

Abstract
The rapidly activating delayed rectifier potassium current, IKr, was studied in guinea-pig ventricular myocytes in the presence of thiopentone, which blocks the more slowly activating component of the delayed rectifier potassium current, IKs, and using whole cell perforated patch clamp or switched voltage clamp with sharp electrodes to minimise intracellular dialysis.
Activation of protein kinase A (PKA) by isoprenaline or forskolin caused an increase in IKr tail currents. Following a 300 ms depolarising step to +20 mV, mean tail current amplitude was increased 47 ± 12% by isoprenaline, and 73 ± 13% by forskolin. No increase in IKr was observed when IKr was studied using whole cell ruptured patch clamp and there was no change in the reversal potential of IKr in the presence of isoprenaline.
The rectification of the current sensitive to E4031, a selective IKr blocker, was markedly reduced in the presence of isoprenaline and the region of negative slope was absent. This is consistent with a reduction in the inactivation of IKr and was supported by the finding that IKr, in the presence of isoprenaline, was somewhat less sensitive to block. E4031 (5 μm) blocked only 81 ± 5% of IKr in the presence of isoprenaline compared to 100 ± 0% in control.
The forskolin- and isoprenaline-induced increases in IKr were inhibited by staurosporine and by the selective protein kinase C (PKC) inhibitor bisindolylmaleimide I. Direct activation of PKC by phorbol dibutyrate increased IKr tail currents by 24 ± 5%. Both the isoprenaline- and forskolin-induced increases in IKr were inhibited when calcium entry was reduced by block of ICa with nifedipine or when myocytes were pre-incubated in BAPTA-AM.
The selective PKA inhibitor KT5720 prevented the isoprenaline-induced increase in IKr only when the increase in ICa was also suppressed.
These data show a novel mechanism of regulation of IKr by PKC and this kinase was activated by β-adrenoceptor stimulation. IKr seems to be enhanced through a reduction in the C-type inactivation which underlies the rectification of the channel and such a mechanism may occur in other channels with this type of inactivation.
Delayed rectifier potassium currents (IK) in the heart play an important role in the initiation of repolarisation and in determining the duration of the cardiac action potential. Two components of IK have been identified in many species: a rapidly activating current, IKr, and a more slowly activating current, IKs, and these can be separated by their activation kinetics and pharmacology (Sanguinetti & Jurkiewicz, 1990). IKr is sensitive to block by methanesulfonanilide class III antiarrhythmic drugs, such as E4031, and shows marked inward rectification which is thought to occur through C-type inactivation (Schönherr & Heinemann, 1996; Smith et al. 1996; Spector et al. 1996). In contrast, the slowly activating component, IKs, is not blocked by methanesulfonanilide drugs and shows no inactivation. These two currents are carried through distinct channels which are important targets for antiarrhythmic drugs and a decrease in IKr caused by class III drugs is thought to help prevent arrhythmias. However, excessive block of IKr can be pro-arrhythmic, causing long-QT syndrome (LQTS), a form of which can also be inherited. Chromosome 7-linked LQTS has recently been shown to be caused by mutations in HERG (human ether-à-go-go-related gene) (Curran et al. 1995), the gene thought to encode the human IKr channel (Sanguinetti et al. 1995). Thus a decrease in IKr magnitude, either by block with drugs or by mutations in HERG, can increase the risk of arrhythmias and these findings demonstrate the pivotal role of IKr in the normal repolarisation of human ventricular myocardium.
Many potassium currents are regulated by neurotransmitters or hormones and this is important in the regulation of cardiac function. IKs has been shown to be regulated by β-adrenoceptor stimulation which leads to PKA activation and phosphorylation of the channel (Walsh & Kass, 1988). Activation of PKC also increased IKs (Walsh & Kass, 1991) and the current can be regulated by calcium even in the absence of adrenoceptor stimulation (Tohse, 1990). In contrast, IKr has not been shown to be regulated by β-adrenoceptor stimulation despite the presence of putative phosphorylation sites for PKA and PKC on the HERG channel protein and a region homologous to the cAMP-binding site of EAG (ether-à-go-go) channels. Thus, there are no reports of a regulatory pathway which increases HERG channel currents or IKr in the heart and thereby increase this cardiac repolarisation current.
Previous electrophysiological studies of IKr have commonly used whole cell ruptured patch techniques in which there is significant dialysis of the myocytes and frequently the intracellular calcium is buffered by calcium chelators or entry is blocked by calcium channel blockers. In the present study we used more physiological methods to study the possible regulation of IKr, namely perforated patch clamp and switched voltage clamp with sharp electrodes and here we show that, under these conditions, IKr is substantially enhanced by activation of PKC and this kinase was activated by isoprenaline and forskolin, compounds usually expected to stimulate the kinase A pathway.
METHODS
Isolation of ventricular myocytes
Single ventricular myocytes were isolated daily from the guinea-pig heart by a technique previously described (Powell et al. 1980). Briefly, male guinea-pigs (0.5-0.7 kg) were killed by stunning followed by cervical dislocation, in accordance with Schedule 1 of the Animals (Scientific Procedures) Act 1986. The heart was rapidly removed, cannulated at the aorta and perfused with a zero calcium solution containing (mM): NaCl 137, KCl 5, NaHCO3 12, glucose 5, sodium pyruvate 1, NaHPO4 0.4, MgCl 1, NaOH 1, EGTA 0.1, pH 7.4, gassed with 95% O2-5% CO2. After 3 min perfusion this was replaced with 50 ml of a solution of the same composition with the exception of the EGTA and to which was added 0.2 mM CaCl2 and 40 mg of collagenase (type I, Worthington Biochemicals) and this was recirculated through the heart for up to 36 min (12 min (g heart weight)−1). During the perfusion, 3 × 20 μl aliquots of CaCl2 (0.1 M) were added at intervals to the perfusate. Following perfusion, the heart was cut down and the atria were removed. The ventricles were roughly chopped and shaken in a water bath (34°C) before filtering through a mesh and being allowed to settle. Cells were stored at room temperature in Dulbecco's modified Eagle's medium (DMEM) (Life Technologies).
Electrophysiology
Ventricular myocytes were allowed to settle on the coverslip base of the bath before being superfused with a physiological salt solution containing (mM): NaCl 118.5, NaHCO3 14.5, KCl 4.2, KH2PO4 1.18, MgSO4 1.18, glucose 11.1, CaCl2 2.5; gassed with 95% O2-5% CO2, pH 7.4. All experiments were carried out at 36°C.
IKr was studied in single cells using three methods. Whole cell perforated patch clamp and switched electrode voltage clamp (SEVC) with sharp electrodes were used to minimise cell dialysis and where dialysis was required, conventional whole cell ruptured patch clamp was used. For perforated patch clamp, pipettes contained (mM): KCl 150, MgCl2 5, Hepes, 3, K2ATP 5 (pH 7.2 with KOH) and amphotericin B (240 μg ml−1), and had resistances of 2–5 MΩ. Following seal formation, access developed within 2–5 min and series resistance was compensated up to 80% (Axopatch 200, Axon Instruments). With the ruptured patch clamp method, similar pipettes were filled with a solution containing (mM): potassium aspartate 140, NaCl 5, MgCl2 5.5, Hepes 5, K2ATP 5, pH 7.2 with KOH. Sharp electrodes used for SEVC contained 1 M KCH3SO4 and 10 mM KCl and had resistances of 30–50 MΩ. Axoclamp 2B was used for SEVC with a switching frequency of 4–5 kHz.
