

Abstract
Direct voltage-gated (voltage-dependent Ca2+ release, VDCR) and Ca2+ influx-gated (Ca2+-induced Ca2+ release, CICR) sarcoplasmic reticulum (SR) Ca2+ release were studied in feline ventricular myocytes. The voltage-contraction relationship predicted by the VDCR hypothesis is sigmoidal with large contractions at potentials near the Ca2+ equilibrium potential (ECa). The relationship predicted by the CICR hypothesis is bell-shaped with no contraction at ECa.
The voltage dependence of contraction was measured in ventricular myocytes at physiological temperature (37 °C), resting membrane potential and physiological [K+]. Experiments were performed with cyclic adenosine 3′,5′-monophosphate (cAMP) in the pipette or in the presence of the β-adrenergic agonist isoproterenol (isoprenaline; ISO).
The voltage-contraction relationship was bell-shaped in Na+-free solutions (to eliminate the Na+ current and Na+-Ca2+ exchange, NCX) but the relationship was broader than the L-type Ca2+ current (ICa,L)-voltage relationship.
Contractions induced with voltage steps from normal resting potentials to -40 mV are thought to represent VDCR rather than CICR. We found that cAMP and ISO shifted the voltage dependence of ICa,L activation to more negative potentials so that ICa,L was always present with steps to -40 mV. ICa,L at -40 mV inactivated when the holding potential was decreased (V½ =−57·8 ± 0·49 mV).
ISO increased inward current, SR Ca2+ load and contraction in physiological [Na+] and a broad bell-shaped voltage-contraction relationship was observed. Inhibition of reverse-mode NCX, decreasing ICa,L and decreasing SR Ca2+ loading all decreased contractions at strongly positive potentials near ECa.
The voltage-contraction relationship in 200 μM cadmium (Cd2+) was bell-shaped, supporting a role of ICa,L rather than VDCR.
All results could be accounted for by the CICR hypothesis, and many results exclude the VDCR hypothesis.
The processes that link sarcolemmal depolarization to the opening of the sarcoplasmic reticulum (SR) Ca2+ release channel (and subsequent SR Ca2+ release) have been well studied in isolated cardiac myocytes but are still not fully understood. The hypothesis with the greatest experimental support is SR Ca2+ release induced by Ca2+ influx (Ca2+-induced Ca2+ release, CICR) (Fabiato & Fabiato, 1975; Fabiato, 1983,1985; Bers & Merrill, 1985; Cannell et al. 1987, 1995; Callewaert et al. 1988; Beuckelmann & Wier, 1988; duBell & Houser, 1989; Näbauer et al. 1989; Valdeolmillos et al. 1989; Näbauer & Morad, 1990; Bers, 1991). Studies of CICR show that SR Ca2+ release occurs when Ca2+ moves into the cell and binds to the SR Ca2+ release channel (ryanodine receptor), causing it to open. Many Ca2+ influx pathways have been shown to be capable of inducing SR Ca2+ release. The most prominent of these is Ca2+ influx through L-type Ca2+ channels, ICa,L (London & Krueger, 1986; Beuckelmann & Wier, 1988; Näbauer et al. 1989; Bers, 1991; Cleemann & Morad, 1991). Alternative Ca2+ influx pathways that can induce SR Ca2+ release include reverse-mode Na+-Ca2+ exchange (NCX) (Leblanc & Hume, 1990; Nuss & Houser, 1992; Levi et al. 1994), T-type Ca2+ channels (Sipido et al. 1998), Ca2+ influx through Na+ channels (‘slip-mode conductance’: Santana et al. 1998) and a tetrodotoxin-sensitive Ca2+ current (Aggarwal et al. 1997). The respective role of each of these pathways in normal excitation-contraction (EC) coupling is yet to be firmly established.
Recently it has been suggested that a voltage-dependent process that is independent of Ca2+ influx causes SR Ca2+ release in cardiac myocytes (Ferrier & Howlett, 1995; Howlett et al. 1998). This voltage-dependent Ca2+ release (VDCR) mechanism would represent a direct physical linkage between a voltage-sensitive protein in the t-tubular membrane and the SR Ca2+ release channel. The putative cardiac VDCR is similar to the EC coupling process in skeletal muscle where it is well established that a sarcolemmal voltage sensor causes the SR Ca2+ release channel to open through a direct physical link (Block et al. 1988).
There are several features that distinguish the putative cardiac VDCR from CICR. VDCR requires a more negative membrane potential (near the normal resting potential) to be fully activated with depolarization and it appears to have a more negative activation threshold than CICR. VDCR is also best observed at physiological temperatures, with normal intracellular and extracellular [K+], and it requires cyclic adenosine 3′,5′-monophosphate (cAMP) (Hobai et al. 1997; Howlett et al. 1998). Another feature that separates VDCR from CICR is that VDCR has a sigmoidal (saturating) voltage dependence, whereas the voltage dependence of ICa,L-induced CICR is more bell-shaped (similar to the ICa,L-voltage relationship). Finally, VDCR is not eliminated by agents that block the L-type Ca2+ current and CICR (Howlett et al. 1998).
The experimental conditions required to observe VDCR can make voltage-clamp experiments technically demanding and, therefore, results are often difficult to interpret with confidence. The negative membrane potentials required for activation of VDCR makes the Na+ current available for activation. Inadequate block or control of the Na+ current during voltage steps near its activation threshold (-70 to -50 mV) can cause voltage escape into the potential range in which ICa,L is activated (∼-50 to -40 mV) thereby causing CICR. Even if Na+ current is controlled, the resulting Na+ influx could accumulate in diffusion-limiting subsarcolemmal ‘fuzzy’ spaces (Lederer et al. 1990) and promote Ca2+ influx via reverse-mode Na+-Ca2+ exchange (Leblanc & Hume, 1990; Lipp & Niggli, 1994). Some previous studies of VDCR have used Ca2+ channel blockers to reduce the possibility that CICR underlies VDCR. Unfortunately, the interpretation of these studies rests on the premise that Ca2+ current is completely blocked. This is especially difficult to prove when K+ channel blockers are not employed because ICa,L is not accurately observed when overlapping K+ currents are present. Another complicating factor is that VDCR is studied in myocytes with large SR Ca2+ loads that are caused by addition of cAMP to the pipette solution or bath application of β-adrenergic agonists (Charteir et al. 1999). When the SR Ca2+ load is large, very small Ca2+ currents can induce SR Ca2+ release (Han et al. 1994; Bassani, 1995). Therefore, it is difficult to determine if SR Ca2+ release induced by large voltage steps to positive potentials (used to document the sigmoidal voltage dependence of VDCR) results from either VDCR or from Ca2+ influx via reverse-mode Na+-Ca2+ exchange and/or ICa,L, both of which could cause CICR.
The objective of the present experiments was to re-explore the VDCR hypothesis under conditions that minimize confounding experimental features of previous studies. Specifically, we studied the voltage dependence of contraction from normal resting membrane potentials in the presence of cAMP or the non-selective β-adrenergic agonist isoproterenol (ISO) in feline ventricular myocytes. In some experiments, Na+-free (intracellular and extracellular) solutions were used to eliminate the Na+ current and Ca2+ fluxes through the NCX. Experiments in Na+-containing bath solutions, like those used in most previous studies of VDCR (Hobai et al. 1997; Howlett et al. 1998; Ferrier et al. 1998) were performed with inhibitors of the Na+ current or NCX, or with Na+-free pipette solution to eliminate Ca2+ influx via reverse-mode NCX. If VDCR is present under our experimental conditions, then contractions should be elicited at negative potentials that do not activate the L-type Ca2+ current and the voltage dependence of contraction should be sigmoidal, i.e. with large contractions at positive potentials approaching the Ca2+ equilibrium potential.
Our results show that contractions caused by depolarizing voltage steps in feline myocytes are always associated with Ca2+ influx via the L-type Ca2+ current or reverse-mode NCX. We further show that even in the presence of high cellular cAMP, the voltage dependence of contraction is bell-shaped if reverse-mode NCX is eliminated or if SR Ca2+ loading is reduced. These findings strongly suggest that CICR is the major mechanism of EC coupling in mammalian cardiac myocytes. We find no evidence for VDCR.
