

Abstract
Tight-seal whole-cell patch clamp experiments were performed to examine the ability of different intracellular Ca2+ mobilising agents to activate the Ca2+ release-activated Ca2+ current (ICRAC) in rat basophilic leukaemia (RBL-1) cells under conditions of weak cytoplasmic Ca2+ buffering.
Dialysis with a maximal concentration of inositol 1,4,5-trisphosphate (IP3) routinely failed to activate macroscopic ICRAC in low buffer (0.1 mM EGTA, BAPTA or dimethyl BAPTA), whereas it activated the current to its maximal extent in high buffer (10 mM EGTA). Dialysis with a poorly metabolisable analogue of IP3, with ionomycin, or with IP3 and ionomycin all failed to generate macroscopic ICRAC in low Ca2+ buffering conditions.
Dialysis with the sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) pump blocker thapsigargin was able to activate ICRAC even in the presence of low cytoplasmic Ca2+ buffering, albeit at a slow rate. Exposure to IP3 together with the SERCA blockers thapsigargin, thapsigargicin or cyclopiazonic acid rapidly activated ICRAC in low buffer.
Following activation of ICRAC by intracellular dialysis with IP3 and thapsigargin in low buffer, the current was very selective for Ca2+ (apparent KD of 1 mM). Sr2+ and Ba2+ were less effective charge carriers and Na+ was not conducted to any appreciable extent. The ionic selectivity of ICRAC was very similar in low or high intracellular Ca2+ buffer.
Fast Ca2+-dependent inactivation of ICRAC occurred at a similar rate and to a similar extent in low or high Ca2+ buffer. Ca2+-dependent inactivation is not the reason why macroscopic ICRAC cannot be seen under conditions of low cytoplasmic Ca2+ buffering.
ICRAC could be activated by combining IP3 with thapsigargin, even in the presence of 100 μM Ca2+ and the absence of any exogenous Ca2+ chelator, where ATP and glutamate represented the only Ca2+ buffers in the pipette solution.
Our results suggest that a threshold exists within the IP3-sensitive Ca2+ store, below which intraluminal Ca2+ needs to fall before ICRAC activates. Possible models to explain the results are discussed.
In rat basophilic leukaemia (RBL-1) cells, an experimental model for mucosal mast cells, calcium (Ca2+) influx is a central event in the secretion of inflammatory mediators (Ali et al. 1990; Kim et al. 1997). Activation of cell-surface receptors that couple to inositol 1,4,5-trisphosphate (IP3) production evokes a biphasic increase in intracellular Ca2+: an initial Ca2+ release phase is followed by a smaller but sustained Ca2+ influx component (Berridge, 1993). In RBL-1 cells, like other non-excitable cells, emptying of the intracellular Ca2+ stores activates a Ca2+ current called ICRAC (Ca2+ release-activated Ca2+ current; Hoth & Penner, 1992; Parekh & Penner, 1997). The relationship between IP3-evoked Ca2+ release and subsequent activation of ICRAC is complex. A partial dissociation between Ca2+ release and store-operated Ca2+ influx has been found in several cells including RBL-1 cells (Parekh et al. 1997; Hartmann & Verkhratsky, 1998; Liu et al. 1998). Despite its importance, the mechanisms underlying this widespread phenomenon are not known.
When ICRAC is studied under conditons of physiological cytoplasmic Ca2+ buffering and IP3 is used to deplete the intracellular stores, the current is not detectable, although intracellular fluorescent dyes reveal modest activation of Ca2+ influx following IP3 elevation (Parekh et al. 1997; Huang et al. 1998). It is widely accepted that the stores are fully depleted under these conditions, and the inability to record any macroscopic ICRAC reflects Ca2+-dependent negative feedback mechanisms that maintain very low channel activity (Huang et al. 1998). Our results demonstrate that this explanation is not correct and instead provide evidence that Ca2+ release in response to a maximal concentration of IP3 is unable to deplete the stores sufficiently to activate macroscopic ICRAC. Instead, a threshold exists below which intraluminal Ca2+ has to fall before ICRAC activates. The results also provide a mechanistic explanation for selective activation of Ca2+-dependent processes including Ca2+ influx-dependent exocytosis in these cells.
METHODS
Rat basophilic leukaemia cells (RBL-1) cells, which were bought from the American Type Culture Collection, were cultured as previously described (Fierro & Parekh, 1999a,b).
Patch-clamp experiments were conducted in the tight-seal whole-cell configuration at room temperature (18–25°C) as previously described (Hamill et al. 1981; Fierro & Parekh, 1999a). Sylgard-coated, fire-polished patch pipettes had DC resistances of 2.5-4 MΩ when filled with standard internal solution containing (mM): caesium glutamate 145, NaCl 8, MgCl2 1, Hepes 10 (pH 7.2 with CsOH). Depending on the experiment (described in the text), the Ca2+ chelators EGTA (Sigma), BAPTA tetracaesium salt (BAPTA) or dimethyl BAPTA tetrapotassium salt (both from Molecular Probes) were added to this solution at the specified concentrations, as sometimes was 30 μm IP3, 2 μM thapsigargin (from three independent sources: Sigma, Calbiochem, Alomone Labs), 100 μM cyclopiazonic acid, or 2 μM thapsigargicin (Calbiochem). All chemicals were purchased from Sigma unless otherwise noted. A correction of +10 mV was applied for the subsequent liquid junction potential that arose from this glutamate-based internal solution. Extracellular solution contained (mM): NaCl 145, KCl 2.8, CaCl2 10, MgCl2 2, CsCl 10, glucose 10, Hepes 10 (pH 7.4 with NaOH). In some experiments, external Ca2+ was simply lowered to the desired concentration. High resolution current recordings were acquired and ICRAC was measured as described previously. Data are presented as means ±s.e.m., and statistical evaluation was carried out using Student's unpaired t test.
