

Abstract
The effects of dopamine (DA) on non-NMDA glutamatergic transmission onto dopaminergic neurones in the ventral tegmental area (VTA) were examined in rat midbrain slices using the whole-cell patch-clamp technique. EPSCs in dopaminergic neurones evoked by focal stimulation within the VTA were reversibly blocked by 5 μm CNQX in the presence of bicuculline (20 μm), strychnine (0.5 μm) and D-amino-5-phosphonopentanoic acid (D-AP5, 25 μm).
Bath application of DA reduced the amplitude of EPSCs up to 65.1 ± 9.52% in a concentration-dependent manner between 0.3–1000 μm (IC50, 16.0 μm) without affecting the holding current at −60 mV measured using a Cs+-filled electrode.
The effect of DA on evoked EPSCs was mimicked by the D2-like receptor agonist quinpirole but not by the D1-like receptor agonist SKF 81297, and was antagonized by the D2-like receptor antagonist sulpiride (KB, 0.96 μm), but not by the D1-like receptor antagonist SCH 23390 (KB, 228.6 μm).
Dopamine (30 μm) reduced the mean frequency of spontaneous miniature EPSCs (mEPSCs) without affecting their mean amplitude, and the DA-induced effect on the mEPSCs was dependent on the external Ca2+ concentration.
These results suggest that afferent glutamatergic fibres which terminate on VTA dopaminergic neurones possess presynaptic D2-like receptors, activation of which inhibits glutamate release by reducing Ca2+ influx.
The mesolimbic dopamine (DA) system, which originates in the ventral tegmental area (VTA) and which projects to the nucleus accumbens and prefrontal cortex, participates in various psychological functions such as locomotion (Koob & Swerdlow, 1988) or the reward aspects of food ingestion (Smith & Schneider, 1988), as well as associated disorders including schizophrenia (Nemeroff & Bissette, 1988) or drug abuse (Mereu et al. 1988). Although multiple DA receptors have been cloned (for review see Missale et al. 1998), functional characterization of DA receptors is still currently limited to the two pharmacologically identifiable families, D1-like receptors, which activate adenylyl cyclase (AC), and D2-like receptors, which inhibit or show no effect on AC (Kebabian & Calne, 1979). Previous electrophysiological studies have demonstrated that activation of D2-like receptors on dopaminergic neurones in the VTA or in the substantia nigra pars compacta (SNc) hyperpolarizes the membrane by increasing inward rectifying potassium conductances (Lacey et al. 1987; Kim et al. 1995). In addition, synaptic potentials or currents mediated by GABAA, GABAB and glutamate receptors have been recorded from dopaminergic neurones in the VTA or SNc (Mereu et al. 1991; Johnson & North, 1992; Cameron & Williams, 1993; Wu et al. 1995; Bonci & Williams, 1997; Wigmore & Lacey, 1998; Bonci & Malenka, 1999; Manzoni & Williams, 1999). Activation of D1-like receptors located on the terminals of GABAergic afferents to VTA dopaminergic neurones augments GABAB receptor-mediated inhibitory postsynaptic potentials (Cameron & Williams, 1993). In contrast, little information has been available regarding the function of DA in the modulation of excitatory synaptic transmission onto VTA dopaminergic neurones, despite the suggested significance of glutamatergic inputs to midbrain dopaminergic neurones in regulating firing pattern in these neurones (Grace & Bunney, 1984; Johnson et al. 1992; Zhang et al. 1994). Therefore, the present study was carried out to elucidate the effect of DA on non-NMDA glutamatergic synaptic transmission onto dopaminergic neurones in the VTA using the whole-cell patch-clamp technique in a thin-slice preparation of the rat brain.
Preliminary results have been published previously in an abstract form (Koga & Momiyama, 1998, 1999).
METHODS
Slice preparation and solutions
All experiments were carried out in accordance with the Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences of the Physiological Society of Japan (1998). Neonatal rats (7–17 days old) were decapitated under deep ether anaesthesia and the brains were removed. Horizontal brain slices (200 μm) containing the VTA were cut using a microslicer (DTK-2000, Dosaka, Kyoto, Japan) in an ice-cold oxygenated low Ca2+, high Mg2+ Krebs solution of the following composition (mm): NaCl, 118; KCl, 3; CaCl2, 0.5; MgCl2, 6; NaHCO3, 25; Hepes, 5; D-glucose, 11. The slices were then transferred to a holding chamber containing standard Krebs solution of the following composition (mm): NaCl, 118; KCl, 3; CaCl2, 2.5; MgCl2, 1.2; NaHCO3, 25; Hepes, 5; D-glucose, 11; pH 7.4 when bubbled with 95% O2-5% CO2. Slices were incubated in the holding chamber maintained at room temperature (21–25°C) for at least 1 h before recording.
Recording and data analysis
For recording, a slice was transferred to the recording chamber, held submerged, and superfused continuously with standard Krebs solution (bubbled with 95% O2-5% CO2) at a rate of 4 ml min−1.
Patch electrodes were pulled from standard-walled borosilicate glass capillaries (1.5 mm outer diameter; Clark Electromedical, Reading, UK) and had resistances of 3–7 MΩ when filled with a caesium chloride-based internal solution of the following composition (mm): CsCl, 140; NaCl, 9; Cs-EGTA, 1; Cs-Hepes, 10; Mg-ATP, 2 (pH adjusted with 1 M CsOH). Whole-cell recordings were made using an Axopatch 1D (Axon Instruments, Foster City, CA, USA) from visually identified large (> 24 μm in diameter) neurones within the VTA viewed with a microscope fitted with a water-immersion lens (Nikon, Tokyo, Japan). Using a low-magnification lens, VTA was clearly visible as a grey area medial to the SNc in the horizontal slice (Fig. 1A), and was separated from the SNc by the medial terminal nucleus of the accessory optic tract or medial lemniscus depending on the plane (Wu et al. 1995; Paxinos & Watson, 1998). The reference electrode was a glass bridge containing 4% agar-saline, one end of which was placed in the recording chamber and the other in the internal solution-containing side chamber connected to the ground via an Ag-AgCl pellet. After establishing the whole-cell configuration, a stimulating electrode made from the same glass capillaries for the whole-cell pipettes and filled with 1 M NaCl was placed within a 50–120 μm radius of the recorded neurone. A voltage pulse (0.2–0.5 ms in duration) of suprathreshold intensity was delivered extracellularly via the stimulation electrode to evoke synaptic current. The position of the stimulating electrode was varied until a stable response was evoked in the recorded cell. The mean distance between the stimulating electrode and the nearest edge of the recorded cell was 73.6 ± 1.66 μm (n = 101 cells). Experiments were carried out at room temperature (21–25°C).
Figure 1. Visual characterization of the recorded neurone in the ventral tegmental area (VTA).
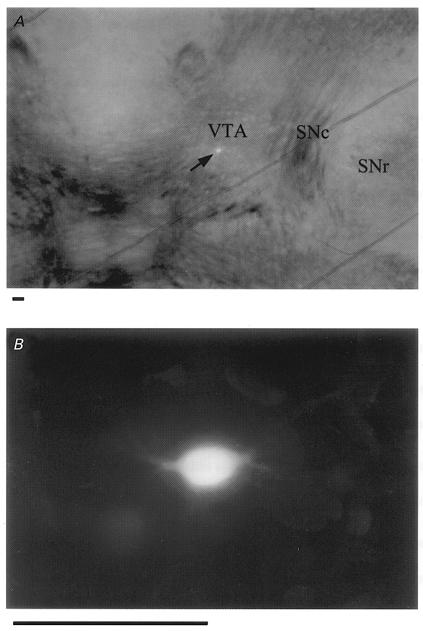
A, photomicrograph of the midbrain region of a horizontal slice obtained from a 15-day-old rat. A putative dopaminergic neurone was filled with Lucifer Yellow (arrow). The caudal part is oriented toward the lower edge of the photograph. SNc, substantia nigra pars compacta; SNr, substantia nigra pars reticulata. The oblique lines represent a grid of nylon threads glued to a platinum frame to fix the slice on the bottom of the recording chamber. B, same neurone as in A at higher magnification. Scale bars, 100 μm.
Data were stored in a DAT recorder (DTR-1204, Biologic, France) (10 kHz). Off-line data acquisition was performed (low-pass filtered at 2–10 kHz with an 8-pole Bessel filter) using pCLAMP 6 software (Axon Instruments) and digitized at 10–20 kHz for computer analysis. Synaptic currents were routinely evoked at 0.2 Hz at a holding potential of −60 mV. For the analysis of mEPSCs, data were digitized at 20 kHz using Axotape software (Axon Instruments) and mEPSCs were detected using software generously provided by Dr Stephan F. Traynelis (Emory University, Atlanta, GA, USA). Curve-fitting was carried out using Origin (Microcal Software, Northampton, MA, USA) software. Data are expressed as means ± s.e.m. Statistical analysis was carried out using Student's t test (two-tailed) or a non-parametrical Mann-Whitney test, where appropriate; P < 0.05 was considered statistically significant.
Drugs
Drugs were stored in frozen stock solutions and dissolved in the perfusion solution just prior to each experiment in the final concentration indicated. All drugs were bath-applied. (±)-6-Chloro-7,8-dihydroxy-1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepine hydrobromide (SKF 81297), trans-(-)-4aR-4,4a,5,6,7,8,8a,9-octahydro-5-propyl-1H-pyrazolo(3,4-g) quinoline hydrochloride (quinpirole), R-(+)-7-chloro-8-hydroxy-3-methyl-1-phenyl-2,3,4,5-tetra-hydro-1H-3-benzazepine hydrochloride (SCH 23390), and S-(-)-5-aminosulfonyl-N-[(1-ethyl-2-pyrrolidinyl)-methyl]-2-methoxybenzamide (sulpiride) were purchased from Research Biochemicals International (Natick, MA, USA). 6-Cyano-7-nitroquinoxalline-2,3-dione (CNQX) and D-(-)-2-amino-5-phosphonopentanoic acid (D-AP5) were from Tocris Cookson (Bristol, UK). Dopamine, bicuculline methochloride and strychnine were from Sigma (St Louis, MO, USA). TTX was from Sankyo (Tokyo, Japan).
RESULTS
General properties of non-NMDA EPSCs in VTA dopaminergic neurones
Whole-cell recordings were made from 561 visually identified neurones within the VTA (Fig. 1A and B). In the present study, 140 of these 561 neurones recorded in the VTA appeared to be dopaminergic on account of a prominent sag in the membrane potential during a large hyperpolarization (Fig. 2A). As reported in a previous study using dopaminergic and non-dopaminergic neurones in the substantia nigra pars reticulata (Radnikow & Misgeld, 1998), the voltage sag was observed even when the recording electrodes were filled with Cs+, similar to studies in dopaminergic neurones using K+-filled electrodes (Häusser et al. 1995; Mercuri et al. 1995). The cell capacitance of these neurones was 42.2 ± 0.97 pF (n = 140). The diameter of the recorded neurones (as measured from the longest axis) was 34.6 ± 0.31 μm (n = 124).
Figure 2. Membrane properties and EPSCs in VTA dopaminergic neurones.
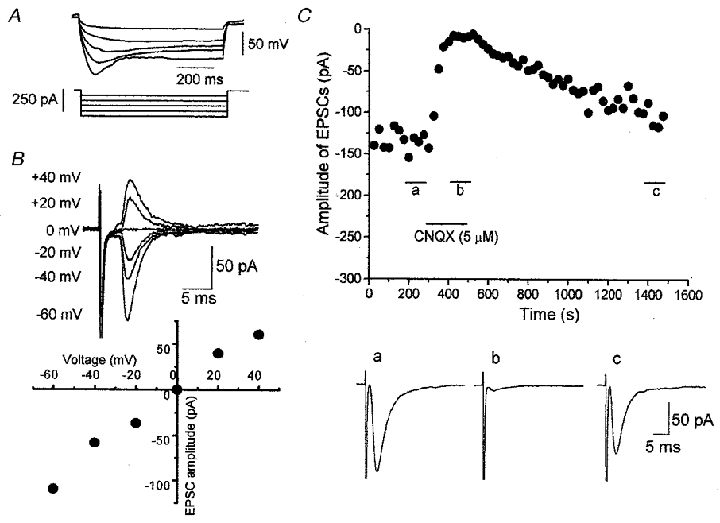
A, superimposed traces of membrane potential elicited by hyperpolarizing current pulses (800 ms in duration). Note the characteristic voltage sag during a large hyperpolarizing pulse. B, current-voltage relationship of EPSCs evoked at 0.2 Hz in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). Each trace is the average of five responses. C, reversible blocking effect of CNQX (5 μM) on the EPSCs at the holding potential of −60 mV. Each point represents the mean amplitude of five consecutive responses evoked at 0.2 Hz. Traces a–c are the averaged traces of 20 responses evoked during the periods indicated in the amplitude-time plot.
Of these 140 putative dopaminergic neurones in the VTA, synaptic currents were evoked in 110 neurones by focal stimulation in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM) to eliminate GABAergic, glycinergic and NMDA glutamatergic components, respectively. The evoked synaptic currents reversed at around 0 mV (Fig. 2B), and were reversibly blocked by bath-applied CNQX (5 μM) (Fig. 2C), suggesting that they are non-NMDA glutamatergic excitatory postsynaptic currents (EPSCs). Access resistance was monitored during the experiments by applying a hyperpolarizing pulse (5 mV, 25 ms) at a holding potential of −60 mV. Some of these neurones were discarded because of the run-down of the evoked EPSCs or the change in access resistance during the control recording, and 77 neurones were used in the analysis of the effects induced by DA, DA receptor agonists, or the effects of DA receptor antagonists on the DA-induced effect. The mean amplitude of these evoked EPSCs was −84.9 ± 4.26 pA (n = 77) at a holding potential of −60 mV. The remaining 30 neurones were used in the analysis of the miniature EPSCs.
Effect of DA on evoked EPSCs
Figure 3A shows the effect of DA on the amplitude of evoked EPSCs in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM) at −60 mV. Bath application of DA produced a gradual decline in the amplitude of the evoked EPSCs, reaching a maximum level 2–4 min after the onset of application. The effect of DA was reversible after 3–10 min washout. The DA-induced inhibitory effect on the evoked EPSCs was concentration dependent between 0.3–1000 μM. Figure 3B depicts the concentration-response curve pooled from 58 neurones. The estimated IC50 value, mean maximum effect and Hill slope value were 16.0 μM, 62.8% and 0.95, respectively. In the present slice preparation and recording condition, no desensitization was observed when DA was applied repeatedly after at least 10 min washout intervals. Dopamine at concentrations up to 1 mm had no effect on the holding current and access resistance at −60 mV under the present recording conditions with Cs+-filled electrodes.
Figure 3. Inhibitory effect of DA on the evoked EPSCs in a VTA dopaminergic neurone.
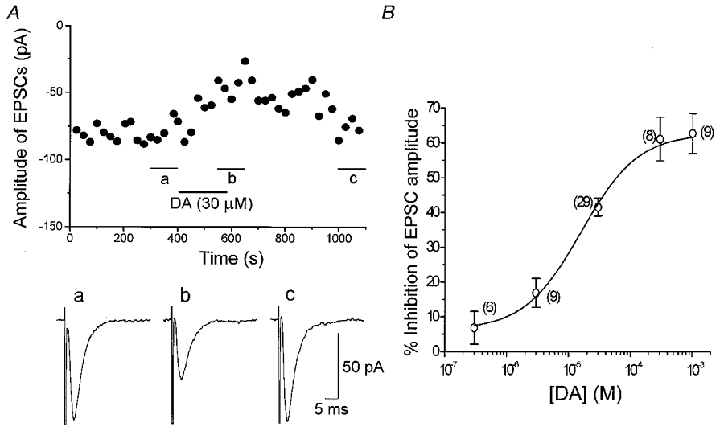
A, effect of DA (30 μM) on the EPSCs evoked at 0.2 Hz in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). The holding potential was −60 mV. Each point represents the mean amplitude of five consecutive responses evoked at 0.2 Hz. Traces a–c are the averaged traces of 30 responses evoked during the period indicated in the amplitude-time plot. B, concentration-response curve of DA-induced effect on the evoked EPSCs. Each point shows the mean ± s.e.m. obtained from the pooled data. The estimated IC50 value, maximum inhibition and Hill slope value were 16.0 μM, 62.8% and 0.95, respectively. The numbers of neurones are shown in parentheses.
DA receptor pharmacology of DA-induced inhibition of evoked EPSCs
To elucidate the DA receptor subtypes mediating DA-induced inhibition of the evoked EPSCs, the effects of DA receptor agonists on the amplitude of evoked EPSCs (Fig. 4) and the effects of DA receptor antagonists on the DA-induced inhibition of the evoked EPSCs (Fig. 5) were investigated.
Figure 4. Effect of DA receptor agonists on the EPSCs.
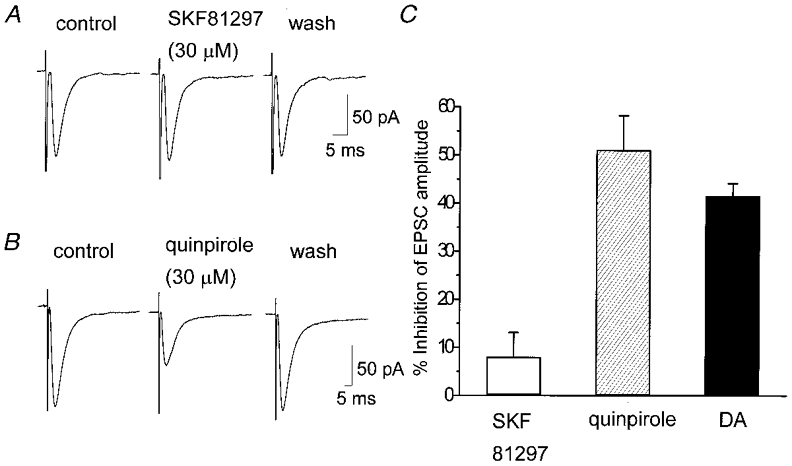
EPSCs were evoked at 0.2 Hz in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). The holding potential was −60 mV. A, lack of effect of the D1-like receptor agonist SKF 81297 (30 μM) on the evoked EPSCs. The traces are the averaged traces of 20 consecutive responses in control, 4 min after the onset of drug application, and 4 min after washout of the drug, respectively. B, inhibitory effect of the D2-like receptor agonist quinpirole (30 μM) on the evoked EPSCs. The traces are the averaged traces of 20 consecutive responses in control, 2 min after the onset of drug application, and 4 min after washout of the drug, respectively. C, summarized histograms showing the means ± s.e.m. of the inhibitory effects of SKF 81297, quinpirole and DA. All agonists were applied at 30 μM. The value of DA was derived from the concentration-response curve in Fig. 3. Values for SKF 81297, quinpirole and DA were 8.04 ± 5.02% (n = 9), 51.1 ± 7.10% (n = 6) and 41.6 ± 2.57% (n = 29), respectively. The difference in the effects between SKF 81297 and quinpirole or SKF 81297 and DA was significant (P < 0.001), whereas the difference between quinpirole and DA was not significant (P = 0.15).
Figure 5. Effects of DA receptor antagonists on DA-induced inhibition of EPSCs.
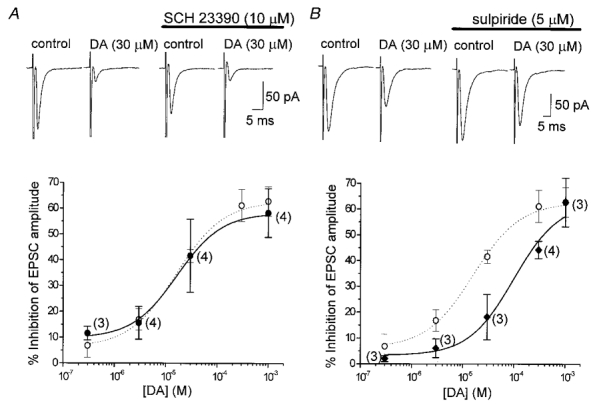
EPSCs were evoked at 0.2 Hz in the presence of bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). Current traces in A and B are the averaged traces of 20 consecutive responses. The holding potential was −60 mV. All points represent means ± s.e.m. The numbers of neurones examined in the presence of each antagonist are given in parentheses. Note that the concentration-response curves of DA alone (○) are the same as in Fig. 3B. All of the neurones examined in the presence of either antagonist are included in the concentration-response curve of DA alone (○). A, lack of effect of the D1-like receptor antagonist SCH 23390 (10 μM) on DA (30 μM)-induced inhibition. SCH 23390 was perfused for 6 min after EPSCs recovered from DA-induced inhibition. Then DA was applied again in the continuous presence of SCH 23390. The concentration-response curve for EPSC inhibition remained unchanged in the presence of SCH 23390 (•) compared with the curve in the absence of SCH 23390 (○). The estimated IC50 value, maximum inhibition and Hill slope were 16.7 μM, 58.3% and 1.00, respectively. B, antagonizing effect of the D2-like receptor antagonist sulpiride (5 μM) on the DA (30 μM)-induced inhibition of EPSCs. Sulpiride was perfused for 6 min before the second application of DA after the EPSCs recovered from DA-induced inhibition. The concentration-response curve was shifted to the right in the presence of sulpiride (♦) from the curve without sulpiride (○), with the estimated IC50 value, maximum effect and Hill slope of 99.7 μM, 62.7% and 1.00, respectively.
Effect of DA receptor agonists
Bath application of the D1-like receptor agonist SKF 81297 at a concentration of 30 μM had little or no effect on the amplitude of evoked EPSCs (8.04 ± 5.02%, n = 9) (Fig. 4A and C). On the other hand, the D2-like receptor agonist quinpirole (30 μM) inhibited the amplitude of evoked EPSCs (Fig. 4B). The quinpirole (30 μM)-induced inhibition of the evoked EPSCs was 51.1 ± 7.10% (n = 6) of the control, which was significantly (P < 0.001) different from SKF81297-induced effect. The effect of quinpirole (30 μM) was rather larger than that of DA (30 μM) (41.6 ± 2.57%, n = 29), but the difference was not significant (P = 0.15) (Fig. 4C).
Effect of DA receptor antagonists on DA-induced inhibition of evoked EPSCs
The effects of the D1-like receptor antagonist SCH 23390, and the D2-like receptor antagonist sulpiride on the DA-induced inhibition of evoked EPSCs were examined. The D1- or D2-like antagonist was perfused for at least 5–10 min after the recovery of evoked EPSCs from DA-induced inhibition, then DA was applied again in the continuing presence of the antagonist. Bath application of SCH 23390 (10 μM) or sulpiride (5 μM) alone had no effect on the amplitude of evoked EPSCs or the holding current at −60 mV. In the presence of SCH 23390, DA showed similar inhibition of the EPSCs to that observed with DA alone (Fig. 5A). The concentration-response curve for DA remained unchanged in the presence of SCH 23390, with an estimated IC50 value of 16.7 μM (16.0 μM in the case of DA alone) (Fig. 5A). On the other hand, DA-induced inhibition of the evoked EPSCs was reduced in the presence of sulpiride (Fig. 5B). The concentration-response curve for DA was shifted to the right in the presence of sulpiride, with an IC50 value of 99.7 μM (Fig. 5B). Assuming that both SCH 23390 and sulpiride behave as competitive antagonists, apparent KB values of 228.6 μM (pKB 3.64) and 0.96 μM (pKB 6.02), respectively, were calculated using the equation KB = [B]/(DR – 1), where [B] is the concentration of the antagonist and DR (dose ratio) is the ratio of the IC50 values for pooled data observed in the presence and absence of antagonist.
Effect of DA on miniature EPSCs
In order to determine whether the DA-induced inhibition of EPSCs is mediated by a presynaptic mechanism, the effect of DA on miniature EPSCs (mEPSCs) was analysed. dopaminergic neurones in the VTA exhibited spontaneous EPSCs in the presence of TTX (0.3 μM) in addition to bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM), and these spontaneous EPSCs were abolished by CNQX (5 μM) (Fig. 6A), so they were mEPSCs mediated by non-NMDA glutamate receptors. The mean frequency of mEPSCs in standard external Ca2+ concentration (2.5 mm) was 0.29 ± 0.05 Hz (n = 8). The frequency was increased to 0.80 ± 0.14 Hz (n = 13) when the external Ca2+ concentration was raised to 7.5 mm, and was decreased to 0.17 ± 0.06 Hz (n = 5) upon replacement of Ca2+ with Mg2+ (5 mm), suggesting that Ca2+ influx contributes to spontaneous glutamate release. Changing the external Ca2+ concentration had little or no effect on the amplitude of mEPSCs. The effect of DA on the mEPSCs was analysed in the external solution containing 7.5 mm Ca2+, 2.5 mm Ca2+ or Ca2+-free solution. When the external Ca2+ concentration was changed, the control recording was started after at least 5 min monitoring of the mEPSCs.
Figure 6. Effect of DA (30 μM) on spontaneous miniature EPSCs (mEPSCs).
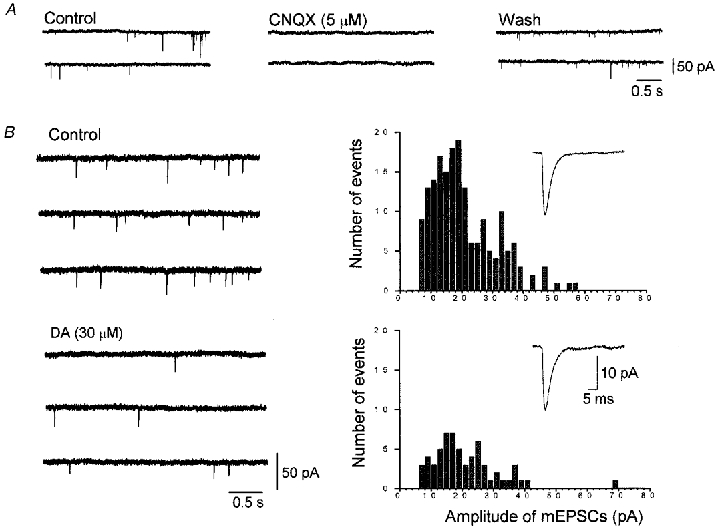
The mEPSCs were recorded in 7.5 mm external CaCl2 Krebs solution containing TTX (0.3 μM), bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). The holding potential was −60 mV. A, consecutive traces taken in control, 2 min after application of CNQX (5 μM), and after 15 min washout of CNQX. The mEPSCs were reversibly abolished by 5 μM CNQX. B, consecutive traces were taken in control and 3 min after application of 30 μM DA. The amplitude histograms were derived from 5 min stretches in control and 2–7 min after the onset of DA application. These histograms show the reduction in the number of events from 180 in control to 61 after application of DA. The inset traces in the histograms are the averaged traces of 180 events in control and 61 events during application of DA, respectively. The mean amplitudes of mEPSCs in control and in the presence of DA were 20.9 and 20.9 pA, respectively.
In 7.5 mm Ca2+ solution, the frequency of mEPSCs was reduced by bath application of DA (30 μM), and reached a steady-state level within 3 min (Fig. 6B). The amplitude histogram shows a reduction in the total number of events but no change in the mean amplitude of mEPSCs (Fig. 6B). Pooled data from twelve VTA neurones showed that DA (30 μM) reduced the frequency of mEPSCs by 48.1 ± 4.60% (Fig. 7B), with no change in mean amplitude (102.3 ± 5.02% of their respective control values). The inhibitory effect of DA on the frequency of mEPSCs was decreased when the external Ca2+ concentration was reduced. In 2.5 mm Ca2+ solution, DA (30 μM) reduced the frequency of mEPSCs by 30.2 ± 6.79% (n = 7) (Fig. 7B) without affecting mean amplitude (96.2 ± 2.69%, n = 7). In Ca2+-free solution, DA (30 μM) had little or no effect on the frequency of mEPSCs (Fig. 7A), the mean reduction being 7.68 ± 3.63% (n = 5) (Fig. 7B). The mean amplitude of mEPSCs was also unaffected by DA (97.9 ± 3.20%, n = 5).
Figure 7. Lack of DA-induced effect on mEPSCs in Ca2+-free solution.

A, the mEPSCs were recorded in Ca2+-free Krebs solution containing TTX (0.3 μM), bicuculline (20 μM), strychnine (0.5 μM) and D-AP5 (25 μM). The holding potential was −60 mV. The consecutive traces were taken from control and 3 min after application of DA (30 μM). The amplitude histograms were derived from 10 min stretches in control (241 events) and 2–12 min after the onset of DA application (231 events). The inset traces in the histograms are the averaged traces of 241 events in control and 231 events during DA application, respectively. The mean amplitudes of mEPSCs in control and during DA application were 21.2 and 23.2 pA, respectively. B, summarized histograms showing the inhibitory effect of DA (30 μM) on the frequency of mEPSCs in Ca2+ concentrations of 7.5, 2.5 and 0 mm. Values for 7.5 mm Ca2+, 2.5 mm Ca2+ and Ca2+-free solutions were 48.1 ± 4.60% (n = 12), 30.2 ± 6.79% (n = 7) and 7.68 ± 3.63% (n = 5), respectively. The differences in the inhibitory effects between 7.5 mm Ca2+ and 2.5 mm Ca2+, 2.5 mm Ca2+ and Ca2+-free, and 7.5 mm Ca2+ and Ca2+-free are all significant (P < 0.04, P < 0.03, and P < 0.0001, respectively).
DISCUSSION
The present results have demonstrated that afferent glutamatergic fibres terminating onto VTA dopaminergic neurones possess presynaptic D2-like receptors, activation of which inhibits glutamate release. They also suggest that this presynaptic action of DA may be mediated mainly by blocking Ca2+ influx, an ionic mechanism different from that underlying D2-like receptor-mediated postsynaptic inhibition of neuronal activity of dopaminergic neurones in the VTA.
A limitation of the approach taken in the present study is that the source of the excitatory afferents cannot be definitively identified. Glutamatergic innervation of the VTA has been reported to arise from three potential sources, the medial prefrontal cortex, pedunculopontine nucleus and subthalamic nucleus (for review see Kalivas, 1993). In addition, an electrophysiological study has recorded glutamatergic excitatory postsynaptic potentials in VTA dopaminergic neurones by stimulation of the habenula efferent fibres of the rat (Matsuda & Fujimura, 1992). Therefore, the EPSCs recorded in the present study may be evoked by stimulation of these fibres. Furthermore, a recent study has demonstrated that dopaminergic neurones in microculture show autaptic glutamatergic EPSCs, which are inhibited by activation of presynaptic D2-like receptors (Sulzer et al. 1998). Therefore, it might be possible that EPSCs evoked in the present study also include those mediated by glutamate released by the stimulation of nearby dopaminergic neurones. Actually, the idea that monoaminergic neurones as a class might release glutamate has been suggested by a morphological study using intact animals (Kaneko et al. 1990). Despite the indefinite source of glutamatergic afferents, the present approach enabled us to demonstrate robust and consistent inhibition of the EPSCs by DA.
The presynaptic locus of DA action on non-NMDA glutamatergic transmission onto dopaminergic neurones in the VTA was indicated by the present finding that DA or quinpirole, the D2-like receptor agonist, did not induce an outward current or had no effect on the access resistance with the caesium chloride internal solution which blocks potassium conductances, and a presynaptic mechanism was confirmed by the finding that DA reduced the frequency of mEPSCs without affecting their mean amplitude.
The inhibitory effect of DA on the frequency of mEPSCs was dependent on the external Ca2+ concentration, suggesting that DA reduced mEPSC frequency mainly by reducing Ca2+ influx, as previously reported in the case of D1-like receptor-mediated presynaptic inhibition of glutamatergic transmission onto magnocellular basal forebrain neurones (Momiyama et al. 1996) or GABAergic transmission onto nucleus accumbens neurones (Nicola & Malenka, 1997), as well as being responsible for the effects of enkephalin or the metabotropic glutamate receptor agonist trans-ACPD at other synapses (Hori et al. 1992; Sladeczek et al. 1993).
The present pharmacological results obtained with DA receptor agonists and antagonists suggest that the inhibitory effect of DA on the EPSCs is mediated by D2-like receptors. Presynaptic D2-like receptor-mediated inhibition of excitatory transmission is not itself a unique mechanism, since previous electrophysiological studies using intracellular recording techniques have reported the involvement of D2-like receptors in the inhibition of excitatory postsynaptic potentials evoked in the nucleus accumbens (O'Donnell & Grace, 1994), striatum (Hsu et al. 1995) and hippocampus (Hsu, 1996). In addition, recent studies using whole-cell patch-clamp technique in brain slices have demonstrated the involvement of presynaptic D1-like receptors in the modulation of excitatory synaptic transmission in several brain regions receiving dopaminergic fibres, including the nucleus accumbens (Harvey & Lacey, 1996; Nicola et al. 1996), basal forebrain nuclei (Momiyama et al. 1996) and striatum (Umemiya & Raymond, 1997). These studies have shown that presynaptic D1- or D2-like receptors located on the terminals of glutamatergic afferents could modulate excitability of neurones receiving dopaminergic projections. The present study has demonstrated that presynaptic D2-like receptors could also modulate dopaminergic neurone excitability. Although no desensitization was observed in the present study after at least 10 min washout intervals, a further detailed analysis will be necessary to elucidate the time course of the desensitization of the present presynaptic D2-like receptors and recovery from it. In apparent contrast to the present results, a previous neurochemical study has reported D1-like receptor-mediated facilitation of glutamate release in the VTA (Kalivas & Duffy, 1995). However, as the authors themselves have speculated in their discussion, the D1-like receptor stimulation may selectively release glutamate from terminals synapsing on GABAergic neurones, which have been reported to inhibit dopaminergic neurones (Johnson & North, 1992). Analysis of DA-induced modulation of glutamatergic transmission onto non-dopaminergic neurones in the VTA will be necessary to clarify the issue. Another apparent discrepancy between the present findings and previous autoradiographic studies with 6-hydroxydopamine (6-OHDA) lesions of the dopaminergic pathway is that D2-like receptors in the VTA or SNc are suggested to be located exclusively on dopaminergic neurones (Araki et al. 1998; for review Missale et al. 1998). One possibility might be that D2-like receptors on the terminals of glutamatergic afferents terminating onto dopaminergic neurones could be down-regulated after 6-OHDA lesions.
Activation of D2-like receptors on midbrain dopaminergic neurones hyperpolarizes the membrane, thereby inhibiting the neuronal activity (Lacey et al. 1987). The apparent KB value of sulpiride, a D2-like receptor antagonist, calculated from the present data (0.96 μM) is higher than that calculated for postsynaptic D2-like receptors on midbrain dopaminergic neurones (11.0–47.3 nM, Lacey et al. 1987). Such a difference, as well as the above-mentioned discrepancies, raises another possibility that the pharmacological and/or molecular biological profiles of D2-like receptors in the glutamatergic terminals terminating onto dopaminergic neurones might be different from those on the dopaminergic neurones themselves. However, this possibility cannot be examined at present, since the pharmacological identification of the receptor is limited due to the lack of available ligands selective for each of the cloned subtypes of D2-like receptors.
It has been shown that glutamatergic innervation to the VTA or SNc plays a crucial role in the switch from the pace-maker-like firing in dopaminergic neurones to burst firing (Grace & Bunney, 1984; Johnson et al. 1992; Zhang et al. 1994; Kang & Futami, 1999). To our knowledge, the exact site of contact of glutamatergic terminals with dopaminergic neurones in the rat VTA remains unclear. However, an ultrastructural study using squirrel monkey brain has shown that most glutamate-enriched terminals contact with dendrites of VTA dopaminergic neurones (Smith et al. 1996). In addition, a potential site for DA release has been demonstrated to be the dendrites of VTA dopaminergic neurones (Nirenberg et al. 1996). Therefore, under physiological conditions, DA released from the dendrites of VTA dopaminergic neurones may act on the D2-like receptors in the nearby terminals of glutamatergic fibres, thereby regulating the firing pattern of VTA dopaminergic neurones by converting the burst firing induced by glutamate to pacemaker-like firing. Dopaminergic neurones in the VTA are thought to play roles in psychopathological behaviours (Nemeroff & Bissette, 1988), and it has been hypothesized that altered glutamatergic transmission contributes to the brain structures and the clinical symptoms of schizophrenia (cf. Tamminga, 1998). Changes in AMPA receptor response in VTA neurones have been demonstrated following adaptation to psychostimulants (Zhang et al. 1997; Bonci & Malenka, 1999). The actions of DA described here might participate in such alterations and provide clues for the elucidation of the underlying mechanisms of these brain functions, as well as for the pharmacological therapeutics of the associated disorders.
Acknowledgments
This work was supported by Grant-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan and the grant from Yamanouchi Foundation for Research on Metabolic Disorders to T.M. The authors are grateful to Professor Y. Matsuda for useful comments and continual encouragement. We are also grateful to Dr Stephen F. Traynelis (Emory University) for the software to analyse miniature EPSCs.
References
- Araki T, Tanji H, Kato H, Itoyama Y. Sequential changes of dopaminergic receptors in the rat brain after 6-hydroxydopamine lesions of the medial forebrain bundle. Journal of the Neurological Sciences. 1998;160:121–127. doi: 10.1016/s0022-510x(98)00248-2. [DOI] [PubMed] [Google Scholar]
- Bonci A, Malenka RC. Properties and plasticity of excitatory synapses on dopaminergic and GABAergic cells in the ventral tegmental area. Journal of Neuroscience. 1999;19:3723–3730. doi: 10.1523/JNEUROSCI.19-10-03723.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonci A, Williams JT. Increased probability of GABA release during withdrawal from morphine. Journal of Neuroscience. 1997;17:796–803. doi: 10.1523/JNEUROSCI.17-02-00796.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron DL, Williams JT. Dopamine D1 receptors facilitate transmitter release. Nature. 1993;366:344–347. doi: 10.1038/366344a0. [DOI] [PubMed] [Google Scholar]
- Grace AA, Bunney BS. The control of firing pattern in nigral dopamine neurons: Burst firing. Journal of Neuroscience. 1984;4:2877–2890. doi: 10.1523/JNEUROSCI.04-11-02877.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey J, Lacey MG. Endogenous and exogenous dopamine depress EPSCs in rat nucleus accumbens in vitro via D1 receptor activation. The Journal of Physiology. 1996;492:143–154. doi: 10.1113/jphysiol.1996.sp021296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Häusser M, Stuart G, Racca C, Sakmann B. Axonal initiation and active dendrite propagation of action potentials in substantia nigra neurons. Neuron. 1995;15:637–647. doi: 10.1016/0896-6273(95)90152-3. [DOI] [PubMed] [Google Scholar]
- Hori Y, Endo K, Takahashi T. Presynaptic inhibitory action of enkephalin on excitatory transmission in superficial dorsal horn of rat spinal cord. The Journal of Physiology. 1992;450:673–685. doi: 10.1113/jphysiol.1992.sp019149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu K-S. Characterization of dopamine receptors mediating inhibition of excitatory synaptic transmission in the rat hippocampal slice. Journal of Neurophysiology. 1996;76:1887–1895. doi: 10.1152/jn.1996.76.3.1887. [DOI] [PubMed] [Google Scholar]
- Hsu K-S, Huang C-C, Yang C-H, Gean P-W. Presynaptic D2 dopaminergic receptors mediate inhibition of excitatory synaptic transmission in rat neostriatum. Brain Research. 1995;690:264–268. doi: 10.1016/0006-8993(95)00734-8. [DOI] [PubMed] [Google Scholar]
- Johnson SW, North RA. Two types of neurone in the rat ventral tegmental area and their synaptic inputs. The Journal of Physiology. 1992;450:455–468. doi: 10.1113/jphysiol.1992.sp019136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SW, Seutin V, North RA. Burst firing in dopamine neurons induced by N-methyl-D-aspartate: Role of electrogenic sodium pump. Science. 1992;258:665–667. doi: 10.1126/science.1329209. [DOI] [PubMed] [Google Scholar]
- Kalivas PW. Neurotransmitter regulation of dopamine neurons in the ventral tegmental area. Brain Research Reviews. 1993;18:75–113. doi: 10.1016/0165-0173(93)90008-n. [DOI] [PubMed] [Google Scholar]
- Kalivas PW, Duffy P. D1 receptors modulate glutamate transmission in the ventral tegmental area. Journal of Neuroscience. 1995;15:5379–5388. doi: 10.1523/JNEUROSCI.15-07-05379.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneko T, Akiyama H, Nagatsu I, Mizuno N. Immunohistochemical demonstration of glutaminase in catcholaminergic and serotonergic neurons of rat brain. Brain Research. 1990;507:151–154. doi: 10.1016/0006-8993(90)90535-j. [DOI] [PubMed] [Google Scholar]
- Kang Y, Futami T. Arrhythmic firing in dopamine neurons of rat substantia nigra evoked by activation of subthalamic neurons. Journal of Neurophysiology. 1999;82:1632–1637. doi: 10.1152/jn.1999.82.3.1632. [DOI] [PubMed] [Google Scholar]
- Kebabian JW, Calne DB. Multiple receptors for dopamine. Nature. 1979;277:93–96. doi: 10.1038/277093a0. [DOI] [PubMed] [Google Scholar]
- Kim K-M, Nakajima Y, Nakajima S. G protein-coupled inward rectifier modulated by dopamine agonists in cultured substantia nigra neurons. Neuroscience. 1995;69:1145–1158. doi: 10.1016/0306-4522(95)00326-e. [DOI] [PubMed] [Google Scholar]
- Koga E, Momiyama T. Dopamine D2-like receptor-mediated inhibition of glutamatergic synaptic transmission onto dopaminergic neurons in the rat ventral tegmental area. Neuroscience Research Supplement. 1998;22:S100. [Google Scholar]
- Koga E, Momiyama T. Presynaptic inhibition of excitatory transmission onto rat ventral tegmental dopaminergic neurons by dopamine. Neuroscience Research Supplement. 1999;23:S76. doi: 10.1111/j.1469-7793.2000.t01-2-00163.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koob GF, Swerdlow NR. The functional output of the mesolimbic dopamine system. Annals of New York Academy of Sciences. 1988;537:216–227. doi: 10.1111/j.1749-6632.1988.tb42108.x. [DOI] [PubMed] [Google Scholar]
- Lacey MG, Mercuri NB, North RA. Dopamine acts on D2 receptors to increase potassium conductance in neurones of the rat substantia nigra zona compacta. The Journal of Physiology. 1987;392:397–416. doi: 10.1113/jphysiol.1987.sp016787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manzoni OJ, Williams JT. Presynaptic regulation of glutamate release in the ventral tegmental area during morphine withdrawal. Journal of Neuroscience. 1999;19:6629–6636. doi: 10.1523/JNEUROSCI.19-15-06629.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuda Y, Fujimura K. Action of habenular efferents on ventral tegmental area neurons studied in vitro. Brain Research Bulletin. 1992;28:743–749. doi: 10.1016/0361-9230(92)90254-u. [DOI] [PubMed] [Google Scholar]
- Mercuri NB, Bonti A, Calabresi P, Stefani A, Bernardi G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. European Journal of Neuroscience. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- Mereu G, Costa E, Armstrong DM, Vicini S. Glutamate receptor subtypes mediate excitatory synaptic currents of dopamine neurons in midbrain slice. Journal of Neuroscience. 1991;11:1359–1366. doi: 10.1523/JNEUROSCI.11-05-01359.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mereu G, Passino N, Caecangiu G, Gessa GL. Electrophysiological evidences for the primary role of dopamine in the central effects of alcohol and other drugs of abuse. In: Nappi G, Hrnykiewics O, Agnoli A, Fariello RG, Clavazza A, editors. Neurodegenerative Disorders. New York: Raven Press; 1988. pp. 287–301. [Google Scholar]
- Missale C, Nash SR, Robinson SW, Jaber M, Caron MG. Dopamine receptors: from structure to function. Physiological Reviews. 1998;78:189–225. doi: 10.1152/physrev.1998.78.1.189. [DOI] [PubMed] [Google Scholar]
- Momiyama T, Sim JA, Brown DA. Dopamine D1-like receptor-mediated presynaptic inhibition of excitatory transmission onto rat magnocellular basal forebrain neurones. The Journal of Physiology. 1996;495:97–106. doi: 10.1113/jphysiol.1996.sp021576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemeroff CB, Bissette G. Neuropeptides, dopamine, and schizophrenia. Annals of the New York Academy of Sciences. 1988;537:273–291. doi: 10.1111/j.1749-6632.1988.tb42113.x. [DOI] [PubMed] [Google Scholar]
- Nicola SM, Kombian SB, Malenka RC. Psychostimulants depress excitatory synaptic transmission in the nucleus accumbens via presynaptic D1-like dopamine receptors. Journal of Neuroscience. 1996;16:1591–1604. doi: 10.1523/JNEUROSCI.16-05-01591.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicola SM, Malenka RC. Dopamine depresses excitatory and inhibitory synaptic transmission by distinct mechanisms in the nucleus accumbens. Journal of Neuroscience. 1997;17:5697–5710. doi: 10.1523/JNEUROSCI.17-15-05697.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nirenberg NJ, Chan J, Liu Y, Edwards RH, Pickel VM. Ultrastructural localization of the vesicular monoamine transporter-2 in midbrain dopaminergic neurons: potential sites for somatodendritic storage and release of dopamine. Journal of Neuroscience. 1996;16:4135–4145. doi: 10.1523/JNEUROSCI.16-13-04135.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Donnell P, Grace AA. Tonic D2-mediated attenuation of cortical excitation in nucleus accumbens neurons recorded in vitro. Brain Research. 1994;634:105–112. doi: 10.1016/0006-8993(94)90263-1. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Watson C. The Rat Brain in Stereotaxic Coordinates. San Diego: Academic Press; 1998. [Google Scholar]
- Radnikow G, Misgeld U. Dopamine D1 receptors facilitate GABAA synaptic currents in the rat substantia nigra pars reticulata. Journal of Neuroscience. 1998;18:2009–2016. doi: 10.1523/JNEUROSCI.18-06-02009.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sladeczek F, Momiyama A, Takahashi T. Presynaptic inhibitory action of a metabotropic glutamate receptor agonist on excitatory transmission in visual cortical neurons. Proceedings of the Royal Society. 1993;B 253:297–303. doi: 10.1098/rspb.1993.0117. [DOI] [PubMed] [Google Scholar]
- Smith GP, Schneider LH. Relationship between mesolimbic dopamine function and eating behavior. Annals of the New York Academy of Sciences. 1988;537:254–261. doi: 10.1111/j.1749-6632.1988.tb42111.x. [DOI] [PubMed] [Google Scholar]
- Smith Y, Charara A, Parent A. Synaptic innervation of midbrain dopaminergic neurons by glutamate-enriched terminals in the squirrel monkey. Journal of Comparative Neurology. 1996;364:231–253. doi: 10.1002/(SICI)1096-9861(19960108)364:2<231::AID-CNE4>3.0.CO;2-6. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Joyce MP, Lin L, Geldwert D, Haber SN, Hattori T, Rayport S. Dopamine neurons make glutamatergic synapses in vitro. Journal of Neuroscience. 1998;18:4588–4602. doi: 10.1523/JNEUROSCI.18-12-04588.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamminga CA. Schizophrenia and glutamatergic transmission. Critical Review of Neurobiology. 1998;12:21–36. doi: 10.1615/critrevneurobiol.v12.i1-2.20. [DOI] [PubMed] [Google Scholar]
- Umemiya M, Raymond LA. Dopaminergic modulation of excitatory postsynaptic currents in rat neostriatal neurons. Journal of Neurophysiology. 1997;78:1248–1255. doi: 10.1152/jn.1997.78.3.1248. [DOI] [PubMed] [Google Scholar]
- Wigmore MA, Lacey MG. Metabotropic glutamate receptors depress glutamate-mediated synaptic input to rat midbrain dopamine neurones in vitro. British Journal of Pharmacology. 1998;123:667–674. doi: 10.1038/sj.bjp.0701662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y-N, Mercuri NB, Johnson SW. Presynaptic inhibition of γ-aminobutyric acidB-mediated synaptic current by adenosine recorded in vitro in midbrain dopamine neurons. Journal of Pharmacology and Experimental Therapeutics. 1995;273:576–581. [PubMed] [Google Scholar]
- Zhang J, Chiod LA, Freeman AS. Influence of excitatory amino acid receptor subtypes on the electrophysiological activity of dopaminergic and nondopaminergic neurons in the substantia nigra. Journal of Pharmacology and Experimental Therapeutics. 1994;269:313–321. [PubMed] [Google Scholar]
- Zhang X-F, Hu X-T, White FJ, Wolf ME. Increased responsiveness of ventral tegmental area dopamine neurons to glutamate after repeated administration of cocaine or amphetamine is transient and selectively involves AMPA receptors. Journal of Pharmacology and Experimental Therapeutics. 1997;281:699–706. [PubMed] [Google Scholar]