

Abstract
Significant Na+ conductance has been described in only a few native and cloned K+ channels, but has been used to characterize inactivation and K+ binding within the permeation pathway, and to refine models of K+ flux through multi-ion pores. Here we use Na+ permeation of the delayed rectifier K+ channel Kv1.5 to study extra- and intracellular K+ ( and , respectively) regulation of conductance and inactivation, using whole-cell recording from human embryonic kidney (HEK)-293 cells.
Kv1.5 Na+ currents in the absence of and were confirmed by: (i) resistance of outward Na+ currents to dialysis by K+-free solutions; (ii) tail current reversal potential changes with with a slope of 55·8 mV per decade; (iii) block by 4-aminopyridine (50 % at 50 μM), and resistance to Cl− channel inhibition.
Na+ currents were transient followed by a small sustained current. An envelope test confirmed that activated Kv1.5 channels conducted Na+, and that rapid current decay reflected C-type inactivation. Sustained currents (≈13 % of peak) represented Na+ flux through inactivated Kv1.5 channels.
could modulate the maximum available Na+ conductance in the stable cell line while channels were closed. Before the first pulse of a train, increasing concentration increased the subsequent Na+ conductance from ≈15 (0 mM ) to 30 nS (5 mM ), with a Kd of 23 μM. Repeated low rate depolarizations in / solutions induced a use-dependent loss of Kv1.5 channel Na+ conductance, distinct from that caused by C-type inactivation. binding that sensed little of the electric field could prevent this secondary loss of available Kv1.5 channels with a Kd of 230 μM. These two effects on conductance were both voltage independent, and had no effect on channel inactivation rate.
concentrations ≥ 0·3 mM slowed the inactivation rate in a strongly voltage-dependent manner. This suggested it could compete for binding at a K+ site or sites deeper in the pore, as well as restoring the Na+ conductance. was able to modulate the inactivation rate but was unable to affect conductance.
Mutation of arginine 487 in the outer pore region of the channel to valine (R487V) greatly reduced C-type inactivation in Na+ solutions, caused loss of channel use dependence, and prevented any conductance increase upon the addition of 0·1 mM . Our results confirm the existence of a high affinity binding site at the selectivity filter that regulates inactivation, and also reveals the presence of at least one additional high affinity outer mouth site that predominantly regulates conductance of resting channels, and protects channels activated by depolarization when they conduct Na+.
Potassium channels are a diverse group of transmembrane proteins which have a large variety of gating mechanisms and kinetics, but which all very selectively conduct K+ rather than Na+. K+ channels have been thought to be generally impermeable to Na+ (Hille, 1992), with only a few native channels conducting Na+, such as delayed rectifier K+ channels in sympathetic and dorsal root ganglion neurons (Zhu & Ikeda, 1993; Callahan & Korn, 1994; Block & Jones, 1996), and the squid axon under extreme conditions (Bezanilla & Armstrong, 1972; French & Wells, 1977). In native cells, the mixture of different channel types makes it difficult to study Na+ permeation through individual channel species in detail, and it is only recently using cloned channels that significant Na+ permeation through K+ channels has been recorded (Korn & Ikeda, 1995; Kiss et al. 1998). In RCK4 and some Shaker channels, conductance appears to collapse in solutions (Pardo et al. 1992; Heginbotham & MacKinnon, 1993; Gómez-Lagunas, 1997; Jäger et al. 1998). However, Na+ currents have been recorded in the absence of K+ in a chimera of Kv2.1 with the pore of Kv1.3 (Kiss et al. 1998; Kiss & Korn, 1998), in a Shaker channel with an A463C mutation in the S6 segment (Ogielska & Aldrich, 1998), and in C-type inactivated or non-conducting Shaker mutant W434F channels (Starkus et al. 1997, 1998), in which tryptophan at position 434 was replaced by phenylalanine. These reports demonstrate that Na+ conductance through K+ channels not only depends on K+ channel subtypes, but also on subtle variations in the amino acid composition of the pore region or adjacent areas of the S6 segment.
The recent chimera experiments of Kiss et al. (1998) and Kiss & Korn (1998) have given valuable insight into permeation and inactivation through K+ channels by examining competition between low concentrations of K+ with conducting Na+ ions. Their model describes a relatively short, narrow region of the pore with a high affinity K+ site located at the pore selectivity filter, flanked by relatively non-selective inner and outer mouth cation binding sites. Binding to these sites regulates both selectivity and inactivation. This kind of model can account for both the high selectivity and flux rates of K+ channels in the absence of ion-ion interactions in a long pore. K+ can also modulate Na+ conductance through both mutant Shaker channels, and C-type inactivated channels (Starkus et al. 1997, 1998; Ogielska & Aldrich, 1998). Other studies have suggested that conductance can be modulated by low in Na+ solutions at outer mouth sites (Pardo et al. 1992; Gómez-Lagunas, 1997), independent of changes in inactivation (Lopez-Barneo et al. 1993).
It has been stated that the Kv1.5 channel has a negligible, but finite permeability to Na+ (Korn & Ikeda, 1995), and also that neither Kv1.3 nor Kv1.5 channels conduct Na+ in the absence of K+ (Ogielska & Aldrich, 1998). In the present study, we have further investigated Na+ currents through WT Kv1.5 channels using a highly expressing stable human embryonic kidney (HEK) cell line and have found a transient Na+ conductance that inactivates almost completely, with a residual current of less than 15 % of peak current that represents Na+ flux through inactivated channels. can modulate the Na+ conductance and both and modulate the Na+ current kinetics. We confirm the existence in Kv1.5 channels of a cation site well within the electric field that regulates both channel selectivity and inactivation as in chimera and mutant channels (Kiss & Korn, 1998). We also describe the existence of a K+-selective outer mouth site disrupted by the mutation R487V that regulates channel entry into inactivated or long-lived non-conducting states.
METHODS
Cells and solutions
Human Kv1.5 channels transiently or stably expressed in HEK-293 cell lines were used in all experiments. Kv1.5 in the plasmid expression vector pCDNA3 was mutagenized using the Quickchange Kit (Stratagene, La Jolla, CA, USA) such that arginine 487 was converted to valine (R487V). HEK-293 cells were transiently or stably transfected with wild-type (WT) hKv1.5 or Kv1.5-R487V cDNAs using LipofectACE reagent (Canadian Life Technologies, Bramalea, ON, Canada) in a 1:10 (w:v) ratio. Patch pipettes used for whole-cell recording contained (mM): NaCl, 130; EGTA, 5; Hepes, 10 and was adjusted to pH 7.2 with NaOH. When NaCl was substituted with KCl or N-methyl-D-glucamine (NMG+), pH was adjusted with KOH or HCl, respectively. The bath solution contained (mM): NMG+, 140; NaCl, 5; Hepes, 10; MgCl2, 1; CaCl2, 1 and was adjusted to pH 7.4 with HCl. For recordings in the presence of different external Na+ or K+ concentrations, the NMG+ based external solution was used and the concentration of NMG+ was reduced as the cation concentration was elevated to maintain constant osmolarity. All chemicals were from Sigma Aldrich Chemical Co. (Mississauga, ON, Canada). The purity of N-methyl-D-glucamine is 99–100.5 % (by HCl titration, M2004). Since the central aim of this work was to examine regulation of K+ channel conductance by small concentrations of and , it was important to reduce K+ contamination as much as possible. For that reason all water used in these experiments was passed through organic filters and two-stage distillation prior to a Milli-Q (Millipore, Canada) de-ionizing system that returned water with ∼20 MΩ resistance. Any contaminating K+ in the water used for the solutions was below detection limits (< 0.25 μM) for coupled plasma optical emission spectroscopy (CANTEST Analytical Services, Vancouver, BC, Canada), and the 140 mM NMG+ solution also had undetectable levels of K+. The 135 mM Na+ solution gave a reading of 9.5 μM K+, due to interference from the high Na+ concentration. 4-Aminopyridine (4-AP) was dissolved in distilled water at a stock concentration of 500 mM and pH adjusted to 7.4 using HCl. Di-isothiocyanatostilbene-N’,N’,N’-disulphonic acid (DIDS) was prepared in a stock solution (1 mM in external medium). 4-AP and DIDS had no effect on the measured pH of extracellular solutions.
Electrophysiological procedures
Coverslips containing cells were removed from the incubator before experiments and placed in a superfusion chamber (volume 250 μl) containing the control bath solution at 22–23°C. The bath solution was exchanged by switching the perfusates at the inlet of the chamber. Complete bath solution changes took 5–10 s. Whole-cell current recording and data analysis were done using an Axopatch-1D or 200A amplifier and pCLAMP6 software (Axon Instruments, Foster City, CA, USA). Patch electrodes were fabricated using thin-walled borosilicate glass (World Precision Instruments, Sarasota, FL, USA). Capacity compensation was routinely used (averaged cell membrane capacitance was 15.4 ± 0.3 pF, mean ±s.e.m., n = 126), but series resistance (Rs) compensation was only used when recording K+ currents. Measured series resistance was between 1–3 MΩ for all recordings (averaged series resistance was 2.18 ± 0.05 MΩ, n = 126). When this changed during the course of an experiment, data were discarded. No difference was observed between results obtained in the absence or presence of Rs compensation when recording Na+ currents. Data were sampled at 10–20 kHz and filtered at 5–10 kHz. The data for analysis and presentation were off-line leak subtracted if necessary, and data were discarded if the leakage conductance was >1 nS. The conductance, g(V), was calculated as: g(V) =I(V)/(V–Erev), where I(V) is the current amplitude, V is the membrane potential, and Erev is the reversal potential. Throughout the paper, conductance values are all from currents measured at +60 mV, except in Fig. 9C. For solutions that contained NMG+ and 5 mM K+, or Na+, measured junctional potentials were -6.2 ± 0.5 mV (mean ±s.e.m., n = 6) against solutions with 130 mM of these cations. When conductance and kinetic calculations were made, membrane potentials have been corrected for this error. Throughout the text data are shown as means ±s.e.m.
