

Abstract
A series of substituted 4,5,6,7-tetrahydrothieno[3,2-c]pyridines (THTPs) was synthesized and evaluated for their human phenylethanolamine N-methyltransferase (hPNMT) inhibitory potency and affinity for the α2-adrenoceptor. The THTP nucleus was suggested as an isosteric replacement for the 1,2,3,4-tetrahydroisoquinoline (THIQ) ring system on the basis that 3-thienylmethylamine (18) was more potent as an inhibitor of hPNMT and more selective towards the α2-adrenoceptor than benzylamine (15). Although the isosterism was confirmed, with similar influence of functional groups and chirality in both systems on hPNMT inhibitory potency and selectivity, the THTP compounds proved, in general, to be less potent as inhibitors of hPNMT than their THIQ counterparts, with the drop in potency being primarily attributed to the electronic properties of the thiophene ring. A hypothesis for the reduced hPNMT inhibitory potency of these compounds has been formed on the basis of molecular modeling and docking studies using the X-ray crystal structures of hPNMT co-crystallized with THIQ-type inhibitors and S-adenosyl-l-homocysteine as a template.
1. Introduction
As one approach to elucidate the role(s) of epinephrine in the CNS, our laboratory has targeted phenylethanolamine N-methyltransferase (PNMT; EC 2.1.1.28). This enzyme catalyzes the terminal step in the biosynthesis of epinephrine (Figure 1).
Figure 1.

The Terminal Step in Epinephrine Biosynthesis.
Compounds based on the 1,2,3,4-tetrahydroisoquinoline (THIQ, 5, Table 1) nucleus have been found to be some of the most potent inhibitors of human PNMT (hPNMT) yet reported. The addition of hydrophilic or lipophilic electron-withdrawing substituents to the 7-position of 5 leads to enhanced hPNMT inhibitory potency,2 as illustrated by compounds 6–9 (Table 1). THIQs having lipophilic 7-substituents, such as halides (6 and 7) are generally more potent at hPNMT than those having hydrophilic groups (8 and 9). However, THIQs bearing lipophilic 7-substituents are less selective than THIQs bearing hydrophilic 7-substituents due to significant affinity for the α2-adrenoceptor.2 The addition of small substituents (e.g., methyl, 10) to the 3-position of THIQ increases potency at hPNMT while decreasing affinity for the α2-adrenoceptor.3,4 The absolute stereochemistry at the 3-position of THIQs is important and is discussed below. A synergistic effect is achieved by adding both 3- and 7-substituents to THIQ, resulting in multiplicative increases in PNMT inhibitory potency and selectivity versus the α2-adrenoceptor compared to the similarly substituted 3- or 7-monosubstituted THIQs.5 This is illustrated by comparing the inhibitory potency and selectivity of disubstituted-THIQs 11 or 12 with their respective monosubstituted analogs 7 and 10 or 9 and 10.
Table 1.
In Vitro Human PNMT (hPNMT) Inhibitory Potency and α2-Adrenoceptor Affinity of Previously Studied Inhibitors.
 | |||
|---|---|---|---|
| Compound | hPNMT Ki (µM) ± SEMa | α2-AdrenoceptorbKi (µM) ± SEMa | Selectivity (α2/PNMT) |
| THIQ (5)c,d | 5.8 ± 0.5e | 0.35 ± 0.11 | 0.060 |
| SK&F 64139 (6)c,f | 0.0031 ± 0.0006g | 0.021 ± 0.005 | 7 |
| 7c,h | 0.056 ± 0.003i | 0.23 ± 0.13 | 77 |
| SK&F 29661 (8)c,j | 0.28 ± 0.02g | 100 ± 10 | 360 |
| 9c,h | 0.12 ± 0.01i | 4.3 ± 0.3 | 36 |
| (±)−10c,k | 1.4 ± 0.01i | 0.76 ± 0.08 | 0.54 |
| (±)−11i | 0.017 ± 0.005 | 1.1 ± 0.1 | 65 |
| (±)−12c,l | 0.072 ± 0.005i | 31 ± 3 | 430 |
| SK&F 7698 [(±)−13]c,m | 0.067 ± 0.006 | 0.20 ± 0.02 | 3.0 |
Standard error of the mean.
In vitro activities for the inhibition of [3H]clonidine binding to the α2- adrenoceptor (rat cortex).
The data for this compound was originally reported for the inhibition of bovine PNMT.
The X-ray crystal structures of hPNMT co-crystallized with THIQs, such as 614 (Figure 2) and 8,15 were consistent with SAR studies at PNMT. 3-Substituents of THIQ are able to occupy a lipophilic channel between the norepinephrine (1) binding site and the S-adenosyl-L-methionine (3) binding site and the 7-substituents are able to occupy an auxiliary binding pocket (Figure 2).
Figure 2.

The active site of hPNMT co-crystallized with SK&F 64139 (6) and S-adenosyl-L-homocysteine (4) and the amino acid residues that form key interactions with 6.14 A Connolly (solvent accessible) surface exposing 6 and 4 is also shown and indicates the presence of a lipophilic channel adjacent to the 3-position of 6 and an auxiliary binding pocket adjacent to the 7-position of 6. A lipophilic potential is mapped on the Connolly surface (areas shown in green are neutral, blue are hydrophilic, and brown are lipophilic). Carbons are shown in white, nitrogen in blue, oxygen in red, chlorine in green, and sulfur in yellow. Hydrogens are not shown for clarity.
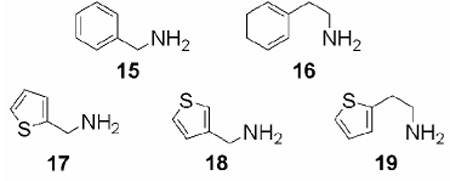
Because the THIQ nucleus can be viewed as either a constrained benzylamine or a constrained 2-phenylethyl-amine and a common isosteric replacement for a benzene ring is a thiophene ring, the commercially available thiophene analogs of benzylamine (15) and 2-phenylethylamine (16) were evaluated at hPNMT and the α2-adrenoceptor (Table 2). As expected, 2-(2-thienyl)ethylamine (19) was found to have hPNMT inhibitory potency that was similar to phenylethylamine 16. The thiophene isostere of benzylamine, with the methylamine group on the 3-position (3-thienylmethylamine, 18), was found to be 3-fold more potent at hPNMT than the isostere with the methylamine group on the 2-position (2-thienylmethylamine, 17). Compared to benzylamine (15), we were pleased to observe that 18 has 50% more hPNMT inhibitory potency and twice the selectivity for hPNMT versus the α2-adrenoceptor. Previous studies have shown that constraining the methylamine side chain of benzylamine (15) to form THIQ (5) increases its hPNMT inhibitory potency 29-fold.16 Because 18 was found to be more potent and selective for hPNMT than 15, we predicted that constraining the side chain of 18 into a 4,5,6,7-tetrahydrothieno[3,2-c]pyridine (THTP, 14) ring system would likely result in superior hPNMT inhibitory potency and selectivity versus 5.
Table 2.
In Vitro hPNMT Inhibitory Potency and α2-Adrenoceptor Affinity of Benzylamine, 2-Phenylethylamine, and their Analogous Thiophenes.

An additional reason for using a thiophene ring is that replacement of the benzene ring of 10 with a benzothiophene ring resulted in a 21-fold increase in hPNMT inhibitory potency (SK&F 7698, 13). Prior to the availability of the crystal structure of hPNMT, it was proposed that the increased potency of 13 versus 10 and that of 6 versus 5 could be due to similar favorable hydrophobic interactions with the enzyme. A molecular overlay of 13 and 6 (not shown) suggested that the thiophene ring of 13 could be binding in the same area of the active site as the benzene ring of 6 and that the benzene ring of 13 could be binding in the same area of the active site as the chlorines of 6. It thus appeared that the isosteric substitution of the benzene ring of THIQ-type inhibitors of hPNMT with a thiophene ring could lead to potent inhibitors of hPNMT. Bioisosteric substitution of a THIQ ring system with a THTP ring system has previously been applied toward the design of potent κ opioid selective analgesics.19
Crystallographic studies suggest that the positions of both the aromatic ring and the 7-substituent of THIQ-type hPNMT inhibitors are important in the binding of these compounds to hPNMT.14,15,20 Also, the preferred configuration is (S) for a 3-methyl substituent [e.g., (S)-12 is 100-fold more potent than (R)-12 (Section 4, Table 3)]. The preferred binding conformer of 3-substituted-THIQs is a half-chair conformation in which the 3-substituent is in an equatorial position and the nitrogen lone pair is axial.21,22 A molecular overlay of (S)-2-bromo-6-methyl-THTP and (S)-7-bromo-3-methyl-THIQ in this energy minimized conformation (Figure 3) shows that 2,6-disubstituted-THTP superimposes well with 3,7-disubstituted-THIQ when the molecules are fit using the respective substituents and the axial lone pair on the nitrogen. As the addition of appropriate substituents to the 3- and 7-positions of THIQ is known to confer both potency and selectivity for PNMT, it was proposed that 2,6-disubstituted-THTPs could be highly potent and selective inhibitors.
Table 3.
In Vitro hPNMT Inhibitory Potency and α2-Adrenoceptor Affinity of 2,6-Disubstituted-THTPs and their Analogous 3,7-Disubstituted-THIQs.
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| R2 for THTPs R7 for THIQs | R6 for THTPs R3 for THIQs | THTP Compd | hPNMT Ki (µM) ± SEMa | α2bKi (µM) ± SEMa | Selectivity (α2/hPNMT) | THIQ Compd | hPNMT Ki (µM) ± SEMa | hPNMT Ki Ratio THTP / THIQ |
| H | H | 14c | 37 ± 1 | 1.5 ± 0.1 | 0.041 | 5 | 5.8 ± 0.5 | 6.3 |
| H | CH3 | 20c | 5.8 ± 0.3 | 29 ± 3 | 5.0 | 10 | 1.4 ± 0.01 | 4.1 |
| H | CF3 | 21 | 40 ± 1 | 1500 ± 100 | 38 | 60d | 23 ± 2e | 1.7 |
| Br | H | 22f | 1.2 ± 0.1 | 0.38 ± 0.04 | 0.32 | 7 | 0.056 ± 0.003c | 21 |
| NO2 | H | 23g | 2.6 ± 0.1 | 18 ± 2 | 16 | 9 | 0.12 ± 0.01 | 22 |
| CN | H | 24 | 15 ± 2 | 16 ± 1 | 1.1 | 61h,i | 1.5 ± 0.1 | 10 |
| (C=O)NH2 | H | 25 | 100 ± 10 | 4.1 ± 0.4 | 0.041 | 62h,i | 72 ± 6 | 1.3 |
| (C=O)CH3 | H | 26 | 61 ± 3 | 9.8 ± 1.0 | 0.16 | 63h,i | 5.0 ± 0.3 | 12 |
| CH3 | H | 27 | 25 ± 1 | 0.49 ± 0.05 | 0.020 | 64h,i | 2.0 ± 0.1 | 13 |
| Br | CH3 | 28 | 0.73 ± 0.04 | 2.3 ± 0.2 | 3.2 | 11 | 0.017 ± 0.005 | 43 |
| Br | CF3 | 29 | 21 ± 1 | > 1000 | > 47 | 65d | 3.2 ± 0.3e | 6.6 |
| NO2 | (R)-CH3 | (R)-30 | 9.6 ± 0.5 | 13 ± 1 | 1.4 | (R)-12 | 1.6 ± 0.1 | 6.0 |
| NO2 | (S)-CH3 | (S)-30 | 0.48 ± 0.04 | 29 ± 3 | 60 | (S)-12 | 0.016 ± 0.001 | 30 |
| CN | (R)-CH3 | (R)-31 | 150 ± 10 | 47 ± 5 | 0.31 | — | — | — |
| CN | (S)-CH3 | (S)-31 | 3.3 ± 0.3 | 130 ± 10 | 39 | — | — | — |
| Compound 32 | 17 ± 1 | > 500 | > 29 | — | — | — | ||
Figure 3.

Overlay of the energy minimized conformations23 of 3,7-disubstituted-THIQ (S)-11 (THIQ nucleus is shown in cyan) and the corresponding 2,6-disubstituted-THTP (S)-28 (THTP nucleus is shown in orange). Hydrogens are not shown for clarity. The overlays show the structural alignment with the aromatic rings of (S)-11 and (S)-28 in the plane of the page (left) and the endview (right). These compounds were fit using the (S)-3-methyl substituent of 11 and the (S)-6-methyl substituent of 28, the 7-bromo substituent of 11 and the 2-bromo substituent of 28, and the end of the axial lone pair (1 Å long, shown in magenta) on the nitrogen.