IKs was blocked in all experiments by the addition of 300 μM thiopentone to all solutions superfusing the cells (Takahashi & Terrar, 1995; Heath & Terrar, 1996). IKr was activated by step depolarisations to a series of potentials (-30 to +40 mV, 10 mV increments) from a holding potential of −40 mV and measured as deactivating tail currents at −40 mV or as the E4031-sensitive current. IKr was also studied using a protocol modified from Carmeliet (1992) and previously used to separate IKr and IKs (see Heath & Terrar, 1996), in which a depolarising step to +40 mV was applied for 600 ms to activate IK and the membrane potential was then repolarised to −10 mV for 1 s before further repolarisation to −40 mV. The current deactivating at −10 mV consists predominantly of IKs and that at −40 mV is IKr. Under the conditions of these experiments, no deactivating current was observed at −10 mV consistent with block of IKs by thiopentone.
Drugs
Isoprenaline was dissolved daily in water. The following were prepared as stock solutions and stored at either 4°C or −20°C as necessary: E4031 (Eisai, Tokyo, Japan) was dissolved in water, forskolin, phorbol dibutyrate, BAPTA-AM (Sigma), staurosporine (Alomone Labs, Israel), bisindolylmaleimide I and KT5720 (Calbiochem) in DMSO and nifedipine (Sigma) in ethanol. The highest final concentration of DMSO in the superfusate was 0.1% and ethanol was 0.025%. Thiopentone was dissolved directly into the physiological salt solution (pH 7.4 with NaOH).
Data analysis
Data were collected using pCLAMP 7 (Axon Instruments) and plotted using Origin 6.0 (Microcal). Data are means ±s.e.m. The voltage-dependent activation of IKr was fitted using the Boltzmann equation and data were analysed using Student's t test for paired or unpaired data as appropriate with P < 0.05 taken to indicate statistical significance.
RESULTS
Effect of isoprenaline and forskolin on IKr
The voltage-dependent activation of IKr studied using SEVC or perforated patch clamp was similar to that previously reported in guinea-pig ventricular cells (Sanguinetti & Jurkiewicz, 1990; Heath & Terrar, 1996) with tail currents reaching a peak amplitude at about +20 mV. Activation of the PKA pathway by isoprenaline to stimulate β-adrenoceptors, or by forskolin to directly activate adenylyl cyclase, resulted in a marked increase in IKr tail currents and the mean data are shown in Fig. 1A–D. The forskolin-induced enhancement of IKr was compared using SEVC and perforated patch clamp techniques. IKr activated by a 300 ms step depolarisation to +20 mV was increased in the presence of 5 μM forskolin by 73 ± 13% (n = 7, P < 0.05, Fig. 1A) when using SEVC and by 59 ± 14% (n = 5, P < 0.05, Fig. 1B) with perforated patch clamp and these effects with the two different methods were not significantly different (n = 5 and 7; P > 0.05).
Figure 1. The effect of forskolin and isoprenaline on IKr using SEVC or perforated patch clamp.
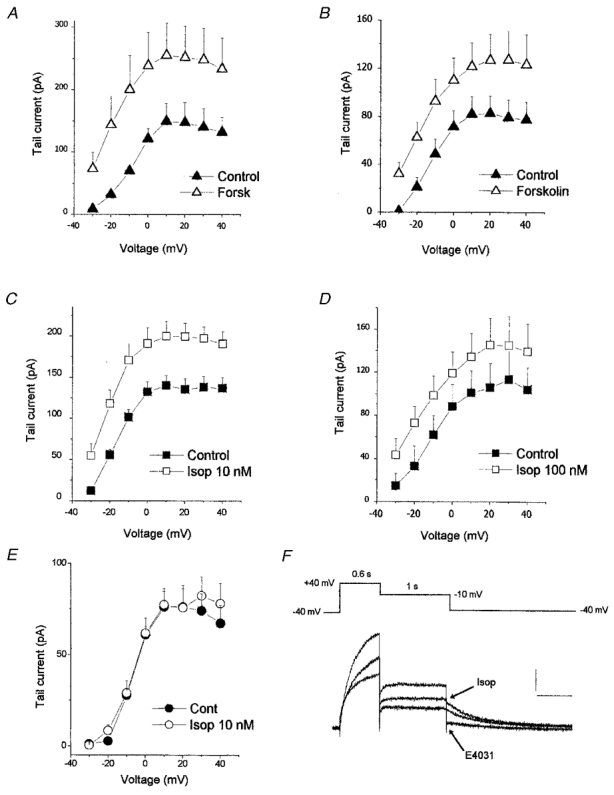
All experiments were carried out in the continuous presence of 300 μM thiopentone and in A–E, IKr was activated by 300 ms step depolarisations to the voltages indicated. A and B: mean IKr tail current amplitudes in control and after exposure to forskolin (5 μM) using SEVC (A) and perforated patch clamp (B). C and D: effect of isoprenaline (10 and 100 nM) on IKr tail current amplitude recorded using SEVC (C) or perforated patch clamp (D). E, mean IKr tail current amplitudes in control and in the presence of 10 nM isoprenaline recorded using the ‘ruptured’ whole cell patch clamp method. F, current records (SEVC) showing in control (middle trace) the absence of IKs (no tail current deactivating at −10 mV) and the presence of IKr (tail current deactivating at −40 mV). The top trace shows the effect of 10 nM isoprenaline, which increased IKr, and this current was reduced by 5 μM E4031 (bottom trace). Calibration bars are 200 pA and 500 ms.
Isoprenaline also caused a substantial increase in IKr when studied with both SEVC and perforated patch clamp techniques. Following a 300 ms step to +20 mV, isoprenaline (10 nM) increased IKr from 135 ± 13 pA (n = 10) in control to 199 ± 17 pA (n = 10, P < 0.05, SEVC), an increase of 47 ± 12% (Fig. 1C). This effect was no greater when 100 nM isoprenaline was applied, which caused a 45 ± 18% (n = 5; P < 0.05; perforated patch clamp) increase in IKr at +20 mV (Fig. 1D). Figure 1E shows the effect of 10 nM isoprenaline on IKr studied using the conventional whole cell ‘ruptured’ patch method and under these conditions there was no increase in IKr (n = 4). Similar results were obtained when IKr was activated using 1 s depolarising pulses and some of these data are shown later. All the following experiments were carried out using SEVC unless otherwise indicated.
