

Abstract
In endothelial cells, release of Ca2+ from endoplasmic reticulum (ER) Ca2+ stores activates Ca2+ influx via the capacitative Ca2+ entry (CCE) pathway. In cultured bovine pulmonary artery endothelial cells, we investigated the relationship between intracellular Ca2+ store load and CCE activity, as well as the kinetics of CCE activation and deactivation, by simultaneously measuring changes in [Ca2+]i and unidirectional manganese (Mn2+) entry through the CCE pathway.
Submaximal concentrations of ATP caused quantal release of Ca2+ from the ER, resulting in a dose-dependent depletion of Ca2+ stores and acceleration of Mn2+ entry. Mn2+ entry rate, as a measure of CCE activity, was graded with the amount of released Ca2+. Maximal activation of CCE did not require complete store depletion.
Slow depletion of the ER by exposure to the ER Ca2+ pump inhibitor cyclopiazonic acid resulted in a delayed activation of CCE, revealing a temporal dissociation between release of Ca2+ from intracellular stores and activation of CCE.
During [Ca2+]i oscillations, at frequencies higher than 0·5 spikes min−1, each Ca2+ spike resulted in a progressive acceleration of CCE without leading to oscillations of Ca2+ entry. In contrast, low frequency [Ca2+]i oscillations were paralleled by transient CCE that was activated and deactivated with each Ca2+ spike, resulting in an oscillatory pattern of Ca2+ entry.
It is concluded that CCE is a rapidly activating process which is graded with store depletion and becomes fully activated before complete depletion. The duration of CCE activation correlates with the degree of store depletion and the time that is required to refill depleted stores. Overall, a mechanism of graded CCE prevents exhaustion of intracellular Ca2+ reserves and provides an efficient way to respond to variable degrees of intracellular store depletion.
In a wide variety of non-excitable cell types, release of Ca2+ from intracellular Ca2+ stores initiates a store depletion-dependent Ca2+ influx mechanism, termed capacitative Ca2+ entry (for review see Berridge, 1995; Putney, 1997; Parekh & Penner, 1997; Holda et al. 1998). This store-operated or Ca2+ release-dependent capacitative Ca2+ entry (CCE) plays a key role in maintaining the Ca2+ load of intracellular stores (endoplasmic reticulum, ER), but has also been implicated in important cellular functions such as secretion, regulation of adenylate cyclase, gene transcription, cell cycle and proliferation, apoptosis and modulation of [Ca2+]i oscillations and [Ca2+]i waves (for review see e.g. Parekh & Penner, 1997). Furthermore, in endothelial cells CCE has been shown to be a preferential Ca2+ source for the regulation of the Ca2+-calmodulin-dependent NO synthase activity (Xu et al. 1994; Wang et al. 1996). Hoth & Penner (1992) were the first to demonstrate a membrane conductance directly related to Ca2+ store depletion, which subsequently was termed Ca2+ release-activated Ca2+ current (ICRAC). A similar membrane current has been identified in vascular endothelial cells (Fasolato & Nilius, 1998). ICRAC is the best characterized store-operated Ca2+ current, although in recent years an increasing number of store-dependent membrane conductances have been characterized in different cell types that differ from the original ICRAC in their biophysical properties and modes of regulation and modulation (see Clapham, 1995; Parekh & Penner, 1997).
Several key questions regarding regulation of CCE have remained unanswered. For example, the signalling mechanism that communicates the degree of store filling to the surface membrane Ca2+-permeable channel remains elusive. Arguments in favour of or against a mechanism based on diffusible signalling messengers or protein-protein interactions between release channel and surface membrane channel are abundant (e.g. Berridge, 1996; Csutora et al. 1999; Patterson et al. 1999; Yao et al. 1999). Other questions relate to the issue of whether CCE is activated in an all-or-none fashion or is graded with the degree of store depletion. Several previous reports have provided evidence that the relationship between store depletion and activation of ICRAC is highly non-linear (Petersen & Berridge, 1994; Mathes & Thompson, 1995; Zweifach & Lewis, 1996; Hofer et al. 1998a; Huang & Putney, 1998; Parekh et al. 1997). Moreover, under physiological conditions additional signalling pathways other than the extent of store filling or depletion might be involved in regulation or modulation of CCE. Results are also contradictory as to whether CCE requires a certain threshold level of store depletion for activation (Parekh et al. 1997; Huang & Putney, 1998), or whether activation can occur even after only marginal drops in store content (Hofer et al. 1998a). In addition, the mechanism which turns off CCE (or ICRAC) has received little consideration and is largely unknown.
