

Abstract
A technique was developed to counteract the changes in threshold to electrical stimuli of large myelinated cutaneous afferents in the human median nerve induced by ischaemia for 13 min. Intermittent application of polarizing currents was used in five subjects, in whom refractoriness, supernormality and the strength-duration time constant (τSD) were tracked to determine whether compensating for the ischaemia-induced changes in threshold also controlled the ischaemic changes in these excitability parameters.
The threshold compensation prevented the ischaemic changes in τSD, an excitability parameter dependent on nodal Na+ channels. Threshold compensation did not prevent the changes in refractoriness and supernormality, whether the compensation began 10, 100 or 200 ms prior to the test stimuli.
In three subjects, continuous polarizing current was injected for 13 min to compensate for the ischaemic change in threshold, thus clamping threshold at the pre-ischaemic level. Again, τSD was effectively controlled, but there were still ischaemic changes in refractoriness and supernormality.
The effective control of τSD suggests that both the intermittent threshold compensation and the continuous threshold clamp effectively controlled membrane potential at the node of Ranvier.
The ischaemic increase in refractoriness when threshold was kept constant could be due to interference with the processes responsible for refractoriness by a metabolic product of ischaemia. The ischaemic change in supernormality during effective compensation probably results from the intrusion of refractoriness into the conditioning-test intervals normally associated with maximal supernormality.
The present results indicate that ischaemia has effects on axonal excitability that cannot be readily explained by changes in membrane potential. Specifically, it is suggested that ischaemic metabolites interfere with the recovery of Na+ channels from inactivation.
Peripheral nerves are commonly subjected to ischaemic insults. The physiological consequences of ischaemic insults and their underlying mechanisms have been the subject of recent investigations in human subjects using threshold-tracking techniques (Bostock et al. 1991a,b, 1994; Mogyoros et al. 1997). During ischaemia, axons depolarize and their excitability increases until conduction block ensues, when they become inexcitable. As axonal excitability increases, there are changes in other properties consistent with axonal depolarization: refractoriness increases, supernormal excitability decreases, and the strength-duration time constant (τSD) increases. Axons hyperpolarize on release of ischaemia but, paradoxically, may develop quite intense ectopic activity, such that subjects experience strong paraesthesiae and fasciculations (Bostock et al. 1991a,b, 1994).
Some of the changes in nerve excitability produced by ischaemia and its release can be adequately reproduced by injecting polarizing current to depolarize or hyperpolarize axons (Bostock et al. 1991a, 1998; Horn et al. 1996). However, ischaemia has effects on refractoriness that cannot be explained merely by the change in membrane potential, while the release of ischaemia has effects on supernormality that are similarly difficult to explain (Grosskreutz et al. 1999). Specifically, during ischaemia there was a disproportionate increase in refractoriness that was dependent on the duration of ischaemia, and was interpreted as the result of interference with the processes underlying refractoriness (presumably recovery of Na+ channels from inactivation) by an accumulating metabolite produced by ischaemia. On release of ischaemia, supernormality was disproportionately enhanced and less responsive to induced changes in axonal excitability, as if paranodal K+ channels (the activity of which influences the extent of supernormal excitability; see Baker et al. 1987; David et al. 1995) were blocked.
In the present study, a technique was developed to control the excitability of human axons, the intention being to keep axonal excitability at the pre-ischaemic control level so that the typical ischaemic and post-ischaemic changes in threshold would not occur. Measurements of other indices of axonal excitability were then undertaken during intermittent threshold compensation or continuous threshold clamp to see whether the ischaemic changes in these indices were similarly controlled. A threshold-clamp technique has previously been used by Bostock & Grafe (1985) to counteract the hyperpolarization and, thereby, conduction block that resulted when acutely demyelinated rat ventral root axons were activated repetitively. However, such a technique has not previously been used in human subjects.
METHODS
Eighteen experiments were performed on five healthy adult volunteers (aged 22–53 years) who gave informed written consent to the experimental procedures, which had the approval of the Committee on Experimental Procedures Involving Human Subjects of the University of New South Wales.
Antidromic recordings of the compound sensory action potential (CSAP) of large myelinated cutaneous afferents were made using ring electrodes around the index finger of the right hand, as described previously (Kiernan et al. 1996; Mogyoros et al. 1996, 1997; Grosskreutz et al. 1999). For stimulation, the cathode was located over the median nerve on the palmar side of the wrist, and the anode was 15–20 cm proximal over the forearm muscle. Using the QTRAC threshold tracking program (copyright, Institute of Neurology, Queen Square, London, UK), stimuli were delivered at 2 Hz, rotating through a sequence of ten different test stimuli, alone or in combination with a conditioning pulse and/or compensating currents as described in detail below. A fixed supramaximal stimulus of 0.2 ms was delivered on one channel (channel 1) to produce a CSAP of maximal amplitude. On the other channels the threshold currents needed to elicit a CSAP with an amplitude of 50 % of maximum were tracked by an IBM compatible computer. ‘Proportional’ tracking was used, such that the extent to which the stimulus current increased or decreased was proportional to the difference between the target and the measured response. On channels 2 and 3, thresholds were recorded using test stimulus durations of 0.1 and 1 ms, respectively, and these were used to calculate the strength-duration time constant (τSD) according to Weiss’ Law (see Bostock & Bergmans, 1994; Mogyoros et al. 1996, 1997; Grosskreutz et al. 1999).