Using the second pulse protocol in which a depolarising step to +40 mV was applied for 600 ms to activate IK and the membrane potential was then repolarised to −10 mV for 1 s before further repolarisation to −40 mV (see Methods), the effect of isoprenaline on IKr was also studied. The current deactivating at −10 mV consists predominantly of IKs and that at −40 mV is IKr. However, in these experiments, there was no current deactivating at −10 mV, consistent with complete block of IKs by thiopentone, and at −40 mV the deactivating tail current consisted entirely of IKr (Fig. 1F, middle trace). In the presence of 10 nM isoprenaline, the tail current deactivating at −40 mV (IKr) was markedly increased and there was still no tail current observed at −10 mV, consistent with the continued block of IKs. Following the addition of 5 μM E4031, a selective blocker of IKr, the current deactivating at −40 mV was reduced (Fig. 1F).
A Boltzmann equation was fitted to the voltage-dependent activation of IKr tail currents and in the presence of isoprenaline and forskolin there was a small shift in V½ to a more negative potential and change in the slope. In control, V½ was −17 ± 1 mV and the slope was 6 ± 0.3 mV, and in the presence of isoprenaline V½ was shifted to −22 ± 1 mV and the slope was 7 ± 0.6 mV (n = 10; P < 0.05; Fig. 2B). Similarly, exposure to 5 μM forskolin shifted V½ of IKr from −12.5 ± 2 mV in control to −22.6 ± 4 mV and changed the slope from 5.9 ± 1 mV in control to 7.5 ± 1 mV (n = 8, P < 0.05; Fig. 2A).
Figure 2. The effect of forskolin and isoprenaline on the voltage-dependent activation of IKr tail currents.
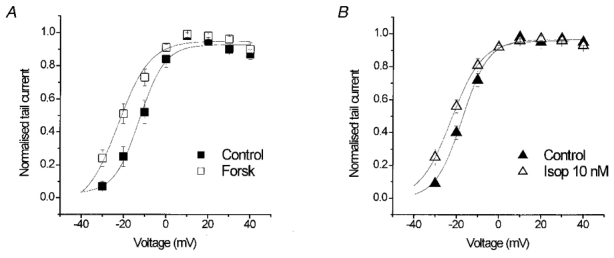
Data were recorded using SEVC and were fitted with Boltzmann equations. A, effect of forskolin (5 μM) on IKr activation (n = 8). B, effect of isoprenaline on IKr activation (n = 10).
Mechanism of IKr enhancement by isoprenaline and forskolin
IKr was also studied as the current sensitive to block by E4031 in order to isolate the current during the depolarising pulse (time-dependent pulse current) from other currents in the cardiac myocytes such as calcium and chloride currents or Na+-Ca2+ exchange current (see Heath & Terrar, 1996). Previous studies have shown that 5 μM E4031 completely blocks IKr in guinea-pig ventricular myocytes (Sanguinetti & Jurkiewicz, 1990) and this concentration was used here to compare the IKr current in control conditions and after enhancement by activation of the PKA pathway by isoprenaline (10 nM). Figure 3A shows the E4031-sensitive currents obtained by subtraction in the absence and presence of 10 nM isoprenaline. These currents are averages of the drug-sensitive current from four or seven cells and in control, the current displays the characteristic inactivation during the depolarising step, which is most prominent during steps to the most positive potentials. In contrast, in the presence of isoprenaline, the current during the pulse shows no such inactivation and is increased in amplitude. The mean data in Fig. 3B show the current-voltage curve for the time-dependent E4031-sensitive current measured at the end of 1 s depolarising steps which in control showed typical rectification with a region of negative slope at potentials positive to approximately 0 mV. However, in the presence of 10 nM isoprenaline, there was no negative slope in the current-voltage curve and instead the current was found to continue increasing at positive potentials without reaching a maximum. Therefore it seems that in the presence of isoprenaline, IKr showed less rectification and this is consistent with a reduction in the inactivation of IKr.
Figure 3. Isoprenaline reduces the rectification of IKr.
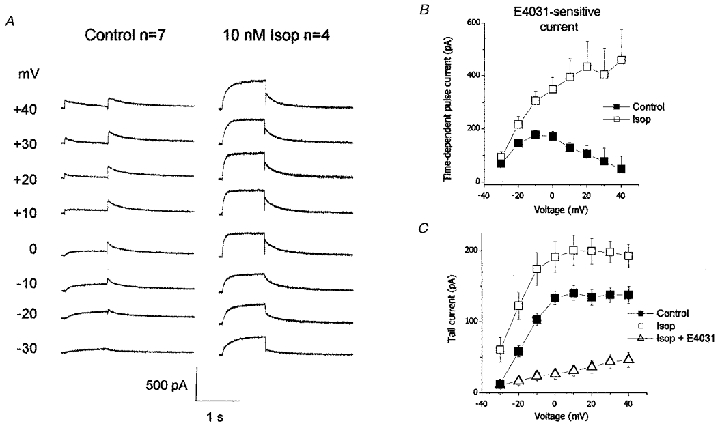
A, E4031-sensitive current (5 μM E4031 throughout) obtained by subtraction in control conditions (left) showing the typical rectification of IKr, particularly at positive potentials, and in the presence of 10 nM isoprenaline (right) showing the reduction in rectification and the increase in current amplitude. Currents were activated by 1 s step depolarisations to the voltage indicated on the left. B, the effect of 10 nM isoprenaline on the E4031-sensitive time-dependent pulse current activated during the depolarising steps (n = 4 or 7) again showing the reduction in rectification of the current. C, tail current amplitudes in control, after 10 nM isoprenaline and in the presence of isoprenaline and E4031, which failed to completely suppress the current (n = 8).
As described above, 5 μM E4031 was used to confirm that the current enhanced by isoprenaline and forskolin was indeed IKr and also to study the drug-sensitive currents. In the absence of kinase activation, 5 μM E4031 completely suppressed IKr at all potentials (100 ± 0%, n = 8), but in the presence of isoprenaline, the increased IKr tail currents were reduced by only 81 ± 5% at +20 mV (n = 8; Fig. 3C). The remaining current in the presence of E4031 was thought to be residual IKr and not IKs since, as shown in Fig. 1F, there was no current deactivating at −10 mV following a step to +40 mV, which would be expected if IKs were present, and previous work has shown that thiopentone is effective at blocking IKs (Heath & Terrar, 1996). Instead it seems possible that the IKr channels modified by isoprenaline and forskolin are somewhat less sensitive to block by E4031.
Since kinase activation also enhances IKs and we are relying on thiopentone to completely suppress IKs under these conditions, we compared the extent of block of IKs by thiopentone in the absence and presence of forskolin. We have previously shown that thiopentone at a concentration of 100 μM completely suppressed IKs in guinea-pig ventricular cells, with no detectable effect on IKr. When total IK tail current amplitude was studied (without blocking IKr) the amplitude of the tail current activated by a 400 ms step to +40 mV was 667 ± 122 pA (n = 6) and this was reduced by 57 ± 7% by 100 μM thiopentone. The remaining current had the characteristics of IKr (e.g. see Fig. 1F in Heath & Terrar, 1996). In a separate set of experiments in the presence of 1 μM forskolin, the amplitude of the total IK tail current was 1407 ± 197 pA (n = 3) and under these conditions 100 μM thiopentone reduced the current by 62 ± 6% (current activated by 400 ms depolarisation to +40 mV from a holding potential of −40 mV using SEVC; data not shown). Therefore it seems likely that the blocking effect of thiopentone was not substantially affected by the enhancement of IKs by forskolin. In the experiments presented here we used thiopentone at a concentration three times that which we have previously shown to completely block IKs to try to ensure maximum block of the current.