It has been shown previously that in vascular endothelial cells CCE represents a major, possible the sole, Ca2+ entry pathway (Madge et al. 1997; Klishin et al. 1998). Moreover, in this cell type CCE is controlled by a subcellularly restricted mechanism (Hüser et al. 1999), i.e. local store depletion results in local activation of CCE. On a slower time scale, however, spatial heterogeneity of store Ca2+ content is effectively counteracted by intraluminal redistribution of Ca2+. In this study we aimed to determine the quantitative relationship between degree of store depletion and activity of CCE in cultured bovine vascular endothelial cells (CPAE cell line), the requirement of a minimum level of store depletion for activation of CCE, and the time course of CCE deactivation. Our results indicate that in the vascular endothelium CCE is graded over a large range with the level of store filling and becomes fully activated before store depletion is complete. Slow depletion of stores revealed that activation of CCE lags behind the rise of [Ca2+]i. Furthermore, deactivation of CCE occurs on a much slower time scale than activation. The time interval during which CCE remains activated after termination of Ca2+ release and return of [Ca2+]i to basal levels correlates with the degree of depletion and directly reflects the time required for the refilling process to replenish intracellular Ca2+ stores.
An initial account of this work has been presented in abstract form (Klishin et al. 1999).
METHODS
Cell culture and solutions
Experiments were performed on calf pulmonary artery endothelial (CPAE) cells in non-confluent cultures. The CPAE cell line was obtained from American Type Culture Collection. The cells were cultured in Eagle's minimum essential medium, supplemented with 20 % fetal bovine serum (Gibco) and L-glutamine (2 mM), and kept at 37°C in an atmosphere of 5 % CO2 and 95 % air. Once per week the cells were dispersed using a Ca2+-free (0.1 % EDTA) 0.25 % trypsin solution, and subcultured onto glass coverslips for later experimentation. Cells from passages 1–6 were used for experimentation. The cells were superfused continuously with a physiological salt solution (standard Tyrode solution), containing (mM): 135 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose and 10 Hepes, titrated to pH 7.3 with NaOH. In nominally Ca2+-free solution, CaCl2 was omitted. In some experiments Ca2+-free solution contained 1 mM EGTA, as indicated. All experiments were carried out at room temperature (20-22°C).
Fluorescence measurements
Spatially averaged [Ca2+]i measurements from single endothelial cells were performed using the fluorescent ratiometric Ca2+ indicator fura-2. Cultured endothelial cells were loaded with fura-2 by exposure to 1 ml standard Tyrode solution containing 5 μM fura-2 acetoxymethyl ester (fura-2 AM; Molecular Probes, Inc.) and 5 μl of 20 % (w/w) Pluronic F-127 (Molecular Probes; solubilized in DMSO) for 20 min at room temperature. Fura-2 fluorescence was excited by alternately illuminating the cells at 360 nm (F360) and 380 nm (F380). Emitted cellular fluorescence was recorded at 510 nm and changes in [Ca2+]i are expressed as changes of the ratio R =F360/F380.
The rate of manganese entry ([Mn2+]o = 100 μM) was inferred from the rate of quenching of fura-2 fluorescence excited at the Ca2+-insensitive (isosbestic) wavelength of 360 nm (F360). Small cell-to-cell differences for the fura-2 isosbestic excitation wavelength for Ca2+ were observed. We used a linear combination of F360 and 5–10 % of F380 to compose a truly Ca2+-insensitive fura-2 signal. This was verified by applying a brief ATP pulse to elicit a [Ca2+]i transient at the beginning of the experiment. After correction, F360 remained unaffected by the rise of [Ca2+]i. Control experiments showed that fura-2 fluorescence declined exponentially during prolonged constant influx of Mn2+. Therefore, the Mn2+-sensitive signal (F360) is presented on a logarithmic scale. Furthermore, despite maintained Mn2+ entry, cellular fura-2 quenching by Mn2+ remained incomplete, presumably due to compartmentalization of fura-2 into subcellular organelles that were inaccessible to Mn2+. This Mn2+-insensitive plateau level of fura-2 fluorescence at 360 nm was measured at the end of each experiment and subtracted from original signal. Fluorescence signals were low-pass filtered at 1 kHz. Excitation wavelengths were changed between 360 nm and 380 nm in less than 350 ms and a fura-2 ratio was calculated at a frequency of 0.5 Hz.