RESULTS
IP3 activates ICRAC in high but not low intracellular Ca2+ buffer
Figure 1a compares the effect of dialysing individual RBL-1 cells with either a low or high concentration of EGTA (0.1 or 10 mM) together with a maximal concentration of IP3 (30 μM) in the patch pipette. Most cells failed to generate any detectable ICRAC in low Ca2+ buffer (IP3+ 0.1 mM EGTA/BAPTA, panel A1 of Fig. 1a and Table 1; we can detect with confidence a macroscopic current of around −0.2 pA pF−1 at a bandwidth of 2.3 kHz) whereas a macroscopic current was routinely activated in the presence of high EGTA (panel A1 of Fig. 1a). Because IP3 can be converted to IP4 in a Ca2+-dependent manner (Shears, 1992), we dialysed cells instead with IP3-F (a poorly metabolisable analogue, Kozikowski et al. 1990) together with 0.1 mM EGTA. ICRAC still routinely failed to be detected (panel A3 of Fig. 1A; Table 1). Dialysis with the Ca2+ ionophore ionomycin (10–15 μM) in 0.1 mM EGTA, alone or together with IP3, also failed to activate any macroscopic current (Fig. 1a panels A4 and A5; Table 1). These results are somewhat unexpected, because numerous previous studies using fluorescent dyes have shown that IP3 substantially reduces the Ca2+ content of the intracellular IP3-sensitive Ca2+ stores in RBL-1 cells (Ali et al. 1995; Parekh et al. 1997; Huang et al. 1998). One would have therefore expected ICRAC to fully activate following exposure to IP3 in low Ca2+ buffer. Because the fluorescence measurements were all done using external Ca2+ concentrations around 2 mM, whereas the ICRAC recordings are carried out in 10 mM Ca2+ (which gives the maximum current), we speculated that it might be harder for IP3 to deplete the stores in 10 mM Ca2+ due to their potentially greater loading. However, IP3 was still unable to activate a detectable ICRAC in low Ca2+ buffering conditions when external Ca2+ was reduced to 2 mM (Table 1).
Figure 1. Block of the SERCA pumps rather than IP3 (IP3)-evoked Ca2+ release is sufficient to observe macroscopic ICRAC under physiological conditions of low cytoplasmic Ca2+ buffering.
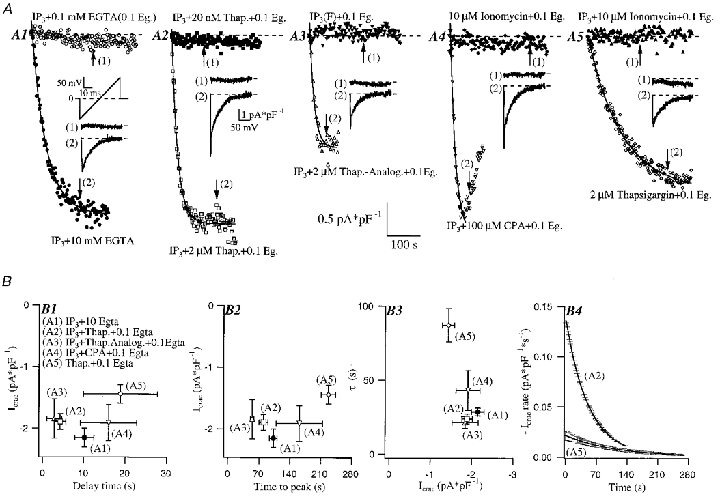
A, the effect of dialysing individual cells with either 0.1 mM (low buffer) or 10 mM EGTA (high buffer) together with 30 μM IP3 and 2 mM ATP is shown. ICRAC amplitude (measured at −80 mV from voltage ramps from −100 to + 100 mV over 50 ms from a holding potential of 0 mV) is plotted against time after the onset of whole-cell recording. Whilst most of the cells dialysed with low buffer and IP3 (A1), IP3-F (A3), ionomycin (A4) or IP3 and ionomycin (A5) failed to generate any detectable ICRAC, the current was routinely activated in the presence of the SERCA pump blocker thapsigargin alone (A5). Dialysis with IP3-F and 10 mM EGTA activated ICRAC rapidly and to the maximal extent and the current was indistinguishable from that seen with IP3 and 10 mM EGTA (data not shown). IP3 interacted synergistically with the structurally distinct SERCA blockers thapsigargin (A2), thapsigargicin (A3) and cyclopiazonic acid (CPA) (A4) to generate ICRAC. The effect of SERCA pump blockers was dose dependent, as is shown in A2. All traces where macroscopic ICRAC is detected are fitted by a single exponential function. Insets show the I–V relationship of the current for each condition. The arrows in the plot of ICRAC development against time indicate where the inset I–V relationships were taken. B, ICRAC amplitude is plotted as a function of delay (B 1), time to peak (B2) and the time constant (τ) of the exponential fits (B3). The different conditions are stated in panel B 1. Each point represents mean ±s.e.m. of more than five cells. Delay was not significantly different between the conditions (P > 0.05). Time to peak and τ were significantly different depending whether (A1-A4) or not (A5) IP3 was present in the dialysing solution (P < 0.0001). In panel B 4, the rate of current development is plotted as a function of time for the conditions where ICRAC was activated by IP3 and thapsigargin (A2, n = 16) or thapsigargin alone (A5, n = 6). The first derivative of the exponential fit for each trace in either condition was calculated, multiplied by −1 and then pooled to calculate mean ±s.d. of the ICRAC rate.
Table 1.
A summary of the several different conditions involving IP3evoked Ca2+ release and/or ionomycin that were used to activate ICRAC is presented
| Condition | + (pA pF−1) | n | — (%) | n | |
|---|---|---|---|---|---|
| (1) | 30 μm IP3−10 mm [Ca2+]o | −1.4 | 1 | 83 | 5/6 |
| (2) | 30 μmIP3+ 2 mm ATP−10 mm [Ca2+]o | −1.1 | 1 | 93 | 14/15 |
| (3) | 30 μm IP3+ 2 mm ATP−2 mm [Ca2+]o | — | — | 100 | 5/5 |
| (4) | 30 μm IP3-F+ 2 mm ATP−10 mm [Ca2+]o | — | — | 100 | 9/9 |
| (5) | 30 μm IP3+ 2 mm ATP + 100 μm [Ca2+]i−10 mm [Ca2+]o | — | — | 100 | 6/6 |
| (6) | 30 μm IP3+ 2 mm ATP + 10 μm ionomycin−10 mm [Ca2+]o | −1.2 | 1 | 89 | 8/9 |
| (7) | 30 μm IP3+ 2 mm ATP + 20 nm thapsigargin−10 mm [Ca2+]o | — | — | 100 | 7/7 |
| (8) | 30 μm IP3+ 2 mm ATP + 10 mm EGTA−10 mm [Ca2+]o | −2.19 ± 0.13 | 16 | 11 | 2/18 |
| (9) | 30 μm IP3+ 2 mm ATP + 2 μm thapsigargin−10 mm [Ca2+]o | −1.96 ± 0.23 | 12 | 14 | 2/14 |
| (10) | 10 μm ionomycin + 2 mm ATP−10 mm [Ca2+]o | −0.9 | 1 | 89 | 8/9 |
For all conditions 0.1 mm EGTA or BAPTA was used unless otherwise is stated. ‘+’ indicates that macroscopic currents could be detected and the corresponding mean ± s.e.m. values (wherever possible) and number of cells (n) are indicated. ‘–’ means that no macroscopic current was detected. The percentage (%) of failures and number of cells (n) are indicated. [Ca2+]o indicates extracellular calcium. No Ca2+ chelator was used in condition (5).
