

Abstract
The effects of chronic pharmacological modulation of L-type Ca2+ channel activity on the cell surface expression of Na+ channels were examined in GH3 cells.
Prolonged inhibition (4–5 days) of L-channels with nimodipine caused a 50–60 % decrease in the peak amplitude of whole-cell Na+ currents recorded with the patch-clamp technique. On the contrary, prolonged exposure to the L-channel agonist Bay K 8644 induced an ≈2.5-fold increase in peak Na+ current. In both cases, there were only minor changes in cell capacitance and no significant changes in Na+ channel gating properties.
Measurements of the specific binding of radiolabelled saxitoxin to intact cells showed that nimodipine treatment reduced the number of cell surface Na+ channels, whereas treatment with Bay K 8664 produced the opposite effect. The dual regulation of Na+ channel abundance explained the mentioned changes in Na+ current amplitude.
Plasma membrane Na+ channels had a half-life of ≈17 h both in control cells and in cells treated with Bay K 8644, as estimated from the rate of decay of peak Na+ current after inhibition of protein synthesis with cycloheximide. Actinomycin D, an inhibitor of gene transcription, and also cycloheximide, occluded the stimulatory effect of Bay K 8644 on Na+ current density when measured over a 24 h period.
These findings indicate that the entry of Ca2+ through L-type channels influences in a positive way the number of functional Na+ channels in GH3 cells, and suggest that Ca2+ influx stimulates either Na+ channel gene expression or the expression of a regulatory protein that promotes translocation of pre-assembled Na+ channels into the plasma membrane.
Voltage-gated Na+ and Ca2+ channels work in concert to generate spontaneous action potentials in most endocrine cell types of the mammalian pituitary (reviewed by Corrette et al. 1995). The biophysical and pharmacological properties of pituitary Na+ channels have been characterized in the GH3 cell line (Dubinsky & Oxford, 1984; Horn & Vandenberg, 1984; Matteson & Armstrong, 1984, 1986; Cota & Armstrong, 1989), which was derived from a rat pituitary tumour (Tashjian, 1979), as well as in primary cultured cells (Cobbett et al. 1987; Mason & Sikdar, 1988; Chen et al. 1990; Horta et al. 1991; Kehl, 1994). However, the cellular signals that control the expression levels of these channels remain largely unexplored. Since increased cytosolic Ca2+ has been shown to reduce the surface density of Na+ channels in cultured skeletal muscle cells (Sherman & Catterall, 1984; Sherman et al. 1985; Brodie et al. 1989), it is reasonable to ask whether the functional expression of Na+ channels in pituitary cells is responsive to changes in Ca2+ channel activity.
Pituitary cells are commonly equipped with both low- and high-threshold Ca2+ channels (Corrette et al. 1995). The low-threshold channels are thought to regulate spike frequency, whereas the high-threshold channels seem better adapted to serve as transducers between action potentials and intracellular Ca2+ transients (Matteson & Armstrong, 1986). In GH3 cells and other prolactin-secreting GH cell lines, the high-threshold Ca2+ current flows in large part through L-type channels, and it is therefore modulated up or down by agonists and blockers of the dihydropyridine (DHP) type such as Bay K 8644 and nimodipine, respectively (Kalman et al. 1988; Simasko et al. 1988; Liévano et al. 1994; Piros et al. 1995). Blocking the L-type channels in GH cells with DHP antagonists abolishes the oscillations in cytosolic Ca2+ evoked by action potential firing (Schlegel et al. 1987; Charles et al. 1999), decreases baseline Ca2+ influx (Mollard et al. 1994), and inhibits the release of prolactin (Enyeart et al. 1985; Charles et al. 1999) as well as the synthesis of this hormone (Enyeart et al. 1987; Hinkle et al. 1988). The DHP agonist Bay K 8644, on the other hand, causes a sustained rise in the time-averaged concentration of cytosolic Ca2+ (Hinkle et al. 1988; Law et al. 1990) and stimulates prolactin production (Enyeart et al. 1987, 1990; Hinkle et al. 1988).
In the present study, GH3 cells were grown in the presence of nimodipine and Bay K 8644 in order to chronically affect the entry of Ca2+ through L-type channels. The patch-clamp technique was then used to evaluate the persistent effects of DHP treatment on the amplitude, time course and voltage dependence of whole-cell Na+ currents. In addition, the abundance of cell surface Na+ channels was estimated by measuring the binding of radioactive saxitoxin ([3H]STX) to intact cells. Our results indicate that the activity of L-type Ca2+ channels is a major determinant of the number of functional Na+ channels in GH3 cells. A preliminary report of this work has appeared in abstract form (Monjaraz et al. 1995).
METHODS
Cell cultures
GH3 cells from the American Type Culture Collection (Rockville, MD, USA) were grown as a monolayer culture at 37°C in a humidified atmosphere of 5 % CO2 and 95 % air. Prior to experimental use, the cells were maintained for 5–15 weeks in standard culture medium, consisting of Ham's F-10 medium supplemented with 15 % horse serum, 2.5 % fetal bovine serum, 2 mm L-glutamine, 100 i.u. ml−1 penicillin and 100 μg ml−1 streptomycin (Life Technologies, Grand Island, NY, USA). The maintenance culture, grown in 25 cm2 polystyrene flasks (Corning Costar, Cambridge, MA, USA), was split once a week by using a mild trypsinization to remove the cells, and by replating at 20 % original density in a new flask. The medium was changed on day 3 after plating, and daily thereafter.
For experimental purposes, cells were seeded into 35 mm culture dishes containing poly-L-lysine-coated glass coverslips (cells for current recordings) or into 100 mm culture dishes (for binding assays) at a cell density of ∼3 × 104 cm−2. In these cases, the culture medium, with or without additions, was replenished on days 1, 3, 4 and 5. The DHP compounds used were the enantiomer of Bay K 8644 that behaves as a pure L-type Ca2+ channel agonist (see Enyeart et al. 1990) and the L-channel blocker nimodipine (both from Research Biochemicals, Natick, MA, USA). The Ca2+ channel modulators were prepared as 10 mm stock solutions in ethanol and added to growth medium at a final concentration of 0.5 μM. Preliminary experiments indicated that ethanol, at the final concentration used (0.005 %), had no effect by itself on Na+ current density. Other drugs employed in this study were the Na+ channel blocker tetrodotoxin (TTX), the protein synthesis inhibitor cycloheximide and the inhibitor of gene transcription actinomycin D (all from Sigma, St Louis, MO, USA). The free Ca2+ concentration in the serum-supplemented Ham's F-10 medium has been found to be close to 0.55 mm (Ramsdell & Tashjian, 1985). In some experiments (see Fig. 5D), EGTA was added to this medium in order to reduce the extracellullar Ca2+ concentration to ∼50 μM.
Figure 5. Effects of different manipulations in culture on sodium current density and sodium channel abundance.
