

Abstract
Brief synaptic stimulation, or exposure to Conus textile venom (CtVm), triggers an afterdischarge in the bag cell neurones of Aplysia. This is associated with an elevation of intracellular calcium ([Ca2+]i) through Ca2+ release from intracellular stores and Ca2+ entry through voltage-gated Ca2+ channels and a non-selective cation channel. The afterdischarge is followed by a prolonged (∼18 h) refractory period during which the ability of both electrical stimulation and CtVm to trigger afterdischarges or elevate [Ca2+]i is severely attenuated. By measuring the response of isolated cells to CtVm, we have now tested the contribution of different sources of Ca2+ elevation to the onset of the prolonged refractory period.
CtVm induced an increase in [Ca2+]i in both normal and Ca2+-free saline, in part by liberating Ca2+ from a store sensitive to thapsigargin or cyclopiazonic acid, but not sensitive to heparin.
In the presence of extracellular Ca2+, the neurones became refractory to CtVm after a single application but recovered following ∼24 h, when CtVm could again elevate [Ca2+]i. However, this refractoriness did not develop if CtVm was applied in Ca2+-free saline. Thus, elevation of [Ca2+]i alone does not induce refractoriness to CtVm-induced [Ca2+]i elevation, but Ca2+ influx triggers this refractory-like state.
CtVm produces a depolarization of isolated bag cell neurones. To determine if Ca2+ influx through voltage-gated Ca2+ channels, activated during this depolarization, caused refractoriness to CtVm-induced [Ca2+]i elevation, cells were depolarized with high external potassium (60 mm), which produced a large increase in [Ca2+]i. Nevertheless, subsequent exposure of the cells to CtVm produced a normal response, suggesting that Ca2+ influx through voltage-gated Ca2+ channels does not induce refractoriness.
As a second test for the role of voltage-gated Ca2+ channels, these channels were blocked with nifedipine. This drug failed to prevent the onset of refractoriness to CtVm-induced [Ca2+]i elevation, providing further evidence that Ca2+ entry through voltage-gated Ca2+ channels does not initiate refractoriness.
To examine if Ca2+ entry through the CtVm-activated, non-selective cation channel caused refractoriness, neurones were treated with a high concentration of TTX, which blocks the cation channel. TTX protected the neurones from the refractoriness to [Ca2+]i elevation produced by CtVm in Ca2+-containing medium.
Using clusters of bag cell neurones in intact abdominal ganglia, we compared the ability of nifedipine and TTX to protect the cells from refractoriness to electrical stimulation. Normal, long-lasting afterdischarges could be triggered by stimulation of an afferent input after a period of exposure to CtVm in the presence of TTX. In contrast, exposure to CtVm in the presence of nifedipine resulted in refractoriness.
Our data indicate that Ca2+ influx through the non-selective cation channel renders cultured bag cell neurones refractory to repeated stimulation with CtVm. Moreover, the refractory period of the afterdischarge itself may also be initiated by Ca2+ entry through this cation channel.
Transient changes in [Ca2+]i produce short- or long-term alterations in ion channels, secretion and gene expression (Partridge & Swandulla, 1988; Latorre et al. 1989; Sobel & Tank, 1994; Clapham, 1995; Ghosh & Greenberg, 1995; Scott et al. 1995; Simpson et al. 1995; Levitan & Kaczmarek, 1997). However, the ability of an elevation in Ca2+ to produce a specific effect depends on the location of the source of Ca2+. For example, the release of classical neurotransmitters is triggered by Ca2+ entry through voltage-gated Ca2+ channels at active zones, which produce a microdomain of high Ca2+ near the mouth of the channels where synaptic vesicles are docked (Stanley, 1997). Recent work has also suggested that changes in gene transcription produced by elevation of [Ca2+]i do not depend on global Ca2+, but can only be induced by Ca2+ entry through specific subtypes of voltage-gated Ca2+ channels, perhaps because the Ca2+-sensitive signalling pathways that translocate to the nucleus are physically coupled to these channels (Deisseroth et al. 1998).
The bag cell neurones of Aplysia are a model system used to study the regulation of prolonged changes in excitability and [Ca2+]i. These neurones, located in the abdominal ganglion in two symmetrical clusters of 200–400 neurones, control a sequence of reproductive behaviours culminating in egg-laying behaviour (Kupfermann, 1967; Kupfermann & Kandel, 1970; Pinsker & Dudek, 1977; Conn & Kaczmarek, 1989). In response to brief stimulation, the normally silent bag cell neurones undergo an ∼30 min afterdischarge, consisting of prolonged depolarization and action potential firing, during which egg-laying hormone and a number of other neuropeptides are secreted (Rothman et al. 1983; Conn & Kaczmarek, 1989). Following an afterdischarge, the bag cells enter an ∼18 h refractory period, during which another lengthy afterdischarge cannot be elicited. This prolonged refractory period is believed to prevent the reinitiation of the sequence of reproductive behaviours.
Prior work suggests that an elevation of [Ca2+]i during the afterdischarge triggers the prolonged refractory period (Kaczmarek & Kauer, 1983). During an afterdischarge, Ca2+ is released from intracellular stores (Fisher et al. 1994), and there is an enhancement of Ca2+ entry through plasma membrane channels, including voltage-dependent Ca2+ channels (Strong et al. 1987) and a non-selective cation channel that appears to be responsible for the maintained depolarization of the cells (Wilson et al. 1996). If Ca2+ is omitted from the external medium, multiple afterdischarges can be evoked, although these are generally much shorter in duration. Moreover, a refractory-like state can be induced artificially by treating the cells with a Ca2+ ionophore (Kaczmarek & Kauer, 1983). However, it is not known whether a specific source of Ca2+ normally contributes to the prolonged refractory period.
The neurotransmitter that triggers afterdischarges is not known. In the isolated CNS, an afterdischarge can be induced in bag cell neurones by stimulating one of the pleuroabdominal connectives, which contain the axons of these neurones. Afterdischarges that appear identical in all aspects to those evoked by electrical stimulation can be induced by venom from the molluscivorous marine snail Conus textile (Wilson et al. 1996). Venom from the snails of the Conus genus has proven extremely useful in the study of ion channels, yielding toxins that act on, for example, voltage-dependent Na+ and Ca2+ channels (Olivera et al. 1990). In cultured bag cell neurones, Conus textile venom (CtVm) depolarizes the cells by activating a slow, voltage-dependent, Ca2+-permeable, non-selective cation channel (which can be blocked by TTX). When applied to the intact cluster, CtVm elicits an afterdischarge that is followed by the normal prolonged refractory period (Wilson et al. 1996).
Using isolated bag cell neurones, we now show that CtVm triggers both Ca2+ entry and the release of Ca2+ from intracellular stores. We also demonstrate that this CtVm-induced [Ca2+]i elevation becomes refractory, for ∼24 h, following a single application. This refractory-like state is produced by Ca2+ entry through the TTX-sensitive, non-selective cation channel, while other pathways of Ca2+ elevation, including voltage-dependent Ca2+ channels and intracellular Ca2+ stores, fail to elicit this refractoriness. In addition, block of this cation channel prevents the onset of true refractoriness to afterdischarge in intact clusters in response to CtVm. Our findings suggest that the non-selective cation channel, that drives afterdischarges, is selectively located in close physical proximity to a Ca2+-sensitive mechanism that produces a very long lasting change in the excitability of the bag cell neurones.
METHODS
Animals and cell culture
Adult Aplysia californica weighing 125–175 g were obtained from the Aplysia Resource Facility (University of Miami, Miami, FL, USA) or Marinus Inc. (Long Beach, CA, USA). Animals were housed in an ∼400 l aquarium containing continuously circulating, aerated Instant Ocean (Aquarium Systems) at 14°C on an ∼12 h-12 h light-dark cycle and fed lettuce twice a week. All experiments were performed at room temperature (18–20°C).
Cell culture and the majority of Ca2+ imaging were performed in normal artificial sea water (nASW) containing (mM): 460 NaCl, 10.4 KCl, 11 CaCl2, 55 MgCl2 and 15 Hepes; plus 100 U ml−1 penicillin and 0.1 mg l−1 streptomycin; pH 7.8 (NaOH). Some Ca2+ imaging was performed in Ca2+-free ASW, which had the same composition as nASW except that the CaCl2 was omitted and 0.5 mM EGTA was added. Salts were obtained from American Bioanalytical, J.T. Baker, Mallinckrodt or Sigma.
Primary cultures of isolated bag cell neurones were prepared essentially as described by Kaczmarek et al. (1979). Animals were anaesthetized by an injection of isotonic MgCl2 (50 % of body weight), the abdominal ganglion was removed and incubated in neutral protease (dispase) dissolved in nASW (13.33 mg ml−1; Boehringer Mannheim 165859) for 18 h at 18–20°C. The ganglion was then transferred to fresh nASW and the bag cell neurone clusters were dissected from their surrounding connective tissue. Using a fire-polished Pasteur pipette and gentle trituration, bag cell neurones were dispersed in nASW onto glass coverslips (VWR 1 48366045) coated with poly-D-lysine (1 μg ml−1, molecular weight 70 000–150 000; Sigma P0899) glued to drilled out 35 mm × 10 mm polystyrene tissue culture dishes (Corning 25000). Cultures were maintained in nASW for 1–3 days in an incubator at 14°C.