Block of SERCA pumps activates ICRAC in low Ca2+ buffer
We considered two explanations for this inability of IP3-evoked Ca2+ release to activate detectable ICRAC despite substantial store depletion. First, the current activates only after the IP3-sensitive stores have been depleted below a certain level, and a sizeable macroscopic current subsequently requires further and rather extensive emptying of the stores or, second, the current rapidly inactivates due to Ca2+-dependent negative feedback mechanisms in the presence of the low Ca2+ chelator concentration in the cytoplasm.
If the first hypothesis is correct, then it predicts that sarco/endoplasmic reticulum Ca2+-ATPases (SERCA)-type blockers (which can, in contrast to IP3, fully empty IP3-sensitive stores in many cells; Tse et al. 1994; Chatton et al. 1995; Montero et al. 1995, 1997) should be able to activate macroscopic ICRAC in physiological cytoplasmic Ca2+ buffering. We therefore dialysed cells with 2 μM thapsigargin, a SERCA pump blocker (Thastrup et al. 1990), and 0.1 mM EGTA. Whereas IP3 and ionomycin had failed to activate ICRAC, we found that 2 μM thapsigargin alone was able to activate a sizeable inward Ca2+ current (Fig. 1a, panel A5). The current developed slowly, but was clearly ICRAC from its biophysical features (ramp inset in panel A5 of Fig. 1A; reversal potential > +50 mV, inward rectification, Ca2+ selective, see below). Addition of IP3 to the 2 μM thapsigargin-0.1 mM EGTA solution generated ICRAC (Fig. 1a panel A2) after a similar delay to that seen with thapsigargin alone. This delay was 9.84 ± 2.23 s (n = 62, data pooled for all cells analysed in Fig. 1B, panel B 1, because there was no difference between them, P > 0.05). The panels B 2-B 4 of Fig. 1B show the striking difference in the rate of development of the current for IP3 and thapsigargin together compared with thapsigargin alone (time to peaks were 92.9 ± 8.5; n = 49, data pooled from all cells in Fig. 1B, panels B 2 and B 3, that were dialysed with IP3 because there was no significant difference between them) vs. 227.85 ± 16.1 (n = 13); activation time constants (τ) were 27.1 ± 2.4 and 87.2 ± 11.3 s (n = 8), respectively (P < 0.0001 for both cases; Fig. 1B, panels B 2 and B 3). Although the current developed much more slowly in thapsigargin alone (panel B 4), its overall extent was not significantly different from that seen in IP3 and thapsigargin (P > 0.05; Fig. 1B). Replacing 0.1 mM EGTA with dimethyl BAPTA or BAPTA did not affect the size or rate of development of the current in response to IP3 and thapsigargin (data not shown).
We could mimic the effects of thapsigargin by using the different SERCA pump blockers cyclopiazonic acid (Inesi & Sagra, 1994; 7/7 cells; Fig. 1a, panel A4) and thapsigargicin (which is almost as potent as thapsigargin but less hydrophobic; Fig. 1a, panel A3). Furthermore, the effect of thapsigargin was concentration dependent because dialysis with IP3 and 20 nM thapsigargin routinely failed to activate a detectable current (Fig. 1a, panel A2; Table 1). By blocking SERCA pumps, thapsigargin will raise intracellular Ca2+ in the presence of a low Ca2+ buffer. However, intracellular dialysis with IP3 and approximately 1 μM free Ca2+ (only exogenous buffers present were 3.3 mM Mg-ATP and 145 mM glutamate in the presence of 100 μM CaCl2 and 1 mM MgCl2) failed to mimic the effect of IP3 and thapsigargin (Table 1, condition (5), 6/6 cells). Block of the SERCA pumps is therefore sufficient for macroscopic ICRAC to be recorded under conditions of physiological Ca2+ buffering.
Comparison of the ionic conductivity profile of ICRAC in low and high intracellular Ca2+ buffer
We next compared the conductivity of ICRAC to different external divalent and monovalent cations in the presence of either high or low cytoplasmic Ca2+ buffering. Figure 2a compares the effects of changing external Ca2+ on the size of ICRAC for the combination of IP3 and thapsigargin in low Ca2+ buffer (0.1 mM EGTA or dimethyl BAPTA) with that seen in high Ca2+ buffer (10 mM EGTA). Reducing external Ca2+ reduced the size of the current for all conditions equally. ICRAC had quite a high affinity for Ca2+ with apparent KD values of 0.84, 1.17 and 1.03 mM for IP3 and 10 mM EGTA, IP3 and thapsigargin in 0.1 mM EGTA, and IP3 and thapsigargin in 0.1 mM dimethyl BAPTA, respectively. These values are very similar and indicate that the current has the same external Ca2+ dependence for the different buffering conditions. The high affinity for Ca2+ in RBL-1 cells is different from a previous report in mast cells and markedly distinct from that in Xenopus oocytes (apparent KD values of 3.3 and 11.5 mM, respectively; Hoth & Penner, 1993; Yao & Tsien, 1997). ICRAC in RBL-1 cells is permeable to Ba2+ and Sr2+, although to lesser extents than Ca2+ (Fierro & Parekh, 1999a). As expected, the current activated by the combination of IP3 and thapsigargin in low cytosolic Ca2+ buffering conditions was also less permeable to Ba2+ and Sr2+ and the conductivity profile for both low and high Ca2+ buffering was Ca2+ > Sr2+≥ Ba2+ (Fig. 2B). We did find that Ba2+ was conducted better than we have previously found for high Ca2+ buffering conditions (Fierro & Parekh, 1999a), although still less so than Ca2+. Furthermore, the current carried by Ba2+ had a more negative reversal potential and approached zero at ≤−35 mV (see ramp in Fig. 2B). This may be related to an anomalous mole fraction effect (Hoth, 1995). There was a tendency for the Sr2+ and Ba2+ currents (relative to Ca2+) to be smaller in low rather than high Ca2+ buffer (Fig. 2B), which might indicate that the channel selectivity depends on the intracellular Ca2+ buffering, as previously noted (Zhang & McCloskey, 1995). However, to address this issue thoroughly will require detailed single-channel recordings, which are not feasible yet for the CRAC channel.