Figure 9. Concentration and voltage dependence of effects on Kv1.5 Na+ currents in 130 mM /5 mM .
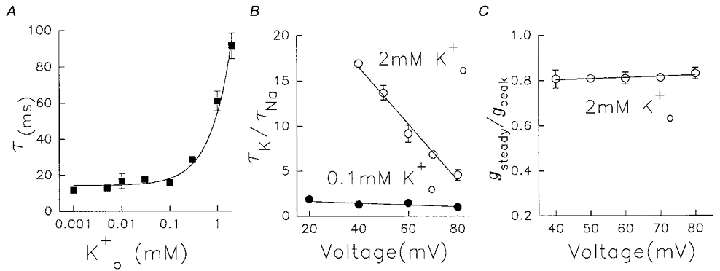
A, time constants of current decay as a function of on a logarithmic scale. Data represent means ±s.e.m. (n = 4-12). B, ratio of decay time constant in K+-containing solution (τK) over that in K+-free solution (τNa) against depolarization voltage. •, 0.1 mM data (n = 5-8); ^, 2 mM data (n = 6–8, means ±s.e.m.). In both cases, before addition of , Na+ currents were at steady state induced by 400 ms pulses from -80 to +60 mV at 0.05 Hz. The straight lines fitted through the data had a slope of -0.01 for 0.1 mM and -0.3 for 2 mM . C, voltage dependence of use dependence of conductance in 2 mM . Cells were pulsed to a range of potentials at 0.05 Hz and steady current conductance was normalized to peak (first pulse) conductance (gsteady/gpeak). Different cells were used for each potential, n = 4–11.
RESULTS
Na+ permeation through Kv1.5 channels
Kv1.5 currents obtained during whole-cell recording (WCR) with K+ in the pipette and bath solutions (130 mM /5 mM ), display a rapidly activating and slowly inactivating time course during 500 ms depolarizations (Fig. 1A). Inactivation occurs in Kv1.5 currents (Fedida et al. 1993) but for depolarizations of this duration inactivation is only partial at potentials ≥+20 mV. In contrast to this, when K+ is replaced by Na+, currents are rapidly inactivating at all positive potentials (Fig. 1B). Na+ permeation through Kv1.5 channels has not been previously reported at any significant level (Korn & Ikeda, 1995), so we took steps to prove that the observed currents did represent Na+ flow through Kv1.5 channels. Experiments with untransfected HEK cells (data not shown), and HEK cells expressing the W472F non-conducting mutant of Kv1.5 (Wang et al. 1999), failed to reveal transient outward ionic currents in the presence of and . However, as the cells had been cultured in standard K+-containing media, the possibility existed that residual K+ currents could be present, or that the remaining could allow Na+ flux through Kv1.5 channels. Theoretically, the time constant of dilution of intracellular K+ into the pipette, based on the formulas provided by Pusch & Neher (1988), is between 3.4 and 25.3 s. The data in Fig. 1C -H demonstrate that relatively complete dialysis and removal of residual was achieved in these experiments with the 130 mM pipette solution. When the pipette contained 130 mM Na+, transient outward currents were very stable over time after the initiation of WCR. Data in Fig. 1C and D illustrate the first Na+ currents recorded from two different cells after 1 and 4 min of whole-cell recording, respectively. When all permeant ions were omitted from the pipette and was used, more slowly inactivating small outward currents were observed at the initiation of WCR (Fig. 1E) that disappeared after 1 min dialysis (Fig. 1F). These data are summarized in Fig. 1G and H. Na+ current amplitude and inactivation time course was stable, independent of cell dialysis time, whereas K+ (presumably) currents disappeared by 1 min WCR. A characteristic and distinguishing feature of the putative K+ currents was their slow decay compared with Na+ currents (Fig. 1H).
Figure 1. Permeation of Na+ through Kv1.5 channels.
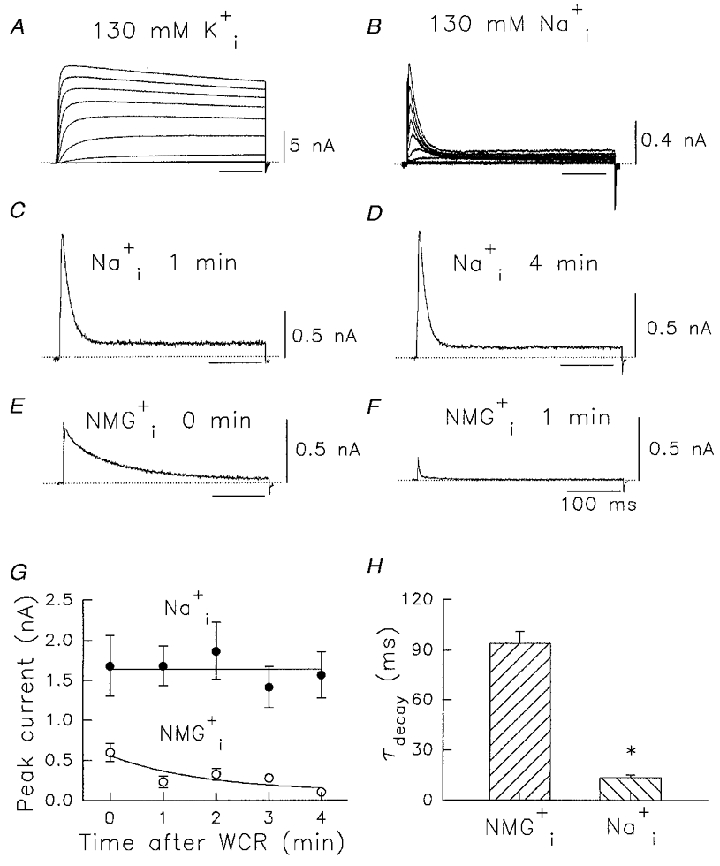
A, currents recorded in 130 mM /5 mM during voltage pulses in 10 mV steps from -60 to +40 mV for 500 ms from a holding potential of -80 mV. B, as for A but in 130 mM /5 mM . C-H, lack of effect of cell dialysis on Na+ currents. C and D, currents elicited by voltage pulses from -80 mV to +60 mV for 400 ms in 130 mM /5 mM after 1 and 4 min WCR, respectively. E and F, currents elicited by the same voltage pulse protocol as in C and D, but NMG+ replaced Na+, after 0 and 1 min WCR, respectively. Dotted lines indicate zero current level in A-F. Time calibration value in F applies also to A–E. G, change in peak recorded currents with time of WCR. •, data recorded under the same conditions as in C and D; ^, data recorded under the same conditions as in E and F. The NMG+ data were fitted to a single exponential decay process with a time constant of 1.8 min. H, time constants of decay of current transients with / or /. data are significantly different from data (*P < 0.001). Data in G and H are means ±s.e.m. (n = 4–10).
Further confirmation that the observed currents were Na+ currents through Kv1.5 channels was obtained from reversal potential measurements at different concentrations of . Representative families of tail currents are shown in Fig. 2A–C at three concentrations of . In each case was 130 mM. Complete instantaneous I–V relations obtained from peak outward or inward tail currents, are shown in Fig. 2D at different concentrations, and the values of measured reversal potentials (Erev) are compared with calculated Erev in Fig. 2E. The experimental measurements of current reversal potential lie along the predicted line, which indicated that currents showed strong Na+ selectivity.
Figure 2. Concentration dependence of Na+ current reversal potentials.
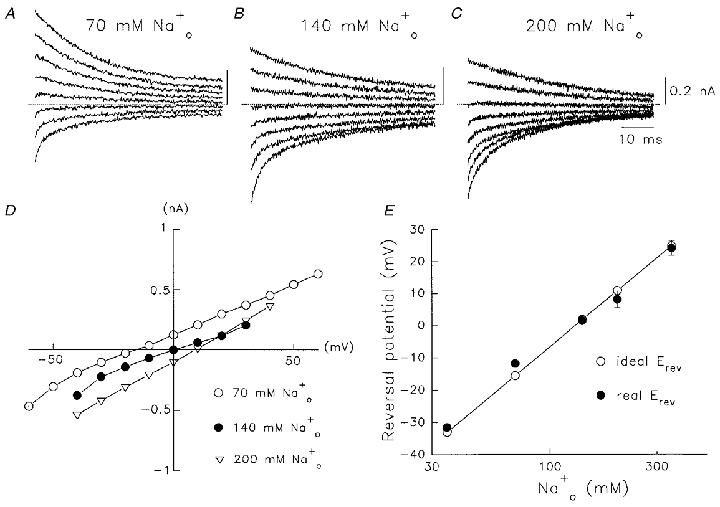
A-C, families of Na+ tail currents through Kv1.5 channels after 15 ms depolarizations to +60 mV from a holding potential of -80 mV. Currents were obtained on repolarization to between +30 mV and -40 mV in 10 mV steps. Dotted lines denote zero current in each panel. was 130 mM in each case, is indicated above the records. The current calibration value and time calibration in C apply also to A and B. D, instantaneous tail I–V relations at the indicated concentrations. The current amplitude was measured immediately on return from +60 mV. E, correlation of measured reversal potentials (real Erev) with predictions from the Nernst equation (ideal Erev). Data are shown as means ±s.e.m. (n = 5–9).