METHODS
Cardiac myocyte isolation
Feline left ventricular myocytes were isolated by a method developed and refined in this laboratory and used in many previous studies (duBell & Houser, 1989; Nuss & Houser, 1991, 1992). In brief, adult cats (2-3 kg) were anaesthetized with a mixture of ketamine (40 mg kg−1) and acepromazine (0.6 mg kg−1) i.m. Heparin (500 U kg−1) was administered i.v. and allowed to circulate for 5 min before an overdose of sodium pentobarbital (100 mg kg−1) was administered i.p. The heart was quickly explanted through a median sternotomy, weighed and cannulated on a constant-flow Langendorff apparatus. The heart was perfused (∼25 ml min−1) with a non-recirculating Ca2+-free Krebs-Henseleit-bicarbonate buffer (KHB-B: composition (mM): 12.5 glucose, 5.4 KCl, 1 lactic acid, 1.2 MgSO4, 130 NaCl, 1.2 NaH2PO4, 25 NaHCO3 and 2 sodium pyruvate; pH 7.4) containing 10 mM taurine for 4–6 min. KHB-B supplemented with 180 U ml−1 collagenase, 10 mM taurine and 0.02 mM CaCl2 was then recirculated for 20 min. When the tissue softened, the heart was removed from the cannula and the atria, right ventricle and septum were separated from the left ventricle. The remaining left ventricular tissue was gently minced and filtered through a 400 μm stainless-steel filter. The cell suspension was gently centrifuged at 75 g for 1 min, and the cell pellet was resuspended in KHB-B solution containing 1 % (w/v) bovine serum albumin, 10 mM taurine and 0.5 mM CaCl2. Initial yields from the cell isolation are consistently 80–90 % rod-shaped (of total cell count). Cells were maintained at room temperature with a 5 % CO2 and 95 % O2 overlay. Throughout the isolation procedure all solutions were equilibrated with 5 % CO2 and 95 % O2 and warmed to 37°C. All experiments were performed within 12 h of isolation. Only quiescent, rod-shaped myocytes with clear sarcomeric cross striations and minimal resting leak currents (< 50 pA) when held at the resting membrane potential were included in the study.
Electrophysiology and contractile measurements
Experiments were performed on the stage of an inverted microscope (Zeiss, Germany). Voltage-contraction relationships were constructed by controlling membrane voltage by the whole-cell patch clamp technique. Micropipettes were fabricated with a Flaming-Brown P-87 Micropipette Puller (Sutter Instruments Company, Novato, CA, USA) and had tip resistances of 2–4 MΩ when filled with the pipette solutions listed in Table 1. Once a gigaohm seal was formed, the patch was ruptured with gentle suction. Experimental protocols were performed after adequate time had been allowed for intracellular dialysis.
Table 1.
Intracellular and extracellular solutions
| Internal pipette solution | External bath solution | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Compound | P1 | P2 | P3 | P4 | P5 | Compound | B1 | B2 | B3 |
| Caesium aspartate | — | — | — | 130 | 130 | 4-AP | — | — | 2 |
| EGTA | — | 0.05 | 0.05 | 5 | 5 | CaCl2 | 1 | 1 | 2 |
| Hepes | 10 | 10 | 10 | 10 | 10 | CsCl | — | — | 5.4 |
| Potassium aspartate | 130 | 130 | 130 | — | — | Glucose | 10 | 10 | 10 |
| K-ATP | 5 | 5 | 5 | — | — | Hepes | 5 | 5 | 5 |
| KCl | 20 | 20 | 20 | — | — | KCl | 5.4 | 5.4 | — |
| MgCl2(H2O)6 | 1 | 1 | 1 | 1 | 1 | MgCl2(H2O)6 | 1.2 | 1.2 | 1.2 |
| NaCl | 10 | — | — | — | — | NaCl | 150 | — | — |
| NMDG | — | 10 | 10 | 10 | 10 | NMDG | — | 150 | 150 |
| TEA-Cl | — | — | — | 20 | 20 | ||||
| Tris ATP | — | — | — | 5 | 5 | ||||
| Tris cAMP | — | — | 0.05 | — | 0.05 | ||||
| pH = 7.2 | pH = 7.4 | ||||||||
All values are millimolar.
Electrophysiological techniques were similar to those previously reported (Hamill et al. 1981; duBell & Houser, 1989). Membrane voltage was controlled by an Axoclamp 2 voltage-clamp amplifier in discontinuous mode (6-8 kHz) (Axon Instruments, Foster City, CA, USA). The sample clock was monitored on a separate oscilloscope to ensure adequate settling of the switch clamp circuit. The amplifier was controlled by pCLAMP 8.0 software and data were acquired by a Digidata 1200 analog-to-digital converter. Once the signal was converted to digital form, it was stored on an IBM PC for off-line analysis. Data were analysed off-line with Clampfit 8.0 analysis software (Axon Instruments).
The intracellular and extracellular solutions used in these experiments are listed in Table 1. Solutions were tailored to the experimental conditions required as discussed in Results. All solutions were heated to 37°C. The flow of the bathing solution was approximately 2–3 ml min−1. All compounds were applied during a 0.5 Hz train to +10 mV to monitor drug effects on the whole-cell current and myocyte shortening.
Myocyte shortening was measured using video-edge detection as previously described (Steadman et al. 1988; Bailey & Houser, 1993). A video camera (Phillips high speed CCD CM800, Philips Components, Slatersville, RI, USA) was placed in the eyepiece of the microscope to acquire the cell image. Cell edges were measured with a video analyser (Crescent Electronics, Salt Lake City, UT, USA). Before experimental protocols were performed, the myocyte resting cell length (RCL) was recorded and all shortening data were referenced with respect to the RCL in order to account for varying cell lengths. This technique has been used in numerous experiments from our laboratory (Nuss & Houser, 1992; Bailey & Houser, 1993).
Experimental protocols
Most experimental protocols were designed to determine the voltage dependence of contraction. In these experiments the holding potential was -80 or -70 mV. To ensure comparable SR Ca2+ loads before each test step, four 500 ms conditioning steps at 0.5 Hz to +10 mV were used in control experiments and two 500 ms steps were used when cAMP was in the pipette solution or when ISO was applied. Test steps were changed in +10 mV increments from -70 to +100 mV. In experiments in which Cd2+ (200 μM) was used to block L-type Ca2+ current, conditioning steps were to +80 mV to promote Ca2+ influx via reverse-mode NCX. Variations from this general protocol are described in specific experiments.
To assess SR Ca2+ loading, the inward sodium-calcium exchange current was measured during rapid administration of caffeine. Myocytes were voltage clamped to -70 mV and paced at 0.5 Hz to +10 mV. When a steady state was achieved, 10 mM caffeine was rapidly applied for 100 ms in place of a voltage step. Caffeine was administered with a picospritzer (Parker, Fairfield, NJ, USA) using a pipette situated close to the cell. Caffeine at this concentration is known to fully release Ca2+ from the SR. Full SR Ca2+ release during the test spritz was verified by failure of a subsequent caffeine spritz to cause an inward sodium-calcium exchange current or contraction.
Statistics
All data in the tables and figures are reported as the mean ±s.e.m. Data were analysed by Student's t test or analysis of variance (ANOVA), where appropriate. A probability level < 0.05 was considered significant.
RESULTS
Na+-free solutions
The first series of voltage-clamp experiments were performed with Na+-free pipette and bath solutions to eliminate the Na+ current and associated voltage control difficulties and to eliminate Ca2+ fluxes through the NCX. These experiments were technically difficult because myocytes easily overload with Ca2+ when it is not removed from the cell by forward-mode NCX (Allen et al. 1983). Therefore, we worked at slow pacing rates (0.2 Hz) to reduce Ca2+ influx. The first series of experiments were conducted in the absence of cAMP. A typical experimental result is shown in Fig. 1A and the average data (n = 5) are shown in Fig. 1B. Contraction magnitude increased with depolarization and reached a maximum near +10 mV. Contractions remained large when voltage steps to values between 0 and +40 mV were used. Steps to values beyond +40 mV induced progressively smaller contractions and revealed the bell-shaped voltage- contraction (V–C) relationship. It is important to note that the bell-shaped V–C relationship observed in Na+-free conditions is much broader than the ICa,L-voltage relationship (see Figs 2C and 3B).