Figure 2. ICRAC activated in the presence of either high or low cytoplasmic Ca2+ buffering is highly selective for Ca2+ ions.
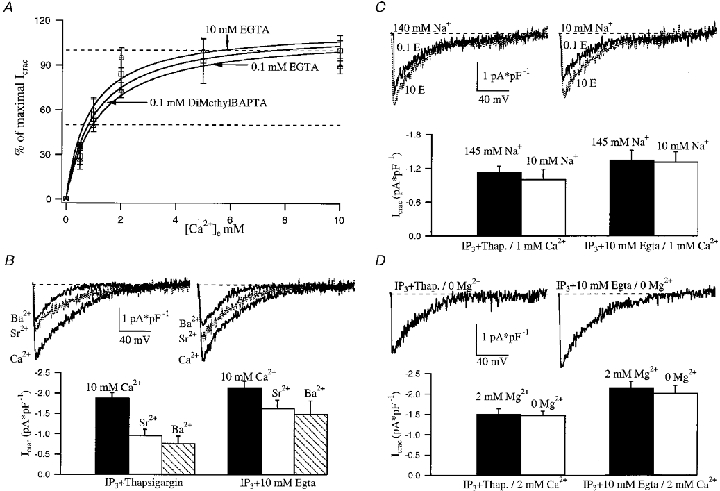
A, effect of extracellular Ca2+ concentration on ICRAC amplitude. Each point represents the mean ±s.e.m. of more than six cells. Data for the three different conditions were fitted with a Michaelis-Menten type equation and a KD of around 1 mM external Ca2+ was obtained for each condition (see text). Cells were directly placed in an external solution containing the desired Ca2+ concentration from the onset. B, ICRAC profile in high and low cytoplasmic Ca2+ buffering is compared for cells bathed with three different extracellular divalent cations (Ca2+, Sr2+, Ba2+). I–V relationships are shown in the upper panel and averaged data below as a histogram. Each bar represents the mean ±s.e.m. of more than seven cells. C, reducing external Na+ 14.5-fold failed to alter the I–V relationship and the size of ICRAC for both high and low cytoplasmic Ca2+ buffering. Each bar represents the mean ±s.e.m. of more than five cells. D, lowering external Mg2+ from 2 mM to 0 (nominally Mg2+ free) did not affect the size of the current (in 2 mM Ca2+) nor its I–V relationship for both Ca2+ buffering conditions. Each bar represents the mean ±s.e.m. of more than four cells.
We then examined whether CRAC channels were permeable to Na+ in the presence of 1 mM external Ca2+ under both low and high Ca2+ buffer conditions. Reducing external Na+ 14.5-fold failed to alter the I–V relationship and the size of the current for either condition (Fig. 2C). Hence ICRAC is very selective for Ca2+, even under physiological Ca2+ buffering conditions. We also tested whether external Mg2+ could affect Ca2+ permeability through ICRAC under the conditions of both low cytosolic Ca2+ buffering and physiological external Ca2+ concentrations. Lowering external Mg2+ from 2 mM to 0 (nominally Mg2+ free) did not affect the size of the current (in 2 mM Ca2+) nor its I–V relationship for both low and high Ca2+ buffering conditions (Fig. 2D). Taken together, the conductivity results demonstrate that ICRAC activated by the combination of IP3 and thapsigargin in low cytoplasmic Ca2+ buffer is very similar to the current seen in the presence of high Ca2+ buffer, and therefore the two are one and the same. The divalent and monovalent cation conductivity profile of ICRAC in native RBL-1 cells is strikingly similar to the profile seen for the recombinant mammalian trp homologue bCCE1 expressed in Chinese hamster ovary cells under conditions of high Ca2+ buffering (Warnat et al. 1999). ICRAC could be related therefore to bCCE1 and the latter might contribute to the CRAC channel pore.
Ca2+-dependent inactivation does not affect ICRAC development in low Ca2+ buffer
Although we have provided evidence to support our first explanation, namely that a threshold exists before ICRAC can activate, we set about testing the currently accepted view that Ca2+-dependent inactivation prevents macroscopic ICRAC from developing in low Ca2+ buffer. ICRAC is subjected to Ca2+-dependent inactivation. A fast process which operates on a millisecond time scale (Hoth & Penner, 1993; Zweifach & Lewis, 1995a; Fierro & Parekh, 1999a) and a slower inactivation occurring over tens of seconds/minutes (Zweifach & Lewis, 1995b; Parekh, 1998) both reduce CRAC channel activity. Fast inactivation is widely believed to account for the inability to measure macroscopic ICRAC under physiological cytosolic Ca2+ buffering during whole-cell recordings (Huang et al. 1998). However, this is an unlikely explanation for several reasons. First, fast inactivation only occurs in RBL-1 cells at potentials negative to −40 mV and, at −80 mV, it reduces the current by less than 40 %; fast inactivation develops with time constants in the range of 10 and 120 ms and recovers with time constants of 34 and 233 s (Fierro & Parekh, 1999a). When the membrane potential is clamped at 0 mV and voltage ramps spanning −100 to +100 mV over 50 ms are applied every 2 s, fast inactivation would therefore not be present during the initial negative portion of the ramp, and hence the CRAC channels should be functional and thus give rise to a clear current. Second, we have found that fast inactivation is reduced when Sr2+ replaces Ca2+ as the charge carrier (Fierro & Parekh, 1999a). However, even with Sr2+ as the charge carrier instead of Ca2+, we generally failed to record a detectable ICRAC in response to IP3 and 0.1 mM EGTA (only 3 of 13 cells responded and these gave small ICRAC; data not shown). Third, we clamped cells at +50 mV (to reduce the driving force for Ca2+ influx) and administered voltage ramps both at a low frequency (1 ramp per 4 s instead of 2 s) and under conditions where less Ca2+ influx would occur during the ramps (now spanning −80 to +80 mV over 20 ms instead of −100 to +100 mV over 50 ms). However, IP3 still routinely failed to activate ICRAC in 0.1 mM EGTA (7/9 cells; data not shown). Taken together, the absence of macroscopic ICRAC in the presence of 0.1 mM chelator and IP3 is unlikely to be due solely to fast inactivation. In Fig. 3a we compared the rate and extent of fast inactivation for ICRAC following activation by IP3 and thapsigargin in 0.1 mM EGTA with that for IP3 and 10 mM EGTA for 2 and 10 mM external Ca2+. The fast and slow rates, as well as the extent of inactivation, were not significantly different for each external Ca2+ concentration for low or high Ca2+ buffer. The extent of inactivation was less in the lower external Ca2+ concentration, in agreement with a previous report from T-cells (Zweifach & Lewis, 1995a). The fact that fast inactivation is the same under the two conditions provides additional evidence that, first, the inability to measure macroscopic ICRAC induced in response to IP3 in low Ca2+ buffer is unlikely to be due to this inactivation mechanism and, second, the channels underlying the current under the two conditions are the same.