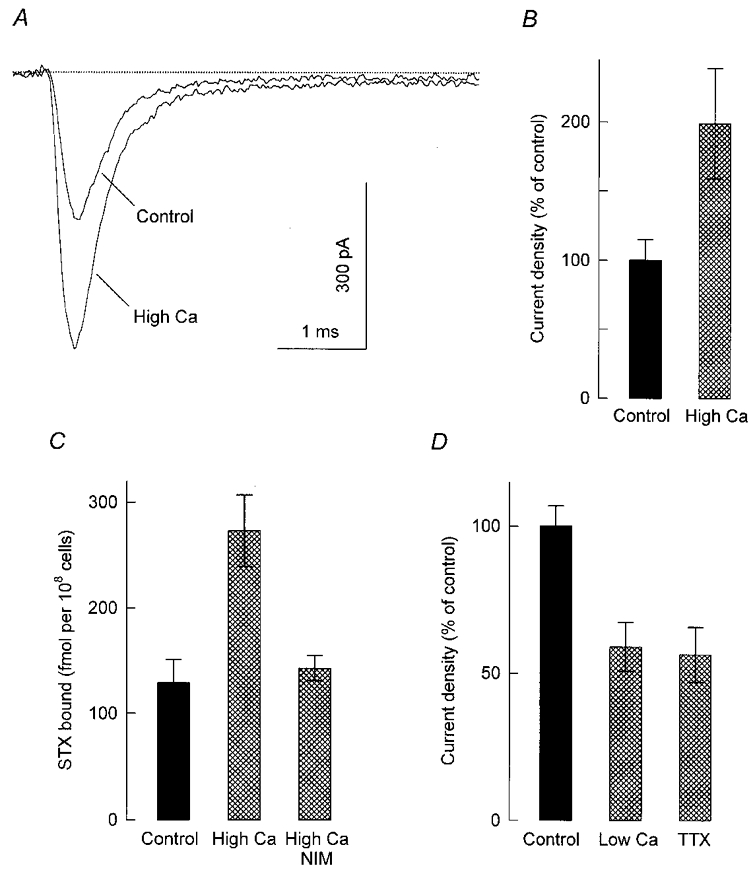
A, representative Na+ currents recorded from a control cell and a cell that was maintained for 24 h in culture medium containing 8 mm Ca2+ and 10 mm K+ (high-Ca2+ condition). The concentration of these ions in the control culture medium was 0.55 and 5 mm, respectively. Whole-cell currents were elicited with step depolarizations to +10 mV from a holding potential of −80 mV. B, peak Na+ current density at +10 mV for control cells (n = 17) and cells that were cultured for 24 h in the high-Ca2+ condition (n = 20). Current density has been converted to a percentage of its mean control value (-24.5 ± 3.7 pA pF−1). C, specific binding of 20 nM [3H]STX to control cells and cells that were exposed to the high-Ca2+ condition for 24 h in the absence or presence of 0.5 μM nimodipine (NIM). Each bar represents the mean of triplicate determinations made in a single experiment. D, Na+ current densities measured from control cells (n = 33) and cells that had been incubated in culture medium supplemented with 0.5 mm EGTA for 72 h (low-Ca2+ condition; n = 15) or 1 μM TTX for 84 h (n = 18). Current density was evaluated as described in B; its average value in control cells was −59.7 ± 4.1 pA pF−1.
All drug treatments and other manipulations were designed to end on day 5 or 6 after plating. At that time, control and treated cells were rinsed with drug-free culture medium, then maintained in this same medium for 60–90 min before Na+ currents or STX binding were measured. In previous studies the existence of discernible variations in ionic current levels from one batch of GH3 cells to another has been noted (Fomina et al. 1993; Cota et al. 1997). Therefore, in each experiment described here, data obtained from treated cells were always compared with measurements performed on control cells from the same donor culture. Although serum-supplemented Ham's F-10 medium was the usual standard culture medium, electrophysiological experiments were also carried out on cells grown in RPMI 1640 medium supplemented with 5 % fetal bovine serum and 2 mm L-glutamine (Life Technologies). This was found not to influence the regulation of Na+ current density by chronic treatment with DHP drugs reported in Results.
Recording of sodium currents
Standard whole-cell patch clamping (Marty & Neher, 1995) was used to record isolated Na+ currents from GH3 cells attached to glass coverslips. The cells were placed in a 0.2 ml experimental chamber mounted on the stage of an inverted microscope and then examined within 10–50 min at room temperature (19–22°C). The bath solution, which was continuously perfused through the chamber at a rate of 0.5 ml min−1, had the following composition (mm): 150 NaCl, 2 CaCl2, 0.5 CdCl2, 10 Hepes, 5 glucose; pH 7.3 with NaOH. Current recording was performed using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, USA) and a data acquisition system that included a TL-1 interface and pCLAMP software (Axon Instruments). Recording electrodes (1.8–3.1 MΩ) consisted of borosilicate glass pipettes filled with (mm): 100 CsCl, 30 NaCl, 10 EGTA, 1 CaCl2, 2 MgCl2, 2 Na2ATP, 0.05 GTP, 10 Hepes, 5 glucose; pH 7.3 with CsOH. Once whole-cell access was established, voltage steps were applied at a frequency of 0.5 Hz from a steady holding potential of −80 mV. Current responses were low-pass filtered at 10 kHz with a four-pole Bessel filter and digitally sampled at intervals of 10–20 μs. Capacitive transients were cancelled with the amplifier circuitry and linear leakage currents were digitally subtracted on-line with scaled pulse routines. The use of the transient cancellation feature on the amplifier provided estimates for cell capacitance and series resistance. The cell capacitance was corroborated by integrating the area under capacitive transients as previously described (Meza et al. 1994). The series resistance was usually in the range 2.2–4.5 MΩ; when appropriate, it was reduced by 40–60 % using the compensation circuit of the amplifier. In most cells, current recording was completed within 3 min of establishing contact between pipette and cytosol.
Under the present recording conditions, brief (7 ms) step depolarizations to +10 mV elicited fast inward ionic currents, which rose to a peak in < 1 ms and spontaneously inactivated. These currents were greatly reduced when TTX (1 μM) was present in the bathing solution. For example, in preliminary experiments carried out on control cells, the peak amplitude of the inward current at +10 mV decreased from −233 ± 17 pA (mean ± s.e.m.; n = 4) in the absence of TTX to −3 ± 1 pA in the presence of the Na+ channel blocker. Bath application of 1 μM TTX also induced a 98–99 % reduction in peak current in cells that had been chronically exposed to Bay K 8644. Before addition of TTX, the current remaining at the end of the 7 ms activating pulses comprised 4–7 % of the respective peak current in both control cells and cells treated with Bay K 8644. Including TTX in the bath resulted in a 40–50 % decrease in such a persistent component of inward current. Thus, the recorded ionic currents were carried almost exclusively through TTX-sensitive Na+ channels.