Statistical analysis
Where appropriate results are reported as means ±s.e.m. for the indicated number (n) of cells. Statistical significance was determined with the Student's t test.
RESULTS
CCE is graded with degree of store depletion
We used Mn2+ as a substitute for Ca2+ to characterize unidirectional ion flux through the store-operated Ca2+-permeable pathway. The rate of ion transport through this pathway was characterized by the rate of quenching of fura-2 fluorescence measured at a Ca2+-independent excitation wavelength (360 nm). Capacitative Ca2+ entry was activated by depletion of intracellular IP3-dependent Ca2+ stores.
Stimulation of single endothelial cells with increasing but submaximal concentrations of ATP (0.2-0.6 μM) caused spike-like transient elevations of [Ca2+]i (Fig. 1A). As shown previously (Holda & Blatter, 1997; Hüser & Blatter, 1997; Holda et al. 1998; Hüser et al. 1999) these ATP-induced [Ca2+]i transients were the result of quantal Ca2+ release from IP3-dependent intracellular Ca2+ stores. Since these experiments were performed in the absence of extracellular Ca2+, the changes of [Ca2+]i were solely due to intracellular Ca2+ release. Receptor stimulation with increasing agonist concentrations resulted in Ca2+ spikes which were paralleled by an increase in the rate of Mn2+ entry ([Mn2+]o = 100 μM).
Figure 1. ATP-induced intracellular Ca2+ release and capacitative Ca2+ entry.
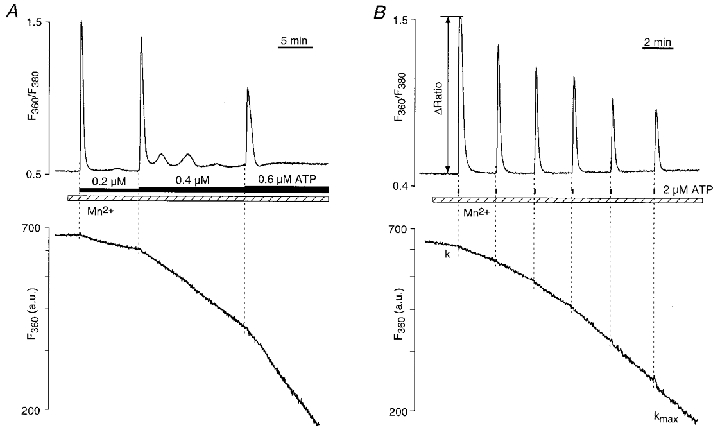
A, changes in [Ca2+]i (top) and rates of Mn2+ entry (bottom) caused by exposure to low concentrations of ATP (0.2-0.6 μM; n = 3). Due to the exponential decay of the fluorescence signal during constant influx of Mn2+ into the cell, the Mn2+-sensitive signal (F360; a.u., arbitrary units) is presented on a logarithmic scale. Ca2+ release triggered by ATP stimulation caused dose-dependent, but constant, rates of Mn2+ entry. B, train of Ca2+ spikes elicited with brief stimulations (5 s) with ATP (2 μM). Each Ca2+ spike was followed by an increased but constant rate (k) of Mn2+ entry. After the 6th Ca2+ spike the rate of Mn2+ entry became maximal (kmax). Experiments in A and B were done in Ca2+-free conditions. [Mn2+]o = 100 μM.
A similar graded activation of CCE was observed during a train of brief (5 s) stimulations with ATP (2 μM) performed in the absence of extracellular Ca2+. As shown in Fig. 1B, each ATP-induced Ca2+ spike resulted in an acceleration of Mn2+ entry, while the amplitude of the [Ca2+]i transients progressively decreased, both a result of a gradual depletion of intracellular Ca2+ stores. To establish a quantitative relationship between store depletion and activation of CCE we measured the rate of Mn2+ entry (k) after each ATP pulse and normalized it to the maximal rate of Mn2+ entry (kmax). With this approach the term k/kmax served as a measure for the degree of activation of CCE caused by loss of Ca2+ from stores. The amplitude of the ATP-induced [Ca2+]i transient (ΔRatio) was taken as a measure of store loading, i.e. a small ΔRatio value indicates a low degree of store loading at the time of ATP stimulation. Figure 2 summarizes the results from nine experiments obtained with ATP concentrations ranging from 1 to 25 μM. For all ATP concentrations tested the graph shows that Mn2+ entry gradually increased with store depletion. The rate of Mn2+ entry (k/kmax), however, became maximal before ΔRatio reached zero, indicating that maximal activation of CCE occurred before complete depletion of Ca2+ stores.