The effect of isoprenaline on the reversal potential of IKr was also studied using E4031 to isolate the current. E4031-sensitive tail currents were measured at test potentials in the range −40 to −100 mV following a 300 ms pre-pulse to +20 mV to activate IKr. The E4031-sensitive current was measured in two separate groups of cells, one in control conditions (n = 4) and one in the presence of 10 nM isoprenaline (n = 4). IKr reversal potential was found to be the same in the absence and presence of 10 nM isoprenaline. In control conditions, the reversal potential was −81.3 ± 3 mV and in isoprenaline, −80.6 ± 3 mV (P > 0.05).
Intracellular pathway for isoprenaline- and forskolin-induced enhancement of IKr
To investigate the intracellular pathway underlying the observed enhancement of IKr described above, the effect of inhibitors of PKA and PKC were tested. Firstly, the effect of staurosporine, a non-selective kinase inhibitor, was tested and when this was applied to the cells prior to exposure to forskolin, it caused a decrease in IKr tail currents. In control, the mean IKr tail current activated by a 300 ms step to +20 mV was 144 ± 27 pA and this was reduced to 75 ± 23 pA following 3 min exposure to 3 μM staurosporine (n = 4; P < 0.05; Fig. 4A). The subsequent addition of 5 μM forskolin did not result in an increase in IKr tails. In fact there was a trend for a further decrease and the mean IKr tail current in forskolin and staurosporine was 35 ± 10 pA, but this further reduction was not statistically significant at all potentials (at +20 mV P > 0.05; n = 4; Fig. 4A).
Figure 4. The role of protein kinases in the forskolin- and isoprenaline-induced increase in IKr.
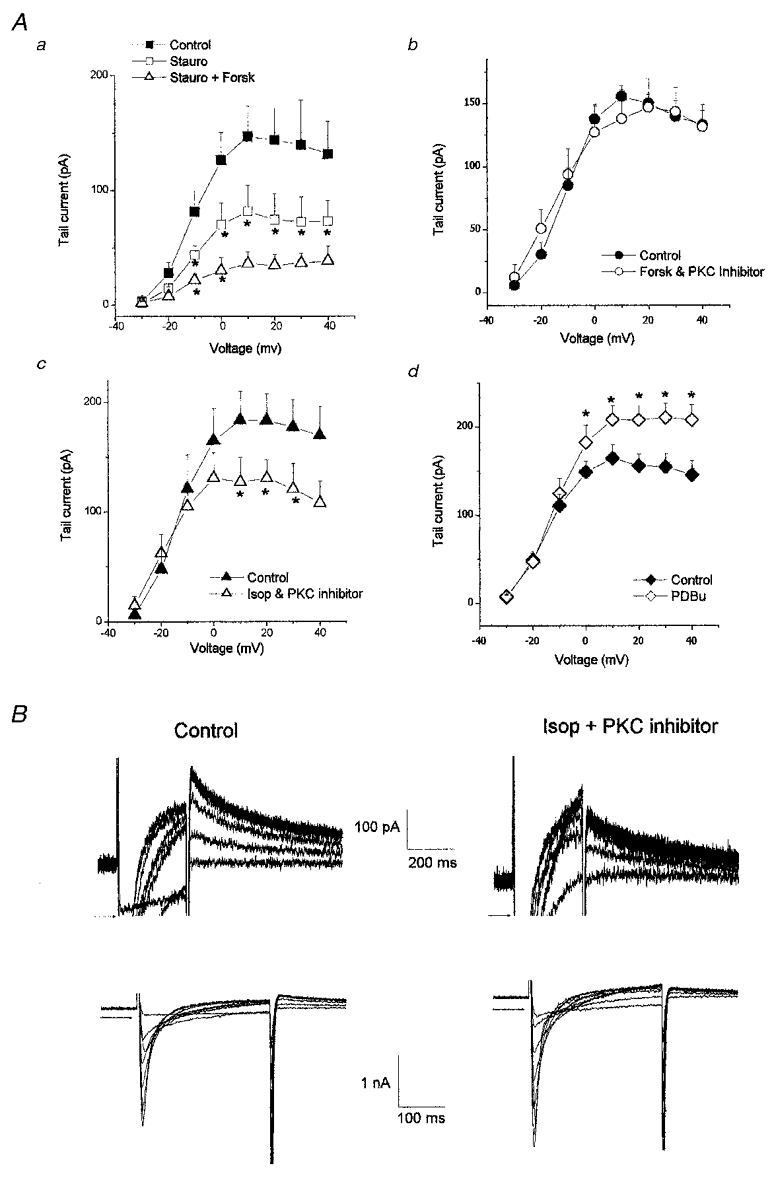
Aa, effect of 3 μM staurosporine on IKr tail current amplitude and the effect of forskolin (5 μM) in the continued presence of staurosporine (n = 4). The protein kinase C inhibitor bisindolylmaleimide I (100 nM) completely inhibited the increase in IKr induced by forskolin (Ab) and isoprenaline (Ac). Ad, phorbol dibutyrate (100 nM) increased IKr tail current amplitude. B, the effect of isoprenaline on IKr (upper traces) and ICa (lower traces) in the presence of bisindolylmaleimide I (100 nM). IKr and ICa in B were recorded from the same cells and the arrows indicate 100 pA above zero current in the upper figures and zero current in the lower figures. *P < 0.05.
These data are consistent with a role for kinases in the isoprenaline- and forskolin-induced enhancement of IKr and to investigate this further we tested the effect of selective inhibitors of PKA and PKC. We found that both the forskolin- and isoprenaline-induced increases in IKr were completely inhibited by the selective PKC inhibitor bisindolylmaleimide I (100 nM). Exposure to forskolin in the presence of bisindolylmaleimide I resulted in no change in IKr at any potential (Fig. 4Ab), and exposure to isoprenaline in the presence of bisindolylmaleimide I actually resulted in a decrease of 28 ± 3% in IKr (+20 mV step, n = 3, P < 0.05; Fig. 4Ac). Under these conditions, the increase in calcium current induced by isoprenaline was maintained and typical calcium currents are shown in Fig. 4B (lower panel) together with IKr tail currents recorded from the same cells (upper panel). These findings are consistent with an essential role for the activation of PKC in the observed enhancement of IKr and to investigate this further, cells were exposed to phorbol dibutyrate (PDBu) to directly activate PKC. In the presence of 100 nM PDBu, IKr tail currents were increased by 24 ± 5% (300 ms depolarisation to +20 mV; n = 8, P < 0.05) and the mean data are shown in Fig. 4Ad.
The current traces in Fig. 4B (upper panel) also show that although there was no increase in IKr tail currents when isoprenaline was applied in the presence of the PKC inhibitor, there was an increase in outward current recorded during the depolarisation (Fig. 4B, upper panel). It is known that PKA activates and increases other currents in the heart such as chloride current and Na+-Ca2+ exchange current (Harvey & Hume, 1989; Perchenet et al. 1998) and a similar effect was seen in experiments described later and shown in Fig. 7. These currents were not blocked in our experiments and therefore were expected to contribute to current during the depolarisation but not to the tail currents or the E4031-sensitive currents.