To monitor refractoriness and supernormality, test stimuli of 0.1 ms duration were preceded by fixed supramaximal conditioning stimuli of 0.2 ms on channels 4 and 5, using conditioning-test intervals of 2 or 7 ms, respectively. For these channels, the test CSAP was measured after the maximal CSAP produced by the conditioning stimulus had been eliminated on-line by subtraction of the CSAP produced by the supramaximal stimulus delivered in isolation (on channel 1). Refractoriness and supernormality were measured as the normalized difference between the thresholds for conditioned and unconditioned test CSAPs.
Intermittent threshold compensation was used to counteract changes during and after ischaemia. In different experiments, DC lasting 30, 120 or 220 ms was used to keep threshold at the pre-ischaemic level. These compensating currents began 10, 100 or 200 ms before the test stimuli on channels 6–10. The strength of the current needed to compensate for the threshold change was determined on channel 6 where a fixed 1 ms test pulse was individually set to the pre-ischaemic threshold current on channel 3. The fixed test pulse on channel 6 was superimposed on the compensating current which was automatically varied using proportional tracking (see above) in the hyper- or depolarizing direction until the fixed 1 ms test pulse elicited the target response. The compensating current was then added to channels 7–10 which were otherwise identical to channels 2–5. Accordingly, computerized tracking was used to set the variable test stimuli on the uncompensated channels 2–5, the variable test pulses on the compensated channels 7–10, and the compensating current on channel 6. The quality of the recording was ensured before the start of the experiment by fine-tuning the strength of the fixed 1 ms test pulse on channel 6 until the polarizing current tracked at approximately zero for at least 5 min. As a result, before ischaemia, the threshold values determined on channels 2–5 and channels 7–10 were the same. Ischaemia was then induced by inflating a sphygmomanometer cuff around the upper arm to 200 mmHg, well above systolic blood pressure in all subjects, and kept at that level for 13 min. Because the compensating current was updated once every 5 s and because of subsequent tracking delays, fast threshold changes, such as those occurring on release of ischaemia, could not be followed.
In three subjects, continuous DC was used to clamp threshold during ischaemia. Only channels 1–5 were used, so that a new threshold value was obtained every 2.5 s for each channel during the continuous threshold clamp. The applied current was manually adjusted to keep the threshold on channel 3 constant at the pre-ischaemic level. Refractoriness, supernormality and τSD were measured, much as in previous studies (Mogyoros et al. 1997; Grosskreutz et al. 1999). Continuous DC was passed for the entire 13 min of ischaemia, so that the change in threshold produced by ischaemia could not be recorded in these experiments (though it could be inferred from the time course of the compensating current). Data were reduced to one measurement every 30 s by averaging 6 adjacent data points for the intermittent threshold compensation and 12 adjacent data points for the continuous threshold clamp.
In all experiments, temperature was monitored continuously by skin sensors at the first metacarpophalangeal joint and at the wrist. It was kept above 32°C by wrapping the arm in blankets and applying radiant heat if necessary.
RESULTS
The excitability of cutaneous afferents in the median nerve was studied during ischaemia produced by inflation of a sphygmomanometer cuff around the upper arm for 13 min in five subjects. Three sets of experiments were performed on each subject using intermittent threshold compensation applied 10, 100 or 200 ms before the test stimuli, and in three subjects a continuous threshold clamp was applied. When the intermittent compensation was used, unconditioned and conditioned thresholds were also recorded without threshold compensation, so that compensated and uncompensated recordings could be directly compared.
Changes in thresholds during ischaemia
The pattern and time course of the changes in threshold produced by ischaemia for 13 min were much as described previously (Bostock et al. 1994; Mogyoros et al. 1997; Grosskreutz et al. 1999). Thresholds decreased during the first 5 min of ischaemia (Fig. 1A) but then reached a plateau. This was accompanied by a marked increase in refractoriness (Fig. 2A, ^), abolition of supernormality (Fig. 3A, ^) and an increase in τSD (Fig. 1D, ^). Restoration of the circulation resulted in an abrupt increase in threshold as axons hyperpolarized, but a depolarizing notch was then superimposed on the hyperpolarization so that threshold approached (but did not reach) the control value before slowly increasing again (Fig. 1A and B). This transient post-ischaemic depolarizing notch has been studied in detail by Bostock et al. (1994) and will not be considered further.
Figure 1. The effect of intermittent threshold compensation on the τSD.