Figure 3. Ca2+-dependent negative feedback mechanisms do not prevent macroscopic activation of ICRAC in physiological cytoplasmic Ca2+ buffering conditons.
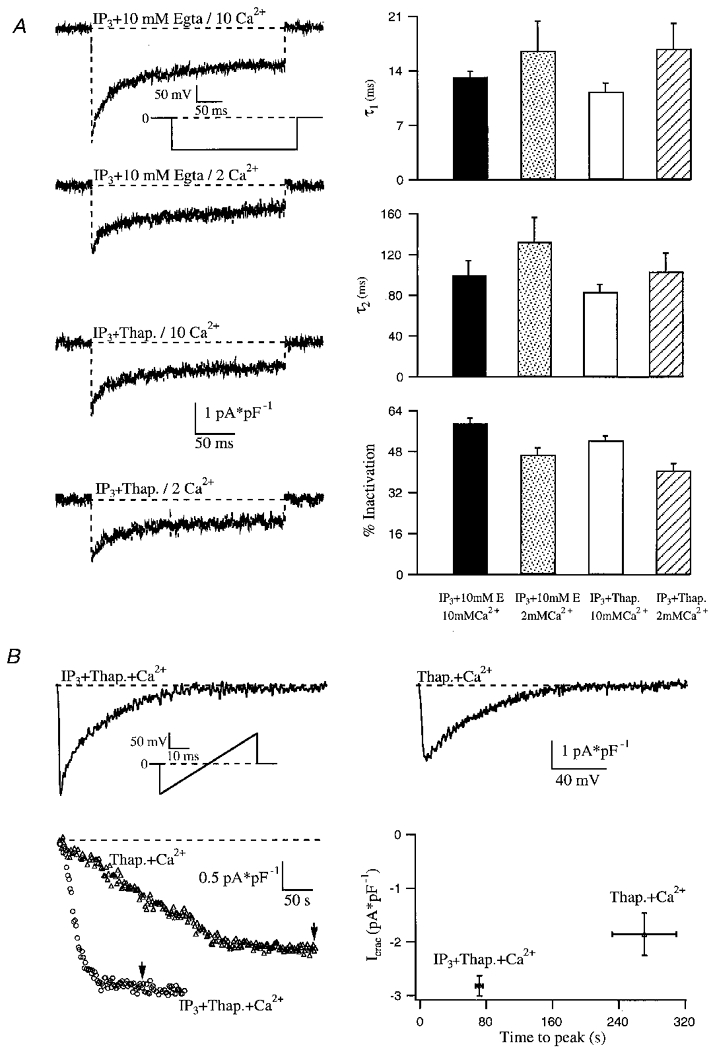
A, Ca2+-dependent fast inactivation of ICRAC is compared under high and low cytoplasmic Ca2+ buffering conditions. The inactivation phase of each trace could be fitted with a double exponential function (not shown). Average time constants from these fits and the extent of inactivation for each condition are shown in the right panel. Each bar represents the mean ±s.e.m. of more than seven cells. B, effects of dialysing cells with 100 μM Ca2+ and no added Ca2+ chelator on ICRAC. ICRAC was activated by inclusion of either IP3 and thapsigargin (left ramp) or thapsigargin alone (right ramp) in the pipette solution. Ca2+ would be weakly buffered by ATP (3 mM total Mg2+), glutamate and the endogenous cytoplasmic Ca2+ buffers. Each point represents the mean ±s.e.m. of more than five cells. Current amplitude (P < 0.05) and time to peak (P < 0.0001) were significantly different for these two groups of cells.
Slow inactivation develops over a time course of several tens of seconds and should not prevent macroscopic development of the current. Furthermore, the combination of IP3 and thapsigargin would raise intracellular Ca2+ to a higher level than IP3 alone and maintain it for longer at an elevated level, yet the current still developed. This argues against activation of Ca2+-dependent inhibitory feedback mechanisms that prevent ICRAC from being measured under low Ca2+ buffering conditions. This also provides evidence against an inhibitory action of IP3 itself or a metabolite thereof, since one would expect such an action to also function in the presence of thapsigargin. In Fig. 3B we examined the intracellular Ca2+ dependence of ICRAC activation. The upper panel shows the current in response to a voltage ramp following dialysis with IP3, thapsigargin, 100 μM Ca2+ and no added Ca2+ chelator. Ca2+ was weakly buffered only, by the ATP and glutamate in the recording pipette. Astonishingly, the current could be activated at the normal rate (Fig. 3B, lower right panel). Dialysis with thapsigargin and 100 μM Ca2+ (in the absence of IP3) also activated ICRAC, with the typical slow kinetics expected for thapsigargin alone. Although we do not know exactly what the free Ca2+ concentration is in the cells under these conditions (due to endogenous Ca2+ buffering/removal), in these latter experiments the slow development of ICRAC would ensure that reasonable dialysis with micromolar Ca2+ concentrations would have occurred as ICRAC activated. Again, both the final extent of the current as well as the time to peak were similar to that seen with thapsigargin and 0.1 mM chelator (compare graphs in Figs 1B and 3B). This unexpected result challenges current views that ICRAC cannot activate when cytosolic Ca2+ is high and that intracellular Ca2+ buffers are required for the current to activate.