Figure 1. The effect of cellular dialysis with cAMP on the voltage dependence of contraction in Na+-free bath and pipette solutions (B2, P2 and P3; Table 1).

A, representative examples of contractions and whole-cell currents from a myocyte in Na+-free (in and out) solutions without cAMP. The holding potential was -80 mV and steps to -40, 0 and +100 mV are shown. Note that at +100 mV the contraction does not start until repolarization. B, voltage-contraction relationship from 5 myocytes in Na+-free solutions. C, representative examples of contractions and whole-cell currents from a myocyte in Na+-free (in and out) solutions with 50 μM cAMP in the pipette. The holding potential was -80 mV and steps to -40, 0 and +100 mV are shown. Note that contraction is present at -40 mV and that contraction at +100 mV occurred with repolarization. D, voltage-contraction relationship from 3 myocytes in Na+-free solutions and 50 μM cAMP in the pipette. Note that the relationship is shifted to the left, and while still bell-shaped, was broader than in B. The dotted line at 0 mV in this and subsequent figures is for reference so that the voltage dependence of contraction can be more easily compared under different conditions.
Figure 2. Effects of bath application of isoproterenol on the voltage dependence of contraction in Na+-free solutions (B2, P2; Table 1).
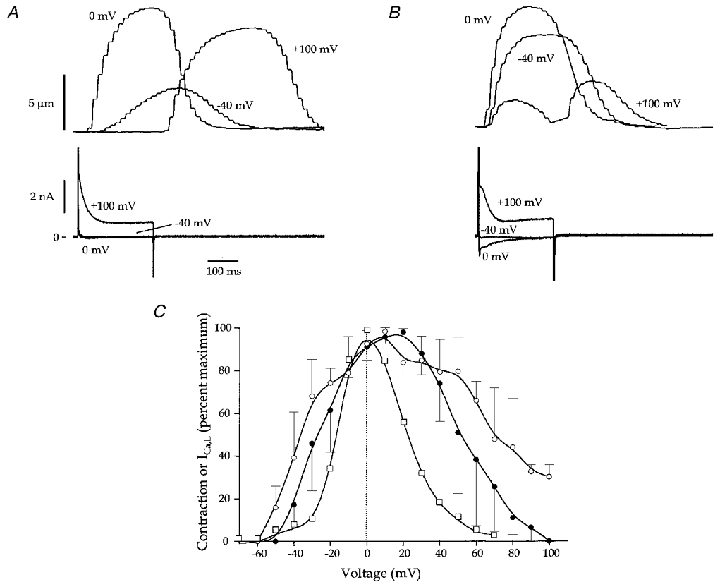
A, representative recordings of contraction and whole-cell current induced by voltage steps from -80 mV to -40, 0 and +100 mV before isoproterenol. B, same cell as in A after exposure to ISO. Note the increase in inward current and larger contraction at -40 mV. There was a small contraction at +100 mV in this cell. C, voltage-contraction relationship from 3 myocytes before and after ISO. ISO broadened the relationship in all 3 myocytes, but it still maintained its bell-shaped features. The voltage dependence of ICa,L (as measured in Fig. 3) is shown for comparison.
Figure 3. Effects of isoproterenol on L-type Ca2+ current recorded in Na+- and K+-free solutions (B3, P4 and P5; Table 1).
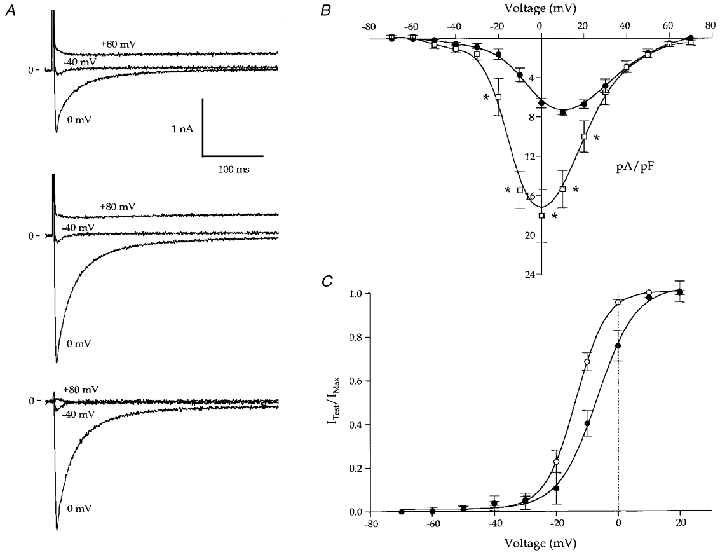
A, representative recordings of whole-cell current before (top trace) and after (middle trace) bath application of ISO. Currents at -40, 0 and +80 mV are shown. The holding potential was -80 mV. Note the small inward currents at -40 mV. The Cd2+-sensitive currents at -40, 0 and +80 mV (after ISO) are shown in the lower panel. B,ICa,L-voltage relationship from 5 myocytes under control conditions (filled symbols) and 3 myocytes with cAMP (open symbols) in the pipette. Currents were significantly greater in the presence of cAMP (*P < 0.05). C, the voltage dependence of ICa,L activation from 5 myocytes before (filled symbols) and after (open symbols) ISO application is shown. The conductance, GCa,L, was calculated from the measured currents and a derived Erev,Ca of +54 mV. The calculations were done with a derivative of Ohm's law: GCa,L =ICa,L/(Emem–Erev,Ca). The results were fitted with a Boltzmann equation: y = (yini–yfin)/(1 + exp(x – x 0/dx) + yfin), where yini and yfin are the initial and final y values, respectively, dx describes the slope factor, and the y value at x0 describes the V1/2 (control: V1/2 =−6.8 ± 1 mV, dx = 6.2 ± 0.2 mV; ISO: V1/2 =−13.7 ± 0.7 mV, dx = 4.9 ± 0.3 mV). There was a significant leftward shift in the relationship (control vs. ISO: V1/2, P < 0.01; dx, P < 0.05).
The putative VDCR mechanism requires cAMP (Hobai et al. 1997). Therefore, the next experiments were performed with either cAMP in the pipette or cAMP generation was induced by bath application of ISO. Contractions were larger and faster when cAMP was included in the Na+-free filling solution and the magnitude of the inward current was increased (Fig. 1C). The bell-shaped V–C relationship was broader in cells dialysed with cAMP than in those in which it was not used (Fig. 1B vs. D). Contractions induced by steps to +10 mV were larger in cells dialysed with versus without cAMP. However, contractions still decreased in size when voltage steps were made to strongly positive potentials approaching the calcium equilibrium potential, ECa (Fig. 1D). Similar results were obtained when ISO (5 × 10−8 M) was used to increase cellular cAMP in Na+-free conditions (Fig. 2A–C). In these experiments ISO broadened the V–C relationship at both negative and positive potentials (Fig. 2C), but contraction size still decreased when voltage steps were increased beyond +40 to +60 mV to strongly positive potentials approaching ECa. These results strongly support the idea that ICa,L, not VDCR, is the trigger for SR Ca2+ release in these experiments.
Ca2+ current at -40 mV
We routinely observed contraction with voltage steps to -40 mV when myocyte cAMP was elevated via the pipette or after bath application of ISO. Previous studies (Ferrier et al. 1998) have used contractions that occur when the membrane voltage is stepped to -40 mV as evidence for VDCR, assuming that ICa,L is not present. To examine the idea that ICa,L, not VDCR, is responsible for contractions induced with voltage steps from -80 to -40 mV we measured ICa,L in the presence of K+ channel blockers, before and after application of ISO. ISO increased ICa,L (Fig. 3A) and caused a leftward shift in the voltage dependence of ICa,L activation (Fig. 3C) and a small cadmium-sensitive inward current was observed at -40 mV in every cell (Fig. 3A, bottom trace). Cells dialysed with pipettes containing cAMP had significantly larger Ca2+ currents than controls (Fig. 3B).
Previous studies that have used contractions at -40 mV as evidence for VDCR have suggested that the ability of depolarizing prepulses to inactivate contractions at -40 mV represents the voltage dependence of inactivation of VDCR (Howlett et al. 1998). An alternative explanation is that contractions at -40 mV are induced by ICa,L and that depolarizing prepulses inactivate this current and thereby reduce contraction magnitude. The voltage dependence of inactivation of ICa,L at -40 mV (in the presence of ISO) was measured in five myocytes and a typical example is shown in Fig. 4A. These experiments show that ICa,L at -40 mV was inactivated by depolarizing prepulses and the half-inactivation voltage was -57.8 ± 49 mV (Fig. 4B). This value is almost identical to the half-inactivation voltage of the putative VDCR mechanism reported previously (-54.1 mV, Ferrier et al. 1998).