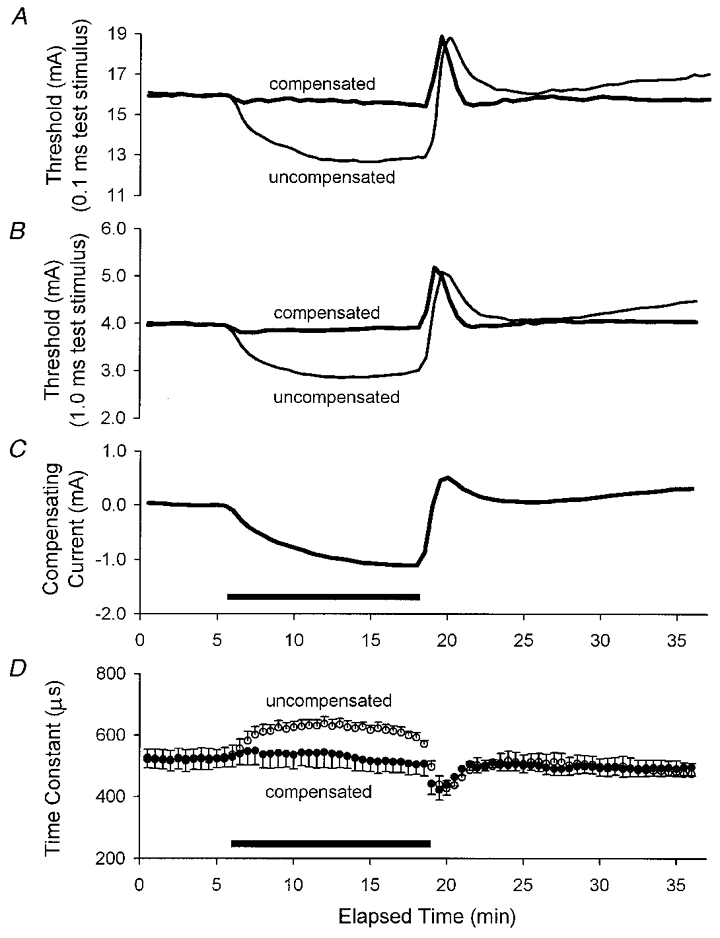
A and B illustrate the threshold changes as measured using test stimuli of 0.1 and 1 ms duration, respectively, produced by ischaemia in the absence of threshold compensation (thin traces) and in its presence (thick traces). The ischaemic change in threshold was effectively controlled except at the onset of the ischaemia and, particularly, at its offset. C shows the profile of the current required to compensate for the ischaemic change in threshold illustrated in B. D illustrates τSD, calculated from the data in A and B. There was little change in τSD during ischaemia. The traces in A-C are the means for five subjects, and the data in D represent the mean ±s.e.m. The compensating current was applied 10 ms before the test stimuli for the data in this figure and in Figs 2 and 3. In this and subsequent figures, the horizontal filled bars indicate the period of ischaemia.
Figure 2. The effects of intermittent threshold compensation on refractoriness, measured as the threshold increase at a conditioning-test interval of 2 ms.
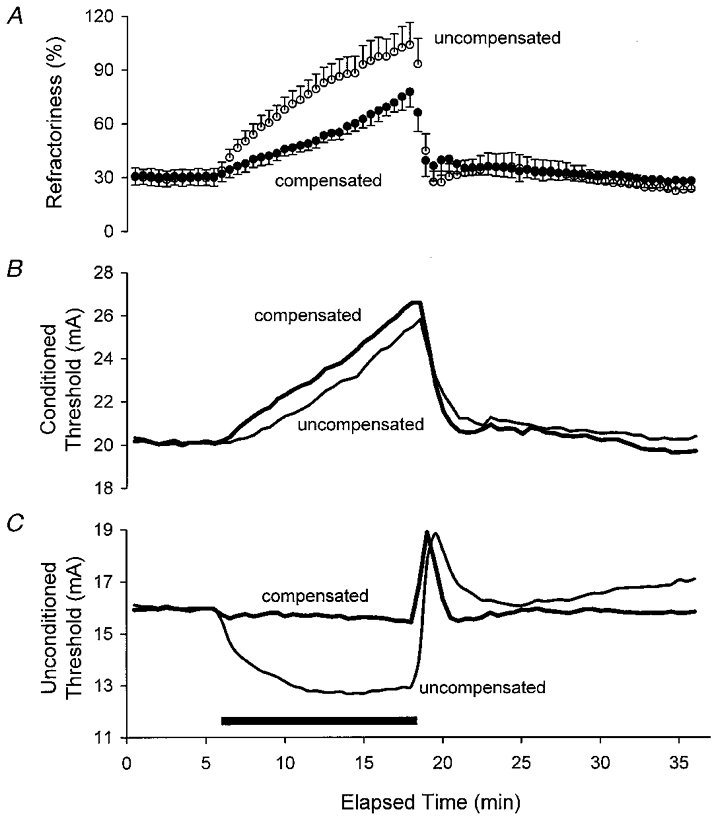
A illustrates mean data ±s.e.m. for five subjects. B and C illustrate, respectively, the mean conditioned thresholds and the mean unconditioned thresholds for the five subjects, i.e. the data used to calculate refractoriness in A. Note that C contains the same data as in Fig. 1A.
Figure 3. The effects of intermittent threshold compensation on supernormality, measured as the threshold decrease at a conditioning-test interval of 7 ms.