Figure 2. Relationship between Mn2+ entry rate and store depletion by ATP-induced Ca2+ release.
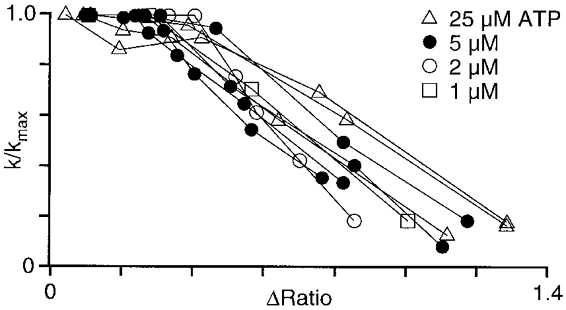
Summary of results obtained from experiments illustrated in Fig. 1B. Gradual store depletion was produced by brief exposure (3-5 s) to 1–25 μM ATP. ΔRatio, amplitude of fura-2 fluorescence ratio of the ATP-induced [Ca2+]i transient (measure for degree of store Ca2+ content); k/kmax, normalized rate of Mn2+ entry (measure for degree of CCE activation).
The results shown in Figs 1 and 2 suggested the following key features of CCE in CPAE cells. The rate of Mn2+ entry (as a measure of degree of CCE activation) appeared to be graded with the level of store depletion. For a given degree of store depletion, however, the rate of Mn2+ entry was constant. Furthermore, a partial depletion of the releasable Ca2+ stores was sufficient to fully activate CCE.
CCE during [Ca2+]i oscillations
Prolonged stimulation with low concentrations of ATP (0.2 μM) in the presence of extracellular Ca2+ (2 mM) often caused oscillatory changes of [Ca2+]i in CPAE cells (Fig. 3A). In contrast, oscillations were damped or absent in Ca2+-free solution (Fig. 1A). Therefore, extracellular Ca2+ was essential to maintain stable cytosolic Ca2+ oscillations in endothelial cells. As shown in Fig. 3A, each Ca2+ spike led to further acceleration of Mn2+ entry as a result of a gradual depletion of intracellular stores. Again, the acceleration of CCE was quantified by normalizing the rate of Mn2+ entry observed after individual Ca2+ spikes (k1…5) to the maximal entry rate (kmax). In the example shown (Fig. 3A), k/kmax increased from 0.12 after the first spike (k1/kmax) to 0.90 after the fifth spike (k5/kmax). Although the rate of Mn2+ entry (and presumably the degree of activation of CCE) accelerated from spike to spike, during the interval between two individual Ca2+ spikes the rate of Mn2+ entry remained constant. During [Ca2+]i oscillations of frequencies of 0.5 spikes min−1 and higher, CCE did not oscillate. The step-like increase in the rate of CCE that coincided with each Ca2+ spike further supported the hypothesis that activation of CCE is graded with the degree of store depletion.
Figure 3. Rates of CCE during [Ca2+]i oscillations.