The 2- and 6-substituents for the proposed THTPs (20–31, below) were selected on the basis of previous SAR data for THIQs.2,5 Methyl and trifluoromethyl groups were added to the 6-position of THTP and while these groups are not likely to be the optimal substituents for the 6-position of THTP, the methyl group is optimal for determining the effect of 6-substitution with minimal steric interference, while the trifluoromethyl group is suitable for determining the influence of the pKa of the THTP amine on α2-adrenoceptor affinity. The decreased affinity for the α2-adrenoceptor associated with reduction in the pKa caused by the addition of fluorinated methyl groups to the 3-position of THIQ is well established.24–26 In addition, 32 (Table 3), in which the methyl group of 13 is replaced with a trifluoromethyl group, was prepared in order to decrease the α2-adrenoceptor affinity of this highly potent, but nonselective, inhibitor.

2. Chemistry
The 4,5,6,7-tetrahydrothieno[3,2-c]pyridine (THTP) ring system can be synthesized in a variety of ways.27–33 For the synthesis of THTPs having nitro, cyano, methyl, acetyl, and carboxamide groups in the 2-position (Scheme 1–Scheme 3), we applied the 2-substituted thiophene synthesis developed by Cagniant and Kirsch34 to the THTP preparation method developed by Matsumura et al.35 This synthetic route was particularly advantageous as it allowed for the enantioselective synthesis of 2,6-disubstituted THTPs. For THTPs having a hydrogen or bromine in the 2-position and the trifluoromethyl analogue of 13, the Pictet-Spengler28 reaction was used to obtain the THTP ring system (Scheme 4).
Scheme 1.

reagents and conditions: (a) BtsCl, dioxane, NaOH (aq); (b) POCl3·DMF; then NaOAc (aq); (c) Na2S·9H2O; then BrCH2R; then NaOH (aq) for the preparation of 40 and 41; (d) PhSH, K2CO3, DMF.
Scheme 3.

reagents and conditions: (a) vinylMgBr, THF, then (S)-α-methylbenzylamine, H2O; (b) H2, Pd(OH)2, THF; then NvocCl, THF, NaOH (aq); (c) POCl3·DMF; then NaOAc (aq); (d) Na2S·9H2O, DMF; then BrCH2NO2 or BrCH2CN; then NaOH (aq); (e) BF3·Et2O, EtSH.
Scheme 4.

reagents and conditions: (a) Na2CO3, thiophene or benzothiophene; (b) trifluoroacetic acid, CH2Cl2; (c) LiAlH4; (d) formaldehyde; then HCl; (e) Br2, AcOH.
The synthesis of THTPs 23–26 is shown in Scheme 1. This reaction sequence required a protecting group for the amine which would be stable in the acidic conditions of step (b), the basic conditions of step (c), and could be removed using conditions which would allow for the preparation of a variety of 2-substituted-THTPs. Also, the use of hydrogenation for the deprotection step was avoided to reduce possible complications of reductive ring opening. The benzothiazole-2-sulfonyl (Bts) group, which is most commonly used in peptide synthesis, met these criteria.36 4-Piperidone (35) was first protected with BtsCl in a 3:1 solution of dioxane and aqueous sodium hydroxide. For optimum yield, it was important to maintain the pH between 10 and 11 with the addition of aqueous 1 M NaOH to neutralize the HCl by-product. The resulting Bts-amine (36) was subjected to Vilsmeyer-Haack formylation to yield chloroformyl derivative 37.37 Compound 37 was reacted with sodium sulfide to generate the thiol in situ. Addition of various alkylbromides resulted in nucleophilic displacement of the bromine by the intermediate thiol, followed by a base catalyzed condensation reaction to form the cyclized THTPs 38–41.34 Removal of the Bts group with PhSH and K2CO3 yielded the desired THTPs 23–26.38
Compound 37 (Scheme 2) was reacted with sodium sulfide to generate the thiol intermediate in situ, which was subsequently reacted with ethyl-2-bromopropionate to afford crude 42, which was cyclized and hydrolyzed under basic conditions, followed by the decarboxylation and condensation of the resulting acid under acidic conditions to yield the protected 2-methyl-THTP (43). Removal of the Bts group with PhSH and K2CO3 yielded THTP 27.38,39
Scheme 2.

reagents and conditions: (a) Na2S·9H2O; then ethyl-2-bromopropionate; (b) EtOH, H2O, NaOH; then HCl; (c) PhSH, K2CO3, DMF.
The enantiospecific synthesis of THTPs 30, 31, 33, and 34 is shown in Scheme 3. Attempts to carry out the reaction sequence in Scheme 3 using Bts, allyloxycarbonyl, benzyloxycarbonyl, 4-nitrobenzyloxycarbonyl, or ethoxycarbonyl protecting groups for the amine were unsuccessful due to an inability to separate the regioisomers formed in step (c). The mixture of regioisomers obtained from step (c) was subjected to step (d), but the products from this reaction were also inseparable. Ultimately, the reaction sequence was successfully completed using the 6-nitroveratryloxy-carbonyl (Nvoc) protecting group. Nvoc-amines are stable under acidic or basic conditions and are most commonly employed due to their photochemical lability.40 The highly crystalline nature of the Nvoc derivatives allowed for the separation of the regioisomers formed in step (d) by fractional recrystallization.
Weinreb amide 44 was reacted with vinylmagnesium bromide in THF to form 1,4-hexadien-3-one in situ, to which (S)-α-methylbenzylamine and water were added. This yielded a mixture of (R)- and (S)-2-methyl-1-((S)-1-phenylethyl)piperidin-4-one (45)41,42 that was separated by flash chromatography. Compound (R,S)- or (S,S)-45 was deprotected by hydrogenation with Pearlman's catalyst at ambient temperature and pressure and the resulting crude amine was reacted with 6-nitroveratryl chloroformate (NvocCl) to yield (R)- or (S)-46. Compound (R)- or (S)-46 was subjected to Vilsmeier conditions to yield a mixture of regioisomers, (R)-47 and (R)-48, or (S)-47 and (S)-48,37 which were separated by fractional crystallization. Compound (R)- or (S)-47 was reacted with sodium sulfide to generate the thiol in situ. Addition of bromonitromethane or bromoacetonitrile resulted in nucleophilic displacement of the bromine by the intermediate thiol, followed by a base catalyzed condensation reaction to afford the cyclized 6-methyl-2-substituted-THTP products (R)- or (S)-49 or (R)-or (S)-50, respectively.34 (R)- or (S)-48 was reacted under the same conditions used to convert 47 to 6-methyl-THTPs 49 and 50, to yield the cyclized 4-methyl-2-substituted-Bioorga THTP products (R)- or (S)-51 or (R)- or (S)-52, respectively. Deprotection of THTPs 49–52 with BF3·Et2O in EtSH afforded THTPs 30, 31, 33, and 34.43 The stereochemistry and regiochemistry of (S)-52 have been verified by X-ray crystallography (see Section 7). This structure was then used to define the stereochemistry and regiochemistry of the final compounds in Scheme 3.
The synthesis of THTPs 20, 21, 28, 29, and 32 is shown in Scheme 4. The reaction of 53 with Na2CO3 yields 1,1,1-trifluoro-2-nitroso-2-propene.44 This heterodiene reacted with thiophene to afford the bicyclic 1,2-oxazine 54, which was converted directly to oxime 55 with trifluoroacetic acid. Reduction of 55 with LiAlH4 yielded amine 56, which was cyclized by a Pictet-Spengler reaction to afford THTP 21. Reaction of 21 or 2045 with bromine in acetic acid yielded 2-bromo-THTPs 29 and 28, respectively. Compound 32 was also prepared from 53 and benzothiophene via the procedure used for the synthesis of 21.
3. Biochemistry
The hPNMT inhibitory potency for compounds 13,13 15,17 and 1618 was originally determined using bovine PNMT. The hPNMT inhibitory potency for these compounds is now reported for comparison purposes. In the current study, human PNMT (hPNMT) with a C-terminal hexahistidine tag was expressed in E. coli.46,47 The radiochemical assay conditions, previously reported for the bovine enzyme,48 were modified to account for the high binding affinity of some inhibitors.9,46 Inhibition constants were determined using four concentrations of phenylethanolamine as the variable substrate, and three concentrations of inhibitor.
α2-Adrenergic receptor binding assays were performed using cortex obtained from male Sprague Dawley rats.49 [3H]Clonidine was used as the radioligand to define the specific binding and phentolamine was used to define the nonspecific binding. Clonidine was used as the ligand to define α2-adrenergic binding affinity to simplify the comparison with previous results.50
4. Results and Discussion
The biochemical data for THTPs 14 and 20–32 are shown in Table 3 along with the data for their corresponding isosteric THIQs for comparison purposes. Unfortunately, the prediction that constraining the methylamine side chain of 18 to form the THTP ring system (14) would result in a large increase in potency, as is observed for benzylamine (15) versus THIQ (5), was incorrect. Only a small increase in hPNMT inhibitory potency (3-fold, as compared to the 29-fold increase in potency of 15 versus 5) is observed, resulting in the parent THTP (14) having 6-fold less potency at hPNMT than the parent THIQ (5). In fact, all of the THTPs examined were found to be significantly less potent as inhibitors of hPNMT than their corresponding THIQs, suggesting that the THIQ nucleus has greater intrinsic affinity for hPNMT than the THTP nucleus. Given the structural similarity of THTPs and THIQs and their molecular overlay in Figure 3, we concluded that the most likely reason for the decreased hPNMT inhibitory potency of the THTPs was fundamental differences between the thiophene ring of THTPs and the benzene ring of THIQs.
4.1. Docking Studies Comparing the Binding of THTPs with THIQs
Docking studies with the crystal structure of hPNMT predict that substituents on the 2- and 6-positions of the THTP nucleus are binding in the same area in the active site as substituents on the 7- and 3-positions of the THIQ nucleus (Figure 4). The 2-bromo substituent of 28 is predicted to form favorable hydrophobic interactions with Val53 in the same way as the 7-bromo substituent of 11 (Figure 4A).10 Docking studies53 (not shown) with 2-nitro-THTPs 23 and 30 predicted that (similarly to 7-nitro-THIQs)10 the nitro group could form hydrogen bonding interactions with Lys57. The 6-methyl group of (S)-28 is predicted to form favorable hydrophobic interaction with Tyr222 in a similar way as the 3-methyl group of (S)-11 (Figure 4A). The interaction between Tyr222 and lipophilic 3-substituents of THIQ is well established.10,24,46
Figure 4.

(S)-7-Bromo-3-methyl-THIQ [(S)-11] and (S)-2-bromo-6-methyl-THTP [(S)-28] docked into the active site of hPNMT [from the X-ray structure of hPNMT co-crystallized with SK&F 64139 (6) and S-adenosyl-l-homocysteine (4)]14 and the amino acid residues that form key interactions with (S)-11 and (S)-28. Nitrogen is blue, oxygen is red, bromine is magenta, sulfur is yellow, carbons of amino acid side chains are gray, carbons of (S)-11 are cyan, and carbons of (S)-28 are orange. Hydrogens are not shown for clarity. (A) Stereoview of an overlay of (S)-11 and (S)-28. The 7-bromo and (S)-3-methyl substituents of (S)-11 are predicted to bind in the same area in the active site as the 2-bromo and (S)-6-methyl substituents of (S)-28. (B) The interaction of (S)-11 with Phe182, the aromatic ring of which is shown in the plane of the paper. The benzene ring of (S)-11 forms a parallel displaced π-stacking interaction with Phe182 at a distance of 3.7 Å. (C) The interaction of (S)-28 with Phe182, the aromatic ring of which is shown in the plane of the paper. The distance between the thiophene ring of (S)-28 and Phe182 is 3.7 Å. The partially negatively charged sulfur of (S)-28 is in the face of Phe182, which results in a less favorable π-stacking interaction as compared to that of (S)-11.
Crystallographic studies have shown that the benzene ring of substrates54 or THIQ-type inhibitors7,14,15,20 is positioned between the side chains of Phe182 and Asn39. The distance between the aromatic ring of 614 (Figure 2) or 815 and the aromatic ring of Phe182 is 3.8 Å or 3.7 Å, respectively, which implies the presence of a π-stacking interaction. Within the hydrophobic environment of a protein, the preferred geometry of a Phe-Phe interaction is in either a T-shaped edge-to-face or a parallel displaced π-stacking orientation, with the latter being more energetically favored.55,56 This is the observed orientation between Phe182 and the benzene ring of 6 (Figure 2) and the predicted orientation between Phe182 and (S)-11 (Figure 4B). However, it is likely that the benzene ring of (S)-11 is able to make a more favorable -stacking interaction with Phe182 than the thiophene ring of (S)-28 (Figure 4C).