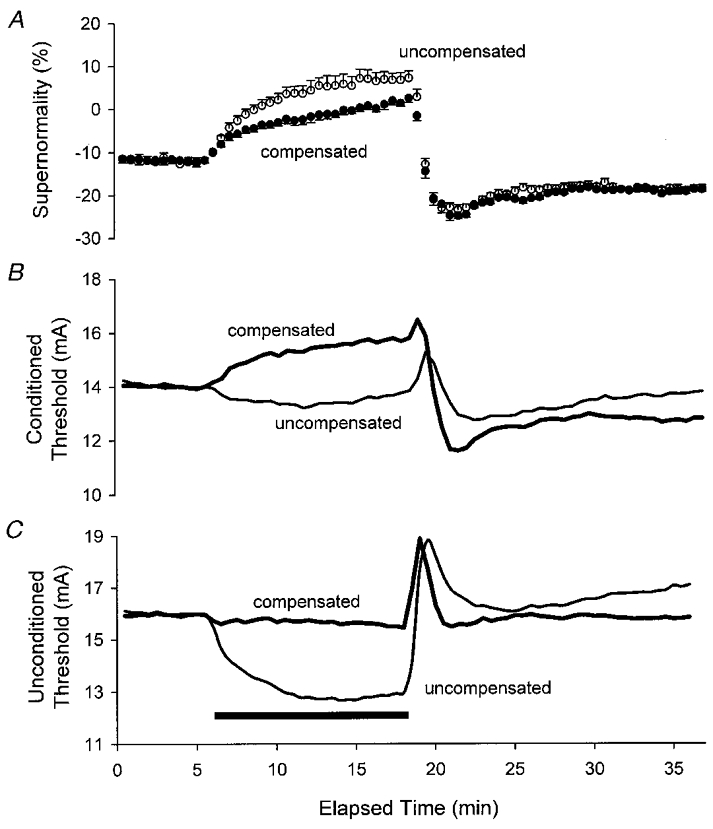
Despite the effective control of threshold (C, same data as in Fig. 1A), there were still changes in the conditioned threshold (measured using a conditioning-test interval of 7 ms, B) and supernormality (A). A illustrates mean data ±s.e.m. for five subjects.
The current required to compensate for the ischaemic change in the 1 ms threshold (Fig. 1C) largely paralleled the ischaemic threshold change recorded simultaneously on the uncompensated channels, but the data diverged at two points. Firstly, the compensating current continued to increase throughout the 13 min period of ischaemia even though the threshold change tended to reach a plateau (compare Fig. 1C with the thin traces in Fig. 1A and B). In this respect, the pattern of the compensating current more closely resembled the increase in refractoriness (Fig. 2A). Secondly, the computer-controlled compensation could not adequately track the rapid hyperpolarizing change in threshold that occurred when ischaemia was terminated.
Control of unconditioned thresholds and τSD
In the uncompensated state, the evolution of threshold measured using test stimuli of 0.1 and 1 ms duration was very similar, except that the absolute change was much less using 1 ms test stimuli (compare Y-axes and thin traces in Fig. 1A and B). However, proportionally the ischaemic change in threshold was greater for the 1 ms stimuli indicating that τSD increased during ischaemia (Fig. 1D, ^).
The threshold compensation was based on the 1 ms threshold and compensated quite accurately for this threshold, except at the onset (slightly) and offset (particularly) of the 13 min episode of ischaemia (Fig. 1B). The same compensating current also effectively controlled the 0.1 ms threshold (Fig. 1A) and, as a result, there was little change in τSD under compensated conditions (Fig. 1D, •). The compensation was equally effective for the unconditioned thresholds and τSD whether it was applied 10 ms (as in Fig. 1), or 100 or 200 ms before the test stimuli (see Fig. 4).
Figure 4. The effects of intermittent compensating currents (D) of different duration on refractoriness (A), supernormality (B) and τSD(C).
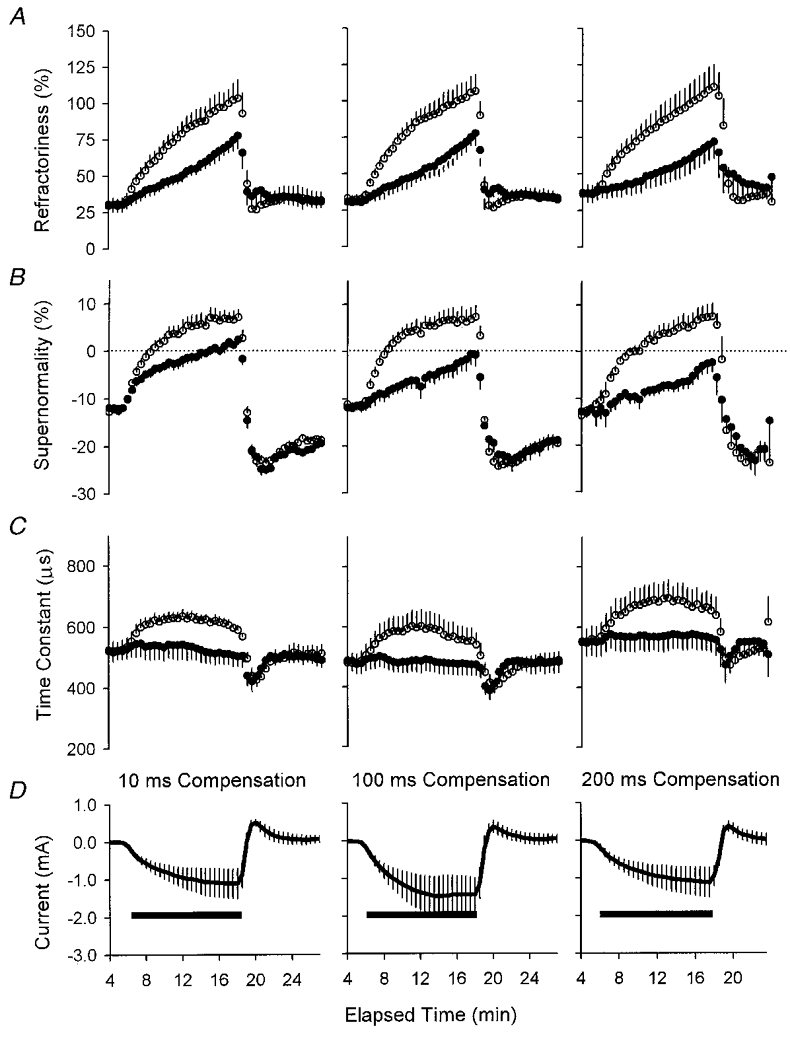
The data in the left-hand column for the 10 ms compensating current are those illustrated in Figs 1–3. ^, uncompensated; •, compensated. Data are means ±s.e.m. for five subjects.