Isolation of Conus textile venom
Conus textile venom lyophilate was provided by Dr B. M. Olivera of the University of Utah. Adult specimens of the molluscivorous snail Conus textile were collected from the ocean around the island of Marinduque in the Philippines. Venom ducts were dissected out of an animal and placed on an ice-cold metal spatula. The duct was then cut into 2 cm sections and the venom extruded out by squeezing with forceps. The venom was then lyophilized in a vacuum centrifuge and stored at −80°C for subsequent extraction. To prepare crude CtVm the lyophilized venom was made up in 0.5 % (v/v) trifluoroacetic acid (TFA) at a concentration of 5 % (w/v). The CtVm was vortexed for 2 min and sonicated for 2 min, in an alternating fashion, for a total of 18 min. The mixture was then centrifuged at 15 000 g for 12 min and the supernatant collected. A second aliquot of TFA was added to the pellet (final concentration 10 % w/v) and the protocol was repeated. The supernatants were pooled and frozen in aliquots at −80°C. All procedures were carried out at 4°C. For experiments, the CtVm aliquots were diluted into 2 mls of either nASW or Ca2+-free ASW for a final protein concentration of ∼100 μg ml−1.
Intracellular Ca2+ imaging
[Ca2+]i was measured by ratiometric imaging of the dye fura-PE3 (K+ salt, Teflabs 0110; Vorndran et al. 1995). Fura-PE3 was pressure injected via sharp electrodes using a General Valve Corporation picospritzer, while simultaneously monitoring the bag cell neuronal membrane potential with a Axoclamp 2B amplifier (Axon Instruments). Microelectrodes were pulled from 1.2 mm internal diameter, borosilicate glass capillaries (World Precision Instruments 1B120F-4) and had a resistance of 30–50 MΩ when the tip was filled with 10 mM fura-PE3 and backfilled with 3 M KCl. Injections usually required ten to fifteen 900 ms pulses to fill the neurones with an optimal amount of dye – estimated to be 50–100 μM. The neurones were then allowed to equilibrate for ∼30 min. For injection, neurones were visualized on a Zeiss IM 35 inverted microscope, equipped with a 16 × Zeiss Plan-Neofluor objective (numerical aperture (NA) = 0.5). Calcium imaging was performed using a Nikon Diaphot inverted microscope equipped with a 40 × Nikon Plan Fluor objective (NA = 1.3) or a 10 × Nikon Fluor objective (NA = 0.5). The illumination system was a 75 W xenon arc lamp, coupled to the microscope via a fibre optic cable and an IBM-compatible computer-controlled grating/monochromator-based excitation device (Photon Technology International). Fluorescent images were acquired with a Hamamatsu C2400 intensified charge coupled device camera. The culture densities were such that one to five bag cell neurones were monitored in a single field when using the 10 × objective. Ratio images of a single field were derived sequentially at 340 and 380 nm excitation wavelength illumination, with an acquisition time (including frame averaging when necessary) for a full frame (256 × 520 pixels) of 1–4 s. The emitted light, detected by the camera, passed through a 520 nm barrier filter. Sampling of [Ca2+]i was performed at 0.5, 1, 2 or 5 min intervals. The camera gain was set based on the initial fluorescence intensity of the cells at the beginning of each experiment and maintained constant thereafter. Between acquisition episodes the excitation illumination was blocked by automatic shutter control. The ratio (R) of the fluorescence intensities (converted to pixel values) from the 340 and 380 nm excitation wavelength-evoked images were used to calculate the free [Ca2+]i from the relationship, [Ca2+]i =QKd(R – Rmin)/(Rmax–R)(Grynkiewicz et al. 1985), where Rmin and Rmax are the fluorescence ratios in the absence and presence of saturationg Ca2+, respectively, and Q is a constant. Values for Rmin, Rmax and Q were determined in intact bag cell neurones by applying 1–10 μM digitonin (Molecular Probes D-8449) in Ca2+-free ASW followed by perfusion with nASW (which contained 11 mM Ca2+). Q was determined from the ratio of 380 nm-evoked fura-PE3 fluorescence in Ca2+-free and 11 mM Ca2+ ASW. Values for Rmin, Rmax and Q ranged from 0.11 to 0.33, 5.1 to 7.5 and 8.7 to 10.2, respectively. Corrections for background fluorescence and camera dark current were carried out as described previously (Knox et al. 1996) and incorporated into the on-line acquisition program. [Ca2+]i was sampled using regions of interests, which were defined over a neurone prior to the start of the experiment and used to gather on-line [Ca2+]i. In some cases, the entire image was saved to disk and [Ca2+]i was determined from analysis off-line.
Whole-cell voltage clamp
Ca2+ current was measured using an EPC-7 amplifier (List Electronics) and the tight-seal, whole-cell method. Microelectrodes were pulled from 1.5 mm internal diameter, borosilicate glass capillaries (World Precision Instruments, TW 150 F-4) and had a resistance of 0.9-1.2 MΩ when filled with intracellular saline. Intracellular saline contained (mM): 570 KCl, 0.595 CaCl2, 1.2 MgCl2, 10 Hepes, 11 glucose, 0.77 EGTA, 10 glutathione, 5 ATP (grade 2, disodium salt; Sigma A3377) and 0.1 GTP (type 3, disodium salt; Sigma G8877); pH = 7.3 (KOH). The estimated free [Ca2+] of the intracellular saline was 300 nM. The extracellular solution was designed to isolate Ca2+ currents and contained (mM): 460 TEA-Cl, 10.4 CsCl, 11 CaCl2, 55 MgCl2 and 15 Hepes; pH 7.8 (CsOH). Pipette junction potentials were nulled immediately before seal formation. Pipette and neuronal capacitive currents were cancelled, the series resistance (range, 3–5 MΩ) was compensated 80–90 % and monitored throughout the experiment. Data were acquired using an IBM-compatible personal computer, a Digidata 1200 analog-to-digital converter (Axon Instruments) and pCLAMP (version 6.02, Axon Instruments) software. Signals were filtered at 1 kHz with a Bessel filter and sampled at 4 kHz. Cells were held at −60 mV and Ca2+ currents were evoked by a series of 10 mV test pulses from −40 to +50 mV. Leak subtraction was performed on line using a P/4 protocol from −40 mV with subpulses of opposite polarity, an inter-subpulse interval of 500 ms and 4 s between actual test pulses.
Extracellular recording
For extracellular recording, abdominal ganglia were set in a 14°C chamber and a wide-bore, fire-polished glass suction electrode placed at the distal end of one connective. A recording suction electrode was placed at the rostral end of the corresponding bag cell neurone cluster. Stimulating current pulses were delivered with a Grass S88 stimulator and isolation unit, while voltage was recorded using a Warner DP-301 differential amplifier. A permanent record of voltage was made on a chart recorder.
Drug application
Most drugs were bath applied by directly pipetting a small volume (< 10 μl) of concentrated stock solution into the Petri dish. Care was taken to pipette the stock near the side of the dish and as far away as possible from the centrally located neurones. This prevents exposing the neurones to high concentrations of drug or vehicle. Heparin (Sigma H3393; 15 mM in the electrode) was co-injected with fura-PE3 to a final estimated concentration in the cell of 75–150 μM. Thapsigargin (Sigma T3250), cyclopiazonic acid (Sigma C1530) and nifedipine (Sigma N7634) required dimethyl sulfoxide (DMSO) as a vehicle. The final concentration of DMSO was 0.01 μM, which in control experiments had no effect on [Ca2+]i. Tetrodotoxin (TTX) was obtained from Sigma (T5651).
Analysis
Unless otherwise noted, the quantitative analysis of [Ca2+]i used steady-state changes. These values were acquired by taking the average value, by eye or with adjacent-averaging, from [Ca2+]i graphs of individual experiments, in regions where the [Ca2+]i had reached an apparent steady-state for 5–10 min. These [Ca2+]i values were then averaged and are presented in the text or in bar graphs as steady-state somatic [Ca2+]i. Statistical analysis was performed using Instat (version 2.01, GraphPad Software). Data are presented as the mean ±s.e.m. Paired and unpaired Student's t test or an analysis of variance (ANOVA) with Bonferroni's multiple comparisons post hoc test were used to test for differences between two or greater than two means, respectively. Data were considered significantly different if the two-tailed P value was < 0.05. Origin (version 4.1, Microcal software) was used to plot line and bar graphs.