Unlike benzene, thiophene has a dipole moment.57,58 The charge distribution of the π cloud of benzene is in the center of the ring, which is surrounded by electron-poor hydrogens. The charge distribution of the π cloud of thiophene, however, is shifted away from the center of the ring toward the sulfur atom, giving the sulfur a partial negative charge. This is consistent with the electrostatic potentials predicted for THIQ and THTP (Figure 5). According to docking studies with (S)-28, the sulfur atom is in the face of Phe182 (Figure 4C). Although π-stacking interactions between benzene and thiophene rings have not been thoroughly investigated, it appears that this π-stacking interaction with the THTP sulfur in the face of Phe182 would be less favorable than the parallel displaced π-stacking orientation observed between the benzene ring of THIQs and Phe182. This is likely the molecular basis for the THIQ nucleus having greater intrinsic affinity for hPNMT than the THTP nucleus.
Figure 5.

Electrostatic potential diagram of unsubstituted THIQ (5, top) and unsubstituted THTP (14, bottom). Electronegative areas are shown in blue and electropositive areas are shown in red (see Section 6.4). Carbon is white, nitrogen is blue, sulfur is yellow and hydrogen is cyan. As discussed in the text, the charge distribution of the π cloud of the benzene ring of 5 is in the center of the ring, and the charge distribution of the π cloud of the thiophene ring of 14 is shifted away from the center of the ring toward the sulfur atom.
4.2. Effects of Substitution on THTP and THIQ
4.2.1. 2-Substituted-THTPs versus 7-Substituted-THIQs
The addition of bromo, nitro, and cyano groups (22–24) to the 2-position of THTP (14) increased the hPNMT inhibitory potency 31-fold, 14-fold, and 2.5-fold, respectively. The addition of the same groups to the 7-position (7, 9, and 61) of THIQ (5) increased the hPNMT inhibitory potency 100-fold, 48-fold, and 4-fold, respectively. Thus, the addition of electron-withdrawing substituents to the 2-position of THTP improved the hPNMT inhibitory potency of 14, but not to the same degree as the addition of the same substituents to the 7-position of 5.
Crystallographic studies with THIQ inhibitors show that the position of both the benzene ring and the 7-substituent are important in the binding of these compounds.14,15,20 Thus, the molecular basis for the less substantial improvement in potency for 2-bromo-, nitro-, and cyano-THTP in comparison to the corresponding 7-substituted-THIQs is either (1) the position of the thiophene ring of the THTPs is adversely affecting the orientation of the 2-substituent in the active site such that it cannot make optimal interactions with key residues, or (2) that the orientation of the 2-substituent is forcing the THTP ring into a position in which the thiophene ring makes less optimal interactions with Phe182 in comparison to the benzene ring of THIQs. Docking studies suggest that the latter reason is more probable because they predict that 2-substituents of THTP are binding in the same area of the active site as 7-substituents of THIQ, which shifts the centroid of the thiophene ring of THTPs relative to the centroid of the benzene ring of THIQs. In Figure 4, the centroid of (S)-28 is 0.5 Å from the centroid of (S)-11). Because these changes in binding orientation are very subtle, co-crystallization of a 2-substituted-THTP and its corresponding 7-substituted-THIQ with hPNMT would be required to verify this conclusion.
4.2.2. 6-Substituted-THTPs versus 3-Substituted-THIQs
The addition of a methyl group to the 6-position of THTPs (14, 22, and 23) results in a similar improvement in the hPNMT inhibitory potency as does the addition of a methyl group to the 3-position of THIQs (5, 7, and 9), with the increase being virtually the same for each corresponding 2- or 7-substituent [e.g., 27 (6-fold) versus 10 (4-fold), 28 versus 11 (2-fold for each), and (S)-31 versus (S)-12 (5-fold for each)]. These observations are supported by the docking results (Figure 4A), which show that the (S)-methyl groups of 11 and 28 occupy the same area in the active site of hPNMT and are able to make the same favorable interactions with Tyr222.
The replacement of the 6-methyl group of 28 with a trifluoromethyl group results in a 29-fold reduction in hPNMT inhibitory potency for 29 and the replacement of the methyl group of SK&F 7698 (13) with a trifluoromethyl group results in a 250-fold reduction in potency for 32. This was not unexpected because previous studies have shown that the replacement of the 3-methyl group of 11 with a trifluoromethyl group results in a 190-fold reduction in hPNMT inhibitory potency for 65 (Table 3).26 In these studies, we attributed the reduction in potency of 3-trifluoromethyl-THIQs versus 3-methyl-THIQs to the increased steric bulk of the trifluoromethyl group, specifically from unfavorable interactions with Tyr222.24,26 Similarly, docking studies (not shown) indicate that the decrease in hPNMT inhibitory potency of the 6-trifluoromethyl-THTPs is most likely due these same factors.
The (S)-enantiomers of 30 and 31 are considerably more potent (20-fold and 45-fold, respectively) at hPNMT than the corresponding (R)-enantiomers. This is consistent with hPNMT data for enantiomers of various 3-substituted-THIQs. For compounds (R)- and (S)-12, the (S)-enantiomer is 100-fold more potent at hPNMT than the (R)-enantiomer. Analysis of the PNMT inhibition data for 3-trifluoromethyl-,26 3-fluoromethyl-,4,21 and 3-hydroxymethyl-THIQs4 shows that the (R)-enantiomers are more potent at hPNMT than the (S)-enantiomers. Although the absolute configuration of the more active enantiomer was opposite for these compounds (due to the Cahn-Ingold-Prelog priority rules), the 3-substituents are oriented in the same region of space as the methyl groups of (S)-30, (S)-31, and (S)-12. Similarly to 3-substituted THIQs,21 docking studies (not shown) on 6-methyl-THTP show that the 6-(S)-methyl substituent and the ring nitrogen can favorably interact with Tyr222 and Glu219, respectively, whereas those groups on 6-(R)-methyl-THTP cannot.
Collectively, the similar effects on hPNMT inhibitory potency from the addition of methyl or trifluoromethyl groups to the 6-position of THTP and the 3-position of THIQ, and the observation that the S-enantiomer is more potent for both 6-methyl-THTPs and 3-methyl-THIQs, indicate that 6-substituents of THTP are binding in the same area of the hPNMT active site as the 3-substituents of THIQ as predicted by the docking studies. Thus, the optimization of THTPs by the addition of appropriate substituents to the 6-position is likely to proceed similarly to the optimization of 3-substituted-THIQs, but as discussed in the previous section, the optimization of THTPs by the addition of substituents to the 2-position is not as effective as the optimization of THIQs at the analogous 7-position.
At the α2-adrenoceptor, the addition of a trifluoromethyl group to the 6-position of THTP causes a dramatic reduction in affinity, most likely due to the decrease in the pKa of the THTP amine. Similar results have previously been reported for 3-difluoromethyl-THIQs,24 3-trifluoromethyl-THIQs,25,26 and fluorinated benzazapines.59 Replacement of the methyl group of 20, 28 or 13 with a trifluoromethyl group results in a 1000-fold (21), >430-fold (29) or >2,500-fold (32) decrease in α2-adrenoceptor affinity for these compounds. Unfortunately, the reduction in hPNMT inhibitory potency that results from the addition of a trifluoromethyl group makes them poor candidates for in vivo investigations.
5. Conclusion
A series of substituted THTPs were shown to have considerably less hPNMT inhibitory potency than their corresponding isosteric THIQs. The observation that unsubstituted THTP (14) is 6-fold less potent than unsubstituted THIQ (5) and the overall data trend of THTPs having diminished hPNMT inhibitory potency versus their corresponding THIQs suggests that the THIQ nucleus has greater intrinsic affinity for hPNMT than does the THTP nucleus. Docking studies in conjunction with analysis of the electrostatic potential of THTPs and THIQs suggest that the benzene ring of THIQs is able to make a more favorable π-stacking interaction with Phe182 than the thiophene ring of THTPs. The analysis of the effects of substitution on THTP and THIQ strongly supports the binding orientation predicted by the docking studies, however, the co-crystallization of one or more of these THTPs with hPNMT would be helpful in verifying our conclusions. While we were able to increase the potency of 14 by adding appropriate substituents to the 2- and 6- positions, the decreased potency of these compounds versus the analogous THIQs did not merit further optimization.
6. Experimental
6.1. General methods
All reagents and solvents were of reagent grade or were purified by standard methods before use. Melting points were determined in open capillary tubes on a Thomas-Hoover melting point apparatus calibrated with known compounds. Proton (1H NMR) and carbon (13C NMR) nuclear magnetic resonance spectra were taken on a Bruker DRX-400, Bruker AM-500, or Bruker AV-800 spectrophotometer. High resolution mass spectra (HRMS) were obtained on a Ribermag R 10-10 mass spectrophotometer. Thin-layer chromatography (TLC) was performed on K6F silica gel 60 Å (Whatman) glass backed plates. Flash chromatography was performed using silica gel 60 (230–400 mesh) supplied by Universal Adsorbents, Atlanta, Georgia.
Anhydrous methanol and ethanol were used unless stated otherwise, and were prepared by distillation over magnesium. Other solvents were routinely distilled prior to use. Anhydrous tetrahydrofuran (THF) and diethyl ether (Et2O) were distilled from sodium-benzophenone ketyl. Hexanes refers to the mixture of hexane isomers (bp 40–70 °C) and brine refers to a saturated solution of NaCl. All reactions that required anhydrous conditions were performed under a positive nitrogen or argon flow, and all glassware was either oven-dried or flame-dried before use. [methyl-3H]AdoMet and [3H]clonidine were obtained from PerkinElmer (Boston, MA).
6.2. Radiochemical Assay of PNMT Inhibitors
A typical assay mixture consisted of 25 µL of 0.5 M phosphate buffer (pH 8.0), 25 µL of 50 µM unlabeled AdoMet, 5 µL of [methyl-3H]AdoMet, containing approximately 3 × 105 dpm (specific activity approximately 15 Ci/mmol), 25 µL of substrate solution (phenylethanolamine), 25 µL of inhibitor solution, 25 µL of enzyme preparation (containing 30 ng hPNMT and 25 µg of bovine serum albumin), and sufficient water to achieve a final volume of 250 µL. After incubation for 30 min at 37 °C, the reaction mixture was quenched by addition of 250 µL of 0.5 M borate buffer (pH 10.0) and was extracted with 2 mL of toluene/isoamyl alcohol (7:3). A 1 mL portion of the organic layer was removed, transferred to a scintillation vial, and diluted with cocktail for counting. The mode of inhibition was ascertained to be competitive in all cases reported in Table 1–Table 4 by examination of the correlation coefficients (r2) for the fit routines as calculated in the Enzyme Kinetics module (version 1.1) in SigmaPlot (version 7.0).32 While all Ki values reported were calculated using competitive kinetics, it should be noted that there was not always a great difference between the r2 values for the competitive model versus the non-competitive model. All assays were run in duplicate with 3 inhibitor concentrations over a 5-fold range. Ki values were determined by a hyperbolic fit of the data using the Single Substrate–Single Inhibitor routine in the Enzyme Kinetics module (version 1.1) in SigmaPlot (version 7.0). For inhibitors with apparent IC50 values less than 0.1 µM (as determined by a preliminary screen of the compounds), the Enzyme Kinetics Tight Binding Inhibition routine was used to calculate the Ki values.
6.3. α2-Adrenoceptor Radioligand Binding Assay
The radioligand receptor binding assay was performed according to the method of U’Prichard et al.29 Male Sprague-Dawley rats were decapitated, and the cortexes were dissected out and homogenized in 20 volumes (w/v) of ice-cold 50 mM Tris/HCl buffer (pH 7.7 at 25 °C). Homogenates were centrifuged thrice for 10 min at 50,000 × g with resuspension of the pellet in fresh buffer between spins. The final pellet was homogenized in 200 volumes (w/v) of ice-cold 50 mM Tris/HCl buffer (pH 7.7 at 25 °C). Incubation tubes containing [3H]clonidine (specific activity approximately 55 Ci/mmol, final concentration 2.0 nM), various concentrations of drugs and an aliquot of freshly resuspended tissue (800 µL) in a final volume of 1 mL were used. Tubes were incubated at 25 °C for 30 min and the incubation was terminated by rapid filtration under vacuum through GF/B glass fiber filters. The filters were rinsed with three 5 mL washes of ice-cold 50 mM Tris buffer (pH 7.7 at 25 °C). The filters were counted in vials containing premixed scintillation cocktail. Non-specific binding was defined as the concentration of bound ligand in the presence of 2 µM of phentolamine. All assays were run in quadruplicate with 5 inhibitor concentrations over a 16-fold range. IC50 values were determined by a log-probit analysis of the data and Ki values were determined by the equation Ki = IC50 / (1 + [Clonidine] / KD), as all Hill coefficients were approximately equal to 1.