DISCUSSION
Our results suggest that partial depletion of the IP3-sensitive stores is not sufficient to activate ICRAC to a detectable level in whole-cell recording. Only when stores are substantially depleted of Ca2+, as seen with the SERCA blockers, can the macroscopic current develop. Although SERCA block alone can activate ICRAC, IP3 accelerates the rate at which the current develops. At first glance, one might be surprised that IP3 or ionomycin is unable to fully deplete the stores whereas SERCA blockers can. Recently, we have found that the SERCA pumps in RBL-1 cells are able to operate efficiently when cytosolic Ca2+ is heavily clamped at < 10 nM, even in the absence of ATP from the pipette solution (Fierro & Parekh, 1999b). In fact, SERCA pumps could fully prevent ICRAC from activating in the presence of both a significant passive leak of Ca2+ from the stores and a cytosolic Ca2+ binding ratio of 6000. Under conditions of low cytosolic Ca2+ buffering, SERCA pump activity will be even higher, both because of the elevated cytosolic Ca2+ following IP3-evoked Ca2+ release and the fact that a fall in luminal Ca2+ strongly stimulates pump activity (Favre et al. 1996; Mogami et al. 1998). In addition, both cytosolic and luminal Ca2+ have been reported to reduce the Ca2+ flux through IP3-gated channels that span the stores, thereby enabling the pumps to partially reload the stores (Berridge, 1993). However, our results are not compatible with the notion that Ca2+ influx through CRAC channels causes sizeable inhibition of the IP3 receptor, thereby favouring store refilling, for two reasons. First, manoeuvres that reduced the extent of Ca2+ influx (clamping at +50 mV, reduced duration and size of voltage ramps) still failed to enable IP3 to activate ICRAC in low Ca2+ buffer. Second, although Sr2+ is an effective charge carrier through CRAC channels (Fierro & Parekh, 1999a), it is at least two orders of magnitude less effective than Ca2+ in inducing inhibition of the IP3 receptor (Hannaert-Merah et al. 1995). One might therefore have expected ICRAC to activate substantially in low Ca2+ buffer in the presence of external Sr2+. However, we routinely failed to detect any macroscopic current under these conditions.
Previous studies have shown that low concentrations of IP3 can partially empty Ca2+ stores yet fail to activate any Ca2+ influx (Parekh et al. 1997; Hartmann & Verkhratsky, 1998; Liu et al. 1998). It has been suggested that either functionally distinct IP3-sensitive stores exist in RBL-1 cells and that the one with lower sensitivity for IP3 activates ICRAC, or there may be one homogeneous IP3-sensitive store, but partial depletion does not partially activate ICRAC (Parekh et al. 1997). Our results using a high dose of IP3 (which would not discriminate between the various IP3-sensitive stores) extends these studies and provides an explanation for this widespread phenomenon. Intraluminal Ca2+ must fall below a certain level in the specific IP3-sensitive store that controls ICRAC before the current can activate. Only when SERCA pumps are blocked is this threshold readily overcome. Our results suggest that ICRAC activation probably requires quite a substantial depletion of the stores because the combination of IP3 and 20 nM thapsigargin failed to generate a macroscopic current, despite this concentration of thapsigargin evoking Ca2+ release in fura 2-AM-loaded cells. Two models that could account for our results are shown in Fig. 4 (see legend for details).
Figure 4. Two models to explain how the threshold can account for the inability of IP3 to activate macroscopic ICRAC in low Ca2+ buffering, and the effectiveness of SERCA blockers under the same conditions.
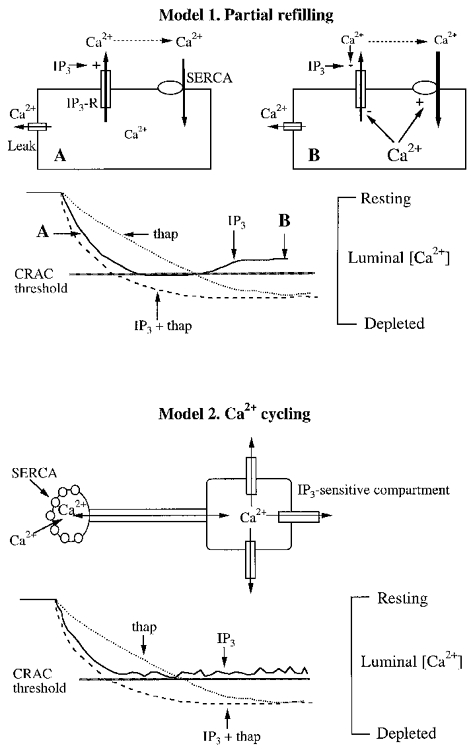
For both models, the rate and extent of store depletion are arbitrary and not based on empirical data. Model 1, 30 μM IP3 is initially quite effective in depleting the stores, but the negative feedback mechanisms start to operate and curtail Ca2+ efflux through the IP3-gated channel. Simultaneously, SERCA pump activity increases. The combination of reduced Ca2+ efflux and enhanced Ca2+ uptake ensure that the threshold is not crossed. Negative feedback mechanisms on the IP3 receptor might include, alone or in combination, the potential effects of a fall in luminal Ca2+ (Berridge, 1993), inhibition by cytosolic Ca2+ directly (Berridge, 1993), or via calmodulin (Missiaen et al. 1999) and inactivation by IP3 itself (Hajnockzy & Thomas, 1994). Such feedback mechanisms might not operate when the potent IP3 receptor agonist adenophostin A is used to deplete the stores (Huang et al. 1998). SERCA pump activity would be enhanced by both the fall in luminal Ca2+ (Favre et al. 1996; Mogami et al. 1998) and the increase in cytosolic Ca2+ in low Ca2+ buffer (see text). Model 2, this model differs from model 1 in that no inhibitory mechanisms are necessary to reduce Ca2+ efflux in response to a supramaximal concentration of IP3. Instead, Ca2+ uptake and release mechanisms are spatially separate. IP3 effectively depletes its own compartment, but a standing Ca2+ gradient exists whereby Ca2+, taken up by the pumps, diffuses to the release sites and then enters the cytosol. The threshold here would reflect to a large extent the Ca2+ in the region of the uptake sites. Only when this falls below a certain level will ICRAC activate. In this scheme, CRAC activation will be determined to a greater extent by the Ca2+ content in the uptake sites. A recent study using differential centrifugation in RBL cells reported that IP3 receptors were not uniformly distributed in the endoplasmic reticulum but were localised instead to certain regions (Vanlingen et al. 1997).