Figure 4. Voltage dependence of inactivation of the Ca2+ current at -40 mV induced by voltage steps from -80 to -40 mV (B3, P4; Table 1).

A, representative current recordings from a myocyte bathed in ISO. The voltage protocol is shown above the data. As the prepulse was progressively depolarized the size of the current with the test step to -40 mV was reduced. B, the relationship between prepulse potential and the size of the Ca2+ current at -40 mV is plotted. The average data of 5 myocytes are shown. The currents at the different prepulse potentials were normalized to the current during the step from -80 to -40 mV and were fitted with a Boltzmann equation: y = (yini–yfin)/(1 + exp(x – x0/dx) + yfin), where dx describes the slope factor and the y value at x0 describes the V1/2. The V1/2 was -57.8 ± 0.49 mV and the dx was 4.13 ± 0.3 mV.
Na+-containing solutions
Almost all previous studies of VDCR have been performed in Na+-containing bath solutions (Hobai et al. 1997; Howlett et al. 1998). It seemed possible that our inability to observe VDCR in Na+-free conditions might be the result of some unknown Na+ requirement of the VDCR mechanism. Therefore, we repeated our experiments in Na+-containing solutions. The voltage dependence of contraction in 150 mM bath Na+ and 10 mM pipette Na+ (no ISO) was more complex than observed in Na+-free solution. The V–C relationship was basically bell-shaped between -60 and +60 mV (Fig. 5A and E). Steps to potentials between +60 and +100 mV caused progressively larger contractions. However, these contractions developed slowly with a delayed onset and were maintained (tonic) for the duration of depolarization (Fig. 5A). ISO application increased the velocity and magnitude of contraction and the inward current (Fig. 5B). In the presence of ISO the V–C relationship broadened and phasic (decaying during the depolarizing step) contractions were observed at strongly positive potentials (Fig. 5C). It is noteworthy that phasic contractions induced with large voltage steps to strongly positive potentials began with a larger delay after depolarization than did those caused by steps that activated large ICa,L (0 mV, Fig. 5C), and that contractions at the most positive steps tested (+100 mV) were smaller than those at the peak of the ICa,L-voltage relationship (0 mV). It should be noted that contractions were routinely observed upon repolarization from strongly positive voltage steps. These ‘tail’ contractions are best explained by SR Ca2+ release induced by Ca2+ influx through open L-type Ca2+ channels (Cannell et al. 1987; Beuckelmann & Wier, 1988).
Figure 5. Effects of isoproterenol on the voltage dependence of contraction in 150 mM bath Na+ and 10 mM pipette Na+ (B1, P1; Table 1).

A, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0 and +100 mV under control conditions (no ISO). Note the large Na+ current at -40 mV and the slowly developing tonic contraction at +100 mV. B, bath application of ISO caused large increases in inward current and contraction of the same myocyte. Voltage step is from -70 to +10 mV. The 1st, 10th and 20th steps after ISO are shown. C, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0 and +100 mV after ISO. Inward and outward currents are larger than in A. Contractions were larger and faster than in control. Note that the contraction at +100 mV is delayed with respect to the others shown but is a phasic contraction. D, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0 and +100 mV in the same cell (in ISO) after the addition of the NCX inhibitor KBR-7943. Note that the phasic contraction at +100 mV was abolished. The ‘tail’ contraction upon repolarization was still present. E, voltage-contraction relationship from 6 myocytes under control conditions (A). Data at strongly positive potentials are the size of the tonic contractions at the end of the test step. When the magnitude of the contraction during the first 300 ms of the test steps was plotted, the relationship was bell-shaped. F, voltage-contraction relationship in the presence of ISO. There was broadening of the bell-shaped portion of the voltage-contraction relationship. G, voltage-contraction relationship in the presence of ISO and KBR-7943. Contractions at strongly positive voltage steps were reduced by KBR-7943.
The broadening of the V–C relationship in 150 and 10 mM Na+ conditions (bath and pipette, respectively) could result from a contribution of reverse-mode NCX to EC coupling. To evaluate this possibility, ISO-treated cells were exposed to the NCX inhibitor KBR-7943 (Iwamoto et al. 1996). KBR-7943 reduced or eliminated both phasic and tonic contractions induced by voltage steps to strongly positive potentials (Fig. 5D and G). It should also be noted that KBR-7943 also blocked a portion of ICa,L. These results suggest that these contractions involve Ca2+ influx via reverse-mode NCX rather than VDCR.
The contribution of reverse-mode NCX to contractions induced by steps to strongly positive potentials was further examined by dialysing myocytes with Na+-free pipette solutions (150 mM Na+ in the bath). In the absence of ISO, the magnitude of contraction was the smallest observed in the present experiments (see Table 2) and the V–C relationship was distinctly bell-shaped (Fig. 6A and D). ISO application increased the magnitude of contraction (Fig. 6B) and broadened the V–C relationship (Fig. 6C and D). However, contractions were very small or absent when the membrane potential was stepped to positive potentials near ECa (Fig. 6C).
Table 2.
Myocyte contractile characteristics under the various experimental conditions

KBR, KBR-7943; RCL, resting cell length.
Figure 6. Effects of ISO on the voltage dependence of contraction in 150 mM bath Na+ and 0 mM pipette Na+ (B1, P2 w/o EGTA; Table 1).

A, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0 and +100 mV under control conditions (no ISO). Note the small size of the contraction at 0 mV and the absence of contractions at -40 or +100 mV. B, bath application of ISO caused large increases in inward current and contraction of the same myocyte. Voltage step is from -70 to +10 mV. The 1st, 10th and 20th steps after ISO are shown. C, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0 and +100 mV after ISO. Contractions are larger at -40 and 0 mV, but are still not present at +100 mV. Note that there is now a ‘tail’ contraction at +100 mV. D, voltage-contraction relationship from 5 myocytes before (filled symbols) and after (open symbols) bath application of ISO. The relationship was distinctly bell-shaped under control conditions. ISO caused a broadening of the relationship.
One of the assumptions of the present experiments is that when myocytes are exposed to ISO, the increase in ICa,L causes SR Ca2+ load to increase. This idea was examined directly in voltage-clamped myocytes (Fig. 7) by rapidly applying 10 mM caffeine for 100 ms (in normal bath solution) 1 s after a voltage step from -70 mV to +10 mV. Caffeine is known to induce SR Ca2+ release (Bailey & Houser, 1993). We measured the forward-mode NCX current that occurs after the SR releases its Ca2+ as an index of SR Ca2+ load (Varro et al. 1993). A typical experiment in which ISO caused a large increase in the size of the caffeine-induced inward NCX current is shown in Fig. 7. A second caffeine application did not cause contraction or inward current (not shown). Similar results were obtained in three other myocytes. These results strongly support the idea that ISO increases SR Ca2+ loading.
Figure 7. The effects of isoproterenol on caffeine-induced inward NCX currents (B1, P1; Table 1).