STX binding assay
A rapid filtration assay was used to measure the high-affinity binding of [3H]STX to intact GH3 cells. Experiments were conducted according to procedures described by Sherman et al. (1983) and Isom et al. (1995). Cells were recovered from the culture dishes with siliconized flame-polished Pasteur pipettes following a brief exposure to 0.34 mm EDTA in phosphate-buffered saline. Harvested cells were collected by centrifugation and resuspended in binding buffer, consisting of (mm): 130 choline chloride, 5.4 KCl, 0.8 MgSO4, 50 Hepes, 5.5 glucose; pH 7.4 with Tris base. To initiate the binding reaction, 250 μl aliquots of the cell suspension (∼6 × 106 cells ml−1) were mixed with equal volumes of binding buffer containing [3H]STX (Amersham; specific activity, 20–40 Ci mmol−1). Unless stated otherwise, the final concentration of [3H]STX in the assay mixture ranged from 0.5 to 50 nM. The cells were then incubated in the absence or presence of 2 μM TTX under continuous gentle shaking at 37°C. After incubation for 45 min, binding was stopped by rapid filtration through glass fibre paper (Whatman GF/B) using a Brandel cell harvester (Gaithersburg, MD, USA), followed by three washes with ice-cold buffer consisting of (mm): 163 choline chloride, 1.8 CaCl2, 0.8 MgSO4, 5 Hepes; pH 7.4 with Tris base. Finally, the filters were transferred to counting vials and radioactivity was measured by liquid scintillation counting. Specific binding, expressed as the amount of ligand bound per 108 cells, was calculated at each concentration of [3H]STX by subtracting the non-specific binding measured in the presence of TTX from the total binding.
Data analysis
All values are given as means ± s.e.m. Electrophysiological data were analysed and plotted by the combined use of pCLAMP software (see above) and SigmaPlot software (SPSS, Chicago, IL, USA). Error bars in figures were plotted only when they exceeded the respective symbol size. Curve fits were made using the non-linear, least-squares fitting procedure included in the SigmaPlot program. The statistical significance of differences between mean values for control and treated cells was evaluated by Student's t tests, with P < 0.05 considered significant.
RESULTS
Chronic treatment with DHP drugs regulates sodium current density
To determine whether chronic modulation of L-type Ca2+ channel activity influences the level of Na+ current expression, GH3 cells that had been grown for 4–5 days in the absence or presence of nimodipine or Bay K 8644 (both at 0.5 μM) were cultured in drug-free medium for 60–90 min and subsequently examined with the patch-clamp technique. The holding potential was set at −80 mV and whole-cell Na+ currents were evoked by voltage steps to a membrane potential (Vm) of +10 mV. As illustrated in Fig. 1A, the presence of nimodipine in the growth medium resulted in Na+ currents smaller than controls, whereas the presence of Bay K 8644 led to the opposite effect. On average, the peak amplitude of the Na+ current decreased to ∼45 % of its control value in the cells that were exposed to nimodipine, and increased by ∼2.5-fold in response to Bay K 8644 (Fig. 1B). These same treatments caused only minor changes in plasma membrane area as suggested by cell capacitance measurements (Fig. 1C).
Figure 1. Whole-cell sodium currents are responsive to chronic treatment with DHP drugs.
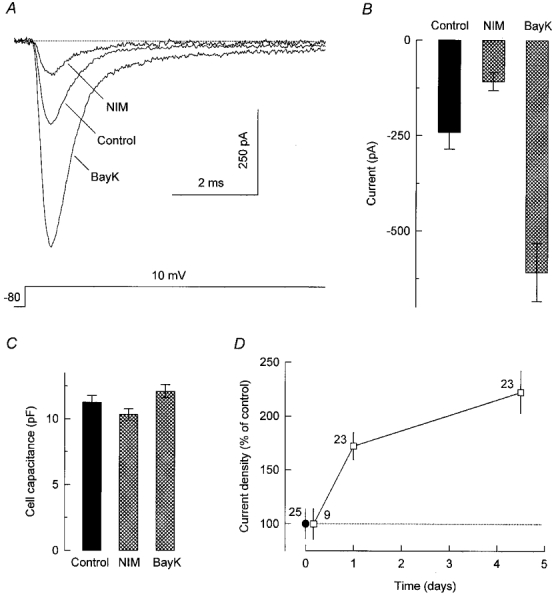
A, Na+ currents evoked during voltage steps to +10 mV from a holding potential of −80 mV. Currents were recorded from GH3 cells that had been grown for 5 days in standard culture medium (Control) or medium supplemented with 0.5 μM nimodipine (NIM) or 0.5 μM Bay K 8644 (BayK). The DHP-containing medium was replaced with drug-free medium 60–90 min before the recordings. A similar recovery period was used in all subsequent experiments. B and C, summary of measurements (means ± s.e.m.) of peak Na+ current at +10 mV (B) and cell capacitance (C) in control cells (n = 21) and cells treated for 4–5 days with nimodipine (n = 19) or Bay K 8644 (n = 19). D, peak Na+ current at +10 mV divided by cell capacitance as a function of time grown (4, 24 or 108 h) in the presence of 0.5 μM Bay K 8644. Current density was converted to a percentage of its average value in control cells (-23.0 ± 3.2 pA pF−1 for these experiments; •). The number of cells examined is indicated near each data point.
Additional current recordings were made from cells that had been incubated with DHP drugs for shorter periods (4 or 24 h). In each cell, the peak amplitude of the Na+ current was normalized to cell capacitance to obtain the current surface density. From the data in Fig. 1D, it can be seen that a 4 h exposure to Bay K 8644 failed to change Na+ current density. However, the stimulatory effect of the DHP agonist was already marked after 24 h of drug treatment. The decrease in Na+ current induced by nimodipine followed a similar temporal pattern (data not shown). Thus, L-type Ca2+ channel modulators effectively regulate Na+ current density when applied chronically to GH3 cells. This long-term effect of DHP drugs does not involve concurrent changes in Na+ current kinetics or voltage dependence of Na+ channel function, as described below.
Sodium channel gating is unaffected by chronic DHP treatment
The Na+ current elicited at +10 mV in control cells reached its peak amplitude within 0.46 ± 0.02 ms (n = 21) after the onset of depolarization and then decayed as the Na+ channels inactivated; the half-decay time was 0.44 ± 0.03 ms. Superimposition of Na+ currents from control and DHP-treated cells suggested similar rates of activation and inactivation (Fig. 2A). Accordingly, neither the time to peak nor the half-decay time of the current were significantly altered by chronic exposure to nimodipine or Bay K 8644 (Fig. 2B).
Figure 2. Sodium current kinetics and voltage dependence of sodium channel inactivation in control and DHP-treated cells.