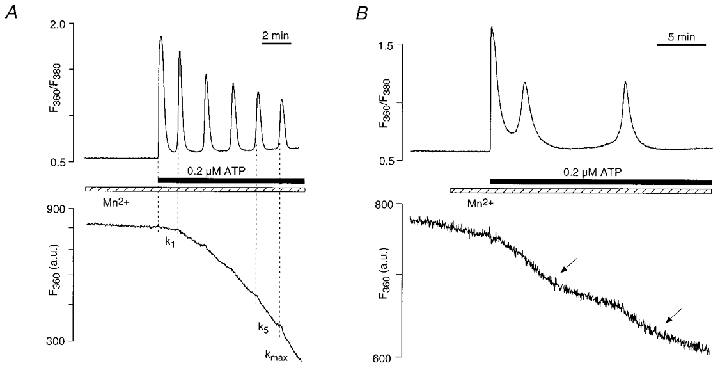
A, [Ca2+]i oscillations and rate of Mn2+ entry elicited by 0.2 μM ATP. A step-like increase in the rate of Mn2+ entry (i.e. CCE activation) was observed after each individual Ca2+ spike. k1, rate of Mn2+ entry after the first Ca2+ spike; k5, rate of Mn2+ entry after the fifth Ca2+ spike; kmax, maximal rate of Mn2+ entry. k1/kmax = 0.12; k5/kmax = 0.90. [Mn2+]o = 100 μM; [Ca2+]o = 2 mM; n = 3. B, periodic changes in the rate of CCE observed during low frequency [Ca2+]i oscillations, elicited by 0.2 μM ATP in the presence of 2 mM extracellular Ca2+. The increase in the rate of CCE was synchronized with Ca2+ spiking whereas the subsequent decrease in CCE activity (arrows) occurred during periods of low and constant [Ca2+]i. [Mn2+]o = 100 μM; n = 2.
As shown in Figs 1 and 3A the rate of Mn2+ entry that followed a given level of store depletion appeared to be rather constant, at least over the time periods observed. Figure 3B shows that during ATP-induced [Ca2+]i oscillations of low frequency, however, Mn2+ entry rates can exhibit oscillatory behaviour, indicating that CCE was activated during the cytosolic Ca2+ spike and deactivated during the ‘inter-spike’ interval. In this particular experiment, exposure to 0.2 μM ATP (in the presence of 2 mM extracellular Ca2+) initially led to two Ca2+ spikes, each followed by an increase in the rate of Mn2+ entry. The second Ca2+ spike was followed by a period of quiescence during which [Ca2+]i recovered completely. During the same period the rate of Mn2+ entry decreased, presumably due to a reduction of CCE as a result of refilling of the Ca2+ stores.
Deactivation kinetics of CCE
We investigated the deactivation kinetics of CCE by comparing the time required for Mn2+ entry to return to the initial rate after different degrees of store depletion. For this purpose cells were stimulated for 200 s with 0.2 and 5 μM ATP in the presence of 2 mM extracellular Ca2+ (Fig. 4). In both cases, [Ca2+]i rapidly decreased after removal of the agonist from the superfusion solution; however, Mn2+ entry continued at a higher rate for some time, indicating that CCE was not immediately turned off after Ca2+ release was terminated. After partial depletion of intracellular Ca2+ stores (0.2 μM ATP; Fig. 4A), the time interval required for the rate of Mn2+ entry (k) to decrease to the value encountered before stimulation with ATP was on average 164 ± 25 s (n = 8). In contrast, after stimulation with a maximal ATP concentration (5 μm) the time interval necessary for complete deactivation of CCE was significantly longer 466 ± 23 s (n = 7; P≤ 0.0001). This observation suggested that refilling of stores is a time consuming process that occurred on a time scale of minutes, and that the duration of CCE activation correlates with the degree of store depletion and the time that is required to refill depleted stores.
Figure 4. Deactivation of CCE after stimulation with low (A) and high (B) concentrations of ATP for 200 s.
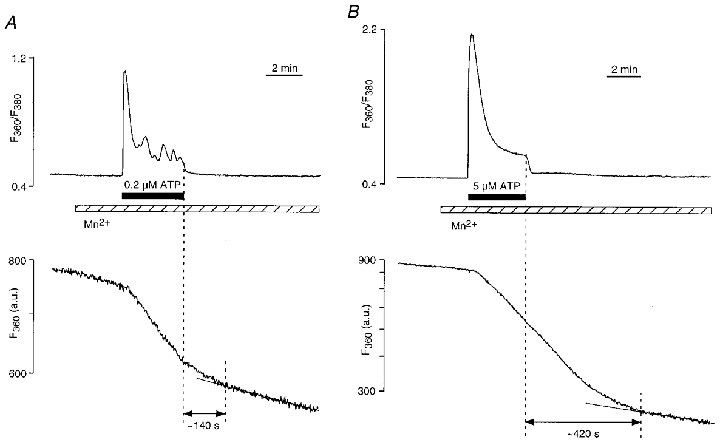
A, partial depletion of intracellular Ca2+ stores with 0.2 μM ATP. After agonist removal, depletion-induced Mn2+ entry continued for ≈140 s to refill the stores. B, complete Ca2+ store depletion with 5 μM ATP resulted in a much longer deactivation time for CCE (≈420 s). [Mn2+]o = 100 μM; [Ca2+]o = 2 mM.