6.4. Molecular Modeling
Connolly surfaces were generated in SYBYL® on a Silicon Graphics Octane workstation.23 Docking of the various inhibitors into the hPNMT active site was performed using AutoDock 3.0.60 The default settings for AutoDock 3.0 were used. The compound to be docked was initially overlayed with the co-crystallized ligand and minimized with the Tripos force field. The docking of inhibitors into the hPNMT active site was performed on the S-enantiomer as the S-enantiomer is preferred over the R-enantiomer in the hPNMT active site. Figure 4 was drawn with Molscript61 and Raster3D.62 For Figure 5, the electrostatic potential was mapped on a Connolly surface generated in SYBYL®.23 Atomic charges were calculated using the Gasteiger-Marsili method.63
6.5. Synthesis
N-Bts-4-piperidone (36)
To a stirred solution of 4-piperidone·H2O·HCl (35, 4.20g, 27.9 mmol), dioxane (15mL), and 0.25 M NaOH (45 mL) at 5 °C, was added 8.50 g (36.3 mmol) of benzothiazole-2-sulfonylchloride. The reaction was stirred for 2 h at 5 °C, and for an additional 10 h at ambient temperature while maintaining the pH between 10 and 11 with the addition of aqueous 1 M NaOH. The reaction was acidified with 1 M HCl and then extracted with CHCl3 (3 × 50 mL). The combined organic extracts were washed with 1M HCl (50 mL) and brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/ether (1:1) to yield 36 (7.25 g, 24.5 mmol, 88%) as a white solid: mp 119–121 °C; 1H NMR (500 MHz, CDCl3) δ 8.20–8.18 (m, 1H), 8.03–8.00 (m, 1H), 7.67–7.59 (m, 2H), 3.85 (t, 4H, J = 6.2 Hz), 2.63 (t, 4H, J = 6.2 Hz); 13C NMR (500 MHz, CDCl3) δ 205.0 164.1, 152.3, 136.1, 127.8, 127.5, 125.1, 122.1, 46.1, 40.6; HRMS (FAB+) m/z calcd for C12H13N2O3S2 (MH+) 297.0368 obsd 297.0357.
N-Bts-4-chloro-1,2,5,6-tetrahydropyridine-3-carbox-aldehyde (37)
DMF (2.70 mL, 35.4 mmol) was dissolved in trichloroethylene (30 mL) and cooled to 5 °C. POCl3 (2.42 mL, 26.0 mmol) was added dropwise and the reaction was warmed to ambient temperature and stirred for 10 min. Compound 36 (3.50 g, 11.8 mmol) was dissolved in trichloroethylene (20 mL) and added dropwise to the POCl3-DMF solution. The reaction was continued for 12 h and then poured into an ice cold solution of sodium acetate (9.0 g) in water (200 mL) and stirred vigorously for 20 min. This mixture was extracted with CH2Cl2 (4 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc/CHCl3 (3:1:1) to yield 37 (3.25 g, 9.48 mmol, 80%) as a pale yellow solid: 1H NMR (500 MHz, CDCl3) δ 10.10 (s, 1H), 8.19–8.17 (m, 1H), 8.02–8.00 (m, 1H), 7.65–7.58 (m, 2H), 4.22 (t, 2H, J = 2.2 Hz), 3.77 (t, 2H, J = 5.8 Hz), 2.90–2.87 (m, 2H); 13C NMR (500 MHz, CDCl3) δ 188.1, 163.5, 152.3, 148.2, 136.1, 129.6, 127.8, 127.5, 125.1, 122.1, 44.3, 43.6, 35.0; HRMS (FAB+) m/z calcd for C13H12ClN2O3S2 (MH+) 342.9978 obsd 342.9962.
N-Bts-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (38)
To a stirred solution of aldehyde 37 (350 mg, 1.02 mmol) in 10 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (368 mg, 1.53 mmol) in water (2 mL) and the reaction was stirred for 15 min. Bromonitromethane (0.285 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (75 mL) and a white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (3:1) to yield a pale yellow solid. Recrystallization from EtOAc/hexanes yielded 38 (175 mg, 0.46 mmol, 45%) as white crystals: mp 170–172 °C; 1H NMR (500 MHz, CDCl3) δ 8.32–8.30 (m, 1H), 8.20–8.18 (m, 1H), 8.01 (s, 1H), 7.72–7.65 (m, 2H), 4.57 (s, 2H), 3.77 (t, 2H, J = 5.8 Hz), 3.03 (t, 2H, J = 5.8 Hz); 13C NMR (500 MHz, CDCl3) δ 164.1, 152.4, 149.0, 143.2, 136.2, 131.7, 128.5, 128.3, 128.3, 125.0, 123.7, 45.7, 43.8, 25.4; HRMS (FAB+) m/z calcd for C14H12N3O4S3 (MH+) 381.9990 obsd 381.9969.
General Procedure for Bts Deprotection. (Selected procedure for 23·HCl)
To a stirred solution of 38 (70 mg, 0.18 mmol) in 2 mL dry DMF was added 37 µL (40 mg, 0.36 mmol) thiophenol and 75 mg (0.54 mmol) K2CO3 at ambient temperature. The suspension was stirred for 1 h and then diluted with water (20 mL) and ether (20 mL). The ether layer was removed and the aqueous layer was extracted with ether (3 × 20 mL). The combined organic extracts were then extracted with 0.5 M HCl (2 × 50 mL). The combined aqueous extracts were made basic (pH ≈ 11) with 1M NaOH and extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over Na2SO4. Removal of the solvent yielded 23 as the free amine, which was redissolved in CHCl3 (20 mL). Dry HCl(g) was bubbled through this solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes to yield 23·HCl52 (40 mg, 0.18 mmol, 96%) as light brown crystals.
2-Nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (23·HCl)
See General Procedure for Bts Deprotection: mp 199–201 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.64 (br, 2H), 8.04 (s, 1H), 4.20 (s, 2H) 3.44 (t, 2H, J = 6.0 Hz), 3.15 (t, 2H, J = 6.0 Hz); 13C NMR (500 MHz, DMSO-d6) δ 149.5, 142.3, 129.6, 128.6, 41.9, 40.5, 22.5; HRMS (FAB+) m/z calcd for C7H9N2O2S (MH+) 185.0385 obsd 185.0371; Anal. calcd for C7H9ClN2O2S: C, 38.10; H, 4.11; N, 12.69. Found: C, 38.44; H, 3.89; N, 12.52.
N-Bts-2-cyano-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (39)
To a stirred solution of aldehyde 37 (310 mg, 0.904 mmol) in 10 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (326 mg, 1.36 mmol) in water (2 mL) and the reaction was stirred for 15 min. Bromoacetonitrile (0.285 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (75 mL) and a white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (3:1) to yield a pale yellow solid. Recrystallization from EtOAc/hexanes yielded 39 (190 mg, 0.53 mmol, 58%) as white crystals: mp 184–186 °C; 1H NMR (500 MHz, CDCl3) δ 8.31–8.30 (m, 1H), 8.19–8.17 (m, 1H), 7.76 (s, 1H), 7.72–7.65 (m, 2H), 4.57 (s, 2H), 3.76 (t, 2H, J = 5.8 Hz), 3.02 (t, 2H, J = 5.8 Hz); 13C NMR (500 MHz,) δ 164.1, 152.4, 142.1, 137.3, 136.1, 132.1, 128.3, 128.2, 125.0, 123.7, 114.8, 106.2, 45.9, 44.0, 25.2; HRMS (FAB+) m/z calcd for C15H12N3O2S3 (MH+) 362.0092 obsd 362.0077.
2-Cyano-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (24·HCl)
Compound 37 (65 mg, 0.18 mmol) was converted to THTP 24 according to the General Procedure for Bts Deprotection. The hydrochloride salt was recrystallized from EtOH/hexanes to yield 24·HCl (35 mg, 0.19 mmol, 97%) as white crystals: mp dec 303–305 °C; 1H NMR (500 MHz, DMSO-d6) δ 9.76 (br, 2H), 7.81 (s, 1H), 4.20 (s, 2H), 3.41 (t, 2H, J = 6.0 Hz), 3.15 (t, 2H, J = 5.9 Hz); 13C NMR (500 MHz, DMSO-d6) δ 141.4, 137.5, 130.1, 114.6, 106.8, 42.0, 40.6, 22.3; HRMS (FAB+) m/z calcd for C8H9N2S (MH+) 165.0486 obsd 165.0468; Anal. calcd for C8H9ClN2S·⅓H2O: C, 46.49; H, 4.71; N, 13.55. Found: C, 46.37; H, 4.37; N, 13.28.
N-Bts-2-carboxamide-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (40)
To a stirred solution of aldehyde 37 (70.0 mg, 0.204mmol) in 3 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (73.6 mg, 0.306 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Bromoacetamide (112 mg, 0.817 mmol) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. To this reaction mixture was added 0.2 M NaOH (15 mL). The reaction mixture became cloudy and after 1 min was poured into a saturated ammonium chloride solution (50 mL). The aqueous mixture was extracted with CH2Cl2 (3 × 30 mL) and the combined organic extracts were washed with brine (25 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with EtOAc to yield a white solid. Recrystallization from acetone yielded 40 (45.0 mg, 0.119 mmol, 58%) as white crystals: mp 214–216 °C; 1H NMR (500 MHz, DMSO-d6) δ 8.31–8.29 (m, 1H), 8.21–8.19 (m, 1H), 7.86 (br, 1H), 7.71–7.65 (m, 2H), 7.46 (s, 1H), 7.34 (br, 1H), 4.52 (s, 2H), 3.73 (t, 2H, J = 5.8 Hz), 2.94 (t, 2H, J = 5.7 Hz); 13C NMR (500 MHz, DMSO-d6) δ 164.3, 163.0, 152.4, 138.2, 137.9, 136.1, 131.2, 128.3, 128.2, 126.8, 125.0, 123.6, 45.9, 44.3, 25.0; HRMS (FAB+) m/z calcd for C15H14N3O3S3 (MH+) 380.0197 obsd 380.0189.
2-Carboxamide-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (25·HCl)
Compound 40 (75.0 mg, 0.198 mmol) was converted to THTP 25 according to the General Procedure for Bts Deprotection. The hydrochloride salt was recrystallized from EtOH/hexanes to yield 25·HCl (32.2 mg, 0.147 mmol, 74%) as white crystals: mp dec 303–305 °C; 1H NMR (500 MHz, CD3OD) δ 7.51 (s, 1H), 4.32 (s, 2H), 3.59 (t, 2H, J = 6.2 Hz), 3.21 (t, 2H, J = 6.2 Hz); 13C NMR (500 MHz, CD3OD) δ 164.4, 138.0, 137.4, 128.2, 126.8, 42.5, 41.4, 21.6; HRMS (FAB+) m/z calcd for C8H11N2OS (MH+) 183.0592 obsd 183.0584; Anal. calcd for C8H11ClN2OS·¼H2O: C, 43.05; H, 5.19; N, 12.55. Found: C, 43.42; H, 5.11; N, 12.17.
N-Bts-2-aceto-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (41)
To a stirred solution of aldehyde 37 (140 mg, 0.408 mmol) in 5 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (147 mg, 0.612 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Chloroacetone (0.130 mL, 1.63 mmol) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. To this reaction mixture was added 0.2 M NaOH (25 mL). The reaction mixture became cloudy and was then diluted to 100 mL with water. This aqueous mixture was extracted with EtOAc (3 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (2:1) to yield a white solid. Recrystallization from EtOAc/hexanes yielded 41 (90 mg, 0.238 mmol, 58%) as white crystals: mp 189–191 °C; 1H NMR (500 MHz, CDCl3) δ 8.17–8.15 (m, 1H), 8.01–7.99 (m, 1H), 7.65–7.57 (m, 2H), 7.37 (s, 1H), 4.65 (s, 2H), 3.84 (t, 2H, J = 5.7 Hz), 3.07 (t, 2H, J = 5.7 Hz), 2.52 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 190.2, 163.8, 152.4, 142.5, 141.9, 136.1, 131.4, 129.7, 127.7, 127.5, 125.1, 122.0, 45.8, 44.1, 26.5, 25.7; HRMS (FAB+) m/z calcd for C16H15N2O3S3 (MH+) 379.0245 obsd 379.0228.