Effect of the intermittent compensation on conditioned thresholds
Prior to ischaemia, the current required to produce the unconditioned 0.1 ms threshold responses was 16.0 ± 3.5 mA (mean ±s.e.m.). Test stimuli of 20.1 ± 3.7 and 14.0 ± 3.1 mA were required when supramaximal stimuli were delivered 2 and 7 ms before the test stimuli (Figs 2B and 3B, respectively). Accordingly, refractoriness was 29.6 ± 4.6 % and supernormality was -12.7 ± 0.3 %. In other words, in order to produce a CSAP of the same size, the test stimulus had to be increased by 30 % when the conditioning-test interval was 2 ms and it had to be decreased by 13 % when the interval was 7 ms.
While the compensating current controlled unconditioned thresholds, it failed to control the conditioned thresholds (Figs 2B and 3B). Indeed, in both instances, the conditioned threshold increased more under compensated conditions than under uncompensated conditions. Accordingly, the threshold compensation failed to control refractoriness and supernormality (Figs 2A and 3A, respectively). The ischaemic changes in refractoriness and supernormality were less prominent with threshold compensation because there was no change in the unconditioned threshold, whereas there was a marked decrease in threshold in the uncompensated recording.
When the duration of the intermittent compensating current was increased from 10 ms to 100 and 200 ms, there was no significant difference in the ability of the compensation to prevent changes in refractoriness during ischaemia (Fig. 4A; Student's two-tailed t test). However, the ischaemic reduction in supernormality was significantly less with the longer compensating currents (P= 0.036, for the 10 and 200 ms data 6 min into ischaemia; Fig. 4B).
Continuous threshold clamp
A continuous threshold clamp was used in three subjects to keep the 0.1 ms threshold constant during ischaemia for 13 min. Because the clamp was continuous, the data cannot be compared with unclamped data for the same sequence.
The clamp effectively controlled the unconditioned thresholds and τSD but not the conditioned thresholds (Figs 5 and 6). Figure 5 shows the data for all three subjects, with four 1 min averages immediately prior to the onset of ischaemia (^), from 9 min into ischaemia (•) and from 10 min after the release of ischaemia (▵). The clustering of the data points along the horizontal axes indicates that the threshold changed little during and after ischaemia, i.e. that the threshold clamp was quite effective in keeping the 0.1 ms threshold constant. In each subject, refractoriness increased by ∼100 % of the control level for that subject. Refractoriness was highly variable between subjects before ischaemia, and remained so during and after ischaemia. Supernormality was abolished during ischaemia in subjects 2 and 3 but only slightly reduced in subject 1, in whom the change in refractoriness was least (see Discussion). τSD did not change in subjects 1 and 2 and increased slightly, by < 10 %, in subject 3.
Figure 5. The effects of a continuous threshold clamp on refractoriness (A), supernormality (B) and τSD (C) for three subjects.
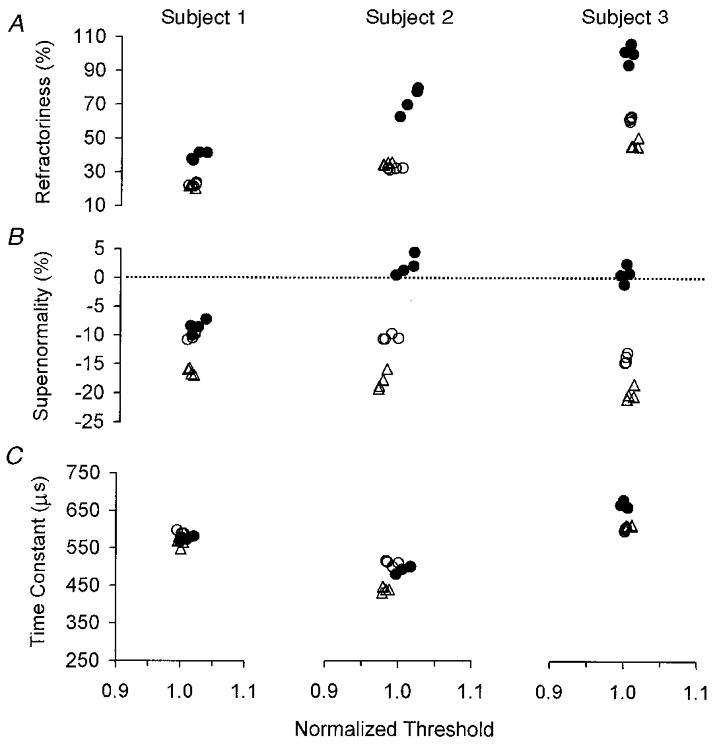
In A-C, the minimal scatter of data along the X-axis indicates the efficacy of the continuous threshold clamp. ^, four 1 min measurements of the appropriate parameter immediately prior to ischaemia; •, four 1 min measurements towards the end of the 13 min period of ischaemia; ▵, four 1 min measurements made after release of ischaemia. Refractoriness more than doubled in each subject during ischaemia under effective threshold-clamp conditions. Supernormality was abolished in subjects 2 and 3 but decreased only slightly in subject 1 (in whom the absolute increase in refractoriness was least). There was little ischaemic change in τSD.