RESULTS
CtVm elevates intracellular Ca2+ in isolated bag cell neurones
We investigated the effects of CtVm on [Ca2+]i levels in the somata of isolated bag cell neurones injected with the Ca2+ indicator dye fura-PE3. Bath application of CtVm in nASW resulted in a significant rise in somatic [Ca2+]i over the course of 10–20 min (n = 16 out of 18; Fig. 1). The time course of the CtVm response in nASW displayed one of two forms: some neurones underwent a rapid rise or ‘peak’ in [Ca2+]i which then declined to a stable level of elevated Ca2+ (Fig. 1a), while other neurones showed a slower, more gradual elevation of [Ca2+]i (Fig. 1B). The majority (9 out of 16) of the neurones that responded to CtVm during initial testing had a peak-type response, while the remainder gave [Ca2+]i elevations that were more gradual in nature and lacked the initial peak. Because the peak-type response was also absent from a portion of neurones used in subsequent sets of experiments, only steady-state changes in [Ca2+]i, rather than peak changes, were used for quantification. The mean change in steady-state somatic [Ca2+]i following CtVm was 114.4 ± 9.7 nM (Fig. 1C) and proved for the most part reversible with [Ca2+]i returning to near control following wash in nASW.
Figure 1. Time course and quantification of the CtVm-induced elevation of somatic [Ca2+]i in bag cell neurones in nASW.
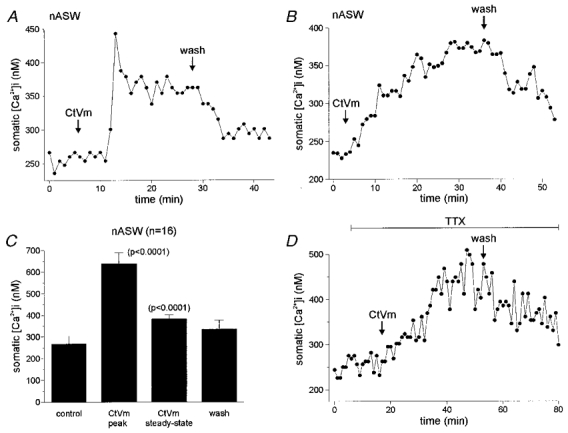
A, time course of a CtVm-induced increase in somatic [Ca2+]i for a bag cell neurone in nASW possessing both peak-type and gradual changes to steady-state levels. Seven minutes after application of 100 μg ml−1 CtVm, there was a rapid and marked increase in [Ca2+]i from a control level of ∼250 nM to a peak of ∼450 nM. The [Ca2+]i subsequently plateaued at a steady-state level of ∼350 nM, and upon wash in nASW, returned to a near-control level of ∼275 nM. B, time course of a CtVm-induced increase in somatic [Ca2+]i displaying only a slow change in steady-state levels and lacking a peak-type response. Four minutes after application of CtVm, a gradual increase in [Ca2+]i developed from a baseline of ∼240 nM to a steady-state level of ∼360 nM. Following wash in nASW, the [Ca2+]i returned to a level of ∼275 nM. C, grouped data of 16 CtVm responses in nASW reveals that the somatic [Ca2+]i during both the peak (when it occurred) and the steady-state periods were significantly greater than that of control (P < 0.0001, paired t test in both cases). The t test is used instead of an ANOVA because the peak-type response occurred only in 9 of the 16 cells that were initially tested. D, the response to CtVm in the presence of 100 μM TTX. On its own, TTX had no effect on [Ca2+]i, furthermore, CtVm was still effective in elevating [Ca2+]i from a baseline of ∼250 nM to a steady-state level of ∼475 nM, which recovered following wash out of CtVm. The CtVm-induced [Ca2+]i increase in TTX lacked a peak-type response and was typically slower in onset as compared with the CtVm response in nASW alone.
Because regulation of [Ca2+]i at the tips of neurites differs from that of the soma (Fink et al. 1988; Knox et al. 1996; Jonas et al. 1997), we also tested the ability of CtVm to elevate [Ca2+]i in the neurites of cultured bag cell neurones (n = 4). The time course of CtVm-induced Ca2+ elevation in the neurites paralleled the response of the soma and, in one instance, showed a peak-type response. The mean elevation of [Ca2+]i in the neurites was a significant (P < 0.05, paired t test) 176.8 ± 57.1 nM above control levels.
CtVm has been shown to activate a Ca2+-permeable cation channel that is blocked by relatively high concentrations of TTX (Wilson et al. 1996). To determine if Ca2+ influx through the cation channel contributes to the CtVm-induced [Ca2+]i elevation, CtVm was applied in the presence of 100 μM TTX. As represented in Fig. 1D, TTX alone had no effect on resting Ca2+ levels (280.0 ± 20.0 nM during control vs. 281.7 ± 19.5 nM in TTX; n = 6); furthermore, application of CtVm significantly elevated somatic [Ca2+]i to 426.8 ± 30.1 nM (repeated measures ANOVA; P < 0.001 Bonferroni's multiple comparisons test). However, there was no peak-type response in the presence of TTX and the time to reach maximal elevation was delayed, being 20–30 min instead of 10–20 min as seen in nASW alone. The lack of a peak-type response and the slowing of the effect are likely to be due to TTX block of Ca2+ influx through the cation channel and block of the CtVm-associated depolarization, which opens voltage-dependent Ca2+ channels (Wilson et al. 1996).
The finding that block of the cation channel by TTX did not impair the ability of CtVm to elevate steady-state [Ca2+]i, suggested that Ca2+ may be liberated from an intracellular store. Therefore, we added CtVm while bathing the neurones in Ca2+-free ASW. Under these conditions, CtVm still elicited a rise in [Ca2+]i, although the kinetics of the response were changed (Fig. 2a) and appeared quite similar to the response seen in the presence of TTX (compare Figs 1D and 2A). Application of CtVm in Ca2+-free ASW resulted in a slow rise (time to maximal elevation 30–40 min) in somatic [Ca2+]i of 102.9 ± 16.1 nM (n = 13 out of 17; Fig. 2B). No peak-type responses similar to those observed in nASW were evoked when CtVm was applied in Ca2+-free ASW (compare panels A in Figs 1 and 2). When the CtVm was washed out and replaced with fresh Ca2+-free ASW, the [Ca2+]i recovered only partially and, for the duration of the recording (up to 2 h), never returned completely to baseline.
Figure 2. Time course and quantification of the CtVm-induced elevation of somatic [Ca2+]i in bag cell neurones in Ca2+-free ASW.
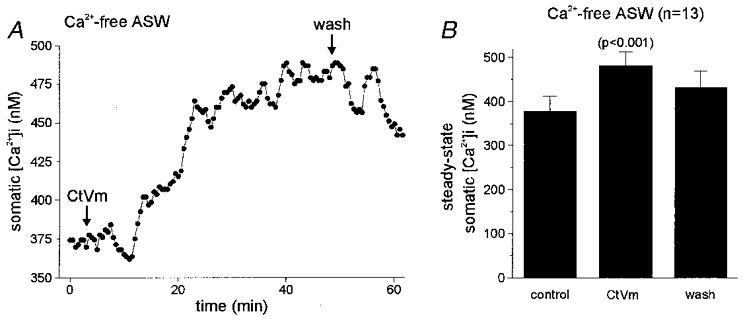
A, the response to CtVm in Ca2+-free ASW displayed a slower time course and lacked a peak-type response. Somatic [Ca2+]i increased within 10 min of 100 μg ml−1 CtVm application, from a baseline of ∼375 nM to a steady-state level of ∼475 nM. Following wash in Ca2+-free ASW, the [Ca2+]i made a partial recovery to ∼440 nM. B, data from 13 preparations responding to CtVm in Ca2+-free ASW reveals that the somatic [Ca2+]i in CtVm is significantly greater than in Ca2+-free ASW alone (standard ANOVA; P < 0.001, Bonferroni's multiple comparisons test).
Contribution of intracellular stores to the CtVm-induced Ca2+ elevation
The persistence of a CtVm-induced Ca2+ elevation in Ca2+-free ASW suggested that an intracellular Ca2+ store plays a role in this response. To examine this possibility, we tested pharmacological agents that perturb the IP3/endoplasmic reticulum Ca2+ release pathway. First, bag cell neurones were injected with either fura-PE3 alone or fura-PE3 with the competitive IP3 receptor blocker heparin (estimated final concentration, 75–150 μM; Ghosh et al. 1988), an agent that effectively prevents IP3-induced Ca2+ elevations in bag cell neurones (Fink et al. 1988; Jonas et al. 1997). However, the CtVm response in Ca2+-free ASW was not significantly reduced in the heparin-injected bag cell neurones (n = 5) as compared with parallel controls (n = 2).