Temporal dissociation between intracellular Ca2+ release and activation of CCE
The increase of [Ca2+]i in CPAE cells elicited by stimulation with ATP is typically rather rapid, even at submaximal agonist concentrations (see e.g. Figs 1, 3 and 4). Therefore, with experimental protocols which used agonist-induced stimulation to elicit store depletion, we were not able to resolve a possible temporal dissociation between a rise of [Ca2+]i and an increase in the rate of Mn2+ entry. To achieve a much more gradual depletion of Ca2+ stores we employed a different strategy. In the absence of extracellular Ca2+ we exposed CPAE cells to cyclopiazonic acid (CPA, 10 μM), an inhibitor of the endoplasmic reticulum Ca2+-ATPase. Inhibition of the ER ATPase results in store depletion through spontaneous Ca2+ leakage from the stores and removal of Ca2+ from the cytoplasm across the surface membrane. As shown in Fig. 5A, treatment with CPA led to a relatively slow increase in [Ca2+]i. The simultaneous measurement of Mn2+ entry revealed that its rate remained unchanged for more than a minute despite a rise of [Ca2+]i resulting from loss of Ca2+ from intracellular stores. In the experiment shown, the rate of Mn2+ entry started to increase approximately 2 min after the CPA-induced rise of [Ca2+]i (arrow in the bottom panel), presumably as a consequence of a now sufficient depletion of Ca2+ stores. Stimulation with a brief pulse of a maximal ATP concentration (5 μM) elicited further Ca2+ release from the partially depleted stores and an acceleration of the Mn2+ entry rate. Subsequent stimulations with ATP triggered a small additional Ca2+ release with no consequences for the Mn2+ entry rate, indicating that CCE was fully activated at this point. The average time interval between the onset of CPA-induced rise of [Ca2+]i and the activation of CCE was 178 ± 36 s (n = 10 cells). This delay was significantly different from zero (P≤ 0.0005), and suggested that CCE activation possibly required a threshold level of store depletion.
Figure 5. Temporal dissociation between Ca2+ store depletion and CCE activation.
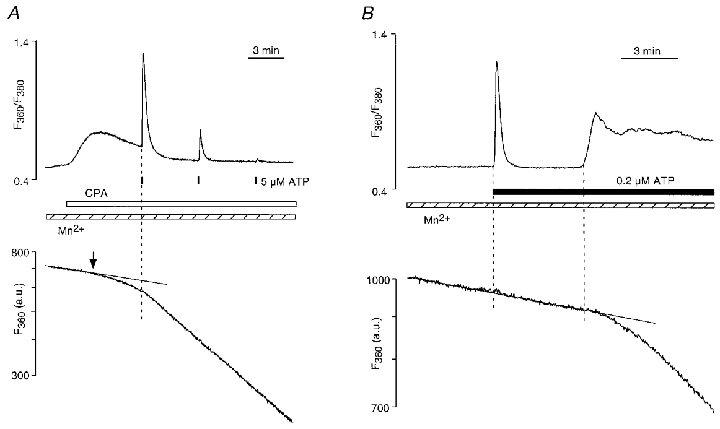
A, cyclopiazonic acid (CPA, 10 μM), an inhibitor of the endoplasmic reticulum Ca2+ pump, caused a slow and very gradual depletion of Ca2+ stores. A detectable change in Mn2+ entry occurred with a delay of ≈2 min (arrow) after the onset of CPA-induced increase of [Ca2+]i. Stimulation with ATP (5 μM) during the CPA-induced elevation of [Ca2+]i elicited a [Ca2+]i transient and subsequently an acceleration of Mn2+ entry. Additional ATP-induced Ca2+ release failed to accelerate Mn2+ entry further. [Mn2+]o = 100 μM; [Ca2+]o = 0. B, delayed activation of Mn2+ entry observed in a cell stimulated with low (0.2 μM) ATP concentration.