Our conclusions differ from those of a previous study in which intraluminal Ca2+ was measured directly using mag-fura (Hofer et al. 1998). It was reported that Ca2+ influx was tightly coupled to the extent of store depletion and no apparent threshold was observed. However, such experiments require that cells are dialysed for several minutes in order to remove non-compartmentalised dye from the cytosol and with a solution in which Ca2+ is strongly buffered close to 100 nM with 10 mM BAPTA, so that ICRAC does not spontaneously activate. Under these conditions, because Ca2+ is continuously cycling across the stores membrane, as suggested by our previous results (Fierro & Parekh, 1999b), then the amount of Ca2+ that is taken back up into the stores by SERCA pumps will be less than in conditions where no external added buffer is present. Assuming a constant Ca2+ leak from the stores (Mogami et al. 1998), then the latter would be slowly but continuously drained of their Ca2+. Over a time course of several minutes, one might expect that the stores content would be reduced to a level close to the threshold where ICRAC activates, so that it would not have been observed. Furthermore, substantial dye loading is required to obtain a resolvable signal from the small Ca2+ organelles and it is likely that the strong intraluminal Ca2+ buffering introduced by the dye also lowered intraluminal Ca2+ close to the threshold.
Our results challenge contemporary views on Ca2+ signalling and have implications for both the physiological role of ICRAC and its activation mechanism. First, the fact that ICRAC can be measured in 1–2 mM external Ca2+ and in the presence of low cytosolic Ca2+ buffering convincingly demonstrates that the CRAC channels are fully functional under physiological conditions and therefore contribute directly to Ca2+-dependent processes. Second, contrary to what is widely thought, the inability of IP3 to activate macroscopic ICRAC in low Ca2+ buffer is not due to Ca2+-dependent feedback mechanisms operating on the current. Instead it seems to reflect incomplete store depletion after Ca2+ release evoked by a maximal concentration of IP3. Third, our characterisation of the ionic selectivity of the CRAC channels under physiological conditions reinforces the notion that ICRAC is highly selective for Ca2+ and the results will be helpful in identifying putative CRAC channel clones. Finally, the threshold phenomenon endows ICRAC with a unique ability to regulate a broad spectrum of Ca2+-dependent processes with different affinities/spatial distributions. Small Ca2+ release events (or larger events at low frequency) fail to reach the threshold and do not activate store-operated Ca2+ influx at all (Parekh et al. 1997). Processes that require only Ca2+ release can therefore be activated separately from those that require Ca2+ entry. Further Ca2+ release that then reaches the threshold activates a small amount of Ca2+ influx through ICRAC and this underlies the plateau seen in most non-excitable cells following receptor stimulation. Changes in membrane potential will play a particularly important role during this Ca2+ plateau by determining the rate and extent of Ca2+ influx through CRAC channels, especially since the latter's I–V relationship is inwardly rectifying (Hoth & Penner, 1992; Parekh & Penner, 1997). The plateau will be able to activate processes with moderate affinity for Ca2+, as well as raising the possibility of creating spatial Ca2+ gradients from the cell periphery. More extensive stimulation (high frequency of large amplitude Ca2+ spikes) will gradually activate ICRAC further (since the released Ca2+ will be extruded across the plasma membrane as well as resequestrated into other stores) and this will result in a large and sustained global Ca2+ increase and hence activation of processes with a low affinity for Ca2+ such as exocytosis. Antigen stimulation generates initially large cytosolic sinusoidal Ca2+ oscillations which develop into a plateau reflecting Ca2+ entry. Exocytosis starts only when the latter occurs (Ali et al. 1990; Kim et al. 1997). Presumably, the initial oscillations are not sufficient to exceed the threshold for significant ICRAC activation. In contrast, thapsigargin generates a plateau quickly, and robust secretion occurs almost immediately (Kim et al. 1997). This model also explains why IP3 alone was unable to evoke capacitance increases in RBL-1 cells (indicative of vesicular fusion) in low Ca2+ buffer whereas the combination of IP3 and thaspigargin was very potent (Artalejo et al. 1998).
Our results, on the other hand, indicate that SERCA pump activity appears to be one of the main determinants of whether or not ICRAC activates under physiological conditions. Therefore they also raise the intriguing possibility that hormones that do not couple to the phosphatidylinositol pathway may still activate ICRAC by inhibiting the pump, for example by protein phosphorylation. The SERCA pumps may therefore represent a means for stimulating Ca2+ influx without IP3-mediated Ca2+ release.
Acknowledgments
This work was supported by a Wellcome Trust Grant (No. 049236/Z/96/Z) to A.B.P. and the Royal Society. A.B.P. is the E.P. Abraham Research Fellow at Keble College, Oxford. We thank Dr Maike Glitsch for comment on the manuscript.