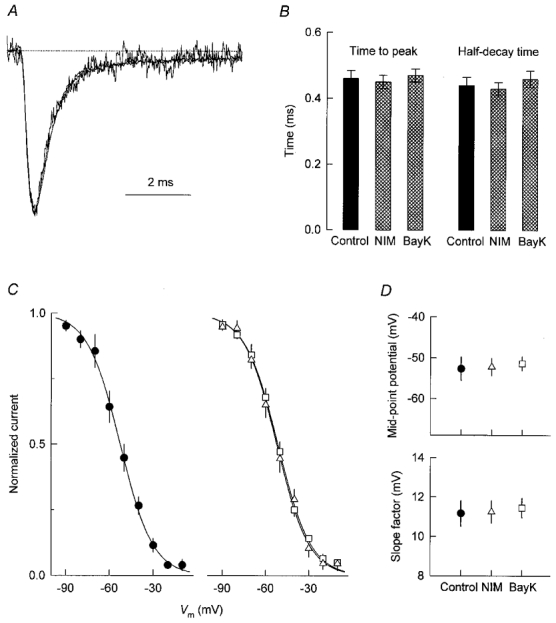
A, overlay of the current traces from Fig. 1A after normalization to the same peak amplitude. B, kinetic properties of the Na+ current at +10 mV. Data were obtained from the same cells as in Fig. 1B, C peak Na+ current at +10 mV, normalized to its maximal value, as a function of voltage during 30 ms prepulses. Currents were measured from control cells (•) and cells treated for 5 days with 0.5 μM nimodipine (▵) or 0.5 μM Bay K 8644 (□); n = 4 in each case. Smooth lines represent fits of eqn (1) to the data points. D, parameters of fitted inactivation curves.
The voltage dependence of inactivation of the Na+ current was measured by applying a constant test pulse to +10 mV after 30 ms prepulses of varying amplitudes. Each data set (a plot of peak Na+ current, INa, during the test pulse versus prepulse voltage) was fitted with a Boltzmann equation of the form:
| (1) |
where Imax is the calculated maximal current, V½ is the mid-point potential (i.e. the potential at which half of the current was inactivated), and k is the slope factor. Data points were then normalized with respect to Imax to obtain the inactivation curves shown in Fig. 2C; the corresponding mid-point potentials and slope factors are compared in Fig. 2D. As is evident, the voltage dependence of Na+ channel inactivation in cells treated with nimodipine or Bay K 8644 was nearly identical to that observed in control cells.
The effect of voltage on Na+ current amplitude was studied using test pulses to Vm values between −50 and +60 mV, as illustrated in Fig. 3A. The average peak Na+ current measured from control cells and cells chronically treated with DHP drugs is plotted as a function of test potential in Fig. 3B. The apparent activation threshold of the current was always between −50 and −40 mV, and the maximal inward current consistently occurred near +10 mV. At more positive voltages, currents became smaller and finally reversed. The interpolated reversal potential (Vrev) in cells exposed to nimodipine and Bay K 8644 was +41.8 ± 0.6 mV (n = 5) and +43.5 ± 0.3 mV (n = 4), respectively, compared with +41.2 ± 1.0 mV in control cells (n = 5). To construct Na+ channel activation curves, the Na+ conductance at each test potential was calculated by dividing peak current amplitude by the respective driving force (Vm–Vrev). Conductance was subsequently normalized to its maximal value and plotted against Vm. The activation curves for control and DHP-treated cells were similar (Fig. 3C) and fits with Boltzmann equations indicated no significant differences in curve parameters (Fig. 3D).
Figure 3. DHP treatment does not alter the voltage dependence of sodium channel activation.
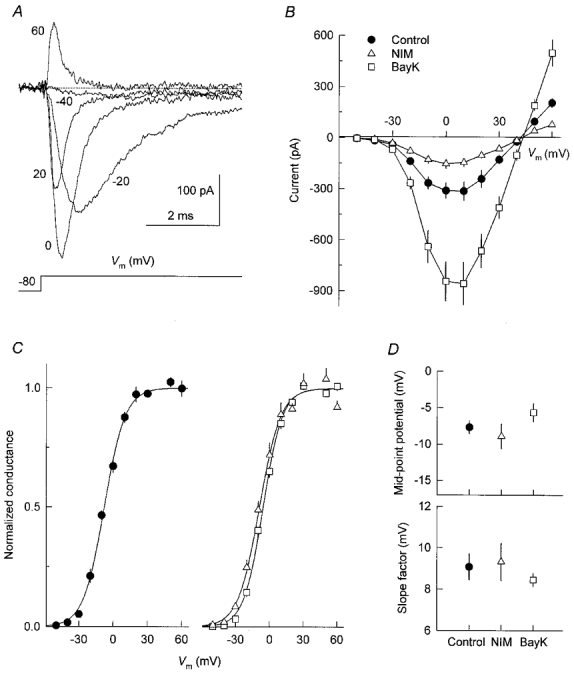
A, typical Na+ currents recorded from a control cell using step depolarizations to the indicated membrane potentials. B, voltage dependence of peak Na+ current in control cells (n = 5) and cells treated for 5 days with 0.5 μM nimodipine (n = 5) or 0.5 μM Bay K 8644 (n = 4). C, activation of normalized Na+ conductance. Same cells and symbols as in B. Conductance was calculated from peak Na+ current measurements (see text) and then normalized to its maximal value. Data points were fitted with smooth lines according to Boltzmann functions. D, parameters of fitted activation curves.
Chronic DHP treatment regulates sodium channel number
Given the similarity of Na+ channel gating in control and treated cells, the DHP-induced changes in whole-cell Na+ current amplitude might be attributed to regulation of the number of Na+ channels in the plasma membrane. Alternatively, they could reflect regulation of the single-channel conductance or regulation of the fraction of plasma membrane Na+ channels that are functionally active. The former hypothesis was investigated by measuring the high-affinity binding of [3H]STX to intact GH3 cells (see Methods). Binding to control cells increased with [3H]STX concentration following a simple saturation curve, as expected for a single class of saturable binding sites (Fig. 4A). The total number of STX binding sites (Bmax) was determined by a non-linear fitting of the data according to the equation:
| (2) |
where B is the amount of toxin bound, KD is the dissociation constant of the toxin-receptor complex, and [T] denotes the concentration of [3H]STX. In three separate experiments performed on control cells, the average KD was 7.3 ± 1.2 nM and the average Bmax was 120 ± 14 fmol per 108 cells. This Bmax value corresponds to a density of 725 ± 85 STX receptors (i.e. Na+ channels) per cell.
Figure 4. Saxitoxin binding to cells grown in the absence or presence of DHP drugs.