Figure 7. Effect of BAPTA on IKr in the absence and presence of isoprenaline.
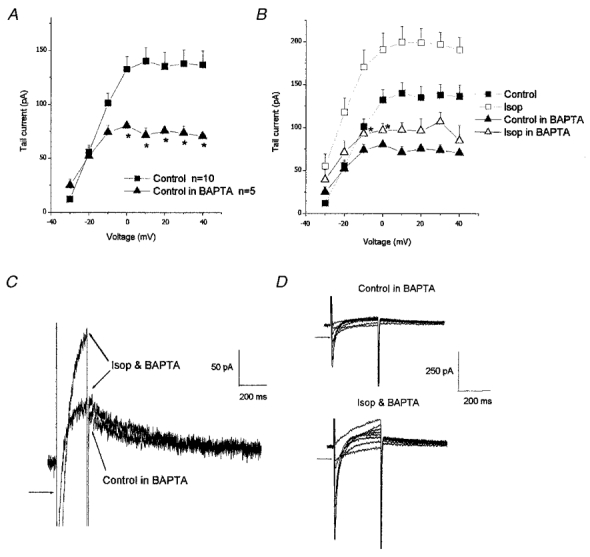
A, comparison of the mean IKr tail current amplitudes in control and in myocytes pre-incubated in BAPTA-AM (10 μM). B, effect of 10 nM isoprenaline on IKr in cells pre-incubated in BAPTA-AM plotted with data from cells not exposed to BAPTA (control data from Fig. 1C). C and D: typical currents showing the effect of 10 nM isoprenaline in the continuous presence of BAPTA on IKr (C) and ICa (D). These currents were recorded from the same cell. Arrow indicates 100 pA above zero current level (C) and zero level in D. *P < 0.05.
Although the effect of isoprenaline and forskolin to enhance IKr was completely prevented by the PKC inhibitor bisindolylmaleimide I, we also investigated the possible role of PKA, which is expected to be activated by isoprenaline and forskolin, using the selective PKA inhibitor KT5720. At 300 nM, KT5720 did not prevent the increase in IKr induced by 10 nM isoprenaline. In the presence of isoprenaline and KT5720, IKr tail currents following a 300 ms step to +20 mV were increased from 129 ± 21 pA to 206 ± 42 pA, an increase of 58 ± 11% (n = 4, P < 0.05). However, at 300 nM, KT5720 also failed to inhibit the isoprenaline-induced increase in the L-type calcium current (ICa) which accompanied the increase in IKr, consistent with incomplete inhibition of PKA. In the presence of 10 nM isoprenaline alone, peak ICa activated by a step to 0 mV was increased by 196 ± 26% (n = 8) and this was not significantly different from the increase of 155 ± 30% recorded in the presence of 300 nM KT5720 (n = 4; P > 0.05). Therefore we used 1 μM KT5720, at which concentration both the increases in ICa and IKr induced by isoprenaline were inhibited. In control, the mean IKr tail current amplitude following a step to +20 mV was 121 ± 15 pA, and in the presence of 10 nM isoprenaline and KT5720 this was unchanged at 142 ± 10 pA (n = 4, P > 0.05). In the same cells the peak ICa recorded during a step to 0 mV was −881 ± 378 pA in control, and in the presence of isoprenaline and KT5720 ICa at 0 mV was −1162 ± 400 pA (n = 4; P > 0.05). These data are summarised in Fig. 5.
Figure 5. Effect of isoprenaline on IKr and ICa in the presence of PKA inhibition.
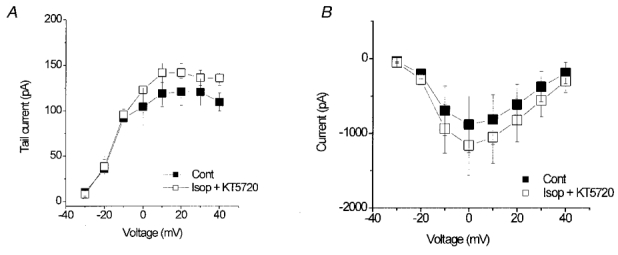
A, mean IKr tail current amplitudes in control and in the presence of 10 nM isoprenaline and 1 μM KT5720 (n = 4). B, mean peak ICa amplitudes in control and in the presence of 10 nM isoprenaline and 1 μM KT5720 (n = 4).
The data presented above show that forskolin and isoprenaline are capable of activating both PKA and PKC in ventricular myocytes. PKC is usually thought to be activated by diacylglycerol and, with some forms of the enzyme, by calcium. Therefore we investigated the mechanism by which PKC activation may occur and one possibility is that PKC was activated simply through an increase in intracellular calcium as a result of the PKA-mediated increase in L-type ICa. This was tested using nifedipine to inhibit L-type ICa and BAPTA to chelate intracellular calcium.
In separate sets of experiments, both forskolin- and isoprenaline-induced increases in IKr were inhibited when the drugs were applied in the continuous presence of nifedipine to block ICa. When cells were exposed to 5 μM forskolin in the presence of 0.2 μM nifedipine there was no significant increase in IKr (n = 3, P > 0.05, data not shown) and similarly, in the continuous presence of 5 μM nifedipine, 10 nM isoprenaline failed to increase IKr (Fig. 6B a). The E4031-sensitive current in control and in the presence of both nifedipine and isoprenaline is compared in Fig. 6A. This shows that there was no change in the rectification of IKr with isoprenaline when nifedipine was present and this contrasts with the currents shown in Fig. 3A. Although the current during the depolarising steps in the presence of nifedipine and isoprenaline shows no reduction in inactivation, the time course is slightly different compared to the control and there may still be some regulation of IKr occurring under these conditions, but without an increase in the current. The mean data showing no change in tail current amplitude are shown in Fig. 6B a.
Figure 6. The effect of isoprenaline on IKr in the presence of ICa block by nifedipine.
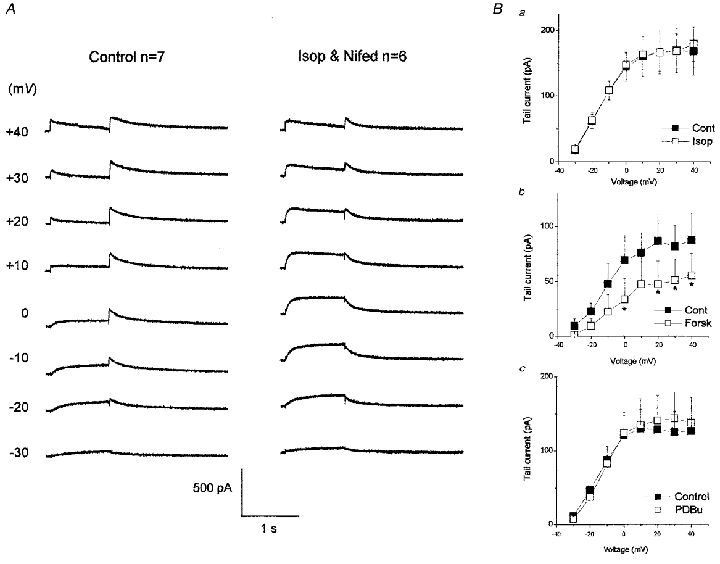
A, E4031-sensitive currents (5 μM) in control conditions (left, same data as Fig. 3A) and in the presence of 10 nM isoprenaline and 5 μM nifedipine. Currents were activated by 1 s step depolarisations to the voltage indicated on the left. B, the effect of 10 nM isoprenaline (a), 5 μM forskolin (b) and 100 nM PDBu (c) on IKr in the presence of 5 μM nifedipine. *P < 0.05.