Proof that these Na+ currents were carried through Kv1.5 channels, rather than any endogenous channels in the HEK cells was obtained using the known high sensitivity of Kv1.5 to 4-aminopyridine (IC50≈ 50 μM, Bouchard & Fedida, 1995). 4-AP (50 μM) effectively blocked Na+ currents in these cells (Fig. 3A) in a reversible manner, whereas the Cl− channel blocking agent DIDS was ineffective (Fig. 3B). Mean data in Fig. 3C confirmed this block in cells exposed to 1 mM 4-AP and 0.5 mM DIDS, when compared with control current amplitude. We also investigated whether the rapid decay of the Kv1.5 Na+ current is a classical inactivation process, by carrying out an envelope test between the amplitude of the outward currents during different duration depolarizations and the corresponding tail current amplitudes. If Na+ ions pass mainly through activated Kv1.5 channels, then the magnitude of the deactivating tail currents should be proportional to the current activated during the depolarizing pulses that elicited those tails (Hodgkin & Huxley, 1952; Snyders et al. 1993). Figure 3D and E shows the current recording in symmetrical 135 mM Na+ solutions during a test pulse to +80 mV from -80 mV to directly compare outward and inward current amplitudes. The Na+ tail currents showed an initial transient inward phase with fast kinetics of decay, and then a rising phase followed by a slower second phase of current decay. With increasing test pulse duration, and putative increased inactivation, the initial fast tail was progressively reduced as the rising phase became apparent. Thus, after a 200 ms test pulse, the fast component almost disappeared (Fig. 3D). The loss of the fast component of the tail corresponded to the time course of outward current decay (Fig. 3E), i.e. inactivation. Therefore, the fast component of tail decay indicates deactivation of activated channels and the slow component of tail decay represents the deactivation of C-type inactivated channels (Starkus et al. 1997). The ratio of the instantaneous amplitude of fast tail current (Itail) to outward current at the end of the pulse (Istep) was plotted as a function of pulse duration on a logarithmic time scale (Fig. 3F), and found to be 1.0, independent of pulse duration and current size. This finding supports the view that Na+ currents through activated Kv1.5 channels induce fast inactivation.
Figure 3. Drug sensitivity of Kv1.5 Na+ currents and envelope of tails test.
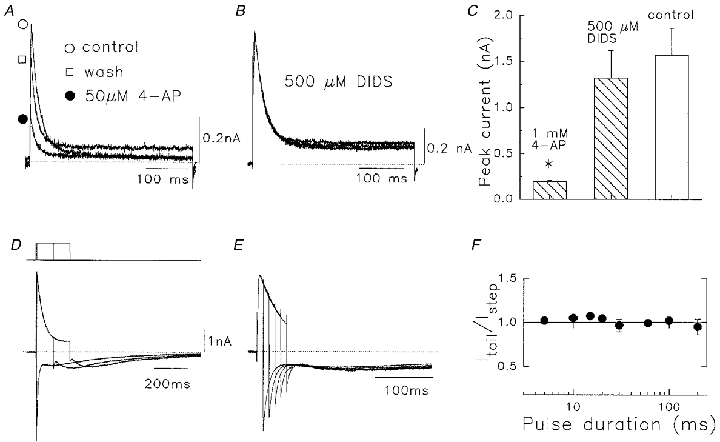
A, effect of 50 μM 4-AP, showing reversibility of action. B, lack of effect of 0.5 mM DIDS on Na+ currents. In both A and B cells were held at -80 mV and pulsed to +60 mV at 0.1 Hz. Pipette and bath solutions contained 130 mM /5 mM . C, mean data for actions of 1 mM 4-AP and 0.5 mM DIDS vs. control, n = 4–6. 4-AP data are significantly different from controls (*P < 0.001). D, superimposed currents during depolarization, and subsequent tail currents. Solutions contained symmetrical 135 mM /. Pulse protocol (top): initial 10 ms depolarization from -80 to +80 mV incremented by 100 ms for each subsequent pulse. E, as for D with initial pulse duration of 10 ms, with 10 ms increment. Note that the amplitude of the fast tail current on repolarization (Itail) was scaled to the preceding outward current amplitude at the end of the pulse (Istep). Dotted lines denote zero current level. F, the Itail/Istep ratio as a function of pulse duration. Data represent means ±s.e.m. (n = 9).
In Shaker channels, transient outward Na+ currents are followed by a sustained current, which is about 50 % of peak INa (Starkus et al. 1997). This sustained current is due to Na+ ions passing through inactivated channels. In Kv1.5 channels, the same Na+ current through inactivated channels could be observed with prolonged depolarization (Figs 1 and 3). However, the amplitude of the sustained current in Kv1.5 channels is smaller than that in Shaker channels. At the end of 1 s depolarizing pulses, the current was 0.13 ± 0.02 (n = 6) of the peak INa. Thus, it is very possible that the progression of conformational changes in Kv1.5 during slow inactivation is different from that in Drosophila Shaker channels.
Rapid reduction of Na+ conductance when channels are used
It was noted early on during experiments that the Na+ conductance through Kv1.5 channels was not constant. When voltage clamp pulses were applied in / solutions after the initiation of WCR, there was a significant initial decrease of peak current amplitude from the first pulse to the second (Fig. 4A). After this reduction, there was only a small further current decrease to the steady peak current level when the pulse rate was less than 0.1 Hz. In the example in Fig. 4A the pulse rate was 0.05 Hz and after the initial decrease in peak current, from the first to second trace, there was little further decline to the steady peak current level. The decrease in peak current was not accompanied by any change in current inactivation rate, and the magnitude of the peak current decline was not affected by the delay after WCR that pulses were given (Fig. 4B). The ratio of steady peak current level to initial peak current was close to 50 %, independent of the frequency of applied pulses up to 0.1 Hz (Fig. 4C) and the duration of the depolarizing pulses up to 400 ms (Fig. 4D). At higher rates than 0.1 Hz, further current decreases were observed that we hypothesized were caused by a simple failure to recover from inactivation between pulses. This idea was tested by measuring the recovery from steady-state inactivation in / solutions.
Figure 4. Use-dependent reduction of peak Na+ current in the absence of K+.
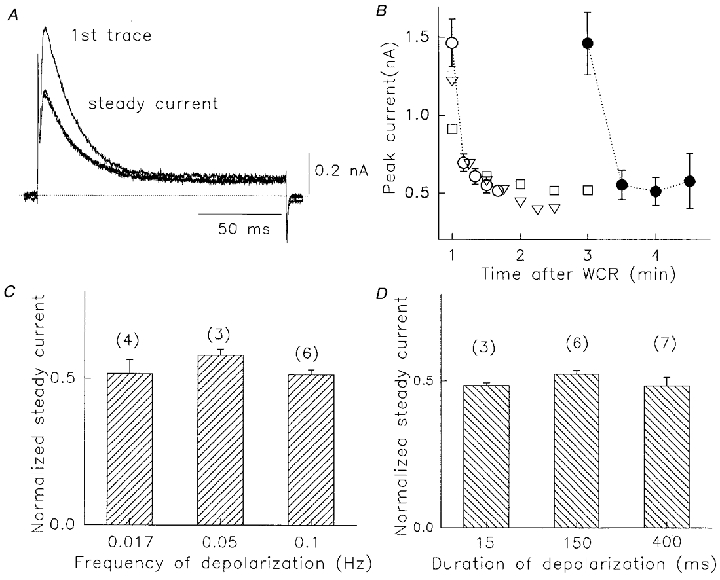
A, Na+ currents recorded 1 min after WCR. Pulses were from -80 to +60 mV at 0.05 Hz. Currents are shown in response to the first three voltage pulses. The second and third pulse current indicated the steady-state level. Dotted line denotes zero current. B, use-dependent decline in peak Na+ currents. Pulses (400 ms) were given 1 min (open symbols, n = 7) or 3 min (filled circles, n = 7) after WCR, at 0.1 (^), 0.067 (▿) or 0.033 (□ and •) Hz. C, ratio of steady-state current to peak current at three pulse rates, 0.017, 0.05 and 0.1 Hz. Pulses were to +60 mV for 400 ms. D, ratio of steady-state current to peak current at three pulse durations, 15, 150 and 400 ms. Rate was 0.05 Hz. In B-D, values are means ±s.e.m. No significant differences were observed.
The recovery from inactivation of Kv1.5 under physiological / conditions is rapid, with a fast time constant of ∼1 s (Fedida et al. 1999). Na+ currents inactivated much more rapidly and recovered more slowly than K+ currents. Recovery was tested using a 400 ms prepulse to +60 mV, and then assessing current recovery at different intervals. Prepulses were given every 30 s to ensure full recovery from inactivation between trials. An example of data obtained using this protocol in 130 mM /5 mM is shown in Fig. 5A. After the prepulse control data, recovery can be seen to occur in a progressive manner for interpulse intervals up to 10 s. Mean data on recovery from inactivation are shown in Fig. 5B and have been fitted to a single exponential recovery function with a time constant of 4.3 ± 0.9 s (n = 8). It seemed unlikely, therefore, that cumulative failure to recover from inactivation is responsible for the decrease in Na+ conductance described in Fig. 4.
Figure 5. Recovery of Kv1.5 Na+ currents from inactivation in 130 mM /5 mM .
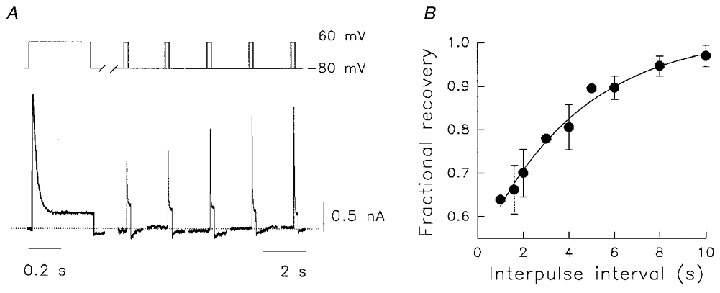
A, the protocol is illustrated above the current data. A 400 ms prepulse to +60 mV from -80 mV conditioned the channels. After repolarization for variable intervals from 1–10 s, current recovery was assessed during 60–200 ms test pulses to +60 mV. Dotted line denotes zero current. Double pulse sequences were given at 30 s intervals to allow full recovery from inactivation between tests. B, mean test pulse data were normalized to the preceding control peak current amplitudes and plotted as a function of the interpulse interval to obtain the fractional recovery from inactivation for Na+ currents. The data were fitted by a single exponential with a recovery time constant of 4.3 ± 0.9 s (n = 8).