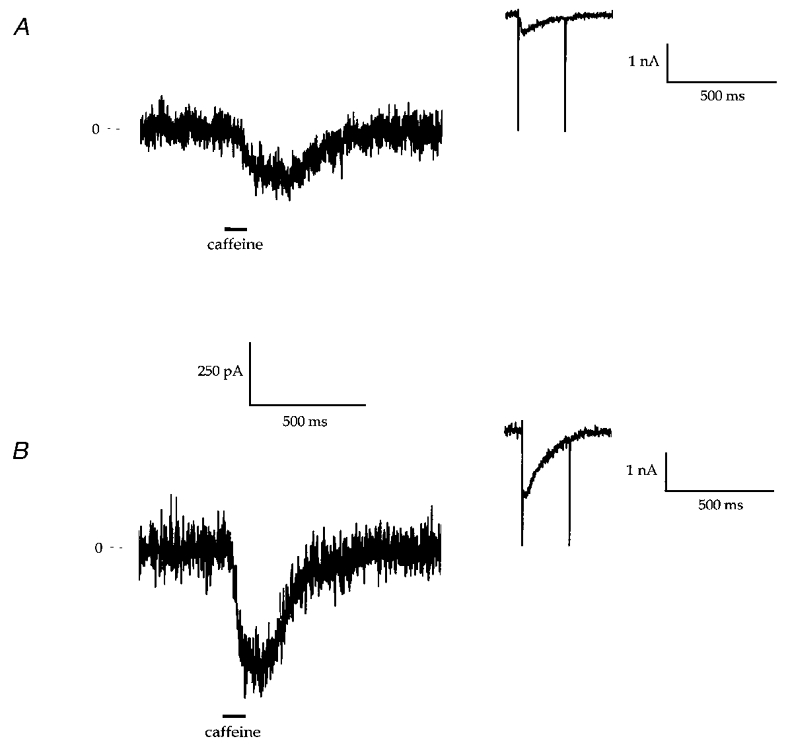
Representative recordings of the inward current induced by rapid application of 10 mM caffeine (in normal bath solution) before (A) and after (B) ISO treatment. The insets show the inward current during conditioning steps from -70 to +10 mV. Note the increase in inward current during the step to +10 mV after ISO. Similar results were observed in 3 other myocytes.
In Na+-containing bath solutions, voltage steps from negative holding potentials cause activation of the Na+ channel that can result in large currents, especially at potentials where the electrochemical driving force for Na+ entry is large. Under our conditions we observed voltage escape with steps from -80 mV to -60 through to 0 mV. At more positive potentials there was little or no voltage escape because Na+ currents are smaller and could be adequately controlled. To define better the role of the Na+ current under our conditions, myocytes were exposed to tetrodotoxin (TTX, 50 μM). These experiments showed that TTX reduced the size of the contractions at steps at which Na+ current-dependent voltage escape was observed (Fig. 8). It is also noteworthy that contractions decreased in magnitude when large voltage steps to potentials approaching ECa were performed (Fig. 8). TTX-treated myocytes that were exposed to ISO responded like those discussed above (Fig. 8). Similar results were obtained in two other myocytes. These experiments show that Na+ current-induced voltage escape influences our results at negative step potentials, most likely by increased activation of ICa,L during voltage escape. The TTX-induced reduction in contraction size at positive potentials suggests that Na+ enters the cell during the rising phase of large positive voltage steps and the associated subsarcolemmal Na+ accumulation promotes reverse-mode NCX.
Figure 8. Effects of tetrodotoxin on contractions and whole-cell current in control and ISO-treated conditions (B1, P1; Table 1).
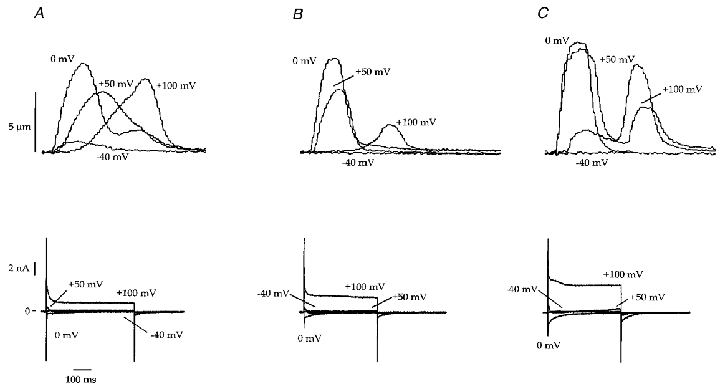
A, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0, +50 and +100 mV under control conditions. The same myocyte is shown throughout. Note that contraction becomes progressively slower and delayed in time after the voltage step as the test potential is increased from 0 to +100 mV. B, representative recordings of contractions and whole-cell currents induced by steps from -70 mV to -40, 0, +50 and +100 mV after the addition of 50 μM TTX. The inward current at -40 mV was decreased. The slow tonic contraction at +100 mV was decreased but the ‘tail’ contraction was still present. C, the same myocyte after bath application of ISO and TTX. Contractions and currents at 0, +50 and +100 were increased but only small contractions were observed at +100 mV.
The results presented above are consistent with the idea that the V–C relationship broadens when cellular cAMP is increased because ICa,L and SR Ca2+ transport rates are increased. This produces an increase in SR Ca2+ load, which then allows a smaller than normal amount of Ca2+ influx to induce full SR Ca2+ release. To explore this hypothesis further, myocytes were first exposed to ISO and then the bath Ca2+ was reduced (in the presence of ISO) to lower the SR Ca2+ load (Fig. 9A–C). If ISO promotes VDCR, then lowering bath Ca2+ should reduce contraction size but not change the shape of the V–C relationship. We found that lowering bath Ca2+ reduced contraction magnitude and caused a leftward shift in the positive portion of the V–C relationship so that contractions were not observed at the most positive voltage steps (Fig. 9C). These experiments strongly support the hypothesis that the voltage dependence and source of the transarcolemmal Ca2+ influx and the state of SR Ca2+ loading determine the shape of the V–C relationship. However, these results are inconsistent with the VDCR hypothesis.
Figure 9. Effect of reducing bath Ca2+ on the voltage dependence of contraction in an isoproterenol-treated myocyte (B1, P1; Table 1).

A, representative recordings of contractions induced by steps from -70 mV to +30, +50 and +100 mV under control conditions. Same myocyte is shown throughout. Note the progressive slowing and delaying of contraction as test steps are made more positive. B, contractions at the same test potentials after bath application of ISO. C, contractions at the same test potentials after lowering bath Ca2+ to 0.25 mM (ISO is still present). Contractions were smaller at all test potentials and were abolished at the most positive test potentials.
The VDCR hypothesis is supported by the observation that contraction (or SR Ca2+ release) can be induced in the presence of Ca2+ channel blockers (Ferrier & Howlett, 1995; Howlett et al. 1998). We examined the effects of Cd2+ (200 μM) on the V–C relationship of ISO-treated feline myocytes. To keep the SR comparably loaded before and after blocking the Ca2+ current we used conditioning steps to +80 mV to promote Ca2+ entry via reverse-mode NCX (Fig. 10A). When Cd2+ was bath-applied during repetitive voltage steps from -70 to +10 mV there was a progressive reduction in contraction size and inward current (other than the Na+ current, Fig. 10B). The voltage dependence of contraction was then measured after the SR was refilled by conditioning steps to +80 mV. Phasic contraction were observed, but only at potentials at which ICa,L is usually seen (Fig. 10C). Therefore, the V–C relationship in the presence of Cd2+ was distinctly bell-shaped (Fig. 10D, n = 5). It is also important to note the ICa,L-related ‘tail’ contraction upon repolarization from the +60 mV voltage step in the presence of Cd2+ (Fig. 10C). Similar results were observed in two cells treated with 10 μM nifedipine (not shown). These results strongly support the idea that in ISO-treated myocytes in which SR Ca2+ loading is maintained, small Ca2+ currents through unblocked L-type Ca2+ channels (current is not clearly visible under our experimental conditions), not VDCR, cause SR Ca2+ release.
Figure 10. Effects of Cd2+ on the voltage dependence of contraction in an isoproterenol-treated myocyte.

A, representative recordings of contraction induced by voltage steps from -70 mV to -10, 0, +10, +20 and +60 mV are shown in a cell bathed in normal solution containing ISO. Whole-cell current at -10, +10 and +60 mV is also shown. B, contractions and current induced by repetitive steps from -70 to +10 mV during the addition of Cd2+ (200 μM) to the bath. Note the progressive reduction of ICa,L and contraction. The step numbers after Cd2+ application are shown. C, contractions induced by voltage steps from -70 mV to -10, 0, +10, +20 and +60 mV in the presence of ISO and Cd2+. SR Ca2+ load was maintained with conditioning steps to +80 mV. Note that the step to +60 mV did not cause contraction but that contraction was induced by repolarization to -70 mV. Whole-cell currents at -10, +10 and +60 mV are also shown. D, voltage-contraction relationship from 5 ISO-treated myocytes before (open symbols) and after (filled symbols) Cd2+. The broad voltage-contraction relationship that was typical of ISO-treated myocytes became distinctly bell-shaped after Cd2+.