2-Aceto-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (26·HCl)
Compound 41 (75.0 mg, 0.198 mmol) was converted to THTP 26 according to the General Procedure for Bts Deprotection. The hydrochloride salt was recrystallized from EtOH/hexanes to yield 26·HCl (36.0 mg, 0.165 mmol, 84%) as white crystals: mp 257–259 °C; 1H NMR (500 MHz, CD3OD) δ 7.71 (s, 1H), 4.35 (s, 2H), 3.59 (t, 2H, J = 6.2 Hz), 3.23 (t, 2H, J = 6.2 Hz), 2.56 (s, 3H); 13C NMR (500 MHz, CD3OD) δ 191.2, 143.2, 141.0, 131.1, 128.9, 42.5, 41.3, 25.0, 22.0; HRMS (FAB+) m/z calcd for C9H12NOS (MH+) 182.0640 obsd 182.0621; Anal. calcd for C9H12ClNOS·⅛H2O: C, 49.14; H, 5.61; N, 6.37. Found: C, 49.03; H, 5.53; N, 6.20.
N-Bts-2-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine (43)
To a stirred solution of aldehyde 37 (280 mg, 0.816 mmol) in 10 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (300 mg, 1.25 mmol) in water (1 mL) and the reaction was stirred for 15 min. Ethyl-2-bromopropionate (0.425 mL, 3.26 mmol) was added dropwise and the reaction was warmed to ambient temperature and stirred for 2 h. The reaction was poured into ice water (50 mL) and a white precipitate formed. The mixture was extracted with ether (3 × 50 mL) and the combined organic extracts were washed with water (4 × 30 mL) and brine (30 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (3:1) to yield the sulfide intermediate (42) as a clear oil. This oil was dissolved in ethanol (30 mL) and cooled to 0 °C. NaOH (1 M, 100 mL) was added to this solution and the reaction was continued for 5 min. The reaction was then made acidic (pH ≈ 1) with 3 M HCl and was stirred for 15 min at ambient temperature. The resulting cloudy reaction mixture was extracted with CH2Cl2 (3 × 50 mL) and the combined organic extracts were washed with brine (25 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (3:1) to yield 43 (195 mg, 0.556 mmol, 68%) as a white solid: mp 138–140 °C; 1H NMR (500 MHz, CDCl3) δ 8.19–8.17 (m, 1H), 8.00–7.98 (m, 1H), 7.64–7.56 (m, 2H), 6.42 (s, 1H), 4.53 (s, 2H), 3.79 (t, 2H, J = 5.7 Hz), 2.94 (t, 2H, J = 5.6 Hz), 2.41 (s, 3H); 13C NMR (500 MHz, CDCl3) δ 164.0, 152.5, 138.2, 136.1, 129.9, 129.7, 127.5, 127.3, 125.1, 122.5, 122.0, 46.1, 44.6, 25.0, 15.1; HRMS (FAB+) m/z calcd for C15H15N2O2S3 (MH+) 351.0296 obsd 351.0280.
2-Methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (27·HCl)
Compound 43 (80.0 mg, 0.228 mmol) was converted to THTP 27 according to the General Procedure for Bts Deprotection. The hydrochloride salt was recrystallized from EtOH/hexanes to yield 27·HCl (40.4 mg, 0.212 mmol, 93%) as white crystals: mp 227–229 °C; 1H NMR (500 MHz, CD3OD) δ 6.59 (s, 1H), 4.21 (s, 2H), 3.53 (t, 2H, J = 6.2 Hz), 3.09 (t, 2H, J = 6.1 Hz), 2.45 (s, 3H); 13C NMR (500 MHz, CD3OD) δ 139.5, 129.1, 127.0, 122.6, 42.8, 41.8, 21.3, 13.6; HRMS (FAB+) m/z calcd for C8H12NS (MH+) 154.0690 obsd 154.0683; Anal. calcd for C8H12ClNS·¼H2O: C, 49.48; H, 6.49; N, 7.21. Found: C, 49.51; H, 6.23; N, 7.15.
(R)- and (S)-2-Methyl-1-((S)-1-phenylethyl)piperidin-4-one [(R,S)-45 and (S,S)-45]
To a stirred solution of 44 (8.00 g, 61.9 mmol) in dry THF (150 mL) at 0 °C, was added 1M vinylmagnesium bromide in THF (68.0 mL, 68.0 mmol) and the reaction was warmed to ambient temperature. The reaction was stirred for 1 h and the disappearance of starting material was observed according to TLC. To the reaction was added [S]-α-methylbenzylamine (16.0 mL, 124 mmol) and water (15 mL). The reaction was stirred for 1 h and the THF was removed under reduced pressure. Water (150 mL) was added and the aqueous mixture was extracted with CH2Cl2 (3 × 100 mL). The combined organic extracts were washed with brine (100 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield a brown oil, which was purified by flash chromatography eluting with hexanes/EtOAc (4:1) to yield the (R,S)-45 (3.72g, 17.1 mmol, 28%, Rf = 0.25 in 4:1 hexanes/EtOAc) as a yellow solid: mp 70–72 °C; [α]D24 = −11.8° (c 0.12, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.45–7.44 (m, 2H), 7.35–7.32 (m, 2H), 7.27–7.24 (m, 1H), 4.03–3.99 (m, 1H), 3.41–3.34 (m, 1H), 2.78–2.72 (m, 1H), 2.69–2.62 (m, 3H), 2.35–2.30 (m, 1H), 2.26–2.17 (m, 2H), 1.34–1.33 (m, 3H), 1.15–1.13 (m, 3H); 13C NMR (500 MHz, CDCl3) δ 210.5, 145.2, 128.3, 127.2, 126.9, 57.6, 52.4, 48.8, 43.9, 41.3, 16.3, 16.2; HRMS (ES+) m/z calcd for C14H19NO (MH+) 219.1545 obsd 219.1537. and (S,S)-45 (3.90g, 17.9 mmol, 29%, Rf = 0.35 in 4:1 hexanes/EtOAc) as a yellow oil: [α]D24 = −4.0° (c 21.4, CH3OH); 1H NMR (500 MHz, CDCl3) δ 7.36–7.31 (m, 4H), 7.28–7.24 (m, 1H), 3.94–3.90 (m, 1H), 3.18–3.13 (m, 1H), 2.99–2.91 (m, 2H), 2.59–2.50 (m, 2H), 2.34–2.29 (m, 1H), 2.12–2.08 (m, 1H), 1.43–1.42 (m, 3H), 1.04 (d, 3H, J = 6.5 Hz); 13C NMR (500 MHz, CDCl3) δ 210.5, 144.0, 128.4, 127.2, 127.1, 58.7, 52.3, 48.2, 43.1, 41.1, 21.8, 14.6; HRMS (ES+) m/z calcd for C14H19NO (MH+) 219.1545 obsd 219.1538.
(R)-N-Nvoc-2-methyl-4-piperidone [(R)-46]
A solution of (R,S)-45 (1.38 g, 6.24 mmol) in dry THF (10 mL) was hydrogenated over Pd(OH)2 (200 mg) for 4 h at ambient temperature and pressure. The reaction mixture was filtered through Celite and washed with THF (2 × 20 mL). To the resulting THF solution (50 mL), was added water (50 mL), NvocCl (2.07 g, 7.49 mmol), and 4 M NaOH (to maintain the pH at ≈ 11). After 1 h, the reaction mixture was partitioned between CH2Cl2 (250 mL) and brine (100 mL). The organic layer was removed and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to yield the crude product as a yellow solid, which was purified by flash chromatography eluting with hexanes/ether/CH2Cl2 (1:1:1) to yield (R)-46 (1.92 mg, 5.44 mmol, 87%) as light yellow crystals: mp 134–136 °C; [α]D24 = −5.0° (c 4.7, CHCl3); 1H NMR (500 MHz, CDCl3) δ 7.68 (s, 1H), 6.97 (s, 1H), 5.57–5.50 (m, 2H), 4.78–4.71 (m, 1H), 4.31–4.27 (m, 1H), 3.96 (s, 3H), 3.95 (s, 3H), 3.44–3.39 (m, 1H), 2.71–2.67 (m, 1H), 2.53–2.47 (m, 1H), 2.40–2.35 (m, 1H), 2.31–2.27 (m, 1H), 1.23 (d, 3H, J = 6.9 Hz); 13C NMR (500 MHz, CDCl3) δ 207.0, 154.7, 153.4, 148.7, 140.8, 127.0, 11.6, 108.6, 64.7, 56.5, 56.4, 48.5, 46.5, 40.5, 38.8, 19.1; HRMS (ES+) m/z calcd for C16H21N2O7 (MH+) 353.1349 obsd 353.1334.
(S)-N-Nvoc-2-methyl-4-piperidone [(S)-46]
Compound (S)-46 was prepared in the same manner as (R)-46 using (S,S)-45 as the starting material. mp 134–136 °C; [α]D24 = −5.2° (c 1.6, CHCl3); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C16H21N2O7 (MH+) 353.1349 obsd 353.1339.
(R)-N-Nvoc-4-chloro-6-methyl-1,2,5,6-tetrahydropyridine-3-carboxaldehyde [(R)-47] and (R)-N-Nvoc-4-chloro-2-methyl-1,2,5,6-tetrahydropyridine-3-carboxaldehyde [(R)-48]
To a stirred solution of DMF (0.542 mL, 7.10 mmol) in CHCl3 (15 mL) at 5 °C, was added POCl3 (0.463 mL, 4.97 mmol) dropwise. The reaction was warmed to ambient temperature and stirred for 10 min. Compound (R)-46 (1.00 g, 2.84 mmol) was dissolved in CHCl3 (8 mL) and added dropwise to the POCl3-DMF solution. The reaction was continued for 10 h and then poured into an ice cold solution of sodium acetate (4.0 g) in water (100 mL) and stirred vigorously for 20 min. This mixture was extracted with CH2Cl2 (4 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was partially purified by flash chromatography eluting with hexanes/CHCl3/ether (1:1:2) to separate the unreacted starting material (R)-46 (151 mg, 0.429 mmol, 15%) from a mixture of (R)-47 and (R)-48 (770 mg, 1.93 mmol, 68%) as a pale yellow solid. The mixture of regioisomers (R)-47 and (R)-48 was separated by fractional crystallization. The mixture was first dissolved in the minimum amount of ether. Hexanes (a volume equal to that of the ether) were added and yellow needles of (R)-47 (460 mg, 1.15 mmol, 41%) formed overnight: mp 153–155 °C; [α]D24 = +79.7° (c 1.0, CHCl3); 1H NMR (500 MHz, CDCl3) δ 10.19 (s, 1H), 7.73 (s, 1H), 7.04 (s, 1H), 5.56 (s, 2H), 4.76–4.72 (m, 2H, major and minor rot.), 4.01 (s, 3H), 3.98 (s, 3H), 3.83 (br, 1H, major rot. 60%), 3.80 (br, 1H, minor rot. 40%), 3.13–3.11 (br, 1H, minor rot. 40%), 3.08–3.05 (br, 1H, major rot. 60%), 2.41 (s, 1H, major rot. 60%), 2.37 (s, 1H, minor rot. 40%), 1.23 (d, 3H, J = 7.0 Hz); 13C NMR (500 MHz, CDCl3) δ 188.4, 154.2, 153.6, 148.4, 147.2, 140.2, 129.5, 127.2, 110.6, 108.5, 64.6, 56.4, 56.4, 45.2, 40.6, 38.6, 17.6; HRMS (ES+) m/z calcd for C17H20ClN2O7 (MH+) 399.0959 obsd 399.0952. The solvent was removed from the filtrate to yield a yellow solid, which was dissolved in the minimum amount of EtOAc. Hexanes were added until the solution became slightly cloudy. Yellow crystals of (R)-48 (291 mg, 0.73 mmol, 26%) formed over 2–3 days: mp 132–134 °C; [α]D23 = −107.4° (c 0.50, CHCl3); 1H NMR (500 MHz, CDCl3) δ 10.14 (s, 1H), 7.73 (s, 1H, major rot. 70%), 7.68 (s, 1H, minor rot. 30%), 7.04 (s, 1H, major rot. 70%), 6.94 (s, 1H, minor rot. 30%), 5.63–5.49 (m, 2H), 5.16–5.10 (m, 1H), 4.37–4.33 (s, 1H, major rot. 70%), 4.19–4.15 (s, 1H, minor rot. 30%), 4.00 (s, 3H), 3.97 (s, 3H), 3.30–3.20 (m, 1H, major and minor rot.), 2.94–2.80 (m, 1H, major and minor rot.), 2.58–2.48 (m, 1H, major and minor rot.), 1.34 (d, 3H, J = 7.0 Hz, major rot. 70%), 1.30 (d, 3H, J = 7.0 Hz, minor rot. 30%); HRMS (ES+) m/z calcd for C17H20ClN2O7 (MH+) 399.0959 obsd 399.0948.