Use-dependent reduction of Na+ conductance is regulated by
The inclusion of small amounts of in the bath solution before opening the channel could reduce or reverse the current decrease. This is shown by data in Figs 6 and 7 where pulses to +60 mV were given every 20 s to avoid accumulating C-type inactivation (Fig. 5), and the bath K+ concentration was varied in each experiment (Fig. 6A–E). There are also changes in inactivation rate at higher concentrations, as will be discussed in relation to Figs 8 and 9. As described in Fig. 4, in the absence of , from the first to the steady-state Na+ current there was a current reduction of ∼50 % (Fig. 6A). The effect on conductance (Fig. 6F) was reduced as concentration was elevated above 0.05 mM (Fig. 6B and C) and appeared to plateau at a conductance ratio of ∼0.85 for steady-state to peak conductance, at 1–2 mM (Fig. 6D–F). The Kd for this effect obtained from a Hill analysis of data in Fig. 6F was 230 μM. It was also of interest to see whether addition of could reverse the current decrease already established in 0 mM . This result is shown in Fig. 7A. In 0 mM , a 50 % initial decrease of current was observed, and subsequently the current remained at a constant level until 0.1 mM was added. This concentration of restored the current to about 80 % of the first pulse level in the next two or three pulses. Once conductance was calculated, mean data indicated a small increase from the first pulse level (Fig. 7B). This implied a second action of , as from Fig. 6F, 0.1 mM should only have had a small effect of at most 5 % on the loss of Na+ conductance in the first few pulses (45 vs. 50 % decrease in 0 ). A second action was confirmed using the experiment illustrated in Fig. 7C. Here the cell was initially in 0.1 mM , and on using the channel there was a ∼45 % decrease in conductance expected from Fig. 6F. However, when this small amount of was removed there was a large further decrease in current in 0 mM (smallest current in Fig. 7C) despite an increase in driving force. The resultant current was only 27 % of the peak current level in the first pulse.
Figure 6. Use-dependent reduction of Na+ conductance during pulsing in various .
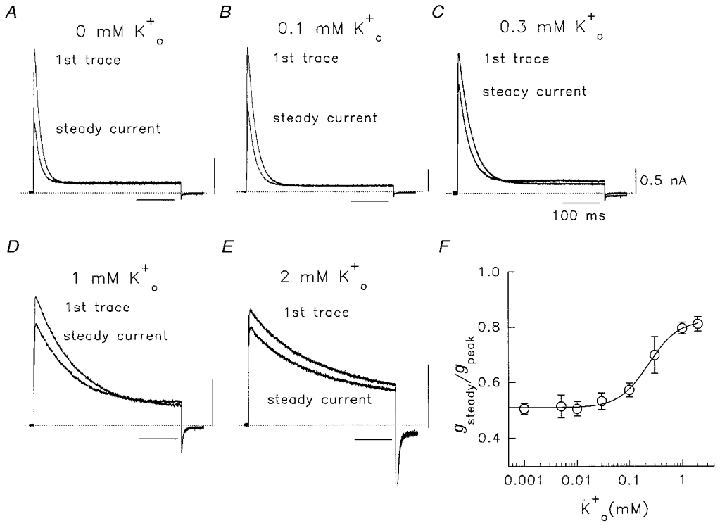
A-E, after a 1 min dialysis period, the cell was pulsed to +60 mV for 400 ms at 0.05 Hz in 130 mM /5 mM plus various concentrations of as indicated. The first current trace and the trace after a steady state was reached (2-3 pulses) are shown. Dotted lines denote zero current. Calibration values in C apply also to A, B, D and E. F, normalized relationship of steady current conductance to peak (first pulse) conductance (gsteady/gpeak) as a function of concentration. Plotted points are from means of 3–22 observations. The line was fitted through data using a Hill equation: f = 1/(1 + (Kd/[K+])nH), where f is the fractional conductance ratio, Kd represents the K+ concentration which produced 50 % of the maximum response and nH is the Hill coefficient. For added , the Kd was 230 μM and nH was 1.55.
Figure 7. Use-dependent reduction of Kv1.5 Na+ currents is modulated by addition or removal of .
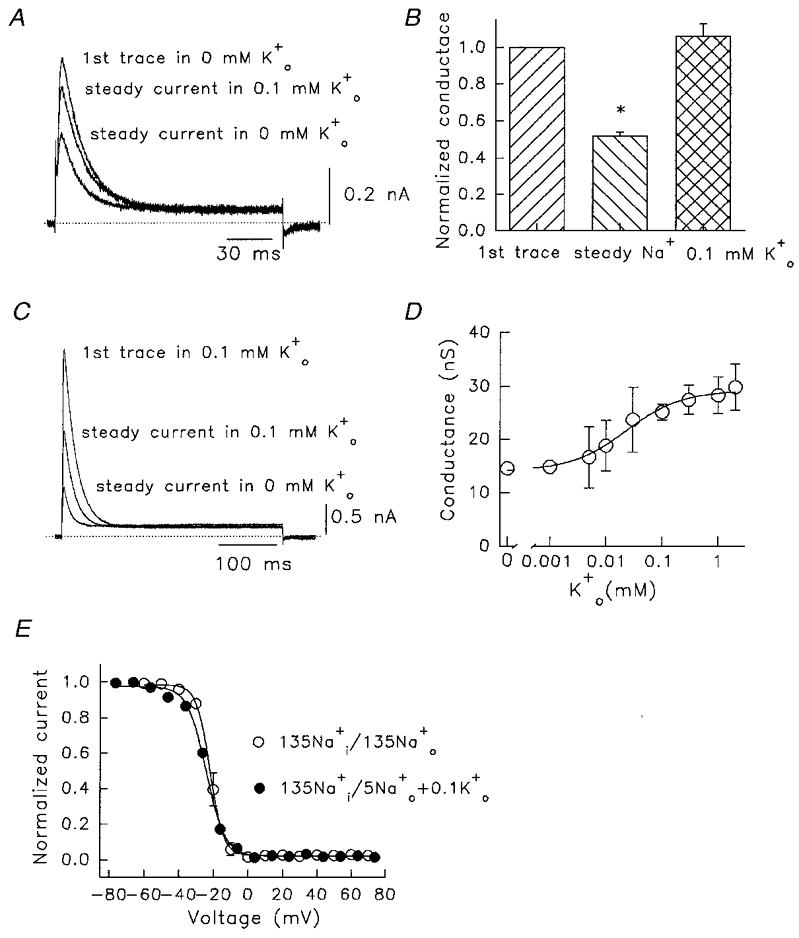
A, after a 1 min dialysis period, the cell was pulsed to +60 mV for 150 ms once every 60 s in 130 mM /5 mM . The first current trace, the trace after a steady state was reached (after 3 pulses), and that after subsequent addition of 0.1 mM are shown. Dotted line denotes zero current. B, mean data illustrating reduction of steady Na+ conductance during constant low rate pulsing (n = 5), and recovery induced by 0.1 mM (n = 6). The middle column is significantly different from both other columns (one-way ANOVA, * P < 0.01). C, reduction of use dependence by inclusion of 0.1 mM in the bath solution. The first current trace in 130 mM /5 mM + 0.1 mM , and the trace after a steady state was reached are shown. The smallest current was obtained after subsequent removal of . Pulses were to +60 mV every 60 s. D, amplitude of first pulse peak conductance as a function of . All data from pulses to +60 mV. For the Hill fit, the Kd was 23 μM and nH was 0.89. Plotted points are mean values from 3–18 observations. E, steady-state inactivation relationships in /, with and without 0.1 mM . In 135 mM /135 mM the V1/2 inactivation was -21.4 ± 1.6 mV and slope factor 3.4 ± 0.2 mV (n = 6), and in 130 mM /5 mM + 0.1 mM , the V1/2 was -24.1 ± 0.7 mV and slope factor was 5.6 ± 0.4 mV (n = 5). No significant differences were observed between the V1/2 and k values for the two relations.
Figure 8. Effect of low concentrations of on outward Na+ currents through Kv1.5 channels.
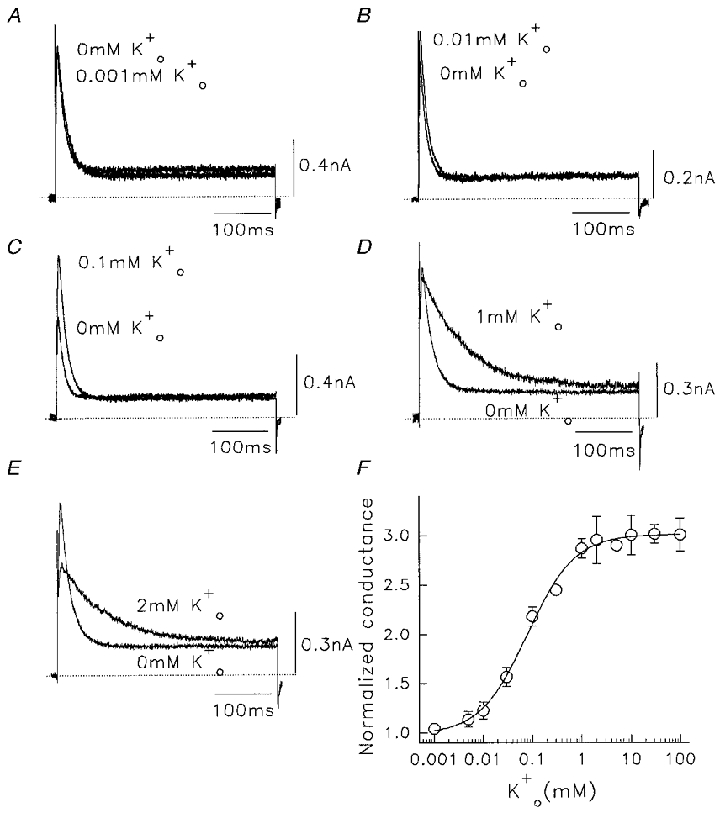
In all cases control solutions were 130 mM /5 mM . A-E, current traces showing effect of addition of different extracellular K+ concentrations. The control trace is labelled 0 mM . Data were obtained at +60 mV, pulses were at 0.05 Hz. Dotted lines denote zero currents. F, relationship between normalized peak Na+ conductance in different added . Plotted points are from a mean of 4–12 cells in each case. From the Hill fit to data points, for added , the Kd was 0.08 ± 0.01 mM and nH was 0.9 ± 0.1.