DISCUSSION
The goal of the present study was to test the hypothesis that SR Ca2+ release in mammalian ventricular myocytes can be induced by a voltage-dependent mechanism that is independent of CICR. Experiments were performed using conditions that have been shown to bring out the putative VDCR process. However, we added conditions and protocols that also helped separate contributions of CICR and VDCR. The major findings of these experiments were that: (1) when no attempts were made to elevate cellular cAMP and the SR Ca2+ load was small (Na+-free pipette solution), the voltage dependence of contraction was distinctly bell-shaped and mirrored the voltage dependence of the L-type Ca2+ current; (2) when cellular cAMP was increased via the pipette or with bath-applied ISO, ICa,L and the SR Ca2+ load increased and the V–C relationship broadened. Importantly, contraction magnitude still decreased as the depolarizing voltage steps were increased from +40 mV to strongly positive potentials approaching ECa; (3) inhibition of NCX or reducing bath Ca2+ in the presence of ISO reduced contraction magnitude at all potentials and increased the bell-shaped nature of the V–C relationship; (4) voltage steps from -80 to -40 mV activated the L-type Ca2+ current when cellular cAMP was increased and this current was reduced in magnitude by depolarizing prepulses; (5) the V–C relationship in the presence of ISO and the L-type Ca2+ channel blocker Cd2+ was bell-shaped and ICa,L-related tail contractions were observed after strongly positive voltage steps. Together, these results suggest that the rate and magnitude of Ca2+ influx and the state of SR Ca2+ loading determine the magnitude of SR Ca2+ release, but that depolarization per se does not directly gate SR Ca2+ release.
Contractions induced by large voltage steps approaching ECa
Previous studies that have examined the VDCR hypothesis have observed a sigmoidal V–C relationship (Ferrier & Howlett, 1995; Howlett et al. 1998). In the present experiments we showed that as voltage steps were increased from those at which ICa,L and contraction reach their maximal values to potentials approaching ECa, contraction magnitudes decreased and were eventually abolished. Voltage steps near ECa only caused contraction upon repolarization. These contractions are thought to result from Ca2+‘tail’ currents (Cannell et al. 1987; Beuckelmann & Wier, 1988). The positive potential beyond which contraction began to decline varied with the conditions employed. Conditions that increased ICa,L and/or SR Ca2+ loading required stronger depolarizing steps to reveal this negative effect of depolarization. These observations are inconsistent with the VDCR hypothesis. Importantly, our results are similar to those in a very large number of previous studies (Bers & Merrill, 1985; London & Krueger, 1986; Barcenas-Ruiz & Weir, 1987; Cannell et al. 1987, 1995; Callewaert et al. 1988; Beuckelmann & Wier, 1988; Näbauer et al. 1989; duBell & Houser, 1989; Cleemann & Morad, 1991; Bers, 1991) that have shown a bell-shaped voltage dependence of contraction. These studies have been criticized by proponents of the VDCR hypothesis (Howlett et al. 1998) because holding potentials that at least partially inactivated the putative VDCR mechanism were employed and non-physiological conditions (Cs2+, TEA, temperature, etc. that somehow inhibited VDCR) were used so that Ca2+ currents could be more faithfully observed. In the present experiments we used physiological holding potentials, normal intra- and extracellular [K+] and increased cellular cAMP. However, the V–C relationship in our experiments was still not sigmoidal if voltage steps were sufficiently positive and good voltage control was maintained (see below). Instead, we observed that when voltage steps were made more positive than +10 to +40 mV, contraction magnitude decreased, and at the most positive potentials near ECa contractions were small or absent. These results are strong evidence against the VDCR hypothesis. The present results suggest that the variable shape of the V–C (or voltage-Ca2+ release) relationships that has been observed in this and previous studies is determined by the state of SR Ca2+ loading, the amount of cellular Ca2+ buffering (usually with Ca2+ indicators), and the size of the L-type Ca2+ current, rather than by the presence or absence of VDCR.
Feline myocytes were used in the present study, while rat, rabbit and guinea-pig myocytes were used in previous studies of VDCR (Hobai et al. 1997; Howlett et al. 1998). Therefore, species differences are a possible explanation for the discrepant results. However, we have obtained results identical to those observed in feline myocytes in both human and rat myocytes (data not shown). Therefore, a species difference is an unlikely possibility. A better explanation for the results of previous experiments thought to represent VDCR is that the contractions observed at positive potentials (+40 to +80 mV) were due to CICR with the Ca2+ influx coming from the L-type Ca2+ current and/or via reverse-mode NCX. Along these lines, some data in previous studies of VDCR (Ferrier et al. 1998) seem more consistent with CICR than VDCR. In these studies (Fig. 9B; Ferrier et al. 1998) the contractions observed at strongly positive potentials (in the presence of cAMP) occurred with a substantial delay after depolarization (when they are compared with those from steps to +10 mV where ICa,L is maximal). This observation is inconsistent with VDCR since strong depolarizations should speed, not slow, the rate at which a voltage-sensitive mechanism produces SR Ca2+ release. A more plausible explanation for these results is that Ca2+ influx via reverse-mode NCX and/or Ca2+ current causes Ca2+ release from a highly loaded SR. Our results are consistent with this idea. A number of other recent studies are also consistent with our findings and contentions (Nuss & Houser, 1992; Han et al. 1994; Bassani et al. 1995; López-López et al. 1995; Hussain & Orchard, 1997; Jing-Song & Palade, 1999). The conclusion of the present study is that when SR Ca2+ release is induced by voltage steps to strongly positive potentials it results from CICR and the source of Ca2+ is ICa,L and reverse-mode NCX.
The idea that Ca2+ influx via reverse-mode NCX can cause SR Ca2+ release has been examined in a number of previous studies (Leblanc & Hume, 1990; Nuss & Houser, 1992; Levi et al. 1994; Litwin et al. 1998) and was not the focus of the present investigation. Our results are consistent with previous reports (Sipido et al. 1997) and suggest that Ca2+ influx via reverse-mode NCX is not a powerful trigger for SR Ca2+ release but can make a contribution to EC coupling when the SR Ca2+ load is increased with cAMP in the pipette or by ISO application. We found that application of a NCX inhibitor (KBR-7943, Iwamoto et al. 1996) reduced or eliminated contractions at strongly positive potentials near ECa causing a leftward shift in the positive portion of the V–C relationship. These results suggest that Ca2+ influx via reverse-mode NCX can cause or contribute to Ca2+ release from a heavily loaded SR when large voltage steps are produced. This conclusion is also supported by the leftward shift of the V–C relationship observed when bath [Ca2+] was lowered after ISO application (Fig. 9) to reduce SR Ca2+ load. These results suggest that Ca2+ influx via reverse-mode NCX (and ICa,L as discussed below) may have been responsible for what was termed VDCR in previous studies.
Large contractions were routinely observed with steps to +10 mV and these contractions remained large over a broad range of potentials (from about +10 to about +60 mV). Voltage steps to more positive potentials (beyond +60 mV) caused smaller contractions. Voltage steps near ECa did not cause contraction until repolarization. Steps to potentials near ECa should cause little or no Ca2+ influx via the L-type Ca2+ channel but there should be large reverse-mode NCX-mediated Ca2+ influx. The fact that very large voltage steps near ECa do not cause contraction even when the SR is highly loaded suggests that Ca2+ influx via the exchanger, in the absence of Ca2+ influx via the L-type Ca2+ channel, is a very weak trigger for SR Ca2+ release. These findings suggest that a likely role for reverse-mode NCX in EC coupling during depolarization to positive potentials is to elevate [Ca2+] in the t-tubular SR junction so that small elevations in Ca2+ produced by the opening of L-type Ca2+ channels can induce the opening of Ca2+ release channels in the SR.