Exposure to 5 μM forskolin in the presence of 5 μM nifedipine not only inhibited the increase in IKr, but caused a decrease in tail current amplitude. IKr tails deactivating following a 300 ms step to +20 mV were decreased by 50 ± 9% (P < 0.05, n = 3) by forskolin under these conditions of complete L-type calcium channel block (Fig. 6B b).
The effect of inhibition of calcium entry on the effect of PDBu to enhance IKr was also tested. In the presence of 5 μM nifedipine, PDBu (100 nM) failed to increase IKr (Fig. 6B c) and this is consistent with the essential requirement for an increase in cytosolic calcium in the activation of the PKC enzyme involved in the forskolin- and isoprenaline-mediated enhancement of IKr.
The role of intracellular calcium was further investigated using the membrane-permeant (acetoxymethyl ester) form of the calcium chelator BAPTA, BAPTA-AM. Ventricular myocytes were incubated in 10 μM BAPTA-AM for at least 2 h prior to experiments and were then superfused with a solution containing the same concentration of BAPTA-AM. Following the incubation in BAPTA-AM, IKr currents were found to be significantly smaller in amplitude compared to cells not exposed to BAPTA-AM (P < 0.05, n = 5–10, unpaired t test; Fig. 7A). Subsequent exposure of these cells to 10 nM isoprenaline resulted in only a small increase in IKr which was statistically significant at only −10 and 0 mV. These data are summarised in Fig. 7B, which also shows the effect of isoprenaline in the absence of BAPTA for comparison (same data as shown in Fig. 1C). Figure 7C shows the effect of isoprenaline on a typical IKr tail current in the continuous presence of BAPTA and it is clear that there was little change in tail current amplitude. Under the conditions of these experiments, the enhancement of ICa by isoprenaline was maintained. Figure 7D shows the calcium current recorded from the same cell as the tail currents shown in panel C and shows the increase in ICa in the presence of isoprenaline and BAPTA.
DISCUSSION
The results of this study demonstrate a novel pathway by which PKC in ventricular myocytes is activated and one consequence of this is a substantial enhancement of IKr through a reduction in C-type inactivation.
The regulation of IKr causing increased current has not been shown before and it is clear that the experimental conditions are an important factor in demonstrating this regulation. Previous work on IKr in native cells has used more conventional ruptured patch clamp methods which allow dialysis of the cells and which probably lead to the washing out of important signalling molecules such as cAMP or protein kinase enzymes. Also, many studies using this technique dialyse the inside of the cell with calcium buffers which, in the light of the experiments with BAPTA presented here, might also suppress the enhancement of IKr. Another common feature of previous studies on IKr is the presence of calcium channel blockers to suppress L-type calcium current, and this was also shown here to inhibit the isoprenaline- and forskolin-induced enhancement of IKr. Therefore, it seems likely that the regulation of IKr which leads to an increase in the current may only be observed by using more physiological methods to study the current.
The enhancement of IKr described in this report was also not observed when the current was studied following HERG channel expression in Xenopus oocytes (Sanguinetti et al. 1995) and two recent studies found PKA activation to cause a decrease in HERG channel currents studied in oocytes (Barros et al. 1998; Kiehn et al. 1998). It is therefore possible that expression in the oocyte somehow inhibits this regulatory pathway. For example, perhaps the oocyte does not have the protein kinase required for HERG phosphorylation or perhaps an increase in intracellular calcium, which we have shown to be required for the activation of PKC, is also required in the oocyte for the enhancement of HERG currents. Alternatively, since the identification of additional channel subunits which associate with HERG, the minK-related peptides (Abbott et al. 1999), it is likely that HERG channels alone do not fully reconstitute native IKr and such additional subunits maybe necessary for the regulation of the channel.
Kiehn et al. (1998) also reported that IKr was decreased by PKA in guinea-pig ventricular cells. These experiments on guinea-pig ventricular cells were carried out using whole cell ruptured patch clamp with EGTA to buffer intracellular calcium and 10 μM nisoldipine to block the calcium current. Therefore it seems likely that this effect, at least in the guinea-pig ventricular cells, was due to the inhibition of PKC activation by the experimental conditions and is similar to the decreases in IKr we observed when we used nifedipine to inhibit ICa (Fig. 6Bb) or when PKC was inhibited by bisindolylmaleimide (Fig. 4Ac). Therefore it seems possible that in cardiac cells there are kinase processes which may regulate IKr to both enhance and decrease the current. However, it seems most likely that β-adrenoceptor stimulation, under physiological conditions, would result in an enhancement of IKr.
It is also possible that even in the absence of kinase activation there is some regulation of IKr in guinea-pig ventricular myocytes since we found that IKr was reduced by staurosporine before exposure to isoprenaline and in cells incubated in BAPTA-AM, the IKr amplitude was significantly smaller than the current recorded from cells not exposed to BAPTA. The exact mechanism of this regulation remains for future study.
The second important finding presented here is the mechanism by which IKr was enhanced by kinase activation. Analysis of the E4031-sensitive currents showed that the typical rectification of the current observed in control conditions was substantially reduced in the presence of isoprenaline and this is likely to have led to the increase in IKr. One interpretation of this reduction in rectification is that isoprenaline caused a decrease in C-type inactivation of the channel. Further support for this comes from the finding that the enhanced current in the presence of forskolin or isoprenaline was less sensitive to block by E4031, a feature which is also consistent with an alteration in C-type inactivation. Previous studies have shown that the class III methanesulfonanilide drugs may interact with the C-type inactivation process (Kiehn et al. 1996; Snyders & Chaudhary, 1996) and studies on mutant HERG channels in which inactivation has been removed have found that these IKr currents were also much less sensitive to block by class III methanesulfonanilide drugs (Wang et al. 1997; Ficker et al. 1998). For example, in the work of Wang et al., mutation of two amino acids (S631C and G628C) in the HERG channel resulted in the removal of inactivation. The currents recorded from these mutant channels following expression in Xenopus oocytes had similar characteristics to those shown here as E4031-sensitive currents in the presence of isoprenaline (Fig. 3A) and were also much less sensitive to block by E4031. Therefore it seems possible that the observed change in rectification and drug sensitivity of IKr in the guinea-pig ventricular cells occurs through a reduction in C-type inactivation.