The results suggest that both the available first pulse conductance, and the amount of conductance loss when the channel is used, are regulated by . This is confirmed by measurements of the initial K+ conductance during the first pulse at different concentrations of (Fig. 7D). Again, a sigmoidal relationship was found with a Kd for this action of 23 μM. Clearly, when is present the initial Na+ conductance is regulated and higher (Fig. 7D). This is in addition to the regulatory action of on the use-dependent decrease of the Na+ conductance when pulses are applied (Fig. 6F). We considered whether small changes in the C-type inactivation properties of the channel could be responsible for the -dependent effects.
The possibility that the time course of recovery from inactivation was altered by very low concentrations of was tested by repeating the same protocol as in Fig. 5 in 130 mM /5 mM plus 0.1 mM in 15 cells. The time constant for recovery from inactivation was 5.2 ± 0.9 s, not significantly different from that in Na+-containing solutions alone. Although more than enough time was available for recovery from inactivation, the possibility remained that subtle changes in the level of steady-state inactivation could be responsible for the initial current decrease and the protective action of . Data in Fig. 7E show steady-state inactivation curves, measured during 200 ms depolarizations, in the presence of symmetrical 135 mM / and in other cells in 130 mM /5 mM plus 0.1 mM . Note that inactivation reached almost 100 % in Na+ solutions, based on our observation that the sustained current at positive potentials reflected Na+ flux through inactivated channels (Fig. 3). This steady-state current was subtracted from residual currents measured during the test pulses to +60 mV. In 135 mM /135 mM the V1/2 inactivation was -21.4 ± 1.6 mV, and with 0.1 mM , the V1/2 was -24.1 ± 0.7 mV. These values are similar to those in K+-containing solutions (Fedida et al. 1999), and at -80 mV small variations in the position of this curve along the potential axis are unlikely to influence the Na+ current level.
Modulation of Na+ current inactivation by
From the data obtained it appears that Kv1.5 channels suffer a decreased whole-cell conductance when depolarized in the absence of . The loss of conductance precedes use of the channel (first pulse conductance, Fig. 7D), and can be augmented by using the channel (Fig. 6F). We do not know if this is the result of some channels becoming unavailable in long-lived closed states, the entry of channels into an inactivated state that exists only in Na+-containing solutions, or a generalized decrease in single channel conductance. The use dependence of this effect suggests the findings of Gómez-Lagunas (1997), where a stable non-conducting state was induced in Shaker B by depolarizations in Na+ solutions lacking . However, in the present experiments, the channels have to be depolarized to inactivating potentials for the effect to be observed. Up to this point we have found little evidence that during the loss of conductance the channels enter a state resembling the C-type inactivated state, although it is possible that multiple states of this type may exist (Loots & Isacoff, 1998).
The two relations described above allow us to understand the action of small quantities of on the amplitude and time course of Na+ currents through Kv1.5 channels (Fig. 8). In this way we have identified extracellular and intracellular control of either the availability of Kv1.5 channels to conduct Na+, or to modulate the rate of inactivation. In all cases, the Na+ currents recorded were initially at a steady current level (Fig. 4) in 0 mM solutions. Low concentrations of between 0.005 mM and 0.5 mM added to the external bath solution increased the amplitude of Na+ currents, with only small changes in the decay rate (Fig. 8A–C). The contaminating K+ concentration in these solutions was measured to be < 1 μM (see Methods). The effects were observed within the first few pulses of exposure to K+-containing solutions, and were maintained for the duration of exposure. Higher concentrations of added to the bath medium suppressed the peak outward Na+ current observed, due to a positive shift of the reversal potential, and also slowed the rate of inactivation in a concentration-dependent manner (Fig. 8D and E). To evaluate the facilitation of Na+ current by independent of changes in driving force, we compared the change of Na+ conductance at different . At ≤ 2 mM , Na+ conductance was calculated by measuring the reversal potentials (Erev) at different concentrations (data not shown). At > 2 mM , depolarization induced an inward K+ current, and so to obtain K+-facilitated Na+ current at +60 mV to calculate conductance, a different method had to be used. Cells were pulsed in different concentration solutions, after which the bath was switched to a K+-free solution. After > 1 min solution exchange, pulses were recommenced. Because the loss of the conductance is use dependent, the Na+ current induced by the first depolarization in K+-free solution should reflect the facilitation by the . The Na+ conductance at different concentrations, normalized to the control value in K+-free bath solution, was plotted against concentration in Fig. 8F. This relationship represents the sum of the two effects of described by the data in Figs 6 and 7. When K+ is added to the bath, there is an increase in the maximum available conductance (Fig. 7D) and the use-dependent decrease is reduced (Fig. 6F). After fitting the Hill equation to these data, a half-maximum dose (Kd) of 0.08 ± 0.01 mM and a Hill coefficient (nH) of 0.91 ± 0.09 were obtained for .
Voltage-dependent action of on Na+ currents
The effects on Na+ currents of at low concentrations (below 0.5 mM) and those seen at higher concentrations are qualitatively different; the former facilitated current conductance and the latter both facilitated current conductance and slowed the time course of inactivation (Figs 6 and 8). In Fig. 9A, the decay time constants (τ) of Na+ currents were plotted against concentration on a logarithmic scale and a line through the data was calculated based on a simple line fit. It shows that the actions of on conductance and inactivation have a different dependence. At 0.1 mM , the peak conductance was almost doubled (Fig. 8), but the inactivation rate was not significantly increased (Fig. 9A). At 1 mM and above the inactivation rate was markedly decreased, without further large effects on peak current (after correction for changes in driving force, Fig. 8F). It is possible that the effects of on conductance and inactivation time course are modulated at the same site(s) and that the effect is dependent on the number of K+ ions binding to the site(s). Another possibility is that different site(s) are responsible for the two actions, and in this case we might expect differential sensitivities of the two effects to changes in the transmembrane electric gradient. To test this possibility we examined the voltage dependence of the action on conductance and inactivation rate.
The membrane potential should significantly affect the competition between K+ and Na+ ions at the outer pore. With K+ only in the bath, an inwardly directed K+ concentration gradient is established and so K+ will try to enter the pore from the outside. More positive intracellular membrane potentials counteract the effect of ion concentration and so decrease entry. Thus, larger positive intracellular potentials will decrease the probability of pore occupancy by externally applied K+ ions. In order to examine the voltage dependence of action on inactivation rate, the ratio of decay time constants of Na+ currents in 0.1 or 2 mM to that in 0 mM has been plotted as a function of pulse potential (Fig. 9B). Decay time constants in alone were essentially voltage independent (Fig. 1B) so changes in the ratio reflect -dependent actions. Straight lines were fitted to the data. At 0.1 mM the current decay was hardly affected and only mildly voltage dependent (slope =−0.01), increasing slightly at the less positive potentials studied. With 2 mM , the decay time constant was strongly voltage dependent and the effect of 2 mM diminished towards +80 mV. The fitted line had a slope of -0.3, which indicates that the action of 2 mM on inactivation sensed a greater fraction of the transmembrane electric field, and was most probably located deeper within the electric field. These observations support the view that the conductance and inactivation are modulated at different site(s). We have also examined the voltage dependence of the action of 2 mM on the use-dependent reduction of peak Na+ conductance (gsteady/gpeak) in Fig. 9C. Like 0.1 mM , the action of 2 mM on peak conductance was not significantly voltage dependent.
Modulation of Na+ current inactivation by
We were interested to see whether small concentrations of could modulate Kv1.5 Na+ currents in the same dual manner as . Compared with zero added solutions, low concentrations of (< 0.5 mM) included in the pipette filling solution apparently had little effect on the amplitude or time course of currents, either during the first pulse or during the steady-state currents (Fig. 10A). There was no significant action of less than 1 mM on the peak current observed during the first pulse, compared with no added (Fig. 10D). Nor was there any effect of (< 1 mM added) on the ratio of steady-state current reached to the initial current during the first pulse (Fig. 10F), which was still ∼0.5 (Fig. 10F). The time course of current inactivation was also unaffected below 0.5 mM added (Fig. 10A and E). These data suggested that concentrations of < 1 mM did not facilitate Na+ currents in the same manner as . Added concentrations of 1 mM and higher slowed inactivation of Na+ currents. This occurred both during the first pulse and in the steady state (Fig. 10B, C and E). The first-pulse peak currents were reduced with 1 mM compared with zero but they were increased with 5 mM (Fig. 10D). The smaller mean first-pulse currents with 1 mM probably reflect an anomalous mole fraction effect caused by the admixture of and . Conversely, the larger mean first-pulse currents with 5 mM added, probably represent K+ flux through Kv1.5 channels augmenting the overall Na+ current. Despite these peak current changes, there were consistent decreases in the ratio of steady-state to peak current amplitude at both 1 mM and 5 mM (Fig. 10F).
Figure 10. Effect of low concentrations of on Na+ currents through Kv1.5 channels.