Second, bag cell neurones were pretreated with the endoplasmic reticulum Ca2+-ATPase blockers thapsigargin (1 μM; Thastrup et al. 1990) or cyclopiazonic acid (10 μM; Seidler et al. 1989) in Ca2+-free ASW, and CtVm was then applied. As shown in Fig. 3a, thapsigargin initially caused a build up of [Ca2+]i, followed by a decrease in [Ca2+]i, probably caused by the pumping of Ca2+ out of the cell (Knox et al. 1996). Once a stable baseline was established, CtVm elicited a small elevation of [Ca2+]i. The CtVm-induced [Ca2+]i elevation in the presence of thapsigargin (n = 15) was significantly smaller than the elevation observed in neurones bathed in Ca2+-free ASW alone (parallel controls, n = 9; Fig. 3C). Pretreatment with cyclopiazonic acid (10 μM) proved even more effective than thapsigargin in reducing the response to subsequent application of CtVm (n = 4; Fig. 3B and C). Prior addition of 5 μM insulin, an agent that stimulates intracellular Ca2+ release from a novel, non-heparin/non-thapsigargin-sensitive store (Jonas et al. 1997), did not affect the ability of CtVm to elevate [Ca2+]i (n = 4; data not shown).
Figure 3. Mechanism of the CtVm-induced elevation in somatic [Ca2+]i.
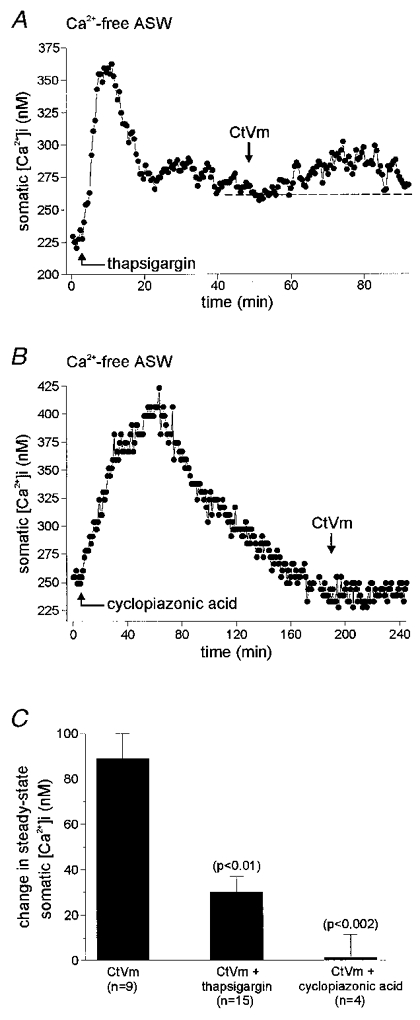
A, when the endoplasmic reticulum Ca2+ store was first depleted by bath application of the Ca2+-ATPase inhibitor thapsigargin (1 μM), subsequent application of CtVm produced only a modest, 35 nM elevation of somatic [Ca2+]i. B, similarly, bath application of a different Ca2+-ATPase inhibitor, cyclopiazonic acid (10 μM), essentially eliminated the response to CtVm. C, the group data for the thapsigargin experiments demonstrate that application of CtVm following endoplasmic reticulum Ca2+ depletion produced a significantly lower response as compared with parallel controls performed in Ca2+-free ASW alone (P < 0.01, paired t test). In addition, prior application of cyclopiazonic acid results in an even further reduction of the CtVm-induced elevation of somatic [Ca2+]i (P < 0.002, paired t test).
CtVm produces a subsequent long-lasting refractory period for [Ca2+]i elevation
Following an afterdischarge of the bag cell neurones in the intact cluster, an ∼18 h refractory period ensues, during which a subsequent afterdischarge cannot be stimulated (Kupfermann & Kandel, 1970; Conn & Kaczmarek, 1989). Similarly, we have found that the depolarizing response to CtVm in cultured bag cell neurones also becomes refractory, as a second application of CtVm fails to excite neurones if they have already undergone a CtVm-induced depolarization (Wilson et al. 1996; authors' unpublished observation). Therefore, we determined whether the CtVm-induced Ca2+ elevation also undergoes this form of refractoriness. Following a Ca2+ response induced by CtVm in nASW, and a subsequent wash with return of [Ca2+]i to near baseline levels, a second application of CtVm (30–40 min after the first) did not produce a second elevation in somatic [Ca2+]i (n = 4; Fig. 4a). While the first application of CtVm caused a significant change in steady-state [Ca2+]i, from a baseline of 239.0 ± 47.5 to 389.7 ± 52.1 nM in CtVm (P < 0.03, paired t test), the second dose of CtVm did not produce a significant change between the new baseline (following wash) of 300.7 ± 47.9 nM and the [Ca2+]i in CtVm of 301.0 ± 49.2 nM (P > 0.05, paired t test). Figure 4B gives the change in steady-state somatic [Ca2+]i for the first and second applications of CtVm. We also performed an additional set of experiments where the time between the two CtVm applications was increased to 2 h, to test the possibility that the intracellular Ca2+ stores contributing to the response may require a longer time period to refill than allotted in the initial experiments. While the first dose of CtVm produced a significant rise in [Ca2+]i (initial baseline: 227.4 ± 21.8 nM; after CtVm: 287.0 ± 26.9 nM; P < 0.002, paired t test), the second application of CtVm again failed to significantly elevate [Ca2+]i, despite the longer waiting period (baseline following wash: 237.6 ± 14.8 nM; after CtVm: 249.8 ± 8.0 nM; P > 0.05, paired t test; data not shown).
Figure 4. Changes in somatic [Ca2+]i in response to repeated applications of CtVm.
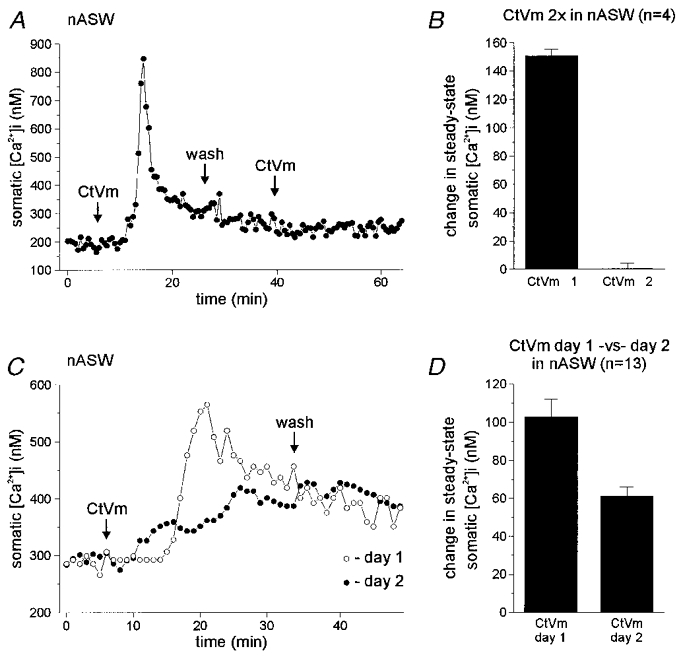
A, dual CtVm exposure in nASW. Application of 100 μg ml−1 CtVm produced a typical elevation of bag cell neurone somatic [Ca2+]i in nASW. Following recovery to steady state and wash in nASW, CtVm was applied again, but had no effect on [Ca2+]i, i.e. the neurone was refractory. B, average data for four such experiments showing that while the initial CtVm exposure (CtVm 1) produced a significant elevation of ∼150 nM in somatic [Ca2+]i, a subsequent exposure (CtVm 2) had essentially no effect on Ca2+ levels. Since the peak-type response was not elicited by the first CtVm exposure in all neurones tested in this manner, only changes in steady-state [Ca2+]i are presented. C, recovery from CtVm-induced refractoriness over 24 h. A bag cell neurone exhibited a standard somatic [Ca2+]i elevation in response to 100 μg ml−1 CtVm in nASW (^). The CtVm was then washed out and the neurone returned to the incubator for ∼24 h. When the CtVm was applied again on day 2, the [Ca2+]i rose to similar steady-state levels seen on day 1 (∼400 nM; •). D, the collective data for 13 experiments shows that CtVm is capable of elevating somatic [Ca2+]i in nASW on both days 1 and 2. If a peak-type response is observed on day 1, it is invariably absent on day 2; consequently, only the steady-state changes in [Ca2+]i are quantified. The steady-state levels reached during day 2 CtVm exposure are typically less than that achieved on day 1.