Occasionally, similar observations of a temporal dissociation between the onset of SR Ca2+ release and activation of CCE were also made in cells stimulated with ATP (4 cells out of 18 tested) (Fig. 5B). In the experiment shown, stimulation with 0.2 μM ATP resulted initially in a Ca2+ spike which was not followed immediately by an increase in the rate of Mn2+ entry, suggesting that the amount of released Ca2+ failed to deplete the stores sufficiently to activate CCE. Subsequently, however, the maintained presence of ATP caused a prolonged elevation of [Ca2+]i which was paralleled by a change in the rate of Mn2+ entry, indicative of eventual activation of CCE.
Quantitative relationship between store depletion and CCE activity
The quantitative relationship between level of store depletion and degree of CCE activation was further investigated in the following set of experiments. In this series of experiments CPAE cells were kept in Ca2+-free conditions (1 mM EGTA) for various periods of time (0-140 min). As we have shown previously, incubation of CPAE cells in Ca2+-free solution led to a very gradual depletion of intracellular Ca2+ stores (Hüser et al. 1999). As with the approach illustrated by Figs 1 and 2, we measured the initial rate of Mn2+ entry (k) and normalized it to the maximal rate of Mn2+ entry (kmax) observed after application of a supramaximal concentration of ATP (5 μM) to achieve complete depletion of the stores (Fig. 6A). With this approach the term k/kmax served as a measure for the degree of activation of CCE and ΔRatio as an estimate for store loading. Figure A shows an example where incubation in Ca2+-free media (10 min) resulted in a moderate degree of store depletion (ΔRatio = 0.65). The initial rate of Mn2+ entry (k) was different from zero, indicating that partial depletion of the Ca2+ stores had already activated CCE to some extent. Subsequent stimulation with 5 μM ATP resulted in a substantial acceleration of Mn2+ entry and full activation of CCE. In contrast, in the experiment shown in Fig. 6B prolonged incubation in a Ca2+-free environment (140 min) resulted in substantial store depletion, as indicated by the much smaller amplitude of the ATP-induced [Ca2+]i transient (ΔRatio = 0.22). Furthermore, the initial rate of Mn2+ entry (k) was clearly higher in this experiment compared to the example in A, indicating that prolonged incubation resulted in a higher degree of CCE activation. Subsequent stimulation with ATP resulted in only a marginal acceleration of Mn2+ entry, suggesting that the degree of store depletion at the time of ATP stimulation was sufficient to almost fully activate CCE (with k/kmax approaching 1). Correlation of k/kmax and ΔRatio revealed (Fig. 6C) that over a large range of store filling (ΔRatio < ∼1.0) the rate of Mn2+ entry (k/kmax), and therefore the activity of CCE, was graded with the degree of store depletion. Furthermore, the graph shows that at higher levels of store loading (ΔRatio > ∼1.0) k/kmax was fairly constant. This suggests that under these conditions the stores did not become sufficiently depleted to activate CCE despite the fact that the amplitude of the ATP-induced [Ca2+]i transient was reduced. Taking ΔRatio as an indicator for store Ca2+ content, a likely interpretation of these experiments is that a certain level of store depletion could occur before a measurable change of k/kmax, and therefore activation of CCE, became observable.
Figure 6. CCE is graded with the degree of Ca2+ store depletion.