Since isoprenaline and forskolin activate the PKA pathway, we expected to find that this pathway was regulating IKr through either a direct effect of cAMP or through activation of PKA. Consistent with a role for kinases in the enhancement of IKr, the effect of forskolin was inhibited by staurosporine, a non-selective kinase inhibitor. Surprisingly, however, both the forskolin- and isoprenaline-induced enhancement of IKr was completely inhibited by a selective inhibitor of PKC. This implies that the PKA pathway, both linked to β-adrenoceptors and downstream of the receptors, can also lead to activation of PKC in these cells. It is conceivable that isoprenaline may also have an effect on α-adrenoceptors which, although only present in a low density in the ventricle (Hescheler et al. 1988), are known to be linked to PKC activation. However, the results obtained with forskolin, which by-passes the receptor to directly activate adenylyl cyclase, were not significantly different from those obtained with isoprenaline and therefore this possibility seems unlikely.
Further evidence to support a role for PKC in this regulation of IKr was obtained in experiments using PDBu to activate PKC directly and in experiments in which increases in intracellular calcium were limited through either block of ICa or buffering of intracellular calcium by BAPTA. Exposure to PDBu enhanced IKr, but not to the same extent as isoprenaline or forskolin. One possible reason for this is that there was no accompanying increase in ICa in the myocytes when PDBu was applied and an increase in intracellular Ca2+ is usually required for the translocation of PKC enzymes from the cytosol to the membrane where the activation process is completed. Therefore, the effect of PDBu may reflect the activation of only those PKC enzymes already located close to the cell membrane. Also, there are many isoforms of PKC and we cannot be sure that PDBu activates the same enzymes as those that seem to be activated by isoprenaline and forskolin.
The mechanism by which PKC is activated by isoprenaline and forskolin is not clear, but the experiments with nifedipine and BAPTA show that an increase in intracellular calcium was essential. The simplest interpretation of this is that following activation of PKA by isoprenaline and forskolin, phosphorylation of L-type Ca2+ channels leads to an increase in intracellular calcium concentration. Such an increase in calcium may then, on its own, lead to the activation of calcium-sensitive PKC enzymes, even in the absence of the production of phospholipid derivatives such as diacylglycerol.
PKC mediates its cellular effects through phosphorylation of target proteins and has been shown previously to phosphorylate a variety of ion channels, thus altering their properties. For example, a recent study has shown that PKC phosphorylation of Kv3.4 channels reduced the N-type inactivation and it was thought that the negative charge of the phosphoserines was important in this process (Beck et al. 1998). C-type inactivation, such as that displayed by the IKr channel, is somewhat different from N-type inactivation and it is thought that the process involves a structural change in the external mouth of the pore (Lui et al. 1996). The amino acid mutations mentioned earlier which remove inactivation in HERG channels are both located in the outer mouth of the pore (Schönherr & Heinemann, 1996; Smith et al. 1996) and therefore, the data presented here are consistent with phosphorylation of either the channel, which has multiple putative PKC phosphorylation sites, or phosphorylation of a related subunit, and this influences the C-type inactivation process of IKr.
The evidence presented here for the regulation of IKr inactivation by phosphorylation also has implications for understanding the mechanism of C-type inactivation in general. As mentioned above, many studies have shown that C-type inactivation involved the closure of the external mouth of the pore and it is mutations in this area, such as the HERG-S631A mutation (Schönherr & Heinemann, 1996; Smith et al. 1996), which have been shown to influence this process. However, there is increasing evidence for the involvement of sites on the intracellular side of the channel. For example, as described above, the removal of C-type inactivation in HERG channels reduces the sensitivity of the channel to block by E4031, suggesting that E4031 interacts with sites involved in this process. However, it is likely that the class III methanesulfonanilide drugs such as E4031, dofetilide and MK-499 block IKr or HERG channels from the inside of the cell. With E4031, evidence for block of IKr channels from the inside of the cells was provided by experiments on rabbit ventricular myocytes in which block of single channel currents was observed with cell-attached recordings (Veldkamp et al. 1993). This is consistent with there being intracellular sites on the channel involved in C-type inactivation in addition to those already identified on extracellular parts of the channels. The findings presented here add further support to this theory since we have shown that phosphorylation of IKr or a related channel subunit can also alter C-type inactivation and such phosphorylation can only occur intracellularly.
Isoprenaline and forskolin may also change the activation of IKr since we observed a negative shift in the voltage-dependence of activation of about 5 mV and change in the slope and this may contribute to the effect of isoprenaline and forskolin to enhance IKr. This shift in activation is similar to the enhancement of IKs by PKA in guinea-pig ventricular cells (Walsh & Kass, 1991). In our experiments, we found that inhibition of PKA with KT5720 also reduced the isoprenaline-induced increase in IKr, but this only occurred when the increase in ICa was also suppressed. Whilst it is possible that PKA was only significantly suppressed when the isoprenaline-induced increase in ICa was also inhibited, it is also possible that it was the suppression of the increase in ICa which prevented the isoprenaline-induced increase in IKr, as shown by the experiments with nifedipine and BAPTA, rather than the inhibition of PKA itself. Because of the dependence of the response to isoprenaline on an increase in ICa, it is impossible to distinguish between these two possibilities based on the experiments with KT5720. However, since we were able to completely suppress the isoprenaline- and forskolin-induced increase in IKr with a selective inhibitor of PKC, it seems likely that the major part of this effect is mediated directly through this protein kinase rather than PKA.
The physiological consequences of an increase in IKr following β-adrenoceptor stimulation are also of great interest. During β-adrenoceptor stimulation, changes in the action potential duration (APD) depend on the balance between the increase in inward current such as L-type calcium current and the opposing increase in outward current such as IKs and now also IKr. The data presented here show that IKr was increased with relatively low concentrations of isoprenaline consistent with this regulation of IKr occurring physiologically in the heart. One might expect the increased IKr to contribute more current during the plateau of the action potential since there will be less rectification at positive potentials and functionally this may contribute to any shortening of the APD. In support of this, one recent study has been carried out on the mutant HERG channel which has reduced inactivation, the HERG-S631A channel, expressed in cultured cells. Hancox et al. (1998b) measured HERG-S631A currents during action potential clamp experiments and found that the voltage and time dependence of HERG-S631A current with reduced inactivation was markedly different to wild-type channel currents (Hancox et al. 1998a). HERG-S631A currents developed much earlier during the action potential and reached a maximum amplitude at much more positive potentials. Therefore it seems likely that β-adrenoceptor stimulation in the heart will result in dramatic changes in the characteristics of IKr and consequently contribute to changes in the APD.
We have also shown that IKr enhanced by β-adrenoceptor stimulation is less sensitive to block by E4031 and one would expect the effect of the class III methanesulfonanilide drugs to be reduced under these conditions. Such an effect has already been described for the mutant HERG channels with reduced inactivation which were found to have a reduced sensitivity to E4031 and dofetilide (Wang et al. 1997; Ficker et al. 1998). More interestingly, a reduced effect of E4031 to prolong APD in the presence of isoprenaline was shown in 1991 by Sanguinetti et al. and this was attributed to an increase in drug-insensitive current such as IKs. In view of the data presented here, it seems likely that the reduced sensitivity of IKr to E4031 will also contribute to this effect.