A hallmark of the bag cell neurone refractory period in the intact cluster is the requirement of a prolonged recovery time (∼18 h) before a long lasting afterdischarge can again be elicited. To determine if the refractoriness of the CtVm-induced Ca2+ increase recovered over a similar time course, CtVm was applied in nASW and then a second dose was delivered ∼24 h later, again in nASW. This was achieved by washing out the initial CtVm dose and placing the Petri dish in a light-tight container, which was stored in the incubator overnight. The neurones remained viable and the loss of fura-PE3 dye was minimal, as determined by a lack of change in the camera gain required to observe maximal fluorescence intensity from day 1 to day 2. The application of CtVm on day 1 resulted in a significant elevation of somatic [Ca2+]i, which in most cases (12 out of 20) involved a peak-type response (Fig. 4C). The resting somatic [Ca2+]i on day 1 was 267.2 ± 9.4 nM, and CtVm elevated this to a steady-state level of 369.8 ± 8.8 nM (P < 0.0001, paired t test). Of the 20 neurones tested on day 1, 13 (65 %) recovered and responded to CtVm with an elevation of [Ca2+]i on day 2, while 7 (35 %) were still refractory on day 2. Only those neurones that recovered were used for quantitative analysis. The response on day 2 was different in that it was smaller, from a baseline of 352.1 ± 17.0 nM to a steady-state level of 412.9 ± 18.7 nM in CtVm (P < 0.0001, paired t test), and regardless of the presence of a peak-type response on day 1, all the CtVm responses on day 2 showed only the more gradual rise in [Ca2+]i. The change in steady-state somatic [Ca2+]i on day 2 was significantly less than that on day 1 (Fig. 4D; P < 0.003, paired t test).
Ca2+ entry induces a refractory period to subsequent [Ca2+]i elevation
Kaczmarek & Kauer (1983) reported that the introduction of extracellular Ca2+ into bag cell neurone clusters with a Ca2+ ionophore produces a state resembling the refractory period. Since CtVm activates a Ca2+-permeable cation channel and depolarizes isolated bag cell neurones, potentially activating voltage-dependent Ca2+ channels, we tested the hypothesis that the entry of Ca2+ from the external medium during the first response to CtVm in nASW may be responsible for initiating the refractory-like period. CtVm was first applied in Ca2+-free ASW, then, after the elevation of [Ca2+]i, the Ca2+-free ASW was replaced by nASW and CtVm was applied again (Fig. 5A; n = 11). The response to CtVm in Ca2+-free ASW was a significant change in somatic [Ca2+]i from a baseline of 202.3 ± 12.6 to 258.9 ± 14.7 nM in CtVm (P < 0.0001, paired t test). The second application of CtVm, now in nASW (Ca2+ containing) also resulted in a significant rise in somatic [Ca2+]i (248.1 ± 14.2 nM in nASW vs. 297.4 ± 18.4 nM in CtVm; P < 0.0001, paired t test), confirming the role of Ca2+ influx in producing refractoriness. Figure 5B gives the change in steady-state somatic [Ca2+]i for the application of CtVm in Ca2+-free ASW and nASW, respectively.
Figure 5. Ca2+ influx through a pathway distinct from voltage-dependent Ca2+ channels causes refractoriness.
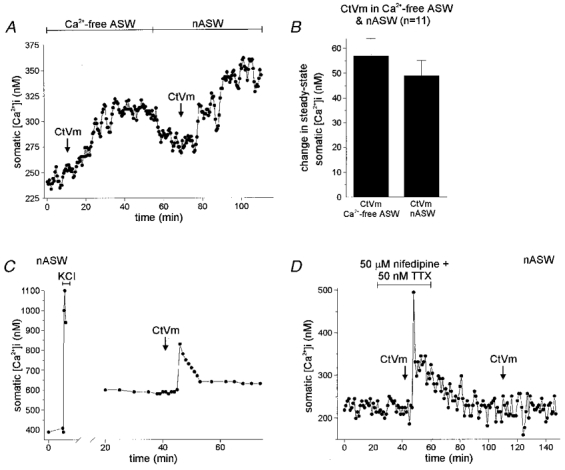
A, CtVm application in Ca2+-free ASW followed by application in nASW. When 100 μg ml−1 CtVm was applied first in Ca2+-free ASW, the somatic [Ca2+]i rose by ∼60 nM. Upon wash in nASW, the Ca2+ levels dropped somewhat, and when CtVm was introduced a second time, the [Ca2+]i increased again by ∼75 nM. B, grouped data for CtVm in Ca2+-free and nASW experiments (n = 11), demonstrates that a lack of extracellular Ca2+ during the first CtVm response results in somatic [Ca2+]i elevation during the second CtVm exposure. C, Ca2+ influx produced by KCl depolarization in nASW does not result in refractoriness to CtVm. Application of 60 mM KCl resulted in a rapid and marked rise in [Ca2+]i, from a baseline of ∼400 nM to a peak of ∼1100 nM. Following wash in nASW and recovery to a new steady state of ∼600 nM, 100 μg ml−1 CtVm was applied and elicited an ∼225 nM peak increase in [Ca2+]i. Thus, the influx of Ca2+ through voltage-dependent Ca2+ channels during the KCl exposure did not make the neurone refractory to CtVm-induced [Ca2+]i elevation. This is representative of three such experiments. Parenthetically, this particular neurone was a rare example where KCl depolarization resulted in a significant shift in baseline [Ca2+]i. However, the subsequent CtVm-induced [Ca2+]i increase shows that elevation of the baseline [Ca2+]i to higher than normal levels does not effect the neurone's response to CtVm. D, block of Ca2+ and Na+ channels does not prevent refractoriness to CtVm-induced [Ca2+]i elevation. In nASW, a bag cell neurone was exposed to both 50 μM nifedipine and 50 nM TTX, to block Ca2+ and Na+ channels, respectively. Application of 100 μg ml−1 CtVm resulted in an elevation of [Ca2+]i from a baseline of ∼225 nM to a peak of ∼500 nM and a plateau of ∼325 nM. The bath was exchanged with fresh nASW, and ∼40 min later, CtVm was applied a second time, but did not result in a significant change in [Ca2+]i.
Ca2+ entry through voltage-gated Ca2+ channels does not induce refractoriness
We investigated whether Ca2+ entry through voltage-gated Ca2+ channels could produce refractoriness to CtVm-induced [Ca2+]i elevation. The first test was to drive Ca2+ into the cells through voltage-gated Ca2+ channels, by K+-mediated depolarization, prior to CtVm application (n = 3). Figure 5C shows an example of such an experiment, where a bag cell neurone was exposed to a brief, 60 mM KCl stimulus in nASW, and the resulting depolarization caused a significant Ca2+ influx through Ca2+ channels (a change in [Ca2+]i from ∼400 to ∼1100 nM). Once the neurone had recovered to a new baseline [Ca2+]i of ∼600 nM, application of CtVm was still capable of eliciting an elevation of [Ca2+]i, despite the prior Ca2+ influx through Ca2+ channels. As a pharmacological test, neurones were exposed to 50 μM nifedipine, a Ca2+ channel blocker (Strong et al. 1987), with 50 nM TTX, to block voltage-gated Na+ channels (n = 7; Fig. 5D). The 50 nM TTX was included because voltage-gated Na+ channels, sensitive to low levels of TTX, have been reported in some cultured bag cell neurones (Fieber, 1995). This cocktail did not prevent either the initial [Ca2+]i elevation to CtVm (baseline of 227.3 ± 13.9 nM vs. a CtVm level of 277.7 ± 13.3 nM; P < 0.0001, paired t test) nor did it protect the cells from refractoriness to subsequent CtVm application (new baseline following wash of 235.4 ± 12.4 nM vs. a [Ca2+]i during the second CtVm exposure of 231.7 ± 12.4 nM (P > 0.05, paired t test).
High concentrations of TTX prevent refractoriness to CtVm-induced Ca2+ elevation
In addition to producing release of Ca2+ from an internal store, CtVm activates a Ca2+-permeable cation channel that is blocked by relatively high concentrations of TTX (80–100 μM; Wilson et al. 1996). To determine if Ca2+ influx through this CtVm-activated cation channel produces refractoriness, CtVm was initially applied in TTX-containing nASW. Following the elevation of [Ca2+]i, the TTX was washed out and CtVm was applied a second time (Fig. 6A; n = 5). In all cases, CtVm caused an elevation of somatic [Ca2+]i during both the first application, in the presence of 100 μM TTX, and during the second application, in the absence of TTX. The CtVm response in 100 μM TTX was a significant elevation in somatic [Ca2+]i from a baseline of 290.0 ± 21.6 nM to 422.2 ± 36.4 nM in CtVm (P < 0.01, paired t test). Introducing CtVm a second time, following wash out of the TTX to nASW alone, produced a significant rise in somatic [Ca2+]i from 307.0 ± 22.0 to 391.0 ± 23.3 nM (P < 0.0001, paired t test). Figure 6B gives the change in steady-state somatic [Ca2+]i for the application of CtVm in TTX containing nASW and nASW alone, respectively.
Figure 6. Preventing Ca2+ entry through the cation channel protects from CtVm-induced refractoriness.