References
- Ali H, Cunha-Melo JR, Saul WF, Beaven MA. Activation of phospholipase C via adenosine receptors provides synergistic signals for secretion in antigen-stimulated RBL-2H3 cells. Journal of Biological Chemistry. 1990;265:745–753. [PubMed] [Google Scholar]
- Ali H, Maeyama K, Sagi-Eisenberg R, Beaven MA. Antigen and thapsigargin promote Ca2+ influx in rat basophilic leukemia cells by ostensibly similar mechanisms that allow filling of inositol 1,4,5-trisphosphate-sensitive and mitochondrial Ca2+ stores. Biochemical Journal. 1995;304:431–440. doi: 10.1042/bj3040431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artalejo AR, Ellory JC, Parekh AB. Ca2+-dependent capacitance increases in rat basophilic leukemia cells following activation of store-operated Ca2+ entry and dialysis with high Ca2+-containing intracellular solution. Pflügers Archiv. 1998;436:934–939. doi: 10.1007/pl00008088. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Chatton JY, Liu H, Stucki JW. Simultaneous measurements of Ca2+ in the intracellular stores and the cytosol of hepatocytes during hormone-induced Ca2+ oscillations. FEBS Letters. 1995;368:165–168. doi: 10.1016/0014-5793(95)00632-j. [DOI] [PubMed] [Google Scholar]
- Favre CJ, Schrenzel J, Jacque J, Lew DP, Krause K-H. Highly supralinear feedback inhibition of Ca2+ uptake by the Ca2+ load of intracellular stores. Journal of Biological Chemistry. 1996;271:14925–14930. doi: 10.1074/jbc.271.25.14925. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Fast calcium-dependent inactivation of calcium release-activated calcium current (CRAC) in RBL-1 cells. Journal of Membrane Biology. 1999a;168:9–17. doi: 10.1007/s002329900493. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC in rat basophilic leukaemia cells. The Journal of Physiology. 1999b;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajnockzy G, Thomas AP. The inositol trisphosphate calcium channel is inactivated by inositol trisphosphate. Nature. 1994;370:474–477. doi: 10.1038/370474a0. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth F. Improved patch clamp techniques for high-resolution current recordings from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hannaert-Merah Z, Combettes L, Coquil J-F, Swillens S, Mauger J-P, Claret M, Champeil P. Characterization of the co-agonist effects of strontium and calcium on myo-inositol trisphosphate-dependent ion fluxes in cerebellar microsomes. Cell Calcium. 1995;18:390–399. doi: 10.1016/0143-4160(95)90054-3. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Verkhratsky A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. The Journal of Physiology. 1998;513:411–424. doi: 10.1111/j.1469-7793.1998.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer A, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal Ca2+ Journal of Cell Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M. Calcium and barium permeation through calcium release-activated calcium (CRAC) channels. Pflügers Archiv. 1995;430:315–322. doi: 10.1007/BF00373905. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Calcium release-activated calcium current in rat mast cells. The Journal of Physiology. 1993;465:359–386. doi: 10.1113/jphysiol.1993.sp019681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Takahashi M, Tanzawa K, Putney JW. Effect of adenophostin A on Ca2+ entry and calcium release-activated calcium current in rat basophilic leukemia cells. Journal of Biological Chemistry. 1998;273:31815–31821. doi: 10.1074/jbc.273.48.31815. [DOI] [PubMed] [Google Scholar]
- Inesi G, Sagra Y. Specific inhibitors of intracellular Ca2+ transport ATPase. Journal of Membrane Biology. 1994;141:1–6. doi: 10.1007/BF00232868. [DOI] [PubMed] [Google Scholar]
- Kim TD, Eddlestone GT, Mahmoud SF, Kuchtey K, Fewtrell C. Correlating Ca2+ responses and secretion in individual RBL-2H3 mucosal mast cells. Journal of Biological Chemistry. 1997;272:31225–31229. doi: 10.1074/jbc.272.50.31225. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Faruq AH, Aksoy IA, Seewald MJ, Powis G. Synthesis of the first optically pure, fluorinated inositol 1,4,5-trisphosphate of myo-inositol stereochemistry and its effects on Ca2+ release in Swiss 3T3 cells. Journal of the American Chemical Society. 1990;112:7403–7404. [Google Scholar]
- Liu K-Q, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. Journal of Experimental Medicine. 1998;187:1721–1727. doi: 10.1084/jem.187.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen L, Parys JB, Weidema AF, Sipma H, Vanlingen S, van Smet P, Callewaert G, De Smedt H. The bell-shaped Ca2+ dependence of the inositol 1,4,5-trisphosphate-induced Ca2+ release is modulated by Ca2+/calmodulin. Journal of Biological Chemistry. 1999;274:13748–13751. doi: 10.1074/jbc.274.20.13748. [DOI] [PubMed] [Google Scholar]
- Mogami H, Tepikin AV, Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO Journal. 1998;17:435–442. doi: 10.1093/emboj/17.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Alvarez J, Scheenen WJJ, Rizzutto R, Meldolesi J, Pozzan T. Ca2+ homeostasis in the endoplasmic reticulum: coexistence of high and low [Ca2+] subcompartments in intact HELA cells. Journal of Cell Biology. 1997;139:601–611. doi: 10.1083/jcb.139.3.601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montero M, Brini M, Marsault R, Alvarez J, Sitia R, Pozzan T, Rizzutto R. Monitoring dynamic changes in the free Ca2+ concentration in the endoplasmic reticulum of intact cells. EMBO Journal. 1995;14:5467–5475. doi: 10.1002/j.1460-2075.1995.tb00233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB. Slow feedback inhibition of calcium release-activated calcium current (CRAC) by calcium entry. Journal of Biological Chemistry. 1998;273:14925–14932. doi: 10.1074/jbc.273.24.14925. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: Nonlinear activation by IP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Shears S. Metabolism of inositol phosphates. Advances in Second Messenger and Phosphoprotein Research. 1992;26:63–92. [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular calcium stores by specific inhibition of the endoplasmic reticulum calcium-ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tse FW, Tse A, Hille B. Cyclic Ca2+ changes in intracellular stores of gonadotropes during gonadotropin-releasing hormone-stimulated Ca2+ oscillations. Proceedings of the National Academy of Sciences of the USA. 1994;91:9750–9754. doi: 10.1073/pnas.91.21.9750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanlingen S, Parys JB, Missiaen L, van Smedt H, Wuytack F, Casteels R. Distribution of inositol 1,4,5-trisphosphate receptor isoforms, SERCA isoforms and Ca2+ binding proteins in RBL-2H3 rat basophilic leukemia cells. Cell Calcium. 1997;22:475–486. doi: 10.1016/s0143-4160(97)90075-0. [DOI] [PubMed] [Google Scholar]
- Warnat J, Philipp S, Zimmer S, Flockerzi V, Cavalie A. Phenotype of a recombinant store-operated channel: highly selective permeation of Ca2+ The Journal of Physiology. 1999;518:631–638. doi: 10.1111/j.1469-7793.1999.0631p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Y, Tsien RY. Calcium current activated by depletion of calcium stores in Xenopus oocytes. Journal of General Physiology. 1997;109:703–715. doi: 10.1085/jgp.109.6.703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Mccloskey MA. Immunoglobulin E receptor-activated calcium conductance in rat mast cells. The Journal of Physiology. 1995;483:59–66. doi: 10.1113/jphysiol.1995.sp020567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Rapid inactivation of depletion-activated calcium current (ICRAC) due to local calcium feedback. Journal of General Physiology. 1995a;105:209–226. doi: 10.1085/jgp.105.2.209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Slow calcium-dependent inactivation of depletion-activated calcium current. Store-dependent and independent mechanisms. Journal of Biological Chemistry. 1995b;270:14445–14451. doi: 10.1074/jbc.270.24.14445. [DOI] [PubMed] [Google Scholar]