Small Ca2+ fluxes through the L-type Ca2+ channel are likely to play a key role in the contractions observed at strongly positive potentials that approach ECa (López-López et al. 1995; Hussain & Orchard, 1997; Jing-Song & Palade, 1999). As discussed above, the present experiments suggest that ISO and cAMP change the shape of the V–C relationship by increasing both the SR Ca2+ load and ICa,L rather than by promoting a voltage-dependent Ca2+ release mechanism. The best evidence for this idea is the observation that when ISO was applied to myocytes bathed in Na+-free solutions or with Na+-free pipette solutions (to eliminate reverse-mode NCX) the V–C relationship still broadened and the shape of the relationship was different to the ICa,L-voltage relationship. However, as voltage steps were increased beyond +40 mV positive (toward ECa), contractions decreased in magnitude and were eventually abolished in every cell studied. These results are inconsistent with the VDCR hypothesis and suggest that Ca2+ influx is the exclusive trigger for SR Ca2+ release in mammalian cardiac ventricular myocytes. The most likely explanation for the broadening of the V–C relationship when cells dialysed with Na+-free solutions were exposed to ISO is that when the SR Ca2+ load is increased, smaller than normal Ca2+ currents can induce full SR Ca2+ release. The ability of ISO to increase ICa,L at all potentials and to shift the voltage dependence of ICa,L activation in the negative direction can explain the broadening of the V–C relationship we observed. This idea is consistent with the findings of a number of recent studies showing that very small Ca2+ currents can induce regenerative SR Ca2+ release when the SR Ca2+ load is near maximal levels (Han et al. 1994; Bassani et al. 1995; Hussain & Orchard, 1997). We conclude that previous studies of VDCR that did not observe a diminution of contraction at positive potentials have not accounted for this possibility (Ferrier et al. 1998).
Technical issues
Voltage control is a critical consideration in experiments using large voltage steps to determine the voltage dependence of SR Ca2+ release. Large voltage steps activate both ICa,L and repolarizing K+ currents. The high resistance microelectrodes and discontinuous voltage-clamp techniques used in most previous studies of VDCR (Howlett et al. 1998) are poorly suited for rapid settling of the membrane potential when large voltage clamp steps are attempted. Poor voltage control will cause slowly rising voltage steps to strongly positive potentials that will allow unrecognized Ca2+ influx via L-type Ca2+ channels to induce SR Ca2+ release. In some of our experiments (not included in this study) in which voltage control was inadequate (settling time of more than 10 ms) large contractions were often observed with steps to positive potentials near ECa. Contractions were not present until repolarization at these positive potentials when rapid voltage control was achieved. This suggests that inadequate control of membrane potential (and associated Ca2+ influx) during large voltage steps was partially responsible for the sigmoidal V–C relationship observed previously (Howlett et al. 1998). This would also cause CICR to be inappropriately termed VDCR.
Contractions at -40 mV
Many previous studies have used contractions or Ca2+ transients induced by voltage steps from negative holding potentials to -40 mV as evidence for VDCR (Howlett et al. 1998; Ferrier et al. 1998). The implicit assumption of these studies is that ICa,L is not activated during these steps. Less ICa,L is activated at -40 mV than at the peak of the ICa,L-voltage relationship. However, it is critical to know if any ICa,L is activated at this potential because, as discussed above, very small Ca2+ currents appear to be capable of inducing a large Ca2+ release when the SR is fully loaded with Ca2+. Our experiments show that after ISO treatment a sizable ICa,L is activated when the membrane potential is stepped from -80 to -40 mV. Therefore, ICa,L not VDCR can explain the contractions induced by steps to -40 mV.
We also showed that depolarizing prepulses reduced the size of the subsequent ICa,L at -40 mV in a voltage-dependent fashion. The V1/2 of this relationship was -57.8 ± 0.49 mV (Fig. 4B). This is almost identical to the value (-54.1 mV) obtained for the voltage dependence of inactivation of the putative VDCR mechanism determined with similar protocols (Ferrier et al. 1998). We conclude that in the presence of ISO, ICa,L is partially inactivated when the resting potential is decreased from -80 to -40 mV and this inactivation will cause associated contractions to be eliminated. Therefore, the voltage dependence of inactivation of VDCR reported previously simply reflects voltage-dependent inactivation of ICa,L and CICR. Raw data in previous studies of VDCR (Fig. 3B in Howlett et al. 1998) show that an inward current is activated with steps to -40 mV and that this current is inactivated with depolarizing prepulses.
Contractions in the presence of L-type Ca2+ channel blockers
Previous studies have shown that after myocytes are exposed to Ca2+ channel blockers depolarizing voltage steps can still induce contraction (Ferrier & Howlett, 1995; Hobai et al. 1997). In our experiments we also found that when SR Ca2+ loading is maintained (using Ca2+ influx via NCX as the source) in ISO-treated myocytes, depolarization can induce contraction in the presence of 200 μM Cd2+ (Fig. 10). This observation is consistent with some previous studies that have either examined the effects of rapid application of Ca2+ channel blockers at one test potential (Hobai et al. 1997) or over a limited voltage range (Ferrier & Howlett, 1995). We examined the voltage dependence of these contractions over a much broader voltage range and found a distinct bell-shaped V–C relationship, similar to that of ICa,L (Fig. 10). In addition, ICa,L-related ‘tail’ contractions were observed upon repolarization from positive (+60 mV and higher) voltage steps that by themselves caused no contraction. These results strongly support the idea that Ca2+ influx through unblocked L-type Ca2+ channels, not VDCR, is the trigger for SR Ca2+ release in our experiments with Cd2+. Previous studies that have examined the effects of Ca2+ channel blockers on SR Ca2+ release at only one test potential or over a narrow voltage range would not have been able to clearly define this role of unblocked ICa,L.
Summary and conclusions
We further tested the VDCR hypothesis, which predicts an activation voltage below ICa,L, a sigmoidal V–C relationship and an insensitivity to L-type Ca2+ channel blockers. Our results show that the threshold for activation of contraction is consistent with the activation of ICa,L. The voltage dependence of contraction was typically bell-shaped when SR Ca2+ loading was low. High SR Ca2+ loads induced by cAMP or ISO caused broadening of the V–C relationship. However, the relationship was not sigmoidal because contraction magnitude always decreased as voltage steps approached ECa. The shape of the V–C relationship could be made more bell-shaped (smaller contractions at potentials approaching ECa) by elimination of reverse-mode NCX and lowering SR Ca2+ loading. The V–C relationship in the presence of Cd2+ was bell-shaped and was best explained by CICR. These results suggest that the changes in the shape of the V–C relationship in the presence of high cellular cAMP result from the fact that when the SR Ca2+ load is near its maximal level, a very small Ca2+ influx (as ICa,L or via ICa,L and NCX) can cause large, regenerative SR Ca2+ release. These results provide strong support for the CICR hypothesis and suggest that VDCR is not present in adult cardiac ventricular myocytes.
Acknowledgments
This study was supported in part by NIH grant HL33920 to S.R.H. KBR-7943 was kindly provided by the New Drug Discovery Research Drug Laboratory, Kanebo Ltd, Osaka, Japan. The authors would like to thank Colleen A. Hefner for technical assistance. Reprint requests should be addressed to Dr S. R. Houser.