(S)-N-Nvoc-4-chloro-6-methyl-1,2,5,6-tetrahydropyridine-3-carboxaldehyde [(S)-47] and (S)-N-Nvoc-4-chloro-2-methyl-1,2,5,6-tetrahydropyridine-3-carboxaldehyde [(S)-48]
Compounds (S)-47 and (S)-48 were prepared in the same manner as (R-)-47 and (R)-48 using (S)-46 as the starting material. (S)-47: mp 153–155 °C; [α]D24 = −75.3° (c 0.50, CHCl3); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C17H20ClN2O7 (MH+) 399.0959 obsd 399.0959. (S)-48: mp 133–135 °C; [α]D24 = +105.0° (c 0.50, CHCl3); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C17H20ClN2O7 (MH+) 399.0959 obsd 399.0952.
(R)-N-Nvoc-6-methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(R)-49]
To a stirred solution of aldehyde (R)-47 (135 mg, 0.339 mmol) in 5 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (98 mg, 0.406 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Bromonitromethane (0.20 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (75 mL) and a white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to yield a pale yellow solid. Recrystallization from EtOAc/hexanes yielded (R)-49 (102 mg, 0.233 mmol, 69%) as light yellow crystals: mp 161–163 °C; [α]D23 = +51.1° (c 0.50, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 1H), 7.68 (s, 1H), 6.99 (s, 1H), 5.60–5.55 (m, 2H), 4.96 (br, 1H, minor rot. 40%), 4.93 (br, 1H, major rot. 60%), 4.19 (br, 1H, major rot. 60%), 4.16 (br, 1H, minor rot. 40%), 3.98 (s, 3H), 3.97 (s, 3H), 3.19–3.17 (m, 1H, minor rot. 40%), 3.16–3.14 (m, 1H, major rot. 60%), 2.72 (s, 1H, major rot. 60%), 2.68 (s, 1H, minor rot. 40%), 1.23 (d, 3H, J = 6.9 Hz); 13C NMR (500 MHz, CDCl3) δ 154.6, 153.3, 150.0, 148.8, 140.8, 140.5, 130.8, 126.7, 126.0, 111.7, 108.6, 64.7, 56.4, 56.3, 45.2, 40.0, 30.8, 17.4; HRMS (ES+) m/z calcd for C18H20N3O8S (MH+) 438.0971 obsd 438.0965.
(S)-N-Nvoc-6-methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(S)-49]
Compound (S)-49 was prepared in the same manner as (R)-49 using S-47 as the starting material. mp 160–162 °C; [α]D23 = −52.3° (c 0.17, CH2Cl2); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C18H20N3O8S (MH+) 438.0971 obsd 438.0963.
General Procedure for Nvoc Deprotection
(Selected procedure for (R)-30·HCl). To a stirred solution of 49 (70 mg, 0.16 mmol) in CH2Cl2 (0.5 mL), was added ethanethiol (1.0 mL) at 0 °C. BF3·Et2O (0.60 mL) was added dropwise and the reaction was warmed to ambient temperature and continued for 10 h. The ethanethiol and CH2Cl2 were blown off with a stream of N2 and the reaction was slowly quenched with H2O (5 mL). This mixture was then diluted with H2O (50 mL), made basic (pH ≈ 12) with 1 M NaOH, and extracted with CH2Cl2 (3 × 25 mL). The combined organic extracts were washed with brine (25 mL) and dried over Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with CH2Cl2/MeOH (10:1) to yield the free base, which was redissolved in CHCl3 (20 mL). Dry HCl(g) was bubbled through this solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes to yield (R)-30·HCl (28.0 mg, 0.119 mmol, 75%) as yellow crystals.
(R)-6-Methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(R)-30·HCl]
See General Procedure for Nvoc Deprotection: mp 210–112 °C; [α]D24 = −72.6° (c 0.46, CH3OH); 1H NMR (500 MHz, CD3OD) α 7.64 (s, 1H), 4.45–4.33 (m, 2H), 3.79–3.72 (m, 1H), 3.38–3.34 (m, 1H), 3.00–2.94 (m, 1H), 1.54 (d, 3H, J = 6.5 Hz); 13C NMR (500 MHz, CD3OD) δ 140.1, 135.5, 128.4, 113.1, 108.7, 49.9, 42.0, 29.0, 16.9; HRMS (ES+) m/z calcd for C8H11N2O2S (MH+) 199.0541 obsd 199.0545; Anal. calcd for C8H11ClN2O2S·¼H2O: C, 40.17; H, 4.85; N, 11.71. Found: C, 39.95; H, 4.67; N, 11.47.
(S)-6-Methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(S)-30·HCl]
Compound (S)-30·HCl was prepared in the same manner as (R)-30 using (S)-49 as the starting material. mp 212–214 °C; [α]D24 = +73.2° (c 0.40, CH3OH); 1H NMR and 13C NMR data were identical.; HRMS (ES+) m/z calcd for C8H11N2O2S (MH+) 199.0541 obsd 199.0547; HRMS (ES+) m/z calcd for C8H11N2O2S (MH+) 199.0541 obsd 199.0545; Anal. calcd for C8H11ClN2O2S: C, 40.94; H, 4.72; N, 11.94. Found: C, 40.68; H, 4.42; N, 11.71.
(R)-N-Nvoc-2-cyano-6-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(R)-50]
To a stirred solution of aldehyde (R)-47 (135 mg, 0.339 mmol) in 5 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (98 mg, 0.406 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Bromoacetonitrile (0.20 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (75 mL) and a white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to yield a pale yellow solid. Recrystallization from EtOAc/hexanes yielded (R)-50 (110 mg, 0.264 mmol, 78%) as white crystals: mp 152–154 °C; [α]D23 = +60.2° (c 0.50, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 1H), 7.35 (s, 1H), 6.98 (s, 1H), 5.60–5.54 (m, 2H), 4.98 (br, 1H, minor rot. 40%), 4.94 (br, 1H, major rot. 60%), 4.20 (br, 1H, major rot. 60%), 4.17 (br, 1H, minor rot. 40%), 3.98 (s, 3H), 3.96 (s, 3H), 3.17–3.15 (m, 1H, minor rot. 40%), 3.14–3.12 (m, 1H, major rot. 60%), 2.74 (s, 1H, major rot. 60%), 2.71 (s, 1H, minor rot. 40%), 1.21 (d, 3H, J = 6.9 Hz); 13C NMR (500 MHz, CDCl3) δ 154.6, 153.3, 148.7, 140.7, 140.1, 134.8, 131.5, 126.8, 114.0, 111.6, 108.6, 107.9, 64.6, 56.4, 56.3, 45.3, 40.1, 30.7, 17.3; HRMS (ES+) m/z calcd for C19H20N3O6S (MH+) 418.1073 obsd 418.1069.
(S)-N-Nvoc-2-cyano-6-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(S)-50]
Compound (S)-50 was prepared in the same manner as (R)-50 using (S)-47 as the starting material. mp 152–154 °C; [α]D23 = −56.1° (c 0.15, CH2Cl2); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C19H20N3O6S (MH+) 418.1073 obsd 418.1069.
(R)-2-Cyano-6-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(R)-31·HCl]
Compound (R)-50 (100 mg, 0.240 mmol) was converted to (R)-31 according to the General Procedure for Nvoc Deprotection. The free amine was purified by flash chromatography eluting with CH2Cl2/MeOH (8:1) and the hydrochloride salt was recrystallized from EtOH/hexanes to yield (R)-31·HCl (43.5 mg, 0.203 mmol, 84%) as yellow crystals: mp 277–279 °C; [α]D24 = −94.5° (c 0.36, CH3OH); 1H NMR (500 MHz, CD3OD) δ 7.64 (s, 1H), 4.45–4.33 (m, 2H), 3.79–3.72 (m, 1H), 3.38–3.34 (m, 1H), 3.00–2.94 (m, 1H), 1.54 (d, 3H, J = 6.5 Hz); 13C NMR (500 MHz, CD3OD) δ 140.1, 135.5, 128.4, 113.1, 108.7, 49.9, 42.0, 29.0, 16.9; HRMS (ES+) m/z calcd for C9H11N2S (MH+) 179.0643 obsd 179.0644; Anal. calcd for C9H11ClN2S: C, 50.34; H, 5.16; N, 13.05. Found: C, 50.14; H, 5.03; N, 12.91.
(S)-2-Cyano-6-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(S)-31·HCl]
Compound (S)-31·HCl was prepared in the same manner as (R)-31·HCl using (S)-50 as the starting material. mp 276–278 °C; [α]D24 = +92.4° (c 0.25, CH3OH); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C9H11N2S (MH+) 179.0643 obsd 179.0648; Anal. calcd for C9H11ClN2S: C, 50.34; H, 5.16; N, 13.05. Found: C, 50.36; H, 5.04; N, 12.97.
(R)-N-Nvoc-4-methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(R)-51]
To a stirred solution of aldehyde (R)-48 (135 mg, 0.339 mmol) in 5 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (98 mg, 0.406 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Bromonitromethane (0.20 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (75 mL) and a white precipitate formed. The mixture was extracted with CH2Cl2 (4 × 50 mL) and the combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to yield a pale yellow solid. Recrystallization from CH2Cl2/hexanes yielded (R)-51 (96 mg, 0.219 mmol, 65%) as light yellow crystals: mp 168–170 °C; [α]D23 = −111.8° (c 0.50, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.71 (s, 1H), 7.66 (s, 1H), 6.99 (s, 1H), 5.61–5.52 (m, 2H), 5.24 (br, 1H, major and minor rot.), 4.48 (br, 1H, major and minor rot.), 3.98 (s, 3H), 3.97 (s, 3H), 3.23–3.17 (m, 1H), 2.98–2.91 (m, 1H, major and minor rot.), 2.84–2.80 (m, 1H, major and minor rot.), 1.47 (d, 3H, J = 6.8 Hz); 13C NMR (CDCl3) δ 154.2, 153.2, 149.8, 148.6, 141.7, 141.1, 137.3, 126.6, 126.3, 111.5, 108.4, 64.5, 56.3, 56.2, 48.9, 36.9, 25.5, 20.2; HRMS (ES+) m/z calcd for C18H20N3O8S (MH+) 438.0971 obsd 438.0971.
(S)-N-Nvoc-4-methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(S)-51]
Compound (S)-51 was prepared in the same manner as (R)-51 using (S)-48 as the starting material. mp 168–170 °C; [α]D23 = +108.1° (c 0.18, CH2Cl2); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C18H20N3O8S (MH+) 438.0971 obsd 438.0975.
(R)-4-Methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(R)-33·HCl]
Compound (R)-51 (85.0 mg, 0.194 mmol) was converted to (R)-33 according to the General Procedure for Nvoc Deprotection. The free amine was purified by flash chromatography eluting with CH2Cl2/MeOH (10:1) and the hydrochloride salt was recrystallized from EtOH/hexanes to yield (R)-33·HCl (37.0 mg, 0.158 mmol, 81%) as yellow crystals: mp 202–204 °C; [α]D24 = +58.4 (c 0.45, CH3OH); 1H NMR (500 MHz, CD3OD) δ 8.00 (s, 1H), 4.63–4.59 (m, 1H), 3.78–3.73 (m, 1H), 3.57–3.51 (m, 1H), 3.26–3.23 (m, 2H), 1.70 (d, 3H, J = 6.9 Hz); 13C NMR (500 MHz, CD3OD) δ 150.8, 140.3, 133.3, 126.2, 50.4, 39.8, 21.9, 17.1; HRMS (ES+) m/z calcd for C8H11N2O2S (MH+) 199.0541 obsd 199.0548; Anal. calcd for C8H11ClN2O2S: C, 40.94; H, 4.72; N, 11.94. Found: C, 40.87; H, 4.58; N, 11.74.
(S)-4-Methyl-2-nitro-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(S)-33·HCl]
Compound (S)-33·HCl was prepared in the same manner as (R)-33 using (S)-51 as the starting material. mp 202–204 °C; [α]D24 = −56.4° (c 0.30, CH3OH); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C8H11N2O2S (MH+) 199.0541 obsd 199.0548; Anal. calcd for C8H11ClN2O2S·½H2O: C, 39.43; H, 4.96; N, 11.49. Found: C, 39.72; H, 4.80; N, 11.41.