Figure 6. The effect of a continuous threshold clamp on different parameters of axonal excitability.
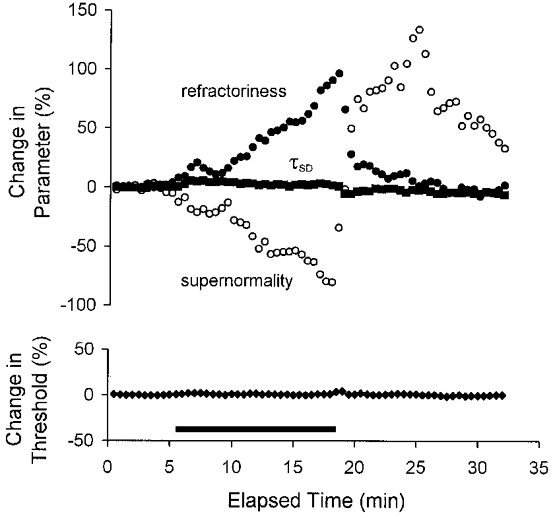
This figure illustrates the mean data for the three subjects during ischaemia, with the change in each parameter being expressed as a percentage of the pre-ischaemic value. Both threshold (lower panel, ♦) and τSD (upper panel, ▪) were effectively controlled, except at the onset and offset of the ischaemic episode. The changes in refractoriness and supernormality mirrored one another during ischaemia but diverged substantially following its release.
Figure 6 presents the mean data for the three subjects, as the percentage change from the pre-ischaemic value. There was little change in τSD (upper panel, ▪), other than that attributable to residual fluctuations in threshold (lower panel, ♦), but there were prominent changes in refractoriness (•) and supernormality (^). During ischaemia the decrease in supernormality largely paralleled the increase in refractoriness (Figs 6 and 7), but following the release of ischaemia these excitability measures diverged (Fig. 6). The reciprocal relationship between refractoriness and supernormality during the development of ischaemia is illustrated in Fig. 7, together with the fact that these changes occurred in the absence of a significant change in threshold.
Figure 7. The relationship between the ischaemic change in supernormality and the ischaemic change in refractoriness during a continuous threshold clamp.
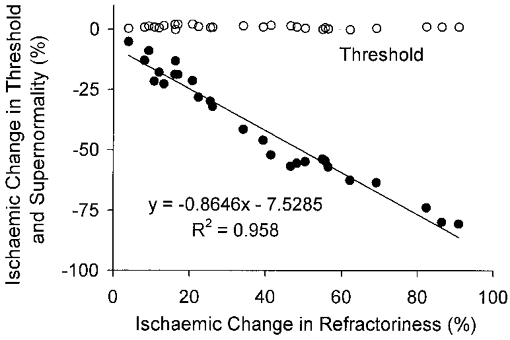
The data from Fig. 6 have been replotted, to demonstrate the close relationship between the changes in supernormality and refractoriness (•). There was little change in threshold during ischaemia under threshold-clamp conditions (^) and there was no relationship between the change in threshold and the change in refractoriness.
DISCUSSION
An effective technique has been used in human subjects to apply a continuous clamp on axonal excitability such that, apart from transients at the beginning and end of ischaemia, the threshold did not change significantly during an ischaemic insult lasting 13 min. In addition, the threshold was intermittently compensated in order to monitor the development of excitability indices when ischaemic alteration of membrane excitability was kept constant. The strength-duration time constant was effectively controlled by both the continuous threshold clamp and the intermittent compensating currents. However, these measures did not prevent ischaemic changes in refractoriness and supernormality, and it will be argued below that these findings indicate that ischaemia interferes with Na+ channel function through a mechanism that is independent of axonal excitability.
Intermittent compensation for the ischaemic changes in threshold
The threshold was compensated for 10, 100 and 200 ms before the test stimuli were delivered and, given an interstimulus interval of 500 ms, there was intermittently no compensation for periods that were longer than the compensated periods. Intermittent compensation was chosen to avoid the risk of electrical burns due to the continuous application of DC for many minutes. However, intermittent compensation would not control processes that had a longer time course than the duration of the compensating current. Specifically, if the membrane depolarization produced by ischaemia activated a process with very slow kinetics, such as ultra-slow Na+ channel inactivation, and if this process contributed significantly to the ischaemic changes in refractoriness (or supernormality), there might still be changes in these indices despite apparently effective threshold compensation. For this reason, continuous DC was used in three subjects to clamp threshold. The results were the same as those with the intermittent compensating currents. τSD was effectively controlled, but refractoriness and supernormality were not. It seems reasonable to conclude that ultra-slow Na+ channel inactivation and similar processes do not contribute significantly to the changes in these indices of axonal excitability. This does not imply that Na+ channels in human cutaneous afferents do not undergo slow inactivation, merely that it does not contribute significantly to refractoriness or to its enhancement during ischaemia.
Strength-duration time constant and Na+ channel function
τSD depends on the passive membrane time constant and a local response due to voltage-dependent conductances active at threshold (Bostock & Rothwell, 1997). The most important threshold conductance is probably a persistent Na+ conductance, and appropriate channels have been identified on neurones in rat dorsal root ganglia (Baker & Bostock 1997, 1998). The voltage dependence of τSD is presumably due to the voltage dependence of this conductance.