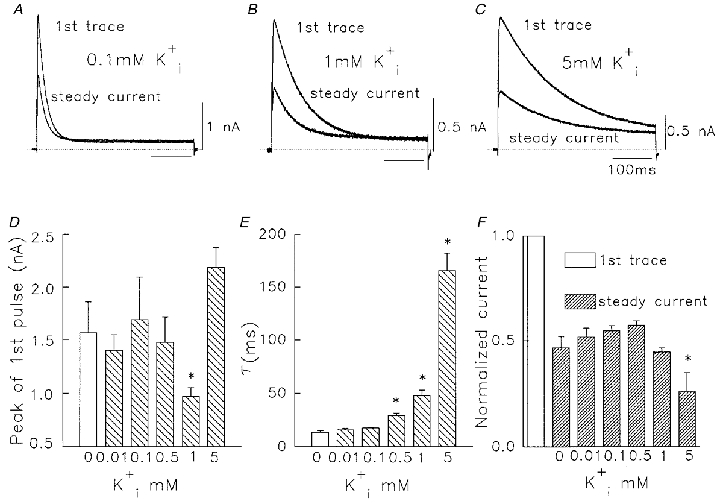
In each panel the pipette contained 130 mM and the stated K+ concentration. The bath contained 5 mM . A-C, first pulse and steady-state currents at different added concentrations of . Pulses were from -80 to +60 mV for 400 ms at 0.05 Hz. Time calibration in C applies also to A and B. D, mean peak Na+ current vs. added . E, time constant of Na+ current inactivation vs. added . F, effect of added on relative amplitude of 1st pulse and steady-state Na+ currents. In D-F, data are means ±s.e.m. (n = 4-7). Columns marked with an asterisk are significantly different from 0 mM column (one-way ANOVA, P < 0.05).
Kv1.5-R487V prevents inactivation and abolishes the actions of low
The modulation of on channel conductance has been examined in different Shaker-related K+ channels. Pardo et al. (1992) reported that removal of external K+ abolished current through rKv1.4 channels, and suggested that the presence of a charged lysine residue (K533) at the outer mouth of the pore was responsible for this -dependent phenomenon. This dependence has been tested in a range of Kv1 channels, Kv1.1-1.4, which have different amino acid residues at the position homologous to K533 in rKv1.4 (Jäger et al. 1998). It was reported that no dependence was observed in Kv1.1 and 1.2, which have uncharged residues (Y in Kv1.1 and V in Kv1.2). Kv1.3 only showed dependence in pH 6.0 solution, suggesting that protonation of H404 accounted for this effect. In Kv1.5, a charged arginine residue (R487) is in the position homologous to K533 in rKv1.4. This residue is located in the outer vestibule (Aiyar et al. 1995; Doyle et al. 1998), participates in the external TEA-binding site (Kavanaugh et al. 1992), and might also account for the dependence in Kv1.5. To test this idea, we replaced R487 in Kv1.5 with valine (V), an uncharged amino acid, present in Kv1.2 which does not show dependence (Jäger et al. 1998).
Replacement of R487 with valine abolished use dependence of Kv1.5 as shown in Fig. 11A when experiments were conducted in 130 mM /5 mM . Unlike the WT channel, no decrease in current level was observed when pulses were initially given in Na+ solutions, and when 0.1 mM was added, no current facilitation was observed. In the presence of 0.1 mM , conductance decreased to 0.72 ± 0.14 (n = 3) of that in Na+ alone. The result confirms that R487 does play a role in modulation of conductance in Kv1.5 channels. A parallel observation during this experiment was that the rapid C-type inactivation of Kv1.5 Na+ currents was prevented in Kv1.5-R487V. Depolarizations (400 ms) induced no significant inactivation (Fig. 10A), in contrast to Na+ current through wild-type Kv1.5 channels (see above). Pulse durations up to 14 s still resulted in only limited inactivation (Fig. 11B), reminiscent of the action of this mutation in K+-conducting Shaker channels (Lopez-Barneo et al. 1993). It was a possibility that R487V mutant channels inactivated extremely rapidly during depolarization, and that the sustained Na+ current was carried through inactivated channels. In Fig. 11B, scaled current recordings show an envelope experiment conducted in symmetrical 135 mM Na+ solutions. All tails recorded at the end of 50 ms, 350 ms and 14 s depolarizing pulses show the same fast kinetics of decay shown in Fig. 3D and E, indicating that the channels deactivated normally and rapidly on repolarization (Fig. 11C). This strongly suggests that sustained Na+ currents in Kv1.5-R487V mutant channels were carried by open channels.
Figure 11. Na+ currents through R487V mutant Kv1.5 channels.
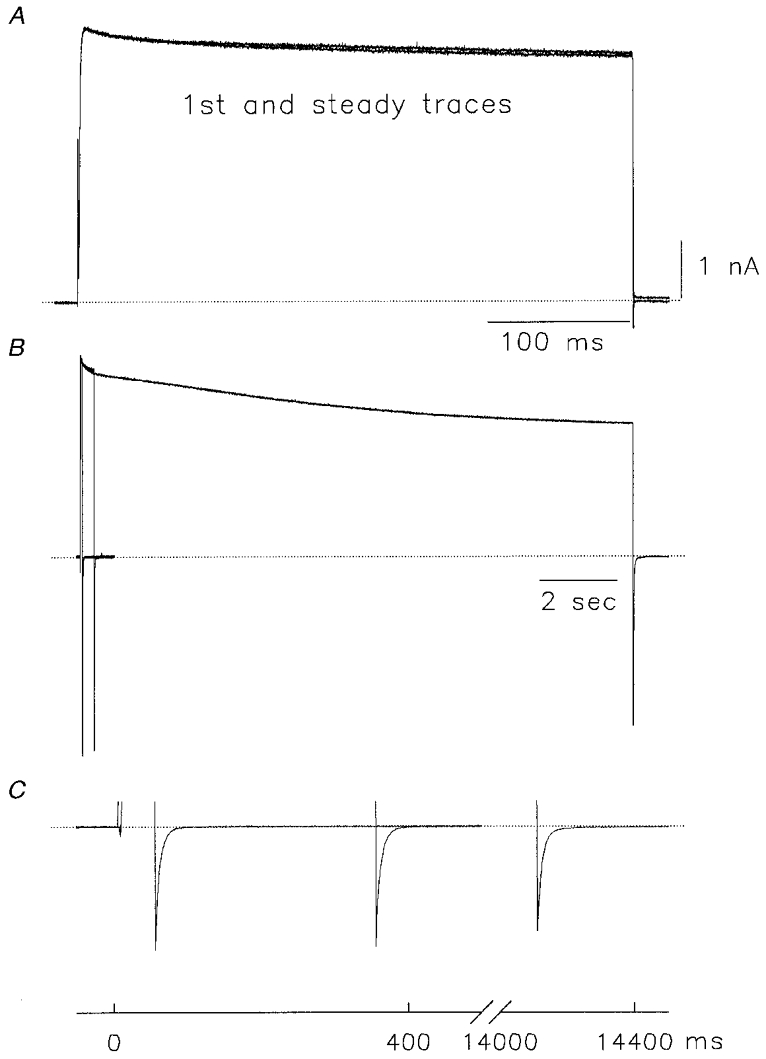
A, whole-cell current recordings from Kv1.5-R487V channels in 130 mM /5 mM . The depolarizing pulse was from -80 mV holding potential to +60 mV for 400 ms at 0.05 Hz. B, envelope of tails experiment conducted in symmetrical 135 mM Na+. Normalized current data during depolarizations from -80 mV to +60 mV for pulse durations of 50 ms, 350 ms and 14 s (data from a different cell). C, expanded scaled tail current recordings from experiment shown in B.
DISCUSSION
Na+ currents through K+ channels
The Na+ currents recorded in transfected HEK cells had the sensitivity to 4-AP expected of Kv1.5 channels from K+ current experiments (Bouchard & Fedida, 1995), with ∼50 % block at 50 μM (Fig. 3). Currents were insensitive to DIDS, a blocker of endogenous Cl− channels, and reversal potential measurements in Fig. 2 confirmed that the current was carried by Na+ through Kv1.5 channels. Although these were unequivocally Na+ currents through Kv1.5 channels, they showed different properties from those seen in Kv2.1 channels (Korn & Ikeda, 1995; Kiss & Korn, 1998), or in Shaker mutant channels (Ogielska & Aldrich, 1998). Like the Kv1.3:Kv2.1 chimera channel used by Kiss et al. (1998), Kv1.5 Na+ currents were rapidly activating and inactivating (Fig. 1), whereas the Kv2.1 channel inactivated slowly when conducting Na+ or K+ (Korn & Ikeda, 1995). Not only was inactivation faster, but the recovery from C-type inactivation was fourfold slower (Fig. 5). A rapid inactivation seems consistent with the accelerated C-type inactivation expected in the absence of and in Kv1 channels (Baukrowitz & Yellen, 1995), and with data that shows modulation of recovery from C-type inactivation by (Levy & Deutsch, 1996). With Na+ as the conducting ion, the WT Shaker channel also inactivates extremely rapidly, showing a transient current followed by a sustained phase due to Na+ passing through the inactivated channels (Starkus et al. 1997). The difference between inactivation rates in Kv1 and Kv2 channels when conducting Na+ or K+ suggests that binding of these two cations to intrapore sites plays different roles in the modulation of C-type inactivation in these channels, or alternatively that additional modulatory sites exist.