(R)-N-Nvoc-2-cyano-4-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(R)-52]
To a stirred solution of aldehyde (R)-48 (160 mg, 0.401 mmol) in 5 mL DMF at 0 °C, was added a solution of sodium sulfide nonahydrate (116 mg, 0.481 mmol) in water (0.5 mL) and the reaction was stirred for 15 min. Bromoacetonitrile (0.168 mL) was added dropwise and the reaction was warmed to ambient temperature and stirred for 1 h. The reaction was poured into ice water (50 mL) and a white precipitate formed. Brine (50 mL) was added and the aqueous mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to yield a pale yellow solid. Recrystallization from EtOAc/hexanes yielded (R)-52 (138 mg, 0.331 mmol, 82%) as white crystals: mp 150–152 °C; [α]D23 = −97.1° (c 0.50, CH2Cl2); 1H NMR (500 MHz, CDCl3) δ 7.70 (s, 1H), 7.33 (s, 1H), 6.98 (s, 1H), 5.60–5.51 (m, 2H), 5.26 (br, 1H, major and minor rot.), 4.33 (br, 1H, major and minor rot.), 3.98 (s, 3H), 3.96 (s, 3H), 3.22–3.16 (m, 1H), 2.95–2.89 (m, 1H, major and minor rot.), 2.85–2.79 (m, 1H, major and minor rot.), 1.45 (d, 3H, J = 6.8 Hz); 13C NMR (CDCl3) δ 154.4, 153.3, 148.7, 141.3, 140.7, 138.1, 135.3, 126.9, 114.0, 111.6, 108.6, 107.8, 64.6, 56.4, 56.3, 49.2, 37.2, 25.5, 20.5; HRMS (ES+) m/z calcd for C19H20N3O6S (MH+) 418.1073 obsd 418.1060.
(S)-N-Nvoc-2-cyano-4-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine [(S)-52]
Compound (S)-52 was prepared in the same manner as (R)-52 using (S)-48 as the starting material. mp 152–154 °C; [α]D23 = +103.4° (c 0.50, CH2Cl2); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C19H20N3O6S (MH+) 418.1073 obsd 418.1071. The structure of (S)-52 has been confirmed by X-ray crystallography (Section 7).
(R)-2-Cyano-4-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(R)-34·HCl]
Compound (R)-52 (100 mg, 0.240 mmol) was converted to (R)-34 according to the General Procedure for Nvoc Deprotection. The free amine was purified by flash chromatography eluting with CH2Cl2/MeOH (8:1) and the hydrochloride salt was recrystallized from EtOH/hexanes to yield (R)-34·HCl (42.2 mg, 0.197 mmol, 82%) as yellow crystals: mp 232–234 °C; [α]D24 = +58.7° (c 0.68, CH3OH); 1H NMR (500 MHz, CD3OD) δ 7.74 (s, 1H), 4.64–4.60 (m, 1H), 3.77–3.72 (m, 1H), 3.55–3.49 (m, 1H), 3.27–3.23 (m, 2H), 1.69 (d, 3H, J = 6.8 Hz); 13C NMR (500 MHz, CD3OD) δ 140.2, 135.5, 134.1, 113.1, 109.6, 50.7, 40.4, 21.8, 17.3; HRMS (ES+) m/z calcd for C9H11N2S (MH+) 179.0643 obsd 179.0646; Anal. calcd for C9H11ClN2S·⅛H2O: C, 49.82; H, 5.23; N, 12.91. Found: C, 49.77; H, 5.12; N, 12.71.
(S)-2-Cyano-4-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride [(S)-34·HCl]
Compound (S)-34·HCl was prepared in the same manner as (R)-34 using (S)-52 as the starting material. mp 234–236 °C; [α]D24 = − 57.5° (c 0.30, CH3OH); 1H NMR and 13C NMR data were identical; HRMS (ES+) m/z calcd for C9H11N2S (MH+) 179.0643 obsd 179.0645; Anal. calcd for C9H11ClN2S·¼H2O: C, 49.31; H, 5.29; N, 12.78. Found: C, 49.31; H, 5.24; N, 12.65.
1,1,1-Trifluoro-3-(thiophen-2-yl)-propan-2-one oxime (55)
3-Bromo-1,1,1-trifluoropropan-2-one oxime (53, 764 mg, 3.71 mmol), thiophene (8.20 mL, 103 mmol), powdered Na2CO3 (5.89 g, 55.6 mmol), and DCE (100 mL) were reacted in a thick-walled pyrex bottle sealed with a Teflon screw cap at 100 °C for 72 h. The reaction was cooled and filtered through Celite, which was washed with CH2Cl2 (3 × 50 mL). The solvent was removed under reduced pressure to yield oxazine 54. This crude product was dissolved in CHCl3 and treated with trifluoroacetic acid (1 mL) at ambient temperature for 1 h. The reaction was diluted with CHCl3 (50 mL) and then extracted with water (50 mL) and brine (50 mL). The organic layer was dried over anhydrous Na2SO4 and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography eluting with hexanes/EtOAc (10:1) to yield 55 (389 mg, 1.86 mmol, 50% for 2 steps) as colorless crystals: mp 43–45 °C; 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.22–7.20 (m, 1H), 6.97–6.95 (m, 2H), 4.04 (s, 2H).
(±)-6-Trifluoromethyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (21·HCl)
To a slurry of LiAlH4 (107 mg, 2.82 mmol) in Et2O (20 mL) was added a solution of 55 (197 mg, 0.942 mmol) in Et2O (8 mL) at 0 °C. The reaction mixture was allowed to warm to ambient temperature and then refluxed at 40 °C for 2.5 h. The reaction was cooled to ambient temperature and was then quenched with the successive dropwise addition of water (0.1 mL), 15% NaOH (0.1 mL), and water (0.3 mL). The reaction mixture was filtered through Celite, which was washed with Et2O (3 × 50 mL). The resulting organic solution was washed with brine (50 mL), dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure. The crude product was purified by flash chromatography eluting with hexanes/EtOAc (9:1) to yield amine 56 (100 mg, 0.512 mmol) as a colorless oil: 1H NMR (400 MHz, CDCl3) δ 7.24 (d, J = 4.9 Hz, 1H), 7.00–6.95 (m, 2H), 3.64–3.60 (m, 1H), 3.49–3.45 (m, 1H), 2.94–2.88 (m, 1H). To this amine was added 20% formaldehyde (0.428 mL) dropwise at 0 °C. This mixture was allowed to warm to ambient temperature and then refluxed at 100 °C for 3 h. The reaction mixture was cooled and then partitioned between Et2O (50 mL) and brine (50 mL) to remove excess formaldehyde. The organic layer was removed, dried over anhydrous Na2SO4, and the solvent was removed under reduced pressure to yield the imine intermediate (120 mg). To this crude product was added 5 M HCl (1 mL) dropwise at 0 °C. The reaction was permitted to warm to ambient temperature and stirred for 3h. The reaction mixture was diluted with water (50 mL), made basic (pH ≈ 10) with 1M NaOH, and extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (6:1) to yield 21 as white crystals. This free base was dissolved in Et2O and dry HCl(g) was bubbled through this solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes to yield 21·HCl (49.5 mg, 0.203 mmol, 22% from 55) as white crystals: mp 183–185 °C; 1H NMR (400 MHz, CD3OD) δ 7.47 (d, J = 4.8 Hz, 1H), 6.97 (d, J = 4.8 Hz, 1H), 4.66–4.57 (m, 1H), 4.53–4.43 (m, 2H), 3.51–3.24 (m, 2H); HRMS (FAB+) m/z calcd for C8H9F3NS (MH+) 208.0408 obsd 208.0388; Anal. calcd for C8H9ClF3NS: C, 39.43; H, 3.72; N, 5.75. Found: C, 39.10; H, 3.17; N, 5.46.
(±)-2-Bromo-6-trifluoromethyl-4,5,6,7-tetrahydrohieno[3,2-c]pyridine hydrochloride (29·HCl)
To a stirred solution of 21 (36.4 mg, 0.176 mmol) in glacial acetic acid (1.5 mL), a solution of bromine (11.8 µL, 0.229 mmol) in glacial acetic acid (1.0 mL) was added dropwise at 0 °C. The reaction mixture was stirred at ambient temperature for 3 h. The reaction mixture was poured into saturated solution of NaHCO3 (50 mL) and then extracted with EtOAc (3 × 50 mL). The combined organic extracts were washed with brine (50 mL) and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure and the crude product was purified by flash chromatography eluting with hexanes/EtOAc (1:1) to yield 29 as white crystals. This free base was dissolved in Et2O and dry HCl(g) was bubbled through this solution to form the hydrochloride salt, which was recrystallized from EtOH/hexanes to yield 29·HCl (35.2 mg, 0.123 mmol, 70%) as white crystals: mp, sublimes above 185 °C; 1H NMR (400 MHz, CD3OD) δ 6.97 (s, 1H), 4.32–4.22 (m, 2H), 3.72–3.64 (m, 1H), 3.19–3.14 (m, 1H), 2.84–2.78 (m, 1H); HRMS (FAB+) m/z calcd for C8H8BrF3NS (MH+) 285.9513 obsd 285.9514; Anal. calcd for C8H8BrClF3NS: C, 29.79; H, 2.50; N, 4.34. Found: C, 29.89; H, 2.23; N, 4.22.
(±)-2-Bromo-6-methyl-4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrobromide (28·HBr)
Compound 28 was prepared from 20 (115 mg, 0.750 mmol) using the same procedure for the conversion of 21 to 29. The crude product was purified by flash chromatography eluting with EtOAc to yield 28 as white crystals. This free base was dissolved in Et2O and dry HBr(g) was bubbled through this solution to form the hydrobromide salt, which was recrystallized from EtOH/hexanes to yield 28·HBr (60.2 mg, 0.224 mmol, 30%) as white crystals: mp 199–201 °C; 1H NMR (400 MHz, CD3OD) δ 6.98 (s, 1H), 4.33–4.23 (m, 2H), 3.74–3.67 (m, 1H), 3.20–3.16 (m, 1H), 2.86–2.79 (m, 1H), 1.51 (d, J = 6.5 Hz, 3H); HRMS (FAB+) m/z calcd for C8H11BrNS (MH+) 231.9795 obsd 231.9766; Anal. calcd for C8H11Br2NS: C, 30.69; H, 3.54; N, 4.47. Found: C, 31.09; H, 3.25; N, 4.38.
3-(Benzo(b)thiophen-2-yl)-1,1,1-trifluoropropan-2-one oxime (58)
Compound 58 was prepared from 53 (678 mg, 3.29 mmol), benzo[b]thiophene (2.0 mL, 18 mmol), powdered Na2CO3 (3.49 g, 32.9 mmol), and CH2Cl2 (25 mL) using the same procedure for the conversion of 53 to 55. Compound 58 (76.3 mg, 29.4 mmol, 9% from 53) was obtained as a white solid: 1H NMR (400 MHz, CDCl3) δ 8.44 (s, 1H), 7.90 (d, J = 7.7 Hz, 1H), 7.84 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 4.10 (s, 2H).
(±)-3-Trifluoromethyl-1,2,3,4-tetrahydrobenzothieno[3,2-c]pyridine hydrochloride (32·HCl)
Compound 32 was prepared from 58 (75.0 mg, 0.289 mmol) using the same procedure for the conversion of 55 to 21. The hydrochloride salt was recrystallized from EtOH/hexanes to yield 32·HCl (26.0 mg, 0.0885 mmol, 31% from 58) as white crystals: mp, sublimes above 195 °C; 1H NMR (500 MHz, CD3OD) δ 7.94–7.92 (m, 1H), 7.83–7.81 (m, 1H), 7.52–7.44 (m, 2H), 4.83–4.79 (m, 1H), 4.77 (s, 2H), 3.60–3.56 (m, 1H), 3.30–3.24 (m, 1H); 13C NMR (500 MHz, CD3OD) δ 139.1, 137.3, 127.3, 125.4, 124.8, 124.6, 123.7 (q, J = 280 Hz), 122.3, 120.9, 53.9 (q, J = 32.6 Hz), 43.4, 20.8; HRMS (FAB+) m/z calcd for C12H11F3NS (MH+) 258.0564 obsd 258.0556. Anal. calcd for C12H11ClF3NS·¼H2O: C, 48.33; H, 3.89; N, 4.70. Found: C, 48.40; H, 3.60; N, 4.55.