References
- Aggarwal R, Shorofsky SR, Goldman L, Balke CW. Tetrodotoxin-blockable calcium currents in rat ventricular myocytes; a third type of cardiac cell sodium current. The Journal of Physiology. 1997;505:353–369. doi: 10.1111/j.1469-7793.1997.353bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen DG, Eisner DA, Lab MJ, Orchard CH. The effects of low sodium solutions on intracellular calcium concentration and tension in ferret ventricular muscle. The Journal of Physiology. 1983;345:391–407. doi: 10.1113/jphysiol.1983.sp014984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey BA, Houser SR. Sarcoplasmic reticulum-related changes in cytosolic calcium in pressure-overload-induced feline LV hypertrophy. American Journal of Physiology. 1993;265:H2009–2016. doi: 10.1152/ajpheart.1993.265.6.H2009. [DOI] [PubMed] [Google Scholar]
- Barcenas-Ruiz L, Wier WG. Voltage dependence of intracellular [Ca2+]i transients in guinea pig ventricular myocytes. Circulation Research. 1987;61:148–154. doi: 10.1161/01.res.61.1.148. [DOI] [PubMed] [Google Scholar]
- Bassani JW, Yuan W, Bers DM. Fractional SR Ca2+ release is regulated by trigger Ca2+ and SR Ca2+ content in cardiac myocytes. American Journal of Physiology. 1995;268:C1313–1319. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Dordrecht, The Netherlands: Kluwer Academic Publishers; 1991. [Google Scholar]
- Bers DM, Merrill DB. The role of Ca2+ influx in cardiac muscle excitation-contraction coupling. Assessment by extracellular Ca2+ microelectrodes. Advances in Myocardiology. 1985;6:49–57. [PubMed] [Google Scholar]
- Beuckelmann DJ, Wier WG. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. The Journal of Physiology. 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block BA, Imagawa T, Campbell KP, Franzini-Armstrong C. Structural evidence for direct interaction between the molecular components of the transverse tubule/sarcoplasmic reticulum junction in skeletal muscle. Journal of Cell Biology. 1988;107:2587–2600. doi: 10.1083/jcb.107.6.2587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callewaert G, Cleemann L, Morad M. Epinephrine enhances Ca2+ current-regulated Ca2+ release and Ca2+ reuptake in rat ventricular myocytes. Proceedings of the National Academy of Sciences of the USA. 1988;85:2009–2013. doi: 10.1073/pnas.85.6.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Chartier D, Moore HM, Howlett S, Ferrier G, Leblanc N. Activation of the voltage-sensitive release mechanism by beta-adrenergic stimulation in dialyzed guinea-pig ventricular myocytes. Biophysical Journal. 1999;76:A458. [Google Scholar]
- Cleemann L, Morad M. Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: evidence from Ca2+ transients and contraction. The Journal of Physiology. 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- duBell WH, Houser SR. Voltage and beat dependence of Ca2+ transient in feline ventricular myocytes. American Journal of Physiology. 1989;257:H746–759. doi: 10.1152/ajpheart.1989.257.3.H746. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. American Journal of Physiology. 1983;245:C1–14. doi: 10.1152/ajpcell.1983.245.1.C1. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. Journal of General Physiology. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Contractions induced by a calcium-triggered release of calcium from the sarcoplasmic reticulum of single skinned cardiac cells. The Journal of Physiology. 1975;249:469–495. doi: 10.1113/jphysiol.1975.sp011026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier GR, Howlett SE. Contractions in guinea-pig ventricular myocytes triggered by a calcium-release mechanism separate from Na+ and L-currents. The Journal of Physiology. 1995;484:107–122. doi: 10.1113/jphysiol.1995.sp020651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrier GR, Zhu J, Redondo IM, Howlett SE. Role of cAMP-dependent protein kinase A in activation of a voltage-sensitive release mechanism for cardiac contraction in guinea-pig myocytes. The Journal of Physiology. 1998;513:185–201. doi: 10.1111/j.1469-7793.1998.185by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Han S, Schiefer A, Isenberg G. Ca2+ load of guinea-pig ventricular myocytes determines efficacy of brief Ca2+ currents as trigger for Ca2+ release. The Journal of Physiology. 1994;480:411–421. doi: 10.1113/jphysiol.1994.sp020371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hobai IA, Howarth FC, Pabbathi VK, Dalton GR, Hancox JC, Zhu JQ, Howlett SE, Ferrier GR, Levi AJ. ‘Voltage-activated Ca2+ release’ in rabbit, rat and guinea pig cardiac myocytes, and modulation by internal cAMP. Pflügers Archiv. 1997;435:164–173. doi: 10.1007/s004240050496. [DOI] [PubMed] [Google Scholar]
- Howlett SE, Zhu J-Q, Ferrier GR. Contribution of a voltage-sensitive calcium release mechanism to contraction in cardiac ventricular myocytes. American Journal of Physiology. 1998;274:H155–170. doi: 10.1152/ajpheart.1998.274.1.H155. [DOI] [PubMed] [Google Scholar]
- Hussain M, Orchard CH. Sarcoplasmic reticulum Ca2+ content, L-type Ca2+ current and the Ca2+ transient in rat myocytes during β-adrenergic stimulation. The Journal of Physiology. 1997;505:385–402. doi: 10.1111/j.1469-7793.1997.385bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto T, Watano T, Shigckawa M. A novel isothiourea derivative selectively inhibits the reverse-mode of Na+/Ca2+ exchange in cells expressing NCX1. Journal of Biological Chemistry. 1996;271:22391–22397. doi: 10.1074/jbc.271.37.22391. [DOI] [PubMed] [Google Scholar]
- Jing-Song F, Palade P. One calcium ion may suffice to open the tetrameric cardiac ryanodine receptor in rat ventricular myocytes. The Journal of Physiology. 1999;516:769–780. doi: 10.1111/j.1469-7793.1999.0769u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leblanc N, Hume JR. Sodium current-induced release of calcium from cardiac sarcoplasmic reticulum. Science. 1990;248:372–376. doi: 10.1126/science.2158146. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- Levi AJ, Spitzer KW, Kohmoto O, Bridge JH. Depolarization-induced Ca2+ entry via Na+-Ca2+ exchange triggers SR release in guinea pig cardiac myocytes. American Journal of Physiology. 1994;266:H1422–1433. doi: 10.1152/ajpheart.1994.266.4.H1422. [DOI] [PubMed] [Google Scholar]
- Lipp P, Niggli E. Sodium current-induced calcium signals in isolated guinea-pig ventricular myocytes. The Journal of Physiology. 1994;474:439–446. doi: 10.1113/jphysiol.1994.sp020035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Litwin SE, Li J, Bridge JH. Na-Ca exchange and the trigger for sarcoplasmic reticulun Ca release: studies in adult rabbit ventricular myocytes. Biophysical Journal. 1998;75:359–371. doi: 10.1016/S0006-3495(98)77520-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London B, Krueger JW. Contraction in voltage-clamped, internally perfused single heart cells. Journal of General Physiology. 1986;88:475–505. doi: 10.1085/jgp.88.4.475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-López JR, Shacklock P, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- Näbauer M, Callewaert G, Cleemann L, Morad M. Regulation of calcium release is gated by calcium current, not gating charge, in cardiac myocytes. Science. 1989;244:800–803. doi: 10.1126/science.2543067. [DOI] [PubMed] [Google Scholar]
- Näbauer M, Morad M. Ca2+-induced Ca2+ release as examined by photolysis of caged Ca2+ in single ventricular myocytes. American Journal of Physiology. 1990;258:C189–193. doi: 10.1152/ajpcell.1990.258.1.C189. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Houser SR. Voltage dependence of contraction and calcium current in severely hypertrophied feline ventricular myocytes. Journal of Molecular and Cellular Cardiology. 1991;23:717–726. doi: 10.1016/0022-2828(91)90981-q. [DOI] [PubMed] [Google Scholar]
- Nuss HB, Houser SR. Sodium-calcium exchange-mediated contractions in feline ventricular myocytes. American Journal of Physiology. 1992;263:H1161–1169. doi: 10.1152/ajpheart.1992.263.4.H1161. [DOI] [PubMed] [Google Scholar]
- Santana LF, Gomez AM, Lederer WJ. Ca2+ flux through promiscuous cardiac Na+ channels: slip-mode conductance. Science. 1998;279:1027–1033. doi: 10.1126/science.279.5353.1027. [DOI] [PubMed] [Google Scholar]
- Sipido KR, Carmeliet E, Van de Werf F. T-type Ca2+ current as a trigger for Ca2+ release from the sarcoplasmic reticulum in guinea-pig ventricular myocytes. The Journal of Physiology. 1998;508:439–451. doi: 10.1111/j.1469-7793.1998.439bq.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na+/Ca2+ exchanger as a trigger for Ca2+ release from the sarcoplasmic reticulum: a comparison between L-type Ca2+ current and reverse-mode Na+/Ca2+ exchange. Circulation Research. 1997;81:1034–1044. doi: 10.1161/01.res.81.6.1034. [DOI] [PubMed] [Google Scholar]
- Steadman BW, Moore KB, Spitzer KW, Bridge JH. A video system for measuring motion in contracting heart cells. IEEE Transactions on Biomedical Engineering. 1988;35:264–272. doi: 10.1109/10.1375. [DOI] [PubMed] [Google Scholar]
- Valdeolmillos M, O'Neill SC, Smith GL, Eisner DA. Calcium-induced calcium release activates contraction in intact cardiac cells. Pflügers Archiv. 1989;413:676–678. doi: 10.1007/BF00581820. [DOI] [PubMed] [Google Scholar]
- Varro A, Negretti N, Hester S B, Eisner DA. An estimate of the calcium content of the sarcoplasmic reticulum in rat ventricular myocytes. Pflügers Archiv. 1993;423:158–160. doi: 10.1007/BF00374975. [DOI] [PubMed] [Google Scholar]