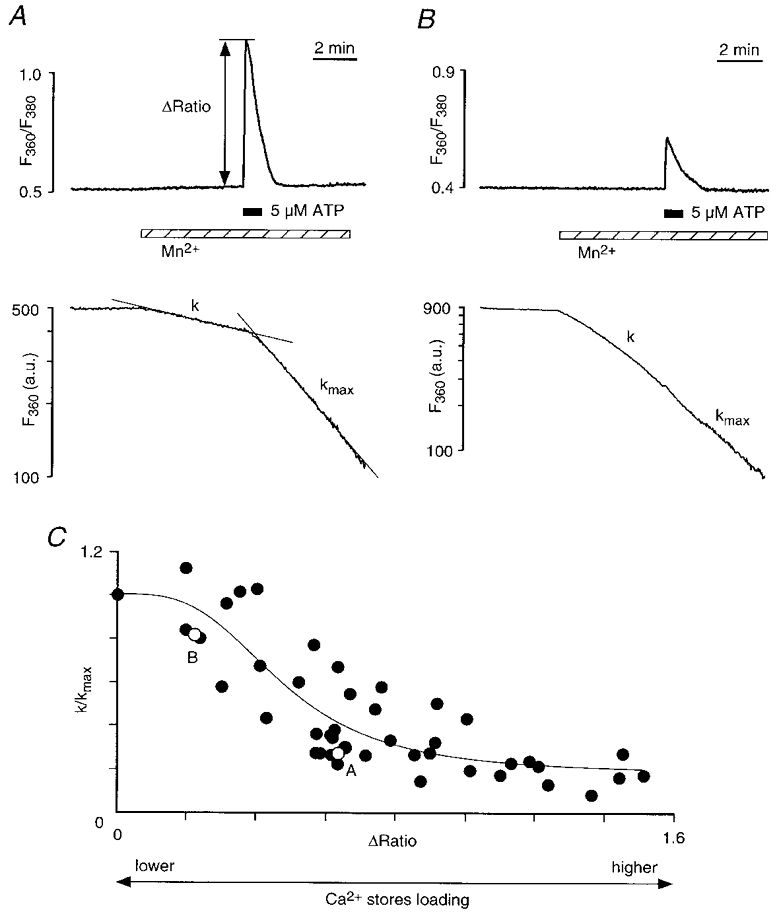
Various degrees of Ca2+ store depletion were achieved by preincubation of endothelial cells in Ca2+-free (1 mM EGTA) Tyrode solution for variable periods of times (0-140 min). A, the initial rate of Mn2+ entry (k) was normalized to the maximal rate of Mn2+ entry (kmax) after application of a supramaximal concentration of ATP (5 μM) to achieve complete depletion of the stores. Example of moderate store depletion (incubation time 10 min; ΔRatio = 0.65; k/kmax = 0.30). [Mn2+]o = 100 μM. B, same protocol applied to a cell with substantially higher degree of store depletion (incubation time 140 min; ΔRatio = 0.22; k/kmax = 0.82). C, plot of k/kmax against the amplitude of the [Ca2+]i transient (ΔRatio of fura-2 fluorescence). The amplitude of the [Ca2+]i transient induced by exposure to supramaximal concentrations of agonist was taken as a measure of the degree of Ca2+ store loading. Each point represents spatially averaged signals from a group of 1–10 adjacent cells (204 cells totally). The data were fitted with the function k/kmax = (1 –a)(1 –ΔRation/(ΔRation+bn)) +a. The best fit was obtained with a = 0.18, b = 0.47 and n = 3.2. The open symbols indicate the examples shown in A and B.
DISCUSSION
Relationship between store content and activity of capacitative Ca2+entry
Endothelial cells typically do not express voltage-gated Ca2+ channels (e.g. Adams, 1994) and therefore have to rely on alternative Ca2+ entry pathways to maintain Ca2+ homeostasis. It has become increasingly clear that store-operated or capacitative Ca2+ entry, where Ca2+ influx is controlled specifically by the filling status of intracellular Ca2+ stores, represents the major avenue of Ca2+ entry in this cell type. Despite the substantial interest CCE has attracted in recent years, many aspects of its regulation have remained elusive. Among those the mechanism of activation of CCE and the relationship between its activity and degree of store depletion have remained rather puzzling and poorly understood issues.
Several reports have provided evidence that the relationship between store content and Ca2+ release-activated membrane current (ICRAC) is non-linear and has complex kinetics (Petersen & Berridge 1994; Mathes & Thompson, 1995; Zweifach & Lewis, 1996; Parekh et al. 1997; Hofer et al. 1998a; Huang & Putney 1998). Parekh et al. (1997) for example showed that IP3-induced activation of ICRAC had all-or-none characteristics in rat basophilic leukaemia cells (RBL-1 cells). Similar conclusions were reached by Huang & Putney (1998) for the same cell type, although in both these studies conditions were identified where ICRAC could be graded with store depletion. Whereas in these two studies a graded response was found to be more of an exception rather than a general feature of store-operated Ca2+ entry, these results clearly contrast with the findings by Hofer et al. (1998a) in RBL-1 cells and hamster kidney BHK-21 fibroblasts. In this particular study it was found that the magnitude of Ca2+ entry correlated closely with the extent of store depletion. Graded Ca2+ influx via ICRAC, despite its inherent all-or-none characteristics, could be the result of additional regulatory mechanisms that modulate CCE. For example, activation of protein kinase C could affect CCE activity and thereby mimic an overall graded response of