The Na+ current through Kv1.5 channels did show a sustained component through inactivated channels like that in Shaker channels. However, the amplitude was only 13 % of peak current, and so smaller than that in Drosophila Shaker channels. Furthermore, in the symmetrical Na+ experiments (in Fig. 3D and E), with the progress of inactivation, a rising phase in the tail appears more apparent than that in Shaker (Starkus et al. 1997). Both results imply a lower Na+ permeability of the inactivated state in Kv1.5 channels than in Shaker channels. The expression of Kv1.5 channels in a monoclonal cell line allows an estimation of the peak whole-cell conductance with different monovalent cations. Currents were measured using Rs compensation, with 130 mM /5 mM ion concentrations, at +60 mV where the channel open probability should approach 1.0. The conductance ratios through Kv1.5 channels were: gNa/gK = 0.062 ± 0.007; gNa/gRb = 0.11 ± 0.013; gNa/gCs = 1.20 ± 0.14. The values for Na+ conductance may be compared with a permeability ratio obtained from bi-ionic measurements of 0.007 for PNa/PK (Snyders et al. 1993), and an inward Na+ conductance of 0.79 ± 0.2 % that of K+ (Korn & Ikeda, 1995). The Na+ conductance in the present experiments is an order of magnitude greater than its permeability in the presence of K+, or its conductance in the presence of . This is probably still an underestimate due to the rapid inactivation kinetics of Na+ currents, and the modulatory actions of (see below) but suggests a relatively higher Na+ conductance of Kv1 channels than has previously been shown.
Modulation of Kv1.5 Na+ current inactivation by
The rapid inactivation of Na+ currents through Kv1.5 channels provided a powerful tool to investigate the modulatory capabilities of various concentrations of intracellular and extracellular K+ on this system. Based on experiments with a chimeric channel, Kiss et al. (1998) presented a pore model which includes a single high affinity cation binding site at the selectivity filter, flanked by lower affinity non-selective sites. The occupancy of the selectivity filter binding site by K+ directly slowed C-type inactivation, and occupancy of the outer site by either K+ or Na+ slowed inactivation indirectly by trapping ions at the pore (Kiss & Korn, 1998). Our data support the applicability of such a model for Kv1.5 at higher concentrations of than 0.3 mM. At these concentrations, external K+ can access the intrapore site(s) to slow inactivation (Figs 8 and 9). The effect on slow inactivation was strongly voltage dependent and depended on the K+ being driven rather deeply into the channel pore (Fig. 9B). slowed inactivation in a similar way to but required two- to tenfold higher concentrations, suggesting a lower affinity for the selectivity site than K+ (data not shown).
Lower binding between 1 μM and 200 μM lacked voltage dependence (Fig. 9B), suggesting an outer mouth site of relatively higher binding affinity (Kd = 20–80 μM), in contrast to the Kiss & Korn (1998) model. Despite evidence for a relatively high affinity action on conductance (Fig. 8F, 80 μM), these concentrations of had little effect on the C-type inactivation rate of currents (Figs 6 and 8). This argued against the ability of outer mouth cation binding to trap ions at the selectivity filter in Kv1.5. It is possible that the outer mouth site may not be directly within the permeation pathway, and so is unable to cause trapping and slow inactivation. Alternatively, the site may be able to bind K+ when the channel is closed, and so regulate conductance, but when the channel opens during depolarization, the high Na+ flux may reduce occupancy of the site by K+. So inactivation may be unaffected at low , whereas at higher , the likelihood of occupancy would increase. Under physiological conditions, when K+ permeates the channel, such a knock-off effect would not occur, and the outer mouth site would normally always be occupied.
Modulation of Kv1.5 Na+ current amplitude and conductance by
Two actions of external were identified that modulated the peak Na+ conductance through Kv1.5 (Figs 6–8), but actions on peak current were concealed at > 0.5 mM due to changes in driving force. had a similar effect but again required higher concentrations, suggesting a lower affinity than (data not shown). These actions are interpreted using the scheme below (Jäger et al. 1998) for closed (C), open (O), and C-type inactivated channels (I):
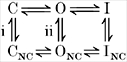 |
Scheme 1 |
where the second layer of channel states represents non-conducting (NC) states occupied in the absence of . The first pulse peak conductance can be modulated by the presence of while the channel is at rest, and even before it has been used. This relationship was defined in Fig. 7D, and suggests that an outer mouth site is accessible when the channel is still closed. It suggests that K+ can regulate transition from CNC to C with a Kd of 23 μM (i). When the channels are then opened, the use-dependent decrease in conductance can also be regulated by , as shown in Fig. 6F, with a Kd of 230 μM. This suggests transition from O to ONC states when the channel is used (ii), and clearly this is also reversible and the number of available channels can be increased or decreased by changes in , as shown in Fig. 7A and C.
This cation action on resting or open channels cannot be explained by access very far into the channel as there was little voltage dependence at 0.1 or 2 mM (Fig. 9). We considered the possibility that current loss might be due to a change in steady-state inactivation and/or a slowed recovery from inactivation in Na+-containing solutions, or after addition of 0.1 mM . However, no significant difference was observed in the steady-state inactivation parameters (V1/2 and k) in these solutions. In most of the experiments shown, we recorded steady-state Na+ current based on an interpulse interval of 20 s. With a 4 s recovery time constant (Fig. 5), or 5.2 s with 0.1 mM , significant accumulation of inactivation should not have occurred. To further test this possibility we prolonged the pulse interval to 1 min (Fig. 7A), but there was the same current reduction between the first and steady-state current traces that was reversible with low . These time-independent properties suggest regulation of the channel conductance in a manner not dependent on the acceleration or prevention of classical C-type channel inactivation.
Mechanism for conductance regulation by
Facilitation of peak Na+ conductance by low cannot easily be attributed to removal of accumulated C-type inactivation, but it is difficult to exclude another inactivation process with an extremely slow recovery course, or with a specific dependence. In the present study we removed inactivation along with the dependence of Kv1.5 in the R487V mutant, suggesting that this arginine residue, which is located in the outer vestibule (Aiyar et al. 1995), might be the binding site, or could affect the binding site for . Mutation of the charged arginine to uncharged valine might alter the conformation of the binding site or affect an adjacent binding site, so the dependence of Kv1.5 is lost. We also observed less inactivation when R487V mutant channels conducted Na+ currents (Fig. 11) without any significant change in K+ current gating kinetics. As reported for the A463C mutant of the Shaker B channel (Ogielska & Aldrich, 1999), changing the charged arginine residue to an uncharged valine may affect the binding affinity of Na+ for the site. As a result, ion interaction in the pore may be decreased and the duration of Na+ binding to intrapore sites prolonged. The mutation may then affect both inactivation rate and dependence, and so it becomes very difficult to separate them.
The data are also consistent with the known entry of K+ channels into long-lived closed states that correspond to a loss of conductance when (Pardo et al. 1992; Lopez-Barneo et al. 1993; Jäger et al. 1998), or and are omitted (Gómez-Lagunas, 1997). Conflicting ideas surround the exact relationship of this state to the C-type inactivated state (Lopez-Barneo et al. 1993), perhaps for reasons discussed above (Jäger et al. 1998). Na+ current facilitation by very low may reflect the movement of channels out of such long-lived states. Binding of releases channels and enables them to conduct Na+. This can explain the increased Na+ current without kinetic changes. Our data suggest that this effect could be mediated at a high affinity K+ site (Hill coefficient is 0.9) in the outer pore mouth, which is consistent with explanations given for the non-conducting closed states (Pardo et al. 1992; Lopez-Barneo et al. 1993; Jäger et al. 1998). The experiments with low revealed that facilitated Na+ current, but we have no direct evidence that this effect also occurs when Kv1.5 channels are conducting K+ ions, as reported previously for other channels (Pardo et al. 1992). One might expect this to be a difficult experiment to carry out because of the very low required for regulation and significant K+ efflux during channel activation (Baukrowitz & Yellen, 1995). Interestingly, the Kv1.3 channel mutant H404R, which is the equivalent residue to that in Kv1.5, does show strong conductance regulation by small changes of (Jäger et al. 1998). This is good evidence that WT Kv1.5 channels should show regulation by very low , and perhaps evidence that the site may not be directly within the permeation pathway.
Modulation of Kv1.5 Na+ current inactivation by
Intracellular application of K+ slowed inactivation in a concentration-dependent manner (Fig. 10), and decreased the peak current at concentrations of ∼1 mM, consistent with an anomalous mole-fraction behaviour. In this situation K+ has a higher affinity for an intrapore selectivity site than Na+. Addition of low concentrations of inhibited Na+ current through Kv1.5 channels, but concentrations of 5 mM and above increased conductance, perhaps due to electrostatic repulsion between multiple ions in the pore. These data support the conclusion above, that K+ binding to a selectivity filter site slows inactivation in Kv1.5 channels as in a Kv2.1 chimera (Kiss & Korn, 1998). In Shaker channels the location of the selectivity filter site was suggested to be near the narrowing of the internal mouth of the channel (Ogielska & Aldrich, 1998). Thus, K+ should access this site more directly from the inside. These results also suggest that intracellular cations might also be able to regulate the rate and onset of inactivation in Kv1.5, as we have recently demonstrated (Fedida et al. 1999). Lower concentrations of had no effect on the peak current amplitude. Thus, the site or sites that cause facilitation of Na+ current may be on the outside of the channel, or at least not readily accessible by from the intracellular side.
Models of the channel permeation pathway
A single-file, multi-ion pore (Hodgkin & Keynes, 1955) with specific binding sites has been the preferred model to explain the high selectivity and rapid ion flux of K+ channels (Hille & Schwarz, 1978; Stampe & Begenisich, 1996). Flux ratio experiments suggest that normally conducting K+ channels are occupied by three to four K+ ions simultaneously, and the high flux occurs partly due to ion-ion electrostatic repulsion (Hille & Schwarz, 1978). The multiple K+ binding sites in the pore have generally been considered to have approximately equal affinities (French & Shoukimas, 1985; Neyton & Miller, 1988; Pérez-Cornejo & Begenisich, 1994). A recent model, proposed for Ca2+ and K+ channels, suggests that a single high affinity site flanked by two lower affinity sites can account for selectivity and high flux rates (Dang & McCleskey, 1998; Kiss et al. 1998). The slowing of Kv1.5 Na+ current inactivation by in our study is consistent with the hypothesis that high affinity K+ binding to the selectivity filter modulates C-type inactivation (Kiss & Korn, 1998). At concentrations of > 0.5 mM our data suggest that K+ is able to penetrate readily into the channel pore from the outside and access the highly selective site for K+. This has the effect of slowing the rate of current inactivation. In the recently published crystal structure of a Streptomyces lividans K+ channel, KscA, two dehydrated permeant ions reside in the selectivity filter region, which extended towards the outer end of the pore (Doyle et al. 1998). A third hydrated ion was located in the water-filled cavity in the internal vestibule of the channel. In this case, both selectivity filter sites are within the permeation pathway, and these multiple binding sites could explain the strong voltage dependence of action at > 1 mM.