The process of C-type inactivation has also been linked to channel selectivity and a recent study has shown that C-type inactivation greatly reduces the permeability of K+ relative to Na+ through C-type inactivated Shaker channels, altering the ion selectivity (Starkus et al. 1997). Therefore, since we found that PKC phosphorylation reduced the C-type inactivation of IKr, we also measured the reversal potential of the current to see if there were any changes in the channel selectivity. However, we found no difference in the reversal potential of the E4031-sensitive current in the absence and presence of isoprenaline, which is consistent with there being no change in channel selectivity. Therefore it seems that regulation of C-type inactivation of IKr by phosphorylation does not involve changes in channel selectivity. In addition, the similarity of the reversal potential of the E4031-sensitive current in the absence and presence of isoprenaline is also consistent with the block by E4031 of only IKr.
In conclusion, these data show a novel mechanism of activation of PKC by elevation of cytosolic calcium following stimulation of β-adrenoceptors in cardiac cells and this causes an increase in IKr. The main mechanism by which IKr is modulated by PKC appears to be through an alteration in the C-type inactivation which underlies the rectification of the channel. This modulation implies a role for intracellular sites which are susceptible to phosphorylation in the process of inactivation and a similar mechanism may occur in other channels with this type of inactivation.
References
- Abbott GW, Sesti F, Splawski I, Buck ME, Lehmann MH, Timothy KW, Keating MT, Goldstein SAN. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmias. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- Barros F, Gómez-Varela D, Viloria CG, Palomero T, Giráldez T, de la Peña P. Modulation of human erg K+ channel gating by activation of a G protein-coupled receptor and protein kinase C. The Journal of Physiology. 1998;511:333–346. doi: 10.1111/j.1469-7793.1998.333bh.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck EJ, Sorenson RG, Slater SJ, Covarrubias M. Interactions between multiple phosphorylation sites on the inactivation particle of a K+ channel. Insights into the molecular mechanism of protein kinase C action. Journal of General Physiology. 1998;112:71–84. doi: 10.1085/jgp.112.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet E. Voltage- and time-dependent block of the delayed potassium current in cardiac myocytes by dofetilide. Journal of Pharmacology and Experimental Therapeutics. 1992;262:809–817. [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- Ficker E, Jarolimik W, Kiehn J, Baumann A, Brown AM. Molecular determinants of dofetilide block of HERG K+ channels. Circulation Research. 1998;82:386–395. doi: 10.1161/01.res.82.3.386. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Levi AJ, Witchel HJ. Time course and voltage dependence of expressed HERG current compared with native ‘rapid’ delayed rectifier K current during the cardiac ventricular action potential. Pflügers Archiv. 1998a;436:843–853. doi: 10.1007/s004240050713. [DOI] [PubMed] [Google Scholar]
- Hancox JC, Witchel HJ, Varghese A. Alteration of HERG current profile during the cardiac ventricular action potential following a pore mutation. Biochemical and Biophysical Research Communications. 1998b;253:719–724. doi: 10.1006/bbrc.1998.9837. [DOI] [PubMed] [Google Scholar]
- Harvey RD, Hume JR. Autonomic regulation of a chloride current in heart. Science. 1989;244:983–985. doi: 10.1126/science.2543073. [DOI] [PubMed] [Google Scholar]
- Heath BM, Terrar DA. Separation of the components of the delayed rectifier K current using selective blockers of IKr and IKs in guinea-pig ventricular myocytes. Experimental Physiology. 1996;81:587–603. doi: 10.1113/expphysiol.1996.sp003961. [DOI] [PubMed] [Google Scholar]
- Hescheler J, Nawrath H, Tang M, Trautwein W. Adrenoceptor-mediated change of excitation and contraction in ventricular heart muscle from guinea-pigs and rabbits. The Journal of Physiology. 1988;397:657–670. doi: 10.1113/jphysiol.1988.sp017024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiehn J, Karle C, Thomas D, Yao X, Brachmann J, Küber W. HERG potassium channel activation is shifted by phorbol esters via protein kinase A-dependent pathways. Journal of Biological Chemistry. 1998;273:25285–25291. doi: 10.1074/jbc.273.39.25285. [DOI] [PubMed] [Google Scholar]
- Kiehn J, Lacerda AE, Wible B, Brown AM. Molecular physiology and pharmacology of HERG: single channel currents and block by dofetilide. Circulation. 1996;94:2572–2579. doi: 10.1161/01.cir.94.10.2572. [DOI] [PubMed] [Google Scholar]
- Lui Y, Jurman ME, Yellen G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- Perchenet L, Hinde AKB, Patel RCR, Pabbathi VK, Hancox JC, Levi AJ. β-Adrenergic stimulation of the Na+-Ca2+ exchange in isolated guinea-pig ventricular myocytes at 37°C. The Journal of Physiology. 1998;509.P:140–141P. [Google Scholar]
- Powell T, Terrar DA, Twist VW. Electrical properties of individual cells isolated from adult rat ventricular myocardium. The Journal of Physiology. 1980;302:131–153. doi: 10.1113/jphysiol.1980.sp013234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:1–20. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK. Two components of delayed rectifier potassium current: differential sensitivity to class III antiarrhythmic agents. Journal of General Physiology. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Jurkiewicz NK, Scott A, Siegl PKS. Isoproterenol antagonises prolongation of refractory period by the class III antiarrhythmic agent E-4031 in guinea-pig myocytes. Circulation Research. 1991;68:77–84. doi: 10.1161/01.res.68.1.77. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. The Journal of Physiology. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–837. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Chaudhary A. High affinity open channel block by dofetilide of HERG expressed in a human cell line. Molecular Pharmacology. 1996;49:949–955. [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. Journal of General Physiology. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD, Heinemann SH. Ion conduction through C-type inactivated Shaker channels. Journal of General Physiology. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi H, Terrar DA. Selectivity of action of thiopentone and propofol on components of delayed rectifier potassium currents in guinea-pig isolated myocytes. British Journal of Pharmacology. 1995;115:P20. [Google Scholar]
- Tohse N. Calcium sensitive delayed rectifier potassium current in guinea-pig ventricular cells. American Journal of Physiology. 1990;258:H1200–1207. doi: 10.1152/ajpheart.1990.258.4.H1200. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Regulation of a heart potassium channel by protein kinase A and C. Science. 1988;242:67–69. doi: 10.1126/science.2845575. [DOI] [PubMed] [Google Scholar]
- Walsh KB, Kass RS. Distinct voltage-dependent regulation of a heart delayed IK by protein kinases A and C. American Journal of Physiology. 1991;261:C1081–1090. doi: 10.1152/ajpcell.1991.261.6.C1081. [DOI] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Lui S, Strauss HJ, Rasmusson RL. Modulation of HERG affinity for E-4031 by [K+]o and C-type inactivation. FEBS Letters. 1997;417:43–47. doi: 10.1016/s0014-5793(97)01245-3. [DOI] [PubMed] [Google Scholar]
- Veldkamp MW, Van Ginneken ACG, Bouman L. Single delayed rectifier channels in the membrane of rabbit ventricular myocytes. Circulation Research. 1993;72:865–878. doi: 10.1161/01.res.72.4.865. [DOI] [PubMed] [Google Scholar]