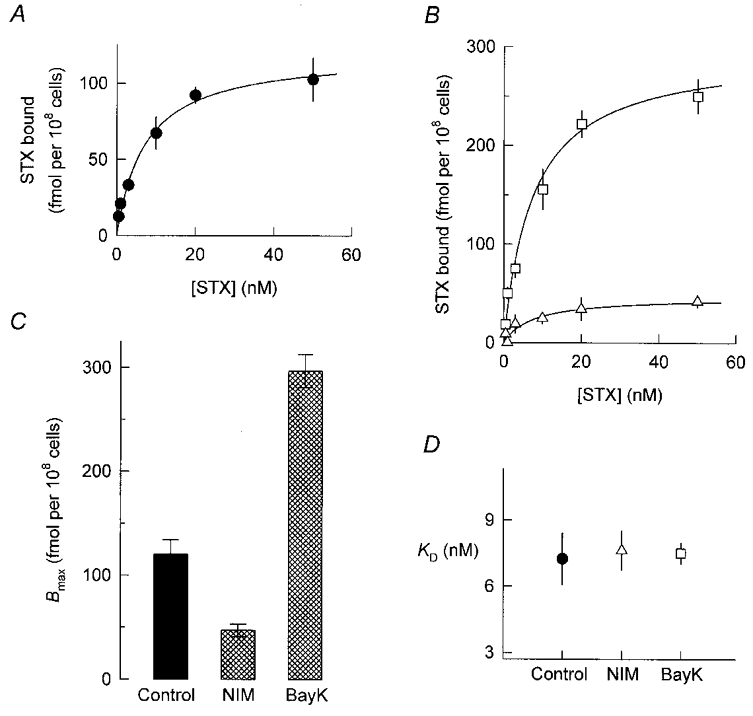
A and B, specific binding of [3H]STX to control cells (A) and cells treated for 5 days with 0.5 μM nimodipine or Bay K 8644 (B;▵ and □, respectively). Aliquots of intact cells were incubated for 45 min with the indicated concentrations of [3H]STX and the amount of toxin bound was then measured by a rapid filtration assay (see Methods). Specific binding was determined as the total binding minus binding in the presence of 2 μM TTX. Data points represent the combined results of three separate experiments. Continuous lines are best-fit hyperbolic functions described by eqn (2). C and D, parameters of fitted saturation curves.
Growth of cells in the presence of nimodipine or Bay K 8644 for 5 days lowered or enhanced, respectively, the specific binding of [3H]STX over the entire range of toxin concentrations tested (Fig. 4B). The analysis of these data revealed a dual modulation of Bmax (Fig. 4C) with no significant variation in KD (Fig. 4D). Thus DHP treatments regulate the abundance of cell surface Na+ channels without affecting their affinity for STX. Furthermore, by comparing Fig. 4C with Fig. 1D, it can be concluded that the changes in Na+ channel number reported by the binding assays are sufficient to fully account for the DHP-induced regulation of Na+ current amplitude.
Effects of other manipulations in culture
The results described above indicate that the number of Na+ channels in the plasma membrane of GH3 cells is critically dependent on the entry of Ca2+ through L-type channels. To test further the role of Ca2+ influx as a determinant of Na+ channel levels, cells were exposed for 24 h to a growth medium containing more than the normal amount of Ca2+ and K+ (8 and 10 mm instead of 0.55 and 5 mm, respectively). Similar to the effect of chronic treatment with Bay K 8644, the sustained elevation of external Ca2+ and K+ (high-Ca2+ condition) resulted in relatively large whole-cell Na+ currents during voltage-clamp depolarizations to +10 mV (Fig. 5A). Peak Na+ current in these experiments increased from −235 ± 36 pA (n = 17) in the control cell group to −462 ± 89 pA (n = 20) in the high-Ca2+ condition; the cell capacitance values were 9.9 ± 0.4 and 10.4 ± 0.4 pF, respectively. Thus cells grown in the high-Ca2+ condition had a ∼2-fold higher Na+ current density than controls (Fig. 5B). Moreover, the stimulation of Na+ current density was associated with an equivalent increase in the specific binding of [3H]STX to intact cells, and this effect was completely blocked by nimodipine (Fig. 5C).
In complementary experiments, the culture medium was supplemented with 0.5 mm EGTA to reduce the concentration gradient for calcium ions across the plasma membrane (see Methods). Figure 5D shows that culturing the cells for 72 h in the presence of the Ca2+ chelator decreased Na+ current density to 59 % of its control value. A significant decrease in Na+ current density was also observed after prolonged treatment with 1 μM TTX (Fig. 5D), which inhibits action potential firing (Biales et al. 1977) and thereby indirectly restrains the opening of high-threshold Ca2+ channels (Matteson & Armstrong, 1986). As mentioned in Methods, a recovery period of 60–90 min in control culture medium was routinely used before recordings. Thus, the inhibitory effects shown in Fig. 5D represent long-lasting reductions in the level of Na+ current expression, similar to those induced by chronic treatments with nimodipine.
Effects of transcription and translation inhibitors
A decrease in the rate of internalization of Na+ channels from the plasma membrane could underlie the positive influence of Ca2+ influx on cell surface levels of these channels. To explore this possibility, the Na+ channel activity of GH3 cells was monitored at different intervals after inhibition of protein synthesis with cycloheximide (35 μM). Filled circles in Fig. 6 show that the addition of cycloheximide to the culture medium of control cells led to a progressive fall in the peak amplitude of Na+ currents evoked at +10 mV. The time-dependent suppression of Na+ current by the translation inhibitor reflected the disappearance of Na+ channels from the plasma membrane, as it was accompanied by a comparable decrease in the number of cell surface STX receptors (see inset in Fig. 6). The time course of loss of Na+ channel activity was well fitted by a single exponential with a time constant of 25 ± 2 h, suggesting that Na+ channels in control cells have a mean half-life of about 17 h. Open squares in Fig. 6 present the results of a similar experiment in which cells that had been treated for 9 days with Bay K 8644 were subsequently incubated with cycloheximide plus the DHP agonist. Again, the application of cycloheximide was followed by an exponential decay of Na+ current magnitude; the corresponding time constant was 24 ± 2 h. Thus, the upregulation of Na+ channels by Bay K 8644 does not seem to result from a decreased rate of channel turnover.
Figure 6. Time course of sodium current inhibition by cycloheximide in control cells and cells treated with Bay K 8644.
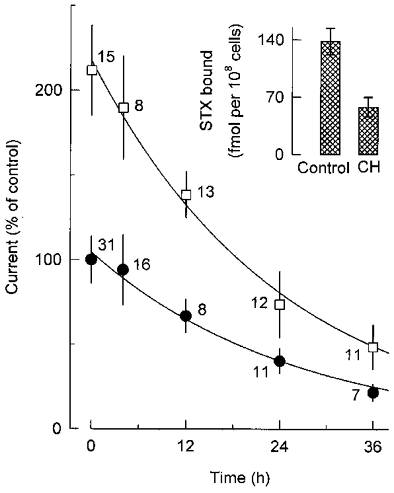
For these determinations, cells were first grown in the absence (•) or presence (□) of 0.5 μM Bay K 8644 for 9 days, then incubated with cycloheximide (35 μM) or cycloheximide plus Bay K 8644, respectively. Shown here is the peak amplitude of the Na+ current at +10 mV as a function of time grown in the presence of cycloheximide. Current data obtained from the indicated number of cells were normalized to the initial control value (-297 ± 41 pA). Smooth lines represent simple exponential functions fitted to the data points; the corresponding time constants are given in the text. Inset, specific binding of 20 nM [3H]STX to control cells and cells exposed to cycloheximide (CH) for 24 h; in each case, data were derived from three separate binding assays.