Our data also suggest the existence of another high affinity K+ site (Hill coefficient suggests one site) in the outer pore mouth (Pardo et al. 1992; Jäger et al. 1998), rather than (or in addition to) a low affinity site for K+ in the outer pore (Kiss et al. 1998). Neither in Kv2.1 nor in chimeric channel studies was a facilitation of Na+ current by low concentrations of reported (Korn & Ikeda, 1995; Kiss et al. 1998; Kiss & Korn, 1998). This site fails to sense the applied potential almost completely, so such high affinity extracellular binding site or sites may not even be within the permeation pathway.
Acknowledgments
Supported by grants from the Heart and Stroke Foundations of British Columbia and Yukon, and the Medical Research Council of Canada to D.F.
References
- Aiyar J, Withka JM, Rizzi JP, Singleton DH, Andrews GC, Lin W, Boyd J, Hanson DC, Simon M, Dethlefs B, Lee CL, Hall JE, Gutman GA, Chandy KG. Topology of the pore-region of a K+ channel revealed by the NMR-derived structures of scorpion toxins. Neuron. 1995;15:1169–1181. doi: 10.1016/0896-6273(95)90104-3. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T, Yellen G. Modulation of K+ current by frequency and external [K+]: A tale of two inactivation mechanisms. Neuron. 1995;15:951–960. doi: 10.1016/0896-6273(95)90185-x. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Negative conductance caused by entry of sodium and cesium ions into the potassium channels of squid axons. Journal of General Physiology. 1972;70:549–566. doi: 10.1085/jgp.60.5.588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Block BM, Jones SW. Ion permeation and block of M-type and delayed rectifier potassium channels. Journal of General Physiology. 1996;107:473–488. doi: 10.1085/jgp.107.4.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard RA, Fedida D. Closed and open state binding of 4-aminopyridine to the cloned human potassium channel Kv1.5. Journal of Pharmacology and Experimental Therapeutics. 1995;275:864–876. [PubMed] [Google Scholar]
- Callahan MJ, Korn SJ. Permeation of Na+ through a delayed rectifier K+ channel in chick dorsal root ganglion neurons. Journal of General Physiology. 1994;104:747–771. doi: 10.1085/jgp.104.4.747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang TX, McCleskey EW. Ion channel selectivity through stepwise changes in binding affinity. Journal of General Physiology. 1998;111:185–193. doi: 10.1085/jgp.111.2.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doyle DA, Cabral JM, Pfuetzner RA, Kuo AL, Gulbis JM, Cohen SL, Chait BT, MacKinnon R. The structure of the potassium channel: Molecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Fedida D, Maruoka N, Lin S. Modulation of slow inactivation in human cardiac Kv1.5 channels by extra- and intra-cellular permeant cations. The Journal of Physiology. 1999;515:315–329. doi: 10.1111/j.1469-7793.1999.315ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedida D, Wible B, Wang Z, Fermini B, Faust F, Nattel S, Brown AM. Identity of a novel delayed rectifier current from human heart with a cloned K+ channel current. Circulation Research. 1993;73:210–216. doi: 10.1161/01.res.73.1.210. [DOI] [PubMed] [Google Scholar]
- French RJ, Shoukimas JJ. An ion's view of the potassium channel. The structure of the permeation pathway as sensed by a variety of blocking ions. Journal of General Physiology. 1985;85:669–698. doi: 10.1085/jgp.85.5.669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French RJ, Wells JB. Sodium ions as blocking agents and charge carriers in the potassium channel of the squid giant axon. Journal of General Physiology. 1977;70:707–724. doi: 10.1085/jgp.70.6.707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Lagunas F. Shaker B K+ conductance in Na+ solutions lacking K+ ions: a remarkably stable non-conducting state produced by membrane depolarizations. The Journal of Physiology. 1997;499:3–15. doi: 10.1113/jphysiol.1997.sp021907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heginbotham L, MacKinnon R. Conduction properties of the cloned Shaker K+ channel. Biophysical Journal. 1993;65:2089–2096. doi: 10.1016/S0006-3495(93)81244-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hille B. Ionic Channels of Excitable Membranes. Sunderland, MA, USA: Sinauer Associates Inc.; 1992. [Google Scholar]
- Hille B, Schwarz W. Potassium ions as multi-ion single file pores. Journal of General Physiology. 1978;72:409–442. doi: 10.1085/jgp.72.4.409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Huxley AF. The components of membrane conductance in the giant axon of Loligo. The Journal of Physiology. 1952;116:473–496. doi: 10.1113/jphysiol.1952.sp004718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgkin AL, Keynes RD. The potassium permeability of a giant nerve fibre. The Journal of Physiology. 1955;128:61–88. doi: 10.1113/jphysiol.1955.sp005291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger H, Rauer H, Nguyen AN, Aiyar J, Chandy KG, Grissmer S. Regulation of mammalian Shaker-related K+ channels: evidence for non-conducting closed and non-conducting inactivated states. The Journal of Physiology. 1998;506:291–301. doi: 10.1111/j.1469-7793.1998.291bw.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kavanaugh MP, Hurst RS, Yakel J, Varnum MD, Adelman JP, North RA. Multiple subunits of a voltage-dependent potassium channel contribute to the binding site for tetraethylammonium. Neuron. 1992;8:493–497. doi: 10.1016/0896-6273(92)90277-k. [DOI] [PubMed] [Google Scholar]
- Kiss L, Immke D, Loturco J, Korn SJ. The interaction of Na+ and K+ in voltage-gated potassium channels – Evidence for cation binding sites of different affinity. Journal of General Physiology. 1998;111:195–206. doi: 10.1085/jgp.111.2.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss L, Korn SJ. Modulation of C-type inactivation by K+ at the potassium channel selectivity filter. Biophysical Journal. 1998;74:1840–1849. doi: 10.1016/S0006-3495(98)77894-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korn SJ, Ikeda SR. Permeation selectivity by competition in a delayed rectifier potassium channel. Science. 1995;269:410–412. doi: 10.1126/science.7618108. [DOI] [PubMed] [Google Scholar]
- Levy DI, Deutsch C. Recovery from C-type inactivation is modulated by extracellular potassium. Biophysical Journal. 1996;70:798–805. doi: 10.1016/S0006-3495(96)79619-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loots E, Isacoff EY. Protein rearrangements underlying slow inactivation of the Shaker K+ channel. Journal of General Physiology. 1998;112:377–389. doi: 10.1085/jgp.112.4.377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Neyton J, Miller C. Discrete Ba2+ block as a probe of ion occupancy and pore structure in the high-conductance Ca2+-activated K+ channel. Journal of General Physiology. 1988;92:569–586. doi: 10.1085/jgp.92.5.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Aldrich RW. A mutation in S6 of shaker potassium channels decreases the K+ affinity of an ion binding site revealing ion-ion interactions in the pore. Journal of General Physiology. 1998;112:243–257. doi: 10.1085/jgp.112.2.243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, Aldrich RW. Functional consequences of a decreased potassium affinity in a potassium channel pore – Ion interactions and C-type inactivation. Journal of General Physiology. 1999;113:347–358. doi: 10.1085/jgp.113.2.347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pardo LA, Heinemann SH, Terlau H, Ludewig U, Lorra C, Pongs O, Stühmer W. Extracellular K+ specifically modulates a rat brain K+ channel. Proceedings of the National Academy of Sciences of the USA. 1992;89:2466–2470. doi: 10.1073/pnas.89.6.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez-Cornejo P, Begenisich T. The multi-ion nature of the pore in Shaker K+ channels. Biophysical Journal. 1994;66:1929–1938. doi: 10.1016/S0006-3495(94)80986-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pusch M, Neher E. Rates of diffusional exchange between small cells and a measuring patch pipette. Pflügers Archiv. 1988;411:204–211. doi: 10.1007/BF00582316. [DOI] [PubMed] [Google Scholar]
- Snyders DJ, Tamkun MM, Bennett PB. A rapidly activating and slowly inactivating potassium channel cloned from human heart. Functional analysis after stable mammalian cell culture expression. Journal of General Physiology. 1993;101:513–543. doi: 10.1085/jgp.101.4.513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stampe P, Begenisich T. Unidirectional K+ fluxes through recombinant Shaker potassium channels expressed in single xenopus oocytes. Journal of General Physiology. 1996;107:449–457. doi: 10.1085/jgp.107.4.449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD, Heinemann SH. Ion conduction through C-type inactivated Shaker channels. Journal of General Physiology. 1997;110:539–550. doi: 10.1085/jgp.110.5.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Starkus JG, Kuschel L, Rayner MD, Heinemann SH. Macroscopic Na+ currents in the ‘nonconducting’Shaker potassium channel mutant W434F. Journal of General Physiology. 1998;112:85–93. doi: 10.1085/jgp.112.1.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Zhang X, Fedida D. Gating current studies reveal both intra- and extra-cellular cation modulation of K+ channel deactivation. The Journal of Physiology. 1999;515:331–339. doi: 10.1111/j.1469-7793.1999.331ac.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Ikeda SR. Anomalous permeation of Na+ through a putative K+ channel in rat superior cervical ganglion neurones. The Journal of Physiology. 1993;468:441–461. doi: 10.1113/jphysiol.1993.sp019781. [DOI] [PMC free article] [PubMed] [Google Scholar]