7. X-ray Crystallography
The structure of compound (S)-52 has been deposited in the Cambridge Crystallographic Data Centre, reference number CCDC 638032.
Supplementary Information
Supplementary Information Available: Biochemical data for the enantiomers of 33 and 34. This material is available free of charge via the Internet. Supplementary Information associated with this article can be found, in the online version, at doi: (please insert DOI)
Supplementary Material
Acknowledgements
This research was supported by National Institutes of Health (NIH) Grant HL 34193. The funding for M.R.S. was also supported by NIH Predoctoral Training Grant GM 07775 and the American Foundation for Pharmaceutical Education. We thank David VanderVelde and Sarah Neuenswander of the University of Kansas Nuclear Magnetic Resonance Laboratory for their assistance. The 500 MHz NMR spectrometer was partially funded by National Science Foundation Grant CHE-9977422. We thank Douglas Powell and Victor Day of the University of Kansas X-ray Crystallography Laboratory, Gerald Lushington of the University of Kansas Molecular Graphics and Modeling Laboratory, and Todd Williams of the University of Kansas Mass Spectrometry Laboratory for their assistance. We also thank Ernst Schönbrunn for assistance with molecular modeling and David Benson for various helpful discussions.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and notes
- 1.Seim Mitchell R. The contents of this paper were taken in large part from the Ph.D. dissertation. University of Kansas; 2006. [Google Scholar]
- 2.Grunewald GL, Dahanukar VH, Jalluri RK, Criscione KR. J. Med. Chem. 1999;42:118. doi: 10.1021/jm980429p. [DOI] [PubMed] [Google Scholar]
- 3.Grunewald GL, Sall DJ, Monn JA. J. Med. Chem. 1988;31:824. doi: 10.1021/jm00399a024. [DOI] [PubMed] [Google Scholar]
- 4.Grunewald GL, Caldwell TM, Li QF, Dahanukar VH, McNeil B, Criscione KR. J. Med. Chem. 1999;42:4351. doi: 10.1021/jm990086a. [DOI] [PubMed] [Google Scholar]
- 5.Grunewald GL, Dahanukar VH, Teoh B, Criscione KR. J. Med. Chem. 1999;42:1982. doi: 10.1021/jm9807252. [DOI] [PubMed] [Google Scholar]
- 6.Sall DJ, Grunewald GL. J. Med. Chem. 1987;30:2208. doi: 10.1021/jm00395a006. [DOI] [PubMed] [Google Scholar]
- 7.Grunewald GL, Seim MR, Regier RC, Martin JL, Gee CL, Drinkwater N, Criscione KR. J. Med. Chem. 2006;49:5424. doi: 10.1021/jm060466d. [DOI] [PubMed] [Google Scholar]
- 8.Pendleton RG, Kaiser C, Gessner G. J. Pharmacol. Exp. Ther. 1976;197:623. [PubMed] [Google Scholar]
- 9.Wu Q, Criscione KR, Grunewald GL, McLeish MJ. Bioorg. Med. Chem. Lett. 2004;14:4217. doi: 10.1016/j.bmcl.2004.06.009. [DOI] [PubMed] [Google Scholar]
- 10.Grunewald GL, Romero FA, Chieu AD, Fincham KJ, Bhat SR, Criscione KR. Bioorg. Med. Chem. 2005;13:1261. doi: 10.1016/j.bmc.2004.11.015. [DOI] [PubMed] [Google Scholar]
- 11.Pendleton RG, Gessner G, Weiner G, Jenkins B, Sawyer J, Bondinell W, Intoccia A. J. Pharmacol. Exp. Ther. 1979;208:24. [PubMed] [Google Scholar]
- 12.Grunewald GL, Sall DJ, Monn JA. J. Med. Chem. 1988;31:433. doi: 10.1021/jm00397a029. [DOI] [PubMed] [Google Scholar]
- 13.Pendleton RG, Kaiser C, Gessner G, Finlay E, Green H. J. Pharmacol. Exp. Ther. 1974;190:551. [PubMed] [Google Scholar]
- 14.Wu Q, Gee CL, Lin F, Tyndall JD, Martin JL, Grunewald GL, McLeish MJ. J. Med. Chem. 2005;48:7243. doi: 10.1021/jm050568o. [DOI] [PubMed] [Google Scholar]
- 15.Martin JL, Begun J, McLeish MJ, Caine JM, Grunewald GL. Structure. 2001;9:977. doi: 10.1016/s0969-2126(01)00662-1. [DOI] [PubMed] [Google Scholar]
- 16.Bondinell WE, Chapin FW, Girard GR, Kaiser C, Krog AJ, Pavloff AM, Schwartz MS, Silvestri JS, Vaidya PD. J. Med. Chem. 1980;23:506. doi: 10.1021/jm00179a007. [DOI] [PubMed] [Google Scholar]
- 17.Fuller RW, Molloy BB, Day WA, Roush BW, Marsh MM. J. Med. Chem. 1973;16:101. doi: 10.1021/jm00260a003. [DOI] [PubMed] [Google Scholar]
- 18.Fuller RW, Mills J, Marsh MM. J. Med. Chem. 1971;14:322. doi: 10.1021/jm00286a012. [DOI] [PubMed] [Google Scholar]
- 19.Vecchietti V, Clarke GD, Colle R, Giardina G, Petrone G, Sbacchi M. J. Med. Chem. 1991;34:2624. doi: 10.1021/jm00112a042. [DOI] [PubMed] [Google Scholar]
- 20.McMillan FM, Archbold J, McLeish MJ, Caine JM, Criscione KR, Grunewald GL, Martin JL. J. Med. Chem. 2004;47:37. doi: 10.1021/jm0205752. [DOI] [PubMed] [Google Scholar]
- 21.Grunewald GL, Romero FA, Criscione KR. J. Med. Chem. 2005;48:1806. doi: 10.1021/jm049594x. [DOI] [PubMed] [Google Scholar]
- 22.Gee CLD, N, Tyndall JDA, Grunewald GL, Wu Q, McLeish MJ, Martin JL. J. Med. Chem. 2007;50 doi: 10.1021/jm0703385. in press. [DOI] [PubMed] [Google Scholar]
- 23.SYBYL® 7.0 Tripos, Inc. 1699 South Hanley Rd. St. Louis, MO 63144, USA: [Google Scholar]
- 24.Grunewald GL, Seim MR, Lu J, Makboul M, Criscione KR. J. Med. Chem. 2006;49:2939. doi: 10.1021/jm051262k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grunewald GL, Caldwell TM, Li QF, Criscione KR. J. Med. Chem. 1999;42:3315. doi: 10.1021/jm980734a. [DOI] [PubMed] [Google Scholar]
- 26.Grunewald GL, Lu J, Criscione KR, Okoro CO. Bioorg. Med. Chem. Lett. 2005;15:5319. doi: 10.1016/j.bmcl.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 27.Herz W. J. Am. Chem. Soc. 1951;73:351. [Google Scholar]
- 28.Gronowitz S, Sandberg E. Arkiv foer Kemi. 1970;32:217. [Google Scholar]
- 29.Eloy F, Deryckere A. Bull. Soc. Chim. Belg. 1970;79:415. [Google Scholar]
- 30.Maffrand JP, Eloy F. J. Heterocycl. Chem. 1976;13:1347. [Google Scholar]
- 31.Warm A. Heteocycles. 1992;34:2263. [Google Scholar]
- 32.Maffrand JP, Biogegrain R. Heteocycles. 1979;12:1479. [Google Scholar]
- 33.Satake K, Imai T, Kimura M, Morosawa S. Heteocycles. 1981;16:1271. [Google Scholar]
- 34.Cagniant P, Kirsch G. Comptes Rendus des Seances de l'Academie des Sciences, Serie C: Sciences Chimiques. 1975;281:35. [Google Scholar]
- 35.Matsumura T, Hirai Y, Nakamoto M, Sasaki T, Yokozuka A, Shinohara T. 86-146356 63002992. Patent JP. 1988:5.
- 36.Vedejs E, Lin S, Klapars A, Wang J. J. Am. Chem. Soc. 1996;118:9796. [Google Scholar]
- 37.Al-Awar RS, Joseph SP, Comins DL. Tetrahedron Lett. 1992;33:7635. [Google Scholar]
- 38.Vedejs E, Kongkittingam C. J. Org. Chem. 2000;65:2309. doi: 10.1021/jo9914115. [DOI] [PubMed] [Google Scholar]
- 39.Kikuchi C, Ando T, Fuji K, Okuno M, Satoh E, Shiiyama M, Ushiroda O, Koyama M, Hiranuma T. 9933804. Patent WO. 1999:139.
- 40.Amit B, Zehavi U, Patchornik A. J. Org. Chem. 1974;39:192. [Google Scholar]
- 41.Grishina GV, Potapov VM, Abdulganeeva SA, Gudasheva TA, Leshcheva IF, Sergeev NM, Kudryavtseva TA, Korchagina EY. Khim. Geterotsikl. Soedin. 1983:1693. [Google Scholar]
- 42.Grishina GV, Potapov VM, Gudasheva TA, Abdulganeeva SA. Khim. Geterotsikl. Soedin. 1985:1378. [Google Scholar]
- 43.Bose DS, Thurston DE. Tetrahedron Lett. 1990;31:6903. [Google Scholar]
- 44.Zimmer R, Reissig HU. J. Org. Chem. 1992;57:339. [Google Scholar]
- 45.Elmore SW, Cooper CS, Schultz CC, Hutchinson DK, Donner PL, Green BE, Anderson DD, Xie Q, Dinges J, Lynch LM. 2001032655. Patent WO. 2001:294.
- 46.Romero FA, Vodonick SM, Criscione KR, McLeish MJ, Grunewald GL. J. Med. Chem. 2004;47:4483. doi: 10.1021/jm0400653. [DOI] [PubMed] [Google Scholar]
- 47.Caine JM, Macreadie IG, Grunewald GL, McLeish MJ. Protein Expression Purif. 1996;8:160. doi: 10.1006/prep.1996.0088. [DOI] [PubMed] [Google Scholar]
- 48.Grunewald GL, Borchardt RT, Rafferty MF, Krass P. Mol. Pharmacol. 1981;20:377. [PubMed] [Google Scholar]
- 49.U’Prichard DC, Greenberg DA, Snyder SH. Mol. Pharmacol. 1977;13:454. [PubMed] [Google Scholar]
- 50.Grunewald GL, Romero FA, Seim MR, Criscione KR, Deupree JD, Spackman CC, Bylund DB. Bioorg. Med. Chem. Lett. 2005;15:1143. doi: 10.1016/j.bmcl.2004.12.013. [DOI] [PubMed] [Google Scholar]
- 51.Halczenko W, Stump CA. 9927929. Application: Patent WO. 1999:141.
- 52.Koike H, Asai F, Sugidachi A, Kimura T, Inoue T, Nishino S, Tsuzaki Y. 2077695. Patent CA. 1993:180.
- 53.These docking studies were performed using the crystal structure of hPNMT co-crystallized with SK&F 29661 (8). See Reference 7.
- 54.Gee CL, Tyndall JD, Grunewald GL, Wu Q, McLeish MJ, Martin JL. Biochemistry. 2005;44:16875. doi: 10.1021/bi051636b. [DOI] [PubMed] [Google Scholar]
- 55.McGaughey GB, Gagne M, Rappe AK. J. Biol. Chem. 1998;273:15458. doi: 10.1074/jbc.273.25.15458. [DOI] [PubMed] [Google Scholar]
- 56.Chelli R, Gervasio FL, Procacci P, Schettino V. J. Am. Chem. Soc. 2002;124:6133. doi: 10.1021/ja0121639. [DOI] [PubMed] [Google Scholar]
- 57.Barton TJ, Roth RW, Verkade JG. J. Am. Chem. Soc. 1972;94:8854. [Google Scholar]
- 58.Marino G. J. Heterocycl. Chem. 1972;9:817. [Google Scholar]
- 59.Grunewald GL, Caldwell TM, Li QF, Criscione KR. J. Med. Chem. 2001;44:2849. doi: 10.1021/jm010147g. [DOI] [PubMed] [Google Scholar]
- 60.Morris GM, Goodsell DS, Halliday RS, Hart WE, Belew RK, Olson AJ. J. Comput. Chem. 1998;19:1639. [Google Scholar]
- 61.Kraulis PJ. J. Appl. Crystallogr. 1991;24:945. [Google Scholar]
- 62.Merrit EA, Bacon DJ. Methods Enzymol. 1997;277:505. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- 63.Gasteiger J, Marsili M. Tetrahedron. 1980;36:3219. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.