That τSD could be effectively controlled when the threshold was returned to the pre-ischaemic level suggests that at least some nodal Na+ channels did not see a change in membrane potential under these conditions. Although it is unsafe to assume that threshold always parallels membrane potential (Baker & Bostock, 1989), the effective clamping of τSD is consistent with effective control of the ischaemic changes in membrane potential at the node of Ranvier.
Refractoriness and Na+ channel function
It would not be expected that a brief compensating current could effectively clamp the internodal membrane, and for this reason the duration of the intermittent current was increased in different series of experiments to 100 and 200 ms. However, there is no good reason to believe that a longer clamp would more effectively control refractoriness, given that internodal factors play little role in the extent of refractoriness (see below), and this proved to be so (Fig. 4). Compensating currents of longer duration did prevent the intra-ischaemic reversal of supernormal excitability into subexcitability, but they were unable to prevent the ischaemic decrease in supernormality. In addition, when a continuous clamp was used in three subjects, there were still changes in refractoriness and supernormality even though threshold was controlled reasonably well and there was little change in τSD.
The major cause of refractoriness is recovery of transient Na+ channels from inactivation (Hodgkin & Huxley, 1952). The increase in refractoriness during ischaemia can be explained by greater inactivation of Na+ channels when the nodal membrane is depolarized by ischaemia, so that fewer channels are available for action potential generation (Mogyoros et al. 1997). However, under threshold-clamp conditions, there was a paradox: one property dependent on nodal Na+ channels (τSD) was effectively controlled, while another (refractoriness) was not. It is therefore possible that some factor (other than membrane potential) was interfering with the recovery of Na+ channels from inactivation. Taken with the results of previous studies (Grosskreutz et al. 1999), the results are consistent with an ischaemia-specific factor that has little or no voltage dependence, perhaps, as previously suggested, an ischaemic metabolite that accumulates during ischaemia.
Brismar (1981) demonstrated that ischaemia shifted the relationship between membrane potential and Na+ channel inactivation to the left, but the reason for this shift was not elucidated. It is possible that the reason is the putative metabolite. Thus, while the extent of refractoriness normally depends on membrane potential (and temperature; see Burke et al. 1999), an additional factor becomes operative during ischaemia. A further implication is that the intra-axonal milieu can influence the function of voltage-dependent channels, again a proposal that has adequate precedent in the literature (e.g. K+ channels: Grafe et al. 1994; persistent Na+ current (INaP): Crill, 1996; inward rectification (Ih): Pape, 1996).
Supernormality
It is perhaps not surprising that a compensating current of long duration was required to minimize the ischaemic change in supernormality, given that the voltage dependence of supernormality depends on paranodal and internodal factors (Barrett & Barrett, 1982; Baker et al. 1987; Bowe et al. 1987; David et al. 1995), and that long current pulses are required to charge the internodal membrane adequately (Barrett & Barrett, 1982; Bostock, 1995; see also Grosskreutz et al. 1999). The major determinant of the voltage dependence of supernormality is the voltage dependence of the depolarizing afterpotential, which is, in turn, largely due to the change in internodal resistance that occurs with the opening or closure of voltage-dependent K+ channels in the paranodal and internodal regions. It is therefore relevant that prolonged anoxia can block K+ channels under hyperglycaemic conditions (Schneider et al. 1992; Grafe et al. 1994).
However, there is probably no necessity to invoke such a mechanism to explain the decrease in supernormality when threshold was controlled using a continuous clamp or a compensating current of long duration, both of which should have been adequate to affect the internodal membrane. In previous studies (Mogyoros et al. 1997), supernormal excitability was replaced during ischaemia by subexcitability at conditioning-test intervals up to 10 ms (the maximum tested), and this was due to both loss of supernormal excitability and dramatic growth of refractoriness in duration as well as extent. Thus, an increase in refractoriness when threshold was clamped could lead to the appearance of subnormal excitability at conditioning-test intervals normally associated with maximal supernormality. The reciprocal nature of the ischaemic changes in supernormality and refractoriness seen in Fig. 7 might then be expected.
In summary, the threshold-clamp and threshold-compensation data in the present study provide further evidence for interference with refractoriness by a metabolic product of ischaemia, independent of membrane potential. The threshold changes at conditioning-test intervals that normally sample supernormality could reflect the ischaemic increase in refractoriness rather than the processes normally responsible for supernormality.
Acknowledgments
This work was supported by the National Health and Medical Research Council of Australia. J. Grosskreutz was supported by the Deutsche Forschungsgemeinschaft.