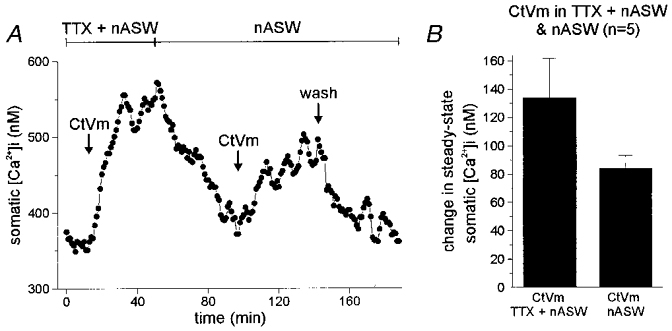
A, CtVm responses in TTX-containing nASW followed by application in nASW alone. Applying 100 μg ml−1 CtVm in nASW containing 100 μM TTX resulted in an ∼175 nM rise in [Ca2+]i. Subsequent wash out of both CtVm and TTX produced a recovery of [Ca2+]i levels to near control. When the neurone was exposed to CtVm again, but now in nASW alone, the [Ca2+]i was elevated a second time, by ∼75 nM. B, the average data for 5 such experiments shows that application of CtVm in TTX protected the bag cell neurones from refractoriness, as the second application of CtVm in nASW consistently produced an elevation of [Ca2+]i, unlike the data of Fig. 4A and B.
To supplement the data from Figs 5 and 6, and eliminate the possibility that TTX was blocking voltage-dependent Ca2+ channels, while also blocking the cation channel, we also tested the effect of TTX on Ca2+ current and Ca2+ influx. TTX, at 100 μM, did not significantly reduce the voltage-gated Ca2+ current recorded from bag cell neurones under whole-cell voltage clamp. The peak Ca2+ current at +20 mV was 1.59 ± 0.28 nA in control vs. 1.46 ± 0.28 nA in TTX (n = 4; P > 0.05, paired t test). Also, the elevation of [Ca2+]i produced by 30 mM KCl depolarization, and monitored with fura-PE3 imaging, was not significantly altered by TTX (653 ± 103 nM in control vs. 571 ± 94 nM in TTX; n = 4; P > 0.05, paired t test).
High concentrations of TTX prevent refractoriness of the afterdischarge
Having established a role for Ca2+ entry through the cation channel in producing refractoriness to [Ca2+]i elevation in cultured bag cell neurones, we next examined what role it might play in refractoriness of the afterdischarge in the intact cluster. Afterdischarges were recorded extracellularly from bag cell neurone clusters in intact abdominal ganglia. Clusters were treated with either TTX or nifedipine for 1 h. CtVm was then added in the continued presence of the blocker (TTX or nifedipine) for a second hour. CtVm did not elicit an afterdischarge in either TTX or nifedipine. After this, both the blocker and CtVm were washed out, and the cluster assayed for refractoriness with electrical stimulation. Following wash of both TTX and CtVm, 5 out of 6 clusters gave robust afterdischarges (duration 31.4 ± 8.0 min; Fig. 7a). In contrast, for the paired experiments of nifedipine and CtVm, only 1 out of 5 clusters gave a modest afterdischarge (duration, 16 min), while the remaining four clusters were refractory following wash out of these agents. A refractory cluster following wash out of nifedipine plus CtVm is shown in Fig. 7B. In addition, regardless of their being refractory or not, all clusters were capable of generating compound action potentials to stimulation following treatment and wash. Finally, when compared with control (n = 6), simply incubating clusters in nifedipine alone (n = 8) and then washing it out did not affect the ability to generate afterdischarges upon electrical stimulation.
Figure 7. Block of the cation channel protects from CtVm-induced refractoriness in the intact bag cell neurone cluster.
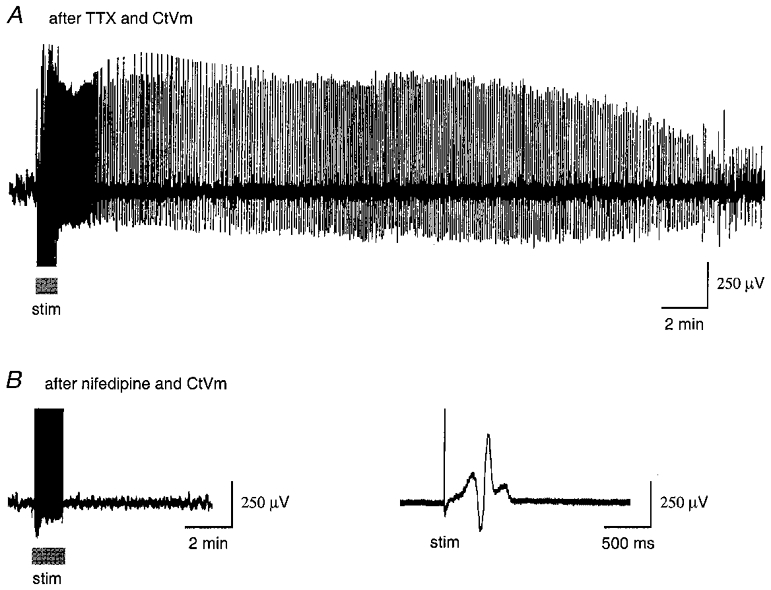
In these experiments, the channel blocker (TTX or nifedipine) was first applied for 1 h, then CtVm was added along with the blocker for a second hour, the preparation was washed thoroughly over the course of a third hour, and finally the afferent connective was stimulated to test for the ability to produce an afterdischarge. A, block of the cation channel with TTX protects from CtVm-induced refractoriness to subsequent stimulation. Following wash of 100 μM TTX plus 100 μg ml−1 CtVm, the cluster was capable of firing an afterdischarge upon electrical stimulation (stim, at bar, 2.5 ms pulses, 6 Hz, 20 V). B, block of voltage-dependent Ca2+ channels did not protect from CtVm-induced refractoriness to subsequent stimulation. Left, following wash of 100 μM nifedipine plus 100 μg ml−1 CtVm, the cluster was refractory to electrical stimulation (stim, at bar). Right, recording from the same cluster at a higher sweep speed shows that while refractory, the cells were fully capable of firing compound action potentials upon stimulation (stim). The action potentials are obscured in the trace shown on the left because of the condensed time scale.
DISCUSSION
We have found that CtVm, an agent that triggers afterdischarges in the intact bag cell neurone cluster and depolarizes isolated bag cell neurones (Wilson et al. 1996), produces an elevation in isolated bag cell neurone [Ca2+]i. CtVm was effective in the presence or absence of extracellular Ca2+, indicating that at least in part, Ca2+ is released from an intracellular store. Fink et al. (1988), showed that IP3 levels increase during the afterdischarge, and that injection of IP3 releases [Ca2+]i from internal stores in bag cell neurones. However, evidence suggests that CtVm may release Ca2+ from a store distinct from those accessed by the IP3 receptor. Previous work has shown that heparin (Ghosh et al. 1988) is a competitive blocker of the IP3 receptor/Ca2+-release channel when injected into bag cell neurones (Jonas et al. 1997), whereas heparin failed to inhibit the CtVm-induced [Ca2+]i increase. In contrast, thapsigargin or cyclopiazonic acid, which deplete intracellular Ca2+ by inhibiting the endoplasmic reticulum Ca2+-ATPase (Seidler et al. 1989; Thastrup et al. 1990), both attenuated the CtVm-induced [Ca2+]i increase. This suggests that CtVm causes the release of Ca2+ from an endoplasmic reticulum-based store, possibly by acting through a membrane-bound receptor that is coupled to the formation of one or more second messengers – as was proposed by Wilson et al. (1996). For instance, CtVm could initiate the production of a second messenger whose receptor is not sensitive to heparin, but can release Ca2+ from a pool that is depleted by thapsigargin and cyclopiazonic acid. Previous work has demonstrated a heparin- and thapsigargin-resistant pool of intracellular Ca2+ that can be mobilized in the bag cell neurones by insulin (Jonas et al. 1997). As is the case for the CtVm-induced response, the insulin-sensitive store is also mobilized at the tips of bag cell neurites, whereas thapsigargin and IP3 appear relatively ineffective in distal neurites. Thus, CtVm may also mobilize this second store, which has been suggested to reside within secretory vesicles.
The plasma membrane cation channel activated by CtVm is Ca2+ permeable and blocked by TTX (Wilson et al. 1996). While this probably contributes somewhat to the overall Ca2+ signal in Ca2+-containing saline, the fact that CtVm potently elevates [Ca2+]i in the presence of TTX suggests that Ca2+ entry through the cation channel has only a small effect on steady-state [Ca2+]i. Furthermore, the CtVm-activated cation channel also depolarizes the neurones, activating voltage-dependent Ca2+ channels. It is quite possible that the ‘peak-type’ response observed in many of the neurones may be due to Ca2+ entry through the cation channel and voltage-dependent Ca2+ channel, while the steady-state response is due more to release of intracellular [Ca2+]i. In addition, we cannot rule out the possibility that one of these routes of Ca2+ entry may initiate Ca2+-induced Ca2+ release (CICR), although prior attempts to alter [Ca2+]i in these neurones with agents known to effect CICR, i.e. ryanodine and cyclic ADP ribose, have been unsuccessful (Jonas et al. 1997).
