

Abstract
ICRAC and intracellular inositol 1,4,5-trisphosphate (InsP3) concentration is complex. In rat basophilic leukaemia (RBL-1) cells dialysed with high intracellular Ca2+ buffer, the relationship is supra-linear with a Hill coefficient of 12 and resembles an apparent ‘all-or-none’ phenomenon. The non-linearity seems to arise from InsP3 metabolism. However, it is not clear which InsP3-metabolising pathway engenders the non-linear behaviour nor whether ICRAC is always activated to its maximal extent by InsP3.
Using the whole-cell patch clamp technique, we dialysed RBL-1 cells with different concentrations of the InsP3 analogue InsP3-F. InsP3-F is broken down by Ins(1,4,5)P3 5-phosphatase but is not a substrate for Ins(1,4,5)P3 3-kinase. The relationship between InsP3-F and ICRAC amplitude was supra-linear and very similar to that with InsP3 but was distinct from the graded relationship seen with the non-metabolisable analogue Ins2,4,5P3.
In the presence of high intracellular Ca2+ buffer, InsP3-F activated ICRAC to its maximal extent. With moderate Ca2+ buffer, however, sub-maximal ICRAC could be obtained to a maximal InsP3-F concentration. Nevertheless, the relationship between the amplitude of ICRAC and InsP3-F concentration was still supra-linear.
Submaximal ICRAC in response to InsP3-F in the presence of moderate Ca2+ buffer was due to partial depletion of the stores, because the size of the current could be increased by thapsigargin.
The data suggest that first, Ins(1,4,5)P3 5-phosphatase is an important factor which contributes to the non-linear relationship between InsP3 concentration and the amplitude of ICRAC and second, InsP3 does not always activate ICRAC to its maximal extent. At moderate buffer strengths, submaximal ICRAC is evoked by maximal InsP3. However, the supra-linear relationship between InsP3 concentration and amplitude of the current still holds.
In a variety of cell types, stimulation of cell-surface receptors that engage the phosphoinositide pathway evokes a biphasic increase in intracellular Ca2+: an initial inositol 1,4,5-trisphosphate (InsP3)-mediated Ca2+ release phase is followed by a smaller but sustained Ca2+ influx component (Berridge, 1993; Parekh & Penner, 1997). In most non-excitable cells, emptying of the intracellular Ca2+ stores activates a selective Ca2+ current called ICRAC (Ca2+ release-activated Ca2+ current; Hoth & Penner, 1992; Parekh & Penner, 1997). Ca2+ entry through CRAC channels is necessary for prolonging the Ca2+ signal following transient Ca2+ release, for refilling the stores as well as for a host of key cellular functions including exocytosis, gene transcription and cell proliferation (Parekh & Penner, 1997).
The relationship between the intracellular InsP3 concentration and activation of the store-operated Ca2+ influx can be graded (Jacob, 1990) or supra-linear (Parekh et al. 1997; Hartmann & Verkhratsky, 1998; Liu et al. 1998) in different non-excitable cells. In rat basophilic leukaemia (RBL-1) cells, a model system for studying ICRAC, the amplitude of the current is steeply related to InsP3 concentration with a Hill coefficient of 12 (Parekh et al. 1997). This highly non-linear behaviour probably reflects metabolism of InsP3 because the non-metabolisable InsP3 analogue Ins2,4,5P3 generates a graded response (Hill coefficient of 1). Stimulation of cell-surface receptors that engage the phosphoinositide pathway also generates ICRAC in a non-linear manner, suggesting that this highly supra-linear behaviour might be of physiological relevance (Parekh et al. 1997).
Two questions arise from this. First, which of the InsP3-metabolising enzymes accounts for the non-linear relationship between InsP3 concentration and activation of ICRAC? Second, is ICRAC invariably activated to its maximal extent by InsP3 (apparent ‘all-or-none’ activation) or can the current be activated submaximally under certain conditions? Whilst graded ICRAC has been reported for both a non-metabolisable InsP3 analogue (Parekh et al. 1997) and following passive depletion of stores (Fierro & Parekh, 1999), these conditions are non-physiological. It is not known whether InsP3 itself activates the current to a submaximal extent. We have designed experiments to address these issues. We have first investigated the biochemical mechanism which engenders supra-linear activation and then examined whether the size of ICRAC is always maximal following a sufficiently high increase in intracellular InsP3. We find that the relationship between InsP3 concentration and the size of ICRAC is set largely by cytoplasmic Ins(1,4,5)P3 5-phosphatase and is complex. ICRAC can be activated maximally or submaximally by a given InsP3 concentration, and this is determined, at least in part, by Ca2+ reuptake into the stores, which operates efficiently at low to moderate concentrations of intracellular Ca2+ buffer. Even when ICRAC is submaximal, the steep relationship between InsP3 concentration and the amplitude of the current still holds.
METHODS
Rat basophilic leukaemia (RBL-1) cells, which were obtained from the American Type Culture Collection, were cultured as described previously (Parekh et al. 1997; Fierro & Parekh, 1999).
Patch clamp experiments were conducted in the tight-seal whole-cell configuration at room temperature (18–25°C) as described previously (Hamill et al. 1981; Parekh et al. 1997; Fierro & Parekh, 1999). Sylgard-coated, fire-polished pipettes had DC resistances of 2.5-3.7 MΩ when filled with standard internal solution that contained (mM): caesium glutamate, 145; NaCl, 8; MgCl2, 1; Hepes, 10; Mg-ATP, 2; pH 7.2 with CsOH. The Ca2+ chelators EGTA (Sigma) or BAPTA tetracaesium salt (Molecular Probes) were added to this solution at the specified concentrations (described in the text). 3-Deoxy-3-fluoro-myo-inositol 4,5-trisphosphate (InsP3-F; added to the pipette solution) was obtained from Calbiochem. Thapsigargin was from Alomone Laboratories. All other chemicals were purchased from Sigma. A correction of +10 mV was applied for the subsequent liquid junction potential that arose from this glutamate-based internal solution. Extracellular solution contained (mM): NaCl, 145; KCl, 2.8; CaCl2, 10; MgCl2, 2; CsCl, 10; glucose, 10; Hepes, 10; pH 7.4 (NaOH). ICRAC was measured by applying voltage ramps (-100 to +100 mV in 50 ms) at 0.5 Hz from the holding potential of 0 mV as described previously (Parekh et al. 1997). Currents were filtered using an 8-pole Bessel filter at 2.5 kHz and digitised at 100 μs. Currents were normalised by dividing the amplitudes (measured from the voltage ramps at −80 mV) by the cell capacitance. Capacitative currents were compensated before each ramp by using the automatic compensation of the EPC 9–2 amplifier. All leak currents were subtracted by averaging the first two to four ramp currents, and then subtracting this from all subsequent currents. Data are presented as means ±s.e.m., and statistical evaluation was carried out using Student's unpaired t test.
RESULTS
InsP3 can be metabolised in mammalian cells through two pathways: an Ins(1,4,5)P3 3-kinase converts InsP3 to InsP4 and Ins(1,4,5)P3 5-phosphatase degrades InsP3 to InsP2 (Shears, 1992). The InsP3 analogue InsP3-F is resistant to Ins(1,4,5)P3 3-kinase activity (Kozikowski et al. 1990; Sarrany et al. 1992) but is readily susceptible to breakdown by the Ins(1,4,5)P3 5-phosphatase (Sarrany et al. 1992). InsP3-F is a full agonist on the InsP3-gated Ca2+ release channels, although it has a slightly lower affinity (Sarrany et al. 1992). We therefore used InsP3-F to dissect out the metabolic pathway that underlay the supra-linear activation of ICRAC.
Figure 1 summarises the effects of dialysing cells with different concentrations of InsP3-F. The pipette solution was strongly buffered with BAPTA and Ca2+ was clamped close to 120 nM in order to prevent spontaneous depletion of stores and hence activation of ICRAC. We constructed a dose-response curve for each coverslip used and Fig. 1a summarises a typical experimental protocol in which cells were dialysed with different concentrations of InsP3-F. The time courses of the currents (obtained from voltage ramps spanning −100 to +100 mV in 50 ms, top right panel of Fig. 1a) are shown on the left and the I–V relationships on the right. The amplitude of the current to 30 μM InsP3-F had a peak value of −3.08 ± 0.21 pA pF−1 (n= 18, Fig. 1B). This was not significantly different to that seen in the presence of 30 μM InsP3+ 10 mM BAPTA (-3.05 ± 0.38 pA pF−1, n= 7) or 30 μM InsP3-F + 10 mM EGTA + 2 μM thapsigargin (-2.88 ± 0.26 pA pF−1, n= 5; we have observed that ICRAC in BAPTA tends to be slightly larger than that in EGTA but this is not significant; Fierro & Parekh, 1999), and indicates that 30 μM InsP3-F activated ICRAC to its maximal extent. The relationship between InsP3-F concentration and the amplitude of ICRAC was clearly supra-linear with a Hill coefficient of 17 (Fig. 1B, ^). This was strikingly similar to that seen with InsP3 (Fig. 1B, ▵; Hill coefficient of 13 obtained with EGTA as the chelator; Parekh et al. 1997) but markedly different from that seen with the non-metabolisable analogue Ins2,4,5P3 (Fig. 1B, •) (Parekh et al. 1997). The lowest concentration of InsP3-F that activated maximal ICRAC (7.5 μM) did so after a delay that was significantly longer than that seen with higher concentrations (P < 0.05, Fig. 1C). However, once the current was activated, both the time constant of activation and time-to-peak were the same irrespective of the concentration used (P > 0.1, Fig. 1C). The supra-linear relationship between InsP3 concentration and activation of ICRAC therefore seems to reflect the activity of cytoplasmic Ins(1,4,5)P3 5-phosphatase. This family of enzymes has a high capacity for InsP3 and KD values in the micromolar range (Shears, 1992). Therefore they can effectively suppress modest increases in intracellular InsP3 levels, preventing macroscopic activation of ICRAC. We predict that the relationship between the size of ICRAC and the InsP3-F concentration would be less steep in the presence of Ins(1,4,5)P3 5-phosphatase block. Specific blockers of this class of enzyme are lacking and agents that inhibit the phosphatase are plagued with additional effects (for example 2,3-diphosphoglycerate (DPG), which is often used to inhibit the Ins(1,4,5)P3 5-phosphatase (Streb et al. 1985), can also interfere with the Ins(1,4,5)P3 3-kinase (Guillemette et al. 1990), inhibit the InsP3 receptor directly (Guillemette et al. 1990), and affect other signalling pathways like phospholipase D (Kanaho et al. 1993). Nevertheless, we tested the effects of DPG and disulfiram (Fowler et al. 1993), another putative Ins(1,4,5)P3 5-phosphatase inhibitor, on the InsP3-F-ICRAC relationship. However, we found that disulfiram (20 μM, added to the bathing medium) seemed to block CRAC channels directly, because the current could not be activated even in the presence of a maximal concentration of InsP3 (30 μM in 10 mM EGTA; 4/4 cells). With 1 mM DPG in the pipette, three of seven cells responded to InsP3 and 10 mM EGTA after a sizeable delay (the other cells behaved normally), consistent with reports that DPG inhibits the InsP3 receptor (Guillemette et al. 1990). We were therefore forced to abandon these drugs.
Figure 1. Highly supra-linear activation of ICRAC by InsP3-F in the presence of high exogenous Ca2+ buffer.
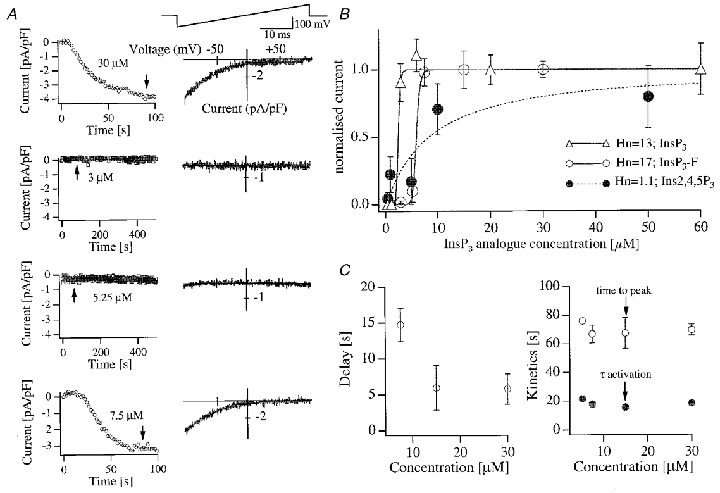
A, patch clamp recordings investigating the effects of dialysing cells from the same coverslip with different concentrations of InsP3-F are shown exactly as the experiment was carried out. The left-hand graphs depict the time course of ICRAC, measured from voltage ramps at −80 mV. The right-hand graphs show the corresponding I–V relationships, taken at the time indicated by the arrows. B shows the overall InsP3-F concentration-ICRAC amplitude curve. Data were fitted using the analysis program IGOR (Wavemetrics, OR, USA). Included in the graph are the corresponding relationships for InsP3 (the physiological isomer) and Ins2,4,5P3 (non-metabolisable analogue). For the InsP3 curve we have included previously published data (Parekh et al. 1997) together with new experiments. The Hill coefficient (Hn) increased from 12 to 13. Our previous Ins2,4,5P3 data have been recently reproduced by Huang & Putney, 1998. Fitting their data yielded a Hill coefficient in the range of 2, reasonably similar to our value of 1. C shows the delay (left) and both the time-to-peak and time constant (τ) of the exponential fits (right). Two cells which gave clear currents to 5.25 μM InsP3-F have been excluded from the left graph (but not the right graph) because the delays (100 and 81 s) would mask the significant difference in delay between 7.5 μM and the higher concentrations, by increasing the range of the y-axis. There were no significant differences between time-to-peak or time constant of activation. For each graph, points represent means ±s.e.m. of 8–18 cells, except for 5.25 μM InsP3-F in C (n= 2). Cells were dialysed with standard internal solution supplemented with 12.5 mM BAPTA and 6.0 mM CaCl2. Free Ca2+ was calculated to be 122 nM and the free BAPTA concentration 6.25 mM. Dialysis with this solution without InsP3 failed to activate ICRAC over a 400 s period (8 cells).
Taken together, these results indicate that ICRAC activates in a supra-linear manner to InsP3 in the presence of high intracellular Ca2+ buffer. Furthermore, the range of InsP3 concentrations over which ICRAC activates is so narrow (2-fold) under these conditions that it approximates to an apparent ‘all-or-none’ process.
We then examined whether first, InsP3 supra-linearly activated ICRAC under more physiological conditions (i.e. weak intracellular Ca2+ buffering) and second, if this was always to its maximal extent. Unfortunately, the current cannot be measured when cells are dialysed with InsP3 in low Ca2+ buffer (Fig. 2a, InsP3-F + 0.1 mM EGTA; see also Huang et al. 1998; Fierro & Parekh, 2000). We therefore determined the lowest concentrations of buffer that would still enable us to reliably measure macroscopic ICRAC and, having found this, we then examined the relationship between InsP3-F concentration and the size of ICRAC. Cells were dialysed with 30 μM InsP3-F and different concentrations of EGTA (range, 0.1-10 mM). For 30 μM InsP3-F, concentrations of EGTA of less than 0.35 mM generally failed to support any macroscopic activation of ICRAC (Fig. 2a). Close to maximum current was seen with around 1 mM EGTA (-2.33 ± 0.19 compared with −2.64 ± 0.31 pA pF−1 for 10 mM EGTA). However, in the presence of 0.6 mM EGTA, we obtained ICRAC that clearly had a submaximal amplitude (-1.39 ± 0.21 pA pF−1, n= 8; Fig. 2a). The relationship between exogenous buffer concentration and ICRAC amplitude in Fig. 2a was supra-linear with a Hill coefficient of 3.5. Figure 2B shows an experiment with a threshold concentration of 3 μM InsP3-F instead of 30 μM (30 cells; 0.1-1 mM EGTA). For comparison, the corresponding data with 30 μM InsP3-F were also included. For EGTA concentrations of 0.6 mM or less, no macroscopic ICRAC was detected. In the presence of 1 mM EGTA and 3 μM InsP3-F, a small current was occasionally seen (6 of 13 cells) but its properties were no different to those of the current evoked by 1 mM EGTA alone (50 % of cells responded to 1 mM EGTA alone compared with 46 % for 1 mM EGTA + 3 μM InsP3-F; their time-to-peak values were 120.1 ± 20.4 and 122.67 ± 44.52 s, respectively, and the overall extent of ICRAC was similar; P > 0.1; Fig. 2B; see also Fierro & Parekh, 1999).
Figure 2. Supra-linear activation of ICRAC in the presence of moderate Ca2+ buffering.
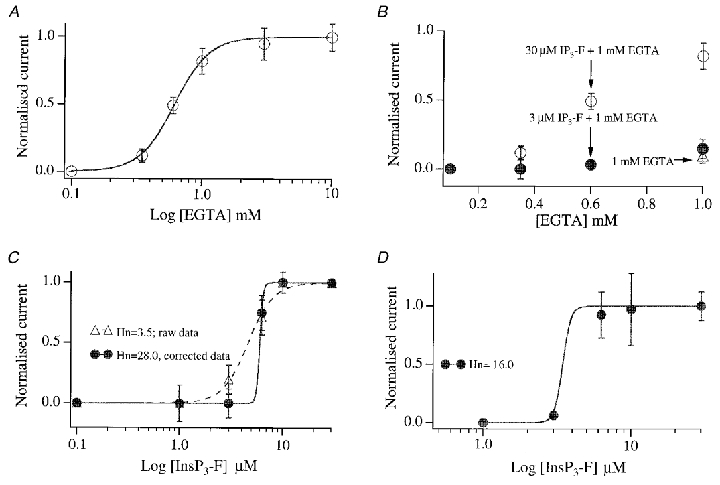
A, relationship between intra-pipette EGTA concentration and the amplitude of ICRAC (evoked by dialysis with 30 μM InsP3-F). Note that ICRAC was clearly submaximal in the presence of 0.6 mM EGTA. Each point is the mean ±s.e.m. of at least 5 cells. B, graph showing the relationship between EGTA concentration and size of ICRAC when the threshold dose of 3 μM InsP3-F (IP3-F) was used. Data for 30 μM have been included for comparison. The EGTA concentration ranged from 0.1 to 1.0 mM (each point is the mean ±s.e.m. of at least 5 cells and error bars are within the circles). The open triangle that overlaps with the filled circle at 1 mM is the size of ICRAC seen following dialysis with 1 mM EGTA and no InsP3-F. C, summary of pooled results when cells were dialysed with different concentrations of InsP3-F in the presence of 1 mM EGTA. Each point is the mean ±s.e.m. of at least 5 cells. Depending on how we treated the response to 3 μM InsP3-F, two different fits were obtained. If we included the size of ICRAC with 1 mM EGTA alone (-0.3 ± 0.11 pA pF−1), and then subtracted this value from the amplitudes seen when different concentrations of InsP3-F were included, we obtained the dashed fit (raw data). However, if we arbitrarily set the amplitude of ICRAC to zero for 3 μM InsP3-F (because there was no significant difference between 1 mM EGTA alone and 3 μM InsP3-F + 1 mM EGTA), and subtracted the value for 3 μM InsP3-F + 1 mM EGTA from the currents seen with higher concentrations of InsP3-F, then we obtained the continuous fit (corrected data). The dashed fit is an underestimate, whereas the continuous one is an overestimate, of the true Hill coefficient (Hn). D, graph summarising the InsP3-F concentration-ICRAC relationship in the presence of 0.6 mM EGTA. Whereas the majority of cells failed to generate a current to 3 μM InsP3-F + 0.6 mM EGTA or 0.6 mM EGTA alone, two cells from one preparation did generate a clear ICRAC to 3 μM InsP3-F (-1.5 and −1.2 pA pF−1). In this same preparation, 0.6 mM EGTA also produced a current rapidly (-1.0 pA pF−1). Because this was so uncharacteristic, we did not include any cells from this preparation.
We then determined the relationship between InsP3-F concentration and activation of ICRAC in the presence of moderate intracellular Ca2+ buffering (1 and 0.6 mM EGTA). Results are summarised in Fig. 2C and D. The relationship between InsP3-F concentration and the amplitude of ICRAC was clearly supra-linear for both cases (Hill coefficient in the range 3.5-28 and apparent KD of 4.1 μM for 1 mM EGTA (see legend to Fig. 2); corresponding values for 0.6 mM EGTA were 16 and 3.4 μM, respectively). Therefore even in the presence of quite moderate amounts of Ca2+ buffer and when the current is submaximal (as in 0.6 mM EGTA), ICRAC is still supra-linearly related to the cytoplasmic InsP3 concentration.
We designed experiments to understand why ICRAC was submaximal in the presence of 0.6 mM EGTA, despite dialysis with a maximal concentration of InsP3-F. We reasoned that stores might refill partially in the presence of such a concentration of Ca2+ buffer and that this would reduce the overall extent of ICRAC activation. If this was the case, then one would predict that the size of ICRAC should increase in the presence of the sarco-endoplasmic reticulum Ca2+-ATPase (SERCA) blocker thapsigargin (Thastrup et al. 1989), since the latter should prevent any Ca2+ reuptake and hence refilling of the stores. The results of Fig. 3a and B show that this was indeed the case. Inclusion of thapsigargin in the pipette solution (with 30 μM InsP3-F and 0.6 mM EGTA) had quite dramatic effects on the properties of ICRAC. The delay before the current activated was substantially reduced (3-fold, P < 0.01; Fig. 3a and B) and, strikingly, the size of the current increased significantly (P < 0.01, Fig. 3a and B). Hence SERCA-mediated reuptake can slow the onset of ICRAC as well as reduce the overall extent of activation. However, the rate of development of ICRAC was the same for both conditions (Fig. 3B). The size of ICRAC in 30 μM InsP3-F + 0.6 mM EGTA + thapsigargin was similar to that seen in the presence of either InsP3+ 10 mM EGTA or InsP3+ 10 mM EGTA + thapsigargin and is therefore the maximal amplitude that can be recorded. Inhibition of SERCA pumps can therefore increase the size of ICRAC evoked by a maximal concentration of InsP3-F in moderate (0.6 mM) but not high (10 mM) EGTA, consistent with enhanced Ca2+-dependent SERCA reuptake in the presence of moderate Ca2+ buffer (Mogami et al. 1998). An alternative explanation for the actions of thapsigargin is that the subsequent elevation of intracellular Ca2+ inhibits the activity of the Ins(1,4,5)P3 5-phosphatase so that InsP3 concentrations in the cytosol are increased and hence ICRAC becomes larger. However, the Ins(1,4,5)P3 5-phosphatase is only weakly sensitive to Ca2+ changes and the most parsimonious explanation of the thapsigargin results is that it reflects reduced Ca2+ reuptake.
Figure 3. Graded ICRAC can be obtained in the presence of moderate Ca2+ buffer because of SERCA uptake and inhibition of the InsP3 receptor.
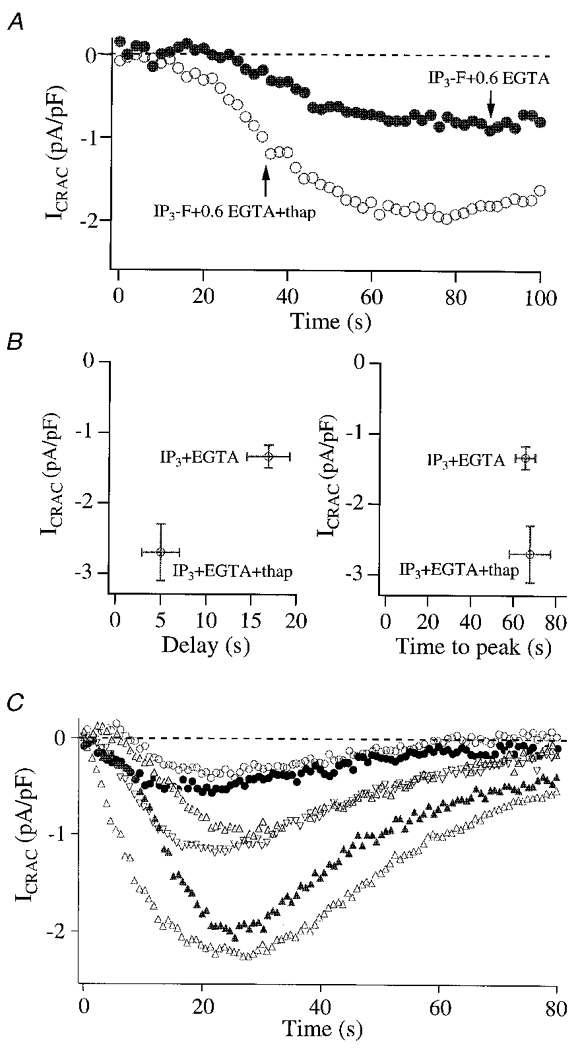
A shows recordings for a cell dialysed with 30 μM InsP3-F and 0.6 mM EGTA (•) and for a cell dialysed with this solution but supplemented with 2 μM thapsigargin (thap), a SERCA blocker. Note the dramatic increase in the size of ICRAC following inhibition of the pumps. B, summary of amplitudes, delays and times to peak for experiments as in A. C, cells dialysed with InsP3 and 1 mg ml−1 heparin (an inhibitor of the InsP3 receptor) can produce a wide range of different ICRAC amplitudes, in spite of the presence of a maximal InsP3 concentration. This experiment will depend on the relative speed of entry of InsP3 and heparin into the cytosol (proportional to series resistance), subsequent diffusion in the cytosol and access to the InsP3 receptors. Cells were dialysed with a solution in which Ca2+ was buffered at 225 nM (3 mM Ca-EGTA and 2 mM EGTA). The decay in the current reflects store refilling because it could be reduced by inclusion of thapsigargin in the pipette (5 cells; data not shown). Dialysis with heparin fails to alter the activation of ICRAC evoked by ionomycin (Parekh & Penner, 1995) or passive depletion (Fierro & Parekh, 1999), indicating that it is not interfering directly with the activation mechanism itself nor with the CRAC channels.
Because inhibition of SERCA-mediated Ca2+ uptake into the stores increases the size of ICRAC in moderate Ca2+ buffer, we examined whether the converse was also true: namely, if inhibition of Ca2+ efflux from the stores, as ICRAC developed, could turn the current off. We reasoned that, in the absence of further InsP3-mediated Ca2+ release, SERCA pumps should rapidly refill the stores, thereby deactivating the current. The experiment of Fig. 3C was designed to test this. We dialysed cells with 30 μM InsP3 and heparin (1 mg ml−1), a competitive antagonist of the InsP3 receptor. Because of its large size, heparin diffuses into the cytosol relatively slowly and hence ICRAC would initially start to activate before appreciable amounts of intracellular heparin would have built up. However, as intracellular heparin levels rise, it should displace InsP3 from its receptor, thereby causing ICRAC to switch off as SERCA pumps refill the stores. Results are summarised in Fig. 3C. We obtained a wide dispersion of ICRAC amplitudes, despite dialysis with a maximally effective InsP3 concentration. Some cells generated a large ICRAC whereas others produced rather small currents. Such pronounced variation was not seen in the absence of heparin. Hence a range of ICRAC amplitudes can be obtained to a fixed InsP3 concentration when Ca2+ release is compromised. The results also indicate that both activation and maintainance of ICRAC in moderate Ca2+ buffering requires the InsP3 receptor to be in an open conducting state in order to compensate SERCA reuptake. Although our results suggest that the InsP3 receptor indirectly controls ICRAC through the Ca2+ content of the stores, it is also conceivable that a more direct regulation involving a physical interaction between the CRAC channels and InsP3 receptors also contributes (Kiselyov et al. 1998). The results of Fig. 3 demonstrate that activation of ICRAC by InsP3 is not a true ‘all-or-none’ phenomenon because, once the current was activated, it did not invariably reach its maximal extent.
DISCUSSION
Our results provide new insights into the activation of ICRAC. The second messenger InsP3 does not invariably activate ICRAC to its maximal extent, because submaximal currents can be observed at moderate levels of intracellular Ca2+ buffering. This seems to reflect a dynamic interplay between Ca2+ release and thapsigargin-sensitive Ca2+ reuptake at the level of the InsP3-sensitive Ca2+ stores. It is interesting that, in moderate buffer, SERCA-mediated reuptake prevents InsP3 from depleting the stores to the extent that ICRAC activates maximally. It seems unlikely that primary active transport (Ca2+ uptake) can match the efflux of Ca2+ through the large conductance InsP3-gated channels. As we have recently suggested (Fierro & Parekh, 2000), the InsP3 receptors on the stores may inactivate, at least partially, hence reducing the extent of Ca2+ movement out of the stores. Furthermore, the fall in intraluminal as well as the increase in cytosolic Ca2+ may both further enhance SERCA-mediated reuptake (Mogami et al. 1998).
The finding that ICRAC activates only partially in response to InsP3 in moderate buffer and that this is due to SERCA activity indicates that regulation of Ca2+ uptake may represent a powerful means for grading the extent of activation of ICRAC in the presence of the weak to moderate levels of intracellular Ca2+ buffering seen in intact cells.
Although ICRAC can be submaximal in the presence of moderate intracellular Ca2+ buffer, the highly supra-linear relationship between InsP3 concentration and activation of the current still holds. However, we cannot be sure that the supra-linear behaviour is valid under conditions of weak buffering, since we and others consistently fail to record activation of the current by InsP3 under these conditions.
The supra-linear relationship between InsP3 concentration and activation of ICRAC seen in moderate to high intracellular Ca2+ buffer probably arises from the cytoplasmic activity of Ins(1,4,5)P3 5-phosphatase. Although this conclusion is drawn from experiments with a phosphatase-resistant InsP3 analogue and the standard pharmacological tools we have employed are not refined enough to selectively interfere with the enzyme, nevertheless this opens up the possibility of new experiments using molecular genetics to test this hypothesis in the future. Although the human Ins(1,4,5)P3 5-phosphatase has been cloned, the corresponding enzyme in the rat has not. Nevertheless, we have tried to reduce expression of the rat inositol 5-phosphatase using antisense oligonucleotides based on the human sequence (supplied by Biognostik, Goettingen, Germany), but without success. Cloning of the rat enzyme should enable us to design more rational antisense oligonucleotides. Interestingly, in certain human malignant haemopoetic cells, the activity of this enzyme is significantly reduced compared with that in normal cells (Mengubas et al. 1994). Because Ca2+ influx through CRAC channels has been linked to cell growth and proliferation (Berridge, 1995), it is tempting to speculate that the abnormal cell growth in these forms of human leukaemia reflects, at least in part, the non-linear relationship between InsP3 concentration and the size of ICRAC.
Acknowledgments
We are grateful to Professors Alison F. Brading and H. Criss Hartzell and Dr Leonardo Fierro for discussion/critical comments on various stages of the manuscript. This work was supported by a grant from the Wellcome Trust to A.B.P. (no. 049236/Z/96/Z). A.B.P. is the Amersham Fellow in Medical Cell Biology, Keble College, Oxford. M.D.G. is a recipient of an international Human Frontiers of Science Programme long-term Fellowship.
References
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Calcium signalling and cell proliferation. Bioessays. 1995;17:491–500. doi: 10.1002/bies.950170605. [DOI] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. On the characterisation of the mechanism underlying passive activation of the Ca2+ release-activated Ca2+ current ICRAC in rat basophilic leukaemia cells. The Journal of Physiology. 1999;520:407–416. doi: 10.1111/j.1469-7793.1999.00407.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fierro L, Parekh AB. Substantial depletion of the intracellular Ca2+ stores is required for macroscopic activation of the Ca2+ release-activated Ca2+ current in rat basophilic leukaemia cells. The Journal of Physiology. 2000;522:247–257. doi: 10.1111/j.1469-7793.2000.t01-1-00247.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fowler CJ, Brannstrom G, Ahlgren PC, Florvall L, Akerman KE. Inhibition of inositol 1,4,5-trisphosphate 5-phosphatase by micromolar concentrations of disulfiram and its analogues. Biochemical Journal. 1993;289:853–859. doi: 10.1042/bj2890853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guillemette G, Favreau I, Lamontagne S, Boulay G. 2,3-Diphosphoglycerate is a nonselective inhibitor of inositol 1,4,5-trisphosphate action and metabolism. European Journal of Pharmacology. 1990;188:251–260. doi: 10.1016/0922-4106(90)90009-m. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hartmann J, Verkhratsky A. Relations between intracellular Ca2+ stores and store-operated Ca2+ entry in primary cultured human glioblastoma cells. The Journal of Physiology. 1998;513:411–424. doi: 10.1111/j.1469-7793.1998.411bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–356. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Huang Y, Putney JW., Jr Relationship between intracellular calcium store depletion and calcium release-activated calcium current in a mast cell-line (RBL-1) Journal of Biological Chemistry. 1998;273:19554–19559. doi: 10.1074/jbc.273.31.19554. [DOI] [PubMed] [Google Scholar]
- Huang Y, Takahashi M, Tanzawa K, Putney JW., Jr Effect of adenophostin A on Ca2+ entry and calcium release-activated calcium current (Icrac) in rat basophilic leukemia cells. Journal of Biological Chemistry. 1998;273:31815–31821. doi: 10.1074/jbc.273.48.31815. [DOI] [PubMed] [Google Scholar]
- Jacob R. Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. The Journal of Physiology. 1990;421:55–77. doi: 10.1113/jphysiol.1990.sp017933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanaho Y, Nakai Y, Katoh M, Nozawa Y. The phosphatase inhibitor 2,3-diphosphoglycerate interferes with phospholipase D activation in rabbit peritoneal neutrophils. Journal of Biological Chemistry. 1993;268:12492–12497. [PubMed] [Google Scholar]
- Kiselyov K, Xu X, Mozhayeva G, Kuo T, Pessah I, Mignery G, Zhu X, Birnbaumer L, Muallem S. Functional interaction between InsP3 receptors and store-operated Htrp3 channels. Nature. 1998;396:478–482. doi: 10.1038/24890. [DOI] [PubMed] [Google Scholar]
- Kozikowski AP, Faruq AH, Aksoy IA, Seewald MJ, Powis G. Synthesis of the first optically pure, fluorinated inositol 1,4,5-trisphosphate of myo-inositol stereochemistry and its effects on Ca2+ release in Swiss 3T3 cells. Journal of the American Chemical Society. 1990;112:7403–7404. [Google Scholar]
- Liu K-Q, Bunnell SC, Gurniak CB, Berg LJ. T cell receptor-initiated calcium release is uncoupled from capacitative calcium entry in Itk-deficient T cells. Journal of Experimental Medicine. 1998;187:1721–1727. doi: 10.1084/jem.187.10.1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mengubas K, Jabbar SAB, Nye KE, Wilkes S, Hoffbrand AV, Wickremasinghe RG. Inactivation of calcium ion-regulating inositol polyphosphate second messengers is impaired in subpopulations of human leukemia cells. Leukemia. 1994;8:1718–1725. [PubMed] [Google Scholar]
- Mogami H, Tepikin AV, Petersen OH. Termination of cytosolic Ca2+ signals: Ca2+ reuptake into intracellular stores is regulated by the free Ca2+ concentration in the store lumen. EMBO Journal. 1998;17:435–442. doi: 10.1093/emboj/17.2.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: Nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Activation of store-operated calcium influx at resting InsP3 levels by sensitization of the InsP3 receptor in rat basophilic leukaemia cells. The Journal of Physiology. 1995;489:377–382. doi: 10.1113/jphysiol.1995.sp021058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Sarrany ST, Wilcox RA, Liu C, Potter BVL, Nahorski SR. 3-Position modification of myo-inositol 1,4,5-trisphosphate: Consequences for intracellular calcium mobilisation and enzyme recognition. European Journal of Pharmacology. 1992;8:265–272. doi: 10.1016/0922-4106(92)90071-3. [DOI] [PubMed] [Google Scholar]
- Shears SB. Metabolism of inositol phosphates. Advances in Second Messenger and Phosphoprotein Research. 1992;26:63–92. [PubMed] [Google Scholar]
- Streb H, Heslop JP, Irvine RF, Schulz I, Berridge MJ. Relationship between secretagogue-induced Ca2+ release and inositol polyphosphate production in permeabilized pancreatic acinar cells. Journal of Biological Chemistry. 1985;260:7309–7315. [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promoter, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+-ATPase. Proceedings of the National Academy of Sciences of the USA. 1989;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]