To gain additional insight into the mechanism underlying Ca2+-mediated stimulation of Na+ channel levels, we examined the possible requirement for RNA and protein synthesis during the onset of the effect of Bay K 8644. Figure 7 summarizes Na+ current measurements from cells that were exposed for 24 h to the DHP agonist in the absence or presence of actinomycin D (an inhibitor of gene transcription) or cycloheximide. As already shown in Fig. 1D, treatment with Bay K 8644 alone increased Na+ current density by a factor of ∼1.7 relative to control cells. In contrast, when the DHP agonist was added in combination with actinomycin D or cycloheximide, the resultant level of Na+ current density corresponded to ∼40 % of the control value. Cells treated for 24 h with actinomycin D or cycloheximide alone also exhibited similarly low Na+ current densities, which is consistent with the rapid turnover of Na+ channels inferred from the data in Fig. 6. Taken together, these observations suggest that the stimulatory effect of Ca2+ influx on cell surface expression of Na+ channels is exerted at the transcriptional level and requires protein synthesis.
Figure 7. Actinomycin D and cycloheximide occlude the Bay K 8644-induced increase in sodium current density.
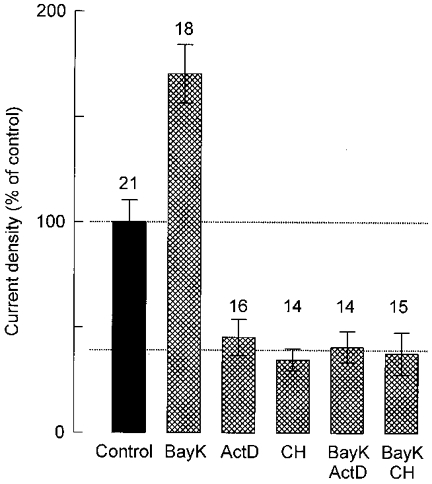
Peak Na+ current density measured at +10 mV from the number of cells given above each bar and expressed as a percentage of the control value (-19.1 ± 2.0 pA pF−1). Before current recording, the cells were grown for 24 h under control conditions or in the presence of 0.5 μM Bay K 8644 (BayK), 40 μM actinomycin D (ActD), 30 μM cycloheximide (CH) or a combination of these drugs.
DISCUSSION
High-threshold Ca2+ channels are known to provide major pathways for voltage-gated Ca2+ influx in excitable pituitary cells (Corrette et al. 1995). The physiological function of these channels has been extensively investigated in the GH3 cell line and its subclones, where they mostly correspond to the L-type (Kalman et al. 1988; Simasko et al. 1988; Liévano et al. 1994; Piros et al. 1995). Previous studies involving incubations of GH cells with DHP blockers and agonists have shown that L-type Ca2+ channels are primarily responsible for the transient increases in cytosolic Ca2+ caused by spontaneous action potentials (Schlegel et al. 1987; Charles et al. 1999), and that such Ca2+ oscillations in turn contribute to sustaining the release and synthesis of prolactin (Enyeart et al. 1985, 1987, 1990; Hinkle et al. 1988; Charles et al. 1999).
In this study, we have used DHP drugs to test whether the cell surface expression of Na+ channels in GH3 cells is dependent on the activity of L-type Ca2+ channels. We have found that growth of cells in the presence of the L-channel blocker nimodipine results in a 50–60 % decrease in the amplitude of whole-cell Na+ currents. The reduced Na+ channel activity of nimodipine-treated cells was due to the loss of Na+ channels from the plasma membrane, as indicated by the concomitant reduction in maximal STX binding to intact cells. When, on the other hand, cells were chronically exposed to the L-channel agonist Bay K 8644, both Na+ current amplitude and the number of STX binding sites markedly increased relative to control values. These effects were not associated with detectable changes in Na+ channel gating or affinity for STX, and were mimicked by chronic alterations in the Ca2+ concentration of the culture medium. We thus conclude that the entry of Ca2+ through L-type channels has a long-term positive impact on the number of Na+ channels located in the plasma membrane of GH3 cells, and so prolonged changes in L-channel activity lead to parallel changes in the level of Na+ current expression.
Our results support the notion that excitable cells can use the influx of Ca2+, and in more general terms the concentration of cytosolic Ca2+, as a feedback signal to adjust the number of functional Na+ channels. This concept was introduced by Sherman & Catterall (1984), who found that chronic blockade of the spontaneous electrical activity of cultured skeletal myotubes from fetal rats increases the number, and probably the sarcolemmal density, of high-affinity binding sites for STX. Sherman & Catterall (1984) also observed that prolonged treatment of myotubes with the Ca2+-specific ionophore A23187 decreases the number of STX receptors; therefore, they proposed that Ca2+ flowing into the cytosol during each action potential tonically restricts the surface density of TTX-sensitive Na+ channels. Subsequent work confirmed this hypothesis and demonstrated that electrical activity and increased cytosolic Ca2+ do not affect the rate of Na+ channel turnover (Sherman et al. 1985; Brodie et al. 1989), but reduce the levels of mRNA encoding the α subunit of the skeletal muscle Na+ channel (Offord & Catterall, 1989; for a review of the molecular composition of Na+ channels, see Marban et al. 1998). A similar Ca2+-mediated downregulation of Na+ channels by electrical activity has been documented in rat cardiac myocytes (Duff et al. 1992; Chiamvimonvat et al. 1995). Our experiments indicate that the number of functional Na+ channels in GH3 cells is also under the control of cytosolic Ca2+. In GH3 cells, however, the regulatory influence of Ca2+ drives the number of channels in the direction opposite to that reported in muscle cells.
Although the precise mechanism by which L-type Ca2+ channel activity increases the number of cell surface Na+ channels in GH3 cells has yet to be defined, we consider that in these cells, like in skeletal myotubes (Sherman et al. 1985), the rate of Na+ channel turnover is Ca2+ independent. The experimental evidence for this view is that plasma membrane Na+ channels had a half-life of ∼17 h both in control cells and in cells treated with Bay K 8644, as inferred from the rate of decay of Na+ channel activity following the addition of cycloheximide to the culture medium. Similar life times have been estimated for TTX-sensitive Na+ channels in neuroblastoma cells (Waechter et al. 1983) and cultured myotubes (Sherman et al. 1985; Brodie et al. 1989). A second consideration is that the onset of the stimulatory effect of Bay K 8644 on Na+ current density appears to require gene transcription and new protein synthesis, since it was occluded by simultaneous treatment with actinomycin D or cycloheximide. This is in agreement with recent studies in neurons and neuroendocrine cells showing that Ca2+ signals initiated at the cell surface by the opening of L-type Ca2+ channels can be propagated to the nucleus, where they elicit changes in gene expression (reviewed by Ghosh & Greenberg, 1995). Moreover, it has been found that the influx of Ca2+ through L-type channels is able to increase the rate of prolactin gene transcription in GH3 cells (Laverrière et al. 1989; Day & Maurer, 1990). Thus an enhanced transcription of Na+ channel subunit genes and the consequent increase in channel production may account for the observed upregulation of Na+ channels by Ca2+ influx. Alternatively, the entry of Ca2+ into GH3 cells could stimulate the expression of a regulatory protein that promotes Na+ channel biosynthesis or the insertion of pre-assembled Na+ channels in the plasma membrane. Measurements of the intracellular abundance of Na+ channel subunits and corresponding mRNAs will be required to begin studying these possibilities. Further experiments are also needed in order to explain the opposing effects of increased cytosolic Ca2+ on Na+ channel levels in myocytes and GH3 cells.
Patch-clamp studies on cultured lactotropes from adult male rats have revealed a greater activity of high-threshold Ca2+ channels in cells that secrete large amounts of prolactin under basal conditions than in cells secreting prolactin at low basal rates (Cota et al. 1990; Horta & Cota, 1993). In keeping with the positive influence of Ca2+ influx on Na+ channel number described here, an extra feature of the former subset of lactotropes is an elevated level of Na+ current density (Horta et al. 1991). More recent experiments have shown that chronic treatment of GH3 cells with epidermal growth factor (EGF) stimulates the functional expression of high-threshold Ca2+ channels, and that this effect is associated with a nearly 2-fold increase in Na+ current density (Meza et al. 1994; Cota et al. 1997). Conversely, the postnatal innervation of rat melanotropes by dopaminergic neurons induces a pronounced suppression of high-threshold Ca2+ current (Gomora et al. 1996), which is accompanied by a 50 % decrease in Na+ current density (López-Santiago et al. 1998). On the basis of our present findings, it could be expected that such regulated changes in Na+ current density are mediated, at least in part, by primary changes in Ca2+ channel activity.
Acknowledgments
We thank Drs R. Felix and U. Meza for help in some experiments, and A. Marin and R. Gonzalez for valuable technical assistance. This work was supported by an International Research Scholars award from the Howard Hughes Medical Institute and a Conacyt grant 26391-N to G.C.
References
- Biales B, Dichter MA, Tischler A. Sodium and calcium action potentials in pituitary cells. Nature. 1977;267:172–174. doi: 10.1038/267172a0. [DOI] [PubMed] [Google Scholar]
- Brodie C, Brody M, Sampson SR. Characterization of the relation between sodium channels and electrical activity in cultured rat skeletal myotubes: regulatory aspects. Brain Research. 1989;488:186–194. doi: 10.1016/0006-8993(89)90708-7. [DOI] [PubMed] [Google Scholar]
- Charles AC, Piros ET, Evans CJ, Hales TG. L-type Ca2+ channels and K+ channels specifically modulate the frequency and amplitude of spontaneous Ca2+ oscillations and have distinct roles in prolactin release in GH3 cells. Journal of Biological Chemistry. 1999;274:7508–7515. doi: 10.1074/jbc.274.11.7508. [DOI] [PubMed] [Google Scholar]
- Chen C, Zhang J, Vincent JD, Israel JM. Sodium and calcium currents in action potentials of rat somatotrophs: their possible functions in growth hormone secretion. Life Science. 1990;46:983–989. doi: 10.1016/0024-3205(90)90021-i. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N, Kargacin ME, Clark RB, Duff HJ. Effects of intracellular calcium on sodium current density in cultured neonatal rat cardiac myocytes. The Journal of Physiology. 1995;483:307–318. doi: 10.1113/jphysiol.1995.sp020587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cobbett P, Ingram CD, Mason WT. Sodium and potassium currents involved in action potential propagation in normal bovine lactotrophs. The Journal of Physiology. 1987;392:273–299. doi: 10.1113/jphysiol.1987.sp016780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrette BJ, Bauer CK, Schwarz JK. Electrophysiology of anterior pituitary cells. In: Scherübl H, Hescheler J, editors. The Electrophysiology of Neuroendocrine Cells. Boca Raton: CRC Press; 1995. pp. 101–143. [Google Scholar]
- Cota G, Armstrong CM. Sodium channel gating in clonal pituitary cells. Journal of General Physiology. 1989;94:213–232. doi: 10.1085/jgp.94.2.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cota G, Hiriart M, Horta J, Torres-Escalante JL. Calcium channels and basal prolactin secretion in single male rat lactotropes. American Journal of Physiology. 1990;259:C949–959. doi: 10.1152/ajpcell.1990.259.6.C949. [DOI] [PubMed] [Google Scholar]
- Cota G, Meza U, Monjaraz E. Regulation of Ca and Na channels in GH3 cells by epidermal growth factor. In: Latorre R, Sáez JC, editors. From Ion Channels to Cell-to-Cell Conversations. New York: Plenum Press; 1997. pp. 185–197. [Google Scholar]
- Day RN, Maurer RA. Pituitary calcium channel modulation and regulation of prolactin gene expression. Molecular Endocrinology. 1990;4:736–742. doi: 10.1210/mend-4-5-736. [DOI] [PubMed] [Google Scholar]
- Dubinsky JM, Oxford GS. Ionic currents in two strains of rat anterior pituitary tumor cells. Journal of General Physiology. 1984;83:309–339. doi: 10.1085/jgp.83.3.309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duff HJ, Offord J, West J, Catterall WA. Class I and IV antiarrhythmic drugs and cytosolic calcium regulate mRNA encoding the sodium channel α subunit in rat cardiac muscle. Molecular Pharmacology. 1992;42:570–574. [PubMed] [Google Scholar]
- Enyeart JJ, Aizawa T, Hinkle PM. Dihydropyridine Ca2+ antagonists: potent inhibitors of secretion from normal and transformed pituitary cells. American Journal of Physiology. 1985;248:C510–519. doi: 10.1152/ajpcell.1985.248.5.C510. [DOI] [PubMed] [Google Scholar]
- Enyeart JJ, Biagi B, Day RN. Opposing actions of Bay K 8644 enantiomers on calcium current, prolactin secretion, and synthesis in pituitary cells. Molecular Endocrinology. 1990;4:727–735. doi: 10.1210/mend-4-5-727. [DOI] [PubMed] [Google Scholar]
- Enyeart JJ, Sheu SS, Hinkle PM. Dihydropyridine modulators of voltage-sensitive Ca2+ channels specifically regulate prolactin production by GH4C1 pituitary tumor cells. Journal of Biological Chemistry. 1987;262:3154–3159. [PubMed] [Google Scholar]
- Fomina AF, Kostyuk PG, Sedova MB. Glucocorticoids modulation of calcium currents in GH3 cells. Neuroscience. 1993;55:721–725. doi: 10.1016/0306-4522(93)90437-k. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Gomora JC, Avila G, Cota G. Ca2+ current expression in pituitary melanotrophs of neonatal rats and its regulation by D2 dopamine receptors. The Journal of Physiology. 1996;492:763–773. doi: 10.1113/jphysiol.1996.sp021344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinkle PM, Jackson AE, Thompson TM, Zavacki AM, Coppola DA, Bancroft C. Calcium channels agonists and antagonists: effects of chronic treatment on pituitary prolactin synthesis and intracellular calcium. Molecular Endocrinology. 1988;2:1132–1138. doi: 10.1210/mend-2-11-1132. [DOI] [PubMed] [Google Scholar]
- Horn R, Vandenberg CA. Statistical properties of single sodium channels. Journal of General Physiology. 1984;84:505–534. doi: 10.1085/jgp.84.4.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horta J, Cota G. Lactotrope subtypes are differentially responsive to calcium channel blockers. Molecular and Cellular Endocrinology. 1993;92:189–193. doi: 10.1016/0303-7207(93)90007-7. [DOI] [PubMed] [Google Scholar]
- Horta J, Hiriart M, Cota G. Differential expression of Na channels in functional subpopulations of rat lactotropes. American Journal of Physiology. 1991;261:C865–871. doi: 10.1152/ajpcell.1991.261.5.C865. [DOI] [PubMed] [Google Scholar]
- Isom LL, Scheuer T, Brownstein AB, Ragsdale DS, Murphy BJ, Catterall WA. Functional co-expression of the β1 and type IIA α subunits of sodium channels in a mammalian cell line. Journal of Biological Chemistry. 1995;270:3306–3312. doi: 10.1074/jbc.270.7.3306. [DOI] [PubMed] [Google Scholar]
- Kalman D, O'Lague PH, Erxleben C, Armstrong DL. Calcium-dependent inactivation of the dihydropyridine-sensitive calcium channels in GH3 cells. Journal of General Physiology. 1988;92:531–548. doi: 10.1085/jgp.92.4.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kehl SJ. Voltage-clamp analysis of the voltage-gated sodium current of the rat pituitary melanotroph. Neuroscience Letters. 1994;165:67–70. doi: 10.1016/0304-3940(94)90711-0. [DOI] [PubMed] [Google Scholar]
- Laverrière JN, Richard JL, Buisson N, Martial JA, Tixier-Vidal A, Gourdji D. Thyroliberin and dihydropyridines modulate prolactin gene expression through interacting pathways in GH3 cells. Neuroendocrinology. 1989;50:693–701. doi: 10.1159/000125301. [DOI] [PubMed] [Google Scholar]
- Law GJ, Pachter JA, Thastrup O, Hanley MR, Dannies PS. Thapsigargin, but not caffeine, blocks the ability of thyrotropin-releasing hormone to release Ca2+ from an intracellular store in GH4C1 pituitary cells. Biochemical Journal. 1990;267:359–364. doi: 10.1042/bj2670359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liévano A, Bolden A, Horn R. Calcium channels in excitable cells: divergent genotypic and phenotypic expression of α1-subunits. American Journal of Physiology. 1994;267:C411–424. doi: 10.1152/ajpcell.1994.267.2.C411. [DOI] [PubMed] [Google Scholar]
- López-Santiago LF, Gomora JC, Marin A, Cota G. Postnatal reduction of sodium current expression in rat melanotropes. Society for Neuroscience Abstracts. 1998;24:1077. [Google Scholar]
- Marban E, Yamagishi T, Tomaselli GF. Structure and function of voltage-gated sodium channels. The Journal of Physiology. 1998;508:647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty A, Neher E. Tight-seal whole-cell recording. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum Press; 1995. pp. 31–52. [Google Scholar]
- Mason WT, Sikdar SK. Characterization of voltage-gated sodium channels in ovine gonadotrophs: relationship to hormone secretion. The Journal of Physiology. 1988;399:493–517. doi: 10.1113/jphysiol.1988.sp017093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteson DR, Armstrong CM. Na and Ca channels in a transformed line of anterior pituitary cells. Journal of General Physiology. 1984;83:371–394. doi: 10.1085/jgp.83.3.371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matteson DR, Armstrong CM. Properties of two types of calcium channels in clonal pituitary cells. Journal of General Physiology. 1986;87:161–182. doi: 10.1085/jgp.87.1.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meza U, Avila G, Felix R, Gomora JC, Cota G. Long-term regulation of calcium channels in clonal pituitary cells by epidermal growth factor, insulin and glucocorticoids. Journal of General Physiology. 1994;104:1019–1038. doi: 10.1085/jgp.104.6.1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollard P, Theler JM, Guérineau N, Vacher P, Chiavaroli C, Schlegel W. Cytosolic Ca2+ of excitable pituitary cells at resting potentials is controlled by steady state Ca2+ currents sensitive to dihydropyridines. Journal of Biological Chemistry. 1994;269:25158–25164. [PubMed] [Google Scholar]
- Monjaraz E, Meza U, Navarrete A, Cota G. Modulation of Na+ current density in clonal pituitary cells by chronic treatment with dihydropyridines. Society for Neuroscience Abstracts. 1995;21:1819. [Google Scholar]
- Offord J, Catterall WA. Electrical activity, cAMP, and cytosolic calcium regulate mRNA encoding sodium channel α subunits in rat muscle cells. Neuron. 1989;2:1447–1452. doi: 10.1016/0896-6273(89)90190-6. [DOI] [PubMed] [Google Scholar]
- Piros E, Prather PL, Loh HH, Law PY, Evans CJ, Hales TG. Ca2+ channel and adenylyl cyclase modulation by cloned μ-opioid receptors in GH3 cells. Molecular Pharmacology. 1995;47:1041–1049. [PubMed] [Google Scholar]
- Ramsdell JS, Tashjian AH. Thyrotropin-releasing hormone and epidermal growth factor stimulate prolactin synthesis by a pathway(s) that differs from that used by phorbol esthers: dissociation of actions by calcium dependency and additivity. Endocrinology. 1985;117:2050–2060. doi: 10.1210/endo-117-5-2050. [DOI] [PubMed] [Google Scholar]
- Schlegel W, Winiger BP, Mollard P, Vacher P, Wuarin F, Zahnd GR, Wollheim CB, Dufy B. Oscillations of cytosolic Ca2+ in pituitary cells due to action potentials. Nature. 1987;329:719–721. doi: 10.1038/329719a0. [DOI] [PubMed] [Google Scholar]
- Sherman SJ, Catterall WA. Electrical activity and cytosolic calcium regulate levels of tetrodotoxin-sensitive sodium channels in cultured rat muscle cells. Proceedings of the National Academy of Sciences of the USA. 1984;81:262–266. doi: 10.1073/pnas.81.1.262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SJ, Chrivia J, Catterall WA. Cyclic adenosine 3′:5′-monophosphate and cytosolic calcium exert opposing effects on biosynthesis of tetrodotoxin-sensitive sodium channels in rat muscle cells. Journal of Neuroscience. 1985;5:1570–1576. doi: 10.1523/JNEUROSCI.05-06-01570.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherman SJ, Lawrence JC, Messner DJ, Jacoby K, Catterall WA. Tetrodotoxin-sensitive sodium channels in rat muscle cells developing in vitro. Journal of Biological Chemistry. 1983;258:2488–2495. [PubMed] [Google Scholar]
- Simasko SM, Weiland GA, Oswald RE. Pharmacological characterization of two calcium currents in GH3cells. American Journal of Physiology. 1988;254:E328–336. doi: 10.1152/ajpendo.1988.254.3.E328. [DOI] [PubMed] [Google Scholar]
- Tashjian AH. Clonal strains of hormone-producing pituitary cells. Methods in Enzymology. 1979;58:527–535. doi: 10.1016/s0076-6879(79)58167-1. [DOI] [PubMed] [Google Scholar]
- Waechter CJ, Schmidt JW, Catterall WA. Glycosylation is required for maintenance of functional sodium channels in neuroblastoma cells. Journal of Biological Chemistry. 1983;258:5117–5123. [PubMed] [Google Scholar]