Once the afterdischarge has occurred, bag cell neurones enter an ∼18 h refractory period, during which a second afterdischarge cannot be elicited (Kupfermann & Kandel, 1970; Conn & Kaczmarek, 1989). The refractory period is thought to prevent a second afterdischarge from disrupting the sequence of ongoing egg laying. Furthermore, the refractory period serves to limit the frequency of egg-laying behaviour. Kaczmarek & Kauer (1983) showed that a refractory-like period can be experimentally produced by incubating bag cell neurone clusters with a Ca2+ ionophore. They concluded that Ca2+ elevation during the afterdischarge may produce refractoriness. In keeping with this, Kaczmarek et al. (1982) reported that, while stimulation of afterdischarges in Ca2+-free ASW attenuated their duration, it did not result in the clusters becoming refractory, again pointing to the conclusion that Ca2+ elevation initiates refractoriness. Our results are also consistent with Ca2+ influx being necessary to produce a refractory-like period in cultured bag cell neurones, i.e. refractoriness to [Ca2+]i elevation by CtVm. Application of CtVm in nASW (11 mM Ca2+) produces an elevation in [Ca2+]i, but subsequent applications fail to produce any further change in [Ca2+]i. The refractoriness to further CtVm-induced [Ca2+]i elevation in nASW is also remarkably similar to that recorded electrophysiologically for bag cell neurones in the intact ganglion, in that it has a lengthy recovery period. When cultured bag cell neurones are initially exposed to CtVm in nASW and then left for ∼24 h to recover, 65 % of the cells show a CtVm-induced elevation in [Ca2+]i on the second day. The general similarity between the intact cluster and the isolated cells in the time required for recovery also suggests that this component of the afterdischarge/refractory mechanism is preserved in culture. This, together with previously reported effects of CtVm on isolated bag cell neurones and intact clusters (Wilson et al. 1996), strengthens the case for making parallel comparisons between the action of CtVm in vitro and electrically stimulated afterdischarges in the intact nervous system.
Preventing Ca2+ entry from the extracellular medium during a first CtVm application, by bathing the neurones in Ca2+-free ASW, allows a second exposure to CtVm to elicit a second rise in [Ca2+]i. This suggests that extracellular Ca2+ entry induces refractoriness to CtVm-induced [Ca2+]i elevation. However, this influx of Ca2+ does not occur through voltage-gated Ca2+ channels, as prior KCl-induced Ca2+ influx does not result in the neurones becoming refractory to subsequent CtVm application. Furthermore, block of Ca2+ channels did not protect the neurones from refractoriness to a second application of CtVm. Work in the intact ganglion has shown that, for both the bag cell neurones of Aplysia (Fisher et al. 1994) and the homologous caudodorsal cells of Lymnaea (Kits et al. 1997), attempts to elevate steady-state [Ca2+]i in refractory clusters, by evoking action potentials, are unsuccessful. Thus, it may possible to extend the concept of refractoriness beyond electrical excitability to processes governing the mobilization of [Ca2+]i.
Evidence suggests that a large part of the depolarizing drive for the afterdischarge arises from the activation of the slow, non-selective cation channel (Kaczmarek & Stumwasser, 1984; Wilson & Kaczmarek, 1993; Wilson et al. 1996). This cation channel is voltage dependent, blocked by relatively high concentrations of TTX, and Ca2+ permeable (Wilson et al. 1996). Similar inward currents have been documented in several mammalian and molluscan neuronal preparations, where they are believed to contribute to bursting and repetitive firing (Wilson & Wachtel, 1974; Green & Gillette, 1983; Stafstrom et al. 1985; Swandulla & Lux, 1985; Alonso & Llinas, 1989). We exploited the ability of TTX to block the bag cell neurone cation channel to investigate whether Ca2+ entry through this conductance contributes to the refractoriness to CtVm-induced [Ca2+]i elevation following a single CtVm exposure. Interestingly, TTX protects the CtVm response from becoming refractory, and consistently allows a second CtVm [Ca2+]i elevation. The route of entry rather than the magnitude of global Ca2+ elevation appears, therefore, to be far more important in triggering refractoriness, suggesting that Ca2+ entry through the cation channel is specifically coupled to a pathway that initiates refractoriness to subsequent CtVm stimulation.
There are very few examples of Ca2+ entry through specific ion channels having an effect on neuronal responsiveness as in the bag cell neurones. In cultured hippocampal neurones, L-type Ca2+ channels are selectively coupled to the opening of small-conductance Ca2+-activated K+ channels, while N-type Ca2+ channels are exclusively coupled to the opening of large-conductance Ca2+-activated K+ channels (Marrion & Tavalin, 1998). This indicates that microdomains of Ca2+ created by different Ca2+ channels may have functional significance. A second study in hippocampal cultures has closer parallels to our work. In this case, Ca2+ entry causes translocation of calmodulin to the nucleus where, upon kinase activation, the transcription factor CREB is phosphorylated (Deisseroth et al. 1998). When stimulated with depolarization, translocation was only achieved when Ca2+ entry occurred through L-type, but not N- or P/Q-type, Ca2+ channels, despite all four types of channels contributing to an elevation of [Ca2+]i. The activation of a transcription factor could conceivably alter the way in which the neurones respond to subsequent stimulation.
In bag cell neurones, the mechanism by which extracellular Ca2+ entry produces refractoriness is unknown. Possibly, Ca2+ entering at the cell membrane binds to an enzyme closely associated with the cation channel, which then alters both the properties of the cation channel and prevents the release of Ca2+ from internal stores. Alternatively, Ca2+ entry may uncouple the receptors that CtVm normally activates. As the magnitude of the global [Ca2+]i elevation produced by cation channel activation is modest, the Ca2+-sensitive mechanism that causes refractoriness must be tightly restricted to the particular microdomain of the cation channel. The ability of an ionophore to induce refractoriness in the intact bag cell neurone cluster is readily explained under the current hypothesis by reasonably assuming that the ionophore elevates Ca2+ globally in the neurone, and that this Ca2+ triggers pathways normally accessed by Ca2+ entering via the cation channel. However, Ca2+ entering via voltage-gated Ca2+ channels does not overlap with the cation channel pathway.
Afterdischarge in the bag cell neurones is an all-or-none event (Conn & Kaczmarek, 1989). Nevertheless, even in the refractory state in the intact nervous system, very prolonged stimulation can sometimes induce afterdischarges, although these are much shorter in duration than the normal first afterdischarge (Kaczmarek et al. 1982). Thus, it is likely that there exists degrees of refractoriness. In the present study, when CtVm was applied in Ca2+-free ASW or in the presence of TTX, the neurones were protected from refractoriness to subsequent CtVm application. In these experiments, however, the second CtVm response was typically the same magnitude or smaller that the first. One may have expected that the second response would be larger than the first, as it reflects the contribution of both Ca2+ entry and release, whereas the initial response reflects Ca2+ release only from internal stores. Therefore, we cannot exclude the possibility that some degree of refractoriness was produced by an initial dose of CtVm, and that more than one independent mechanism contributes to refractoriness. However, it is clear that neurones were not refractory in the strictest sense of the word, i.e. they consistently displayed a second response to CtVm, while those neurones exposed initially to CtVm in Ca2+-containing medium did not respond to a second application of CtVm.
Our experiments with intact clusters of bag cell neurones indicate that, in addition to its effect on [Ca2+]i elevation, Ca2+ entry through the cation channel may play a role in producing the overall refractory period during which electrical stimulation fails to trigger a second, long-lasting afterdischarge. This channel is regulated in a complex manner by several, closely associated protein kinases and phosphatases (Wilson & Kaczmarek, 1993; Wilson et al. 1998). Its putative role in producing refractoriness complements its primary function of providing the depolarizing drive for the afterdischarge.
Acknowledgments
N.S.M. and R.J.K. contributed equally to this work. The authors are very grateful to Dr B.M. Olivera for providing lyophilized Conus textile venom and Ms N.M. Magoski for critical evaluation of earlier drafts of the manuscript. The work was supported by a Human Frontiers Science Program and Medical Research Council of Canada postdoctoral fellowships to N.S.M. and a National Institutes of Health operating grant to L.K.K.
References
- Alonso A, Llinas RR. Subthreshold Na+-dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:175–177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- Clapham DE. Calcium signalling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Conn PJ, Kaczmarek LK. The bag cell neurons of Aplysia. Molecular Neurobiology. 1989;3:237–273. doi: 10.1007/BF02740607. [DOI] [PubMed] [Google Scholar]
- Deisseroth K, Heist EK, Tsien RW. Translocation of calmodulin to the nucleus supports CREB phosphorylation in hippocampal neurons. Nature. 1998;392:198–202. doi: 10.1038/32448. [DOI] [PubMed] [Google Scholar]
- Fieber LA. Characterization and modulation of Na+ and Ca2+ currents underlying the action potential in bag cells of two species of Aplysia. Journal of Experimental Biology. 1995;198:2337–2347. doi: 10.1242/jeb.198.11.2337. [DOI] [PubMed] [Google Scholar]
- Fink LA, Connor JA, Kaczmarek LK. Inositol triphosphate releases intracellularly stored calcium and modulates ion channels in molluscan neurons. Journal of Neuroscience. 1988;8:2544–2555. doi: 10.1523/JNEUROSCI.08-07-02544.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher T, Levy S, Kaczmarek LK. Transient changes in intracellular calcium associated with a prolonged increase in excitability in neurons of Aplysia californica. Journal of Neurophysiolology. 1994;71:1254–1257. doi: 10.1152/jn.1994.71.3.1254. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Calcium signalling in neurons: molecular mechanisms and cellular consequences. Science. 1995;286:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Ghosh TK, Eis PS, Mullaney JM, Ebert CL, Gill DL. Competitive, reversible, and potent antagonism of inositol 1,4,5-triphosphate-activated calcium release by heparin. Journal of Biological Chemistry. 1988;263:11075–11079. [PubMed] [Google Scholar]
- Green DJ, Gillette R. Patch- and voltage-clamp analysis of cyclic AMP-stimulated inward current underlying neurone bursting. Nature. 1983;306:784–785. doi: 10.1038/306784a0. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of calcium indicators with greatly improved fluorescent properties. Journal of Biological Chemistry. 1985;260:3440–3448. [PubMed] [Google Scholar]
- Jonas EA, Knox RJ, Smith TCM, Wayne NL, Connor JA, Kaczmarek LK. Regulation by insulin of a unique neuronal Ca2+ pool and neuropeptide secretion. Nature. 1997;385:343–346. doi: 10.1038/385343a0. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK, Finbow M, Revel JP, Strumwasser F. The morphology and coupling of Aplysia bag cells within the abdominal ganglion and in cell culture. Journal of Neurobiology. 1979;10:535–550. doi: 10.1002/neu.480100604. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK, Jennings K, Strumwasser F. An early sodium and a late calcium phase in the afterdischarge of peptide-secreting neurons of Aplysia. Brain Research. 1982;238:105–115. doi: 10.1016/0006-8993(82)90774-0. [DOI] [PubMed] [Google Scholar]
- Kaczmarek LK, Kauer JA. Calcium entry causes a prolonged refractory period in peptidergic neurons of Aplysia. Journal of Neuroscience. 1983;3:2230–2239. doi: 10.1523/JNEUROSCI.03-11-02230.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczmarek LK, Strumwasser F. A voltage-clamp analysis of currents underlying cyclic AMP-induced membrane modulation in isolated peptidergic neurons of Aplysia. Journal of Neurophysiology. 1984;52:340–349. doi: 10.1152/jn.1984.52.2.340. [DOI] [PubMed] [Google Scholar]
- Kits KS, Dreijer AMC, Lodder JC, Borgdorff A, Wadman WJ. High intracellular calcium levels during and after electrical discharges in molluscan peptidergic neurons. Neuroscience. 1997;79:275–284. [PubMed] [Google Scholar]
- Knox RJ, Jonas EA, Kao L-S, Smith PJS, Connor JA, Kaczmarek LK. Ca2+ influx and activation of a cation current are coupled to intracellular Ca2+ release in peptidergic neurons of Aplysia californica. The Journal of Physiology. 1996;494:627–693. doi: 10.1113/jphysiol.1996.sp021520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kupfermann I. Stimulation of egg laying: possible neuroendocrine function of bag cells of abdominal ganglion of Aplysia californica. Nature. 1967;216:814–815. doi: 10.1038/216814a0. [DOI] [PubMed] [Google Scholar]
- Kupfermann I, Kandel ER. Electrophysiological properties and functional interconnections of two symmetrical neurosecretory clusters (bag cells) in abdominal ganglion of Aplysia. Journal of Neurophysiology. 1970;33:865–876. doi: 10.1152/jn.1970.33.6.865. [DOI] [PubMed] [Google Scholar]
- Latorre R, Oberhauser A, Labarca P, Alvarez O. Varieties of calcium-activated potassium channels. Annual Review of Physiology. 1989;51:385–399. doi: 10.1146/annurev.ph.51.030189.002125. [DOI] [PubMed] [Google Scholar]
- Levitan IB, Kaczmarek LK. The Neuron: Cell and Molecular Biology. New York: Oxford University Press; 1997. [Google Scholar]
- Marrion NV, Tavalin SJ. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 1998;395:900–905. doi: 10.1038/27674. [DOI] [PubMed] [Google Scholar]
- Olivera BM, River J, Clark C, Ramilo CA, Corpuz GP, Abogadie FC, Mena EE, Woodward SR, Hillyard DR, Cruz LJ. Diversity of Conus neuropeptides. Science. 1990;249:257–263. doi: 10.1126/science.2165278. [DOI] [PubMed] [Google Scholar]
- Partridge LD, Swandulla D. Calcium-activated non-specific cation channels. Trends in Neurosciences. 1988;11:69–72. doi: 10.1016/0166-2236(88)90167-1. [DOI] [PubMed] [Google Scholar]
- Pinsker HM, Dudek FE. Bag cell control of egg laying in freely behaving Aplysia. Science. 1977;197:490–493. doi: 10.1126/science.197.4302.490. [DOI] [PubMed] [Google Scholar]
- Rothman BS, Weir G, Dudek FE. Egg-laying hormone: direct action on the ovotestis of Aplysia. General Comparative Endocrinology. 1983;52:134–141. doi: 10.1016/0016-6480(83)90166-1. [DOI] [PubMed] [Google Scholar]
- Scott RH, Sutton KG, Griffin A, Stapleton SR, Currie KPM. Aspects of calcium-activated chloride currents: a neuronal perspective. Pharmacological Therapeutics. 1995;66:535–565. doi: 10.1016/0163-7258(95)00018-c. [DOI] [PubMed] [Google Scholar]
- Seidler NW, Jona I, Vegh M, Martonosi A. Cyclopiazonic acid is a specific inhibitor of the Ca2+-ATPase of sarcoplasmic reticulum. Journal of Biological Chemistry. 1989;264:17816–17823. [PubMed] [Google Scholar]
- Simpson PB, Challiss RAJ, Nahorski SR. Neuronal Ca2+ stores: activation and function. Trends in Neurosciences. 1995;18:299–306. doi: 10.1016/0166-2236(95)93919-o. [DOI] [PubMed] [Google Scholar]
- Sobel EC, Tank DW. In vivo Ca2+ dynamics in a cricket auditory neuron: an example of chemical computation. Science. 1994;263:823–826. doi: 10.1126/science.263.5148.823. [DOI] [PubMed] [Google Scholar]
- Stafstrom CE, Schwindt PC, Chubb MC, Crill WE. Properties of persistent sodium conductance and calcium conductance of layer V neurons from cat sensorimotor cortex in vitro. Journal of Neurophysiology. 1985;53:153–170. doi: 10.1152/jn.1985.53.1.153. [DOI] [PubMed] [Google Scholar]
- Stanley EF. The calcium channel and the organization of the presynaptic transmitter release face. Trends in Neurosciences. 1997;21:404–409. doi: 10.1016/s0166-2236(97)01091-6. [DOI] [PubMed] [Google Scholar]
- Strong JA, Fox AP, Tsien RW, Kaczmarek LK. Stimulation of protein kinase C recruits covert calcium channels in Aplysia bag cell neurons. Nature. 1987;325:714–717. doi: 10.1038/325714a0. [DOI] [PubMed] [Google Scholar]
- Swandulla D, Lux HD. Activation of a nonspecific cation conductance by intracellular Ca2+ elevation in bursting pacemaker neurons of Helix pomatia. Journal of Neurophysiology. 1985;54:1430–1443. doi: 10.1152/jn.1985.54.6.1430. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorndran C, Minta A, Poenie M. New fluorescent calcium indicators designed for cytosolic retention or measuring calcium near membranes. Biophysical Journal. 1995;69:2112–2124. doi: 10.1016/S0006-3495(95)80082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GF, Kaczmarek LK. Mode-switching of a voltage-gated cation channel is mediated by a protein kinase A-regulated tyrosine phosphatase. Nature. 1993;366:433–438. doi: 10.1038/366433a0. [DOI] [PubMed] [Google Scholar]
- Wilson GF, Magoski NS, Kaczmarek LK. Modulation of a calcium-sensitive nonspecific cation channel by ATP via a closely-associated protein kinase. Proceedings of the National Academy of Sciences of the USA. 1998;95:10938–10943. doi: 10.1073/pnas.95.18.10938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson GF, Richardson FC, Fisher TE, Olivera BM, Kaczmarek LK. Identification and characterization of a Ca2+-sensitive nonspecific cation channel underlying prolonged repetitive firing in Aplysia neurons. Journal of Neuroscience. 1996;16:3661–3671. doi: 10.1523/JNEUROSCI.16-11-03661.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson WA, Wachtel H. Negative resistance characteristic essential for the maintenance of slow oscillations in bursting neurons. Science. 1974;186:932–934. doi: 10.1126/science.186.4167.932. [DOI] [PubMed] [Google Scholar]