References
- Baker M, Bostock H. Depolarization changes the mechanism of accommodation in rat and human motor axons. The Journal of Physiology. 1989;411:545–561. doi: 10.1113/jphysiol.1989.sp017589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker MD, Bostock H. Low-threshold, persistent sodium current in rat large dorsal root ganglion neurons in culture. Journal of Neurophysiology. 1997;77:1503–1513. doi: 10.1152/jn.1997.77.3.1503. [DOI] [PubMed] [Google Scholar]
- Baker MD, Bostock H. Inactivation of macroscopic late Na+ current and characteristics of unitary late Na+ currents in sensory neurons. Journal of Neurophysiology. 1998;80:2538–2549. doi: 10.1152/jn.1998.80.5.2538. [DOI] [PubMed] [Google Scholar]
- Baker M, Bostock H, Grafe P, Martius P. Function and distribution of three types of rectifying channel in rat spinal root myelinated axons. The Journal of Physiology. 1987;383:45–67. doi: 10.1113/jphysiol.1987.sp016395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett EF, Barrett JN. Intracellular recording from vertebrate myelinated axons: mechanism of the depolarizing afterpotential. The Journal of Physiology. 1982;323:117–144. doi: 10.1113/jphysiol.1982.sp014064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H. Mechanisms of accommodation and adaptation in myelinated axons. In: Waxman SG, Kocsis JD, Stys PK, editors. The Axon. New York: Oxford University Press; 1995. pp. 311–327. [Google Scholar]
- Bostock H, Baker M, Grafe P, Reid G. Changes in excitability and accommodation of human motor axons following brief periods of ischaemia. The Journal of Physiology. 1991a;441:513–535. doi: 10.1113/jphysiol.1991.sp018765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Baker M, Reid G. Changes in excitability of human motor axons underlying post-ischaemic fasciculations: evidence for two stable states. The Journal of Physiology. 1991b;441:537–557. doi: 10.1113/jphysiol.1991.sp018766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Bergmans J. Post-tetanic excitability changes and ectopic discharges in a human motor axon. Brain. 1994;117:913–928. doi: 10.1093/brain/117.5.913. [DOI] [PubMed] [Google Scholar]
- Bostock H, Burke D, Hales JP. Differences in behaviour of sensory and motor axons following release of ischaemia. Brain. 1994;117:225–234. doi: 10.1093/brain/117.2.225. [DOI] [PubMed] [Google Scholar]
- Bostock H, Cikurel K, Burke D. Threshold tracking techniques in the study of human peripheral nerve. Muscle and Nerve. 1998;21:137–158. doi: 10.1002/(sici)1097-4598(199802)21:2<137::aid-mus1>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- Bostock H, Grafe P. Activity-dependent excitability changes in normal and demyelinated rat spinal root axons. The Journal of Physiology. 1985;365:239–257. doi: 10.1113/jphysiol.1985.sp015769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bostock H, Rothwell JC. Latent addition in motor and sensory fibres of human peripheral nerve. The Journal of Physiology. 1997;498:277–294. doi: 10.1113/jphysiol.1997.sp021857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowe CM, Kocsis JD, Waxman SG. The association of the supernormal period and the depolarizing afterpotential in myelinated frog and rat sciatic nerve. Neuroscience. 1987;21:585–593. doi: 10.1016/0306-4522(87)90144-8. [DOI] [PubMed] [Google Scholar]
- Brismar T. Potential clamp analysis of the effect of anoxia on the nodal function of rat peripheral nerve fibres. Acta Physiologica Scandinavica. 1981;112:495–496. doi: 10.1111/j.1748-1716.1981.tb06851.x. [DOI] [PubMed] [Google Scholar]
- Burke D, Mogyoros I, Vagg R, Kiernan MC. Temperature dependence of excitability indices of human cutaneous afferents. Muscle and Nerve. 1999;22:51–60. doi: 10.1002/(sici)1097-4598(199901)22:1<51::aid-mus9>3.0.co;2-q. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium current in mammalian central neurons. Annual Review of Physiology. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- David G, Modney B, Scappaticci KA, Barrett JN, Barrett EF. Electrical and morphological factors influencing the depolarizing after-potential in rat and lizard myelinated axons. The Journal of Physiology. 1995;489:141–157. doi: 10.1113/jphysiol.1995.sp021037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grafe P, Bostock H, Schneider U. The effects of hyperglycaemic hypoxia on rectification in rat dorsal root axons. The Journal of Physiology. 1994;480:297–307. doi: 10.1113/jphysiol.1994.sp020360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosskreutz J, Lin C, Mogyoros I, Burke D. Changes in excitability indices of cutaneous afferents produced by ischaemia in human subjects. The Journal of Physiology. 1999;518:301–314. doi: 10.1111/j.1469-7793.1999.0301r.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. The Journal of Physiology. 1952;117:500–544. doi: 10.1113/jphysiol.1952.sp004764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horn S, Quasthoff S, Grafe P, Bostock H, Renner R, Schrank B. Abnormal axonal inward rectification in diabetic neuropathy. Muscle and Nerve. 1996;19:1268–1275. doi: 10.1002/mus.880191002. [DOI] [PubMed] [Google Scholar]
- Kiernan MC, Mogyoros I, Burke D. Differences in the recovery of excitability in sensory and motor axons of human median nerve. Brain. 1996;119:1099–1105. doi: 10.1093/brain/119.4.1099. [DOI] [PubMed] [Google Scholar]
- Mogyoros I, Kiernan MC, Burke D. Strength-duration properties of human peripheral nerve. Brain. 1996;119:439–447. doi: 10.1093/brain/119.2.439. [DOI] [PubMed] [Google Scholar]
- Mogyoros I, Kiernan MC, Burke D, Bostock H. Excitability changes in human sensory and motor axons during hyperventilation and ischaemia. Brain. 1997;120:317–325. doi: 10.1093/brain/120.2.317. [DOI] [PubMed] [Google Scholar]
- Pape HC. Queer current and pacemaker: the hyperpolarization-activated cation current in neurons. Annual Review of Physiology. 1996;58:299–327. doi: 10.1146/annurev.ph.58.030196.001503. [DOI] [PubMed] [Google Scholar]
- Schneider U, Jund R, Nees S, Grafe P. Differences in sensitivity to hyperglycemic hypoxia of isolated rat sensory and motor nerve fibers. Annals of Neurology. 1992;31:605–610. doi: 10.1002/ana.410310607. [DOI] [PubMed] [Google Scholar]