15.Ozawa T, Takano H, Onodera O, Kobayashi H, Ikeuchi T, Koide R, Okuizumi K, Shimohata T, Wakabayashi K, Takahashi H, Tsuji S. No mutation in the entire coding region of the alpha-synuclein gene in pathologically confirmed cases of multiple system atrophy. Neurosci Lett. 1999;270:110–2. doi: 10.1016/s0304-3940(99)00475-9. [DOI] [PubMed] [Google Scholar]
- 16.Ozawa T, Healy DG, Abou-Sleiman PM, Ahmadi KR, Quinn N, Lees AJ, Shaw K, Wullner U, Berciano J, Moller JC, Kamm C, Burk K, Josephs KA, Barone P, Tolosa E, Goldstein DB, Wenning G, Geser F, Holton JL, Gasser T, Revesz T, Wood NW. The alpha-synuclein gene in multiple system atrophy. J Neurol Neurosurg Psychiatry. 2006;77:464–7. doi: 10.1136/jnnp.2005.073528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Healy DG, Abou-Sleiman PM, Quinn N, Ahmadi KR, Ozawa T, Kamm C, Wullner U, Oertel WH, Burk K, Dupont E, Pellecchia MT, Tolosa E, Gasser T, Holton JL, Revesz T, Goldstein DB, Lees AJ, Wood NW. UCHL-1 gene in multiple system atrophy: a haplotype tagging approach. Mov Disord. 2005;20:1338–43. doi: 10.1002/mds.20575. [DOI] [PubMed] [Google Scholar]
- 18.Bandmann O, Sweeney MG, Daniel SE, Wenning GK, Quinn N, Marsden CD, Wood NW. Multiple-system atrophy is genetically distinct from identified inherited causes of spinocerebellar degeneration. Neurology. 1997;49:1598–604. doi: 10.1212/wnl.49.6.1598. [DOI] [PubMed] [Google Scholar]
- 19.Iwahashi K, Miyatake R, Tsuneoka Y, Matsuo Y, Ichikawa Y, Hosokawa K, Sato K, Hayabara T. A novel cytochrome P-450IID6 (CYPIID6) mutant gene associated with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;58:263–4. doi: 10.1136/jnnp.58.2.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bandmann O, Wenning GK, Quinn NP, Harding AE. Arg296 to Cys296 polymorphism in exon 6 of cytochrome P-450-2D6 (CYP2D6) is not associated with multiple system atrophy. J Neurol Neurosurg Psychiatry. 1995;59:557. doi: 10.1136/jnnp.59.5.557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Morris HR, Vaughan JR, Datta SR, Bandopadhyay R, Rohan De Silva HA, Schrag A, Cairns NJ, Burn D, Nath U, Lantos PL, Daniel S, Lees AJ, Quinn NP, Wood NW. Multiple system atrophy/progressive supranuclear palsy: alpha-Synuclein, synphilin, tau, and APOE. Neurology. 2000;55:1918–20. doi: 10.1212/wnl.55.12.1918. [DOI] [PubMed] [Google Scholar]
- 22.Farrer MJ. Genetics of Parkinson disease: paradigm shifts and future prospects. Nat Rev Genet. 2006;7:306–18. doi: 10.1038/nrg1831. [DOI] [PubMed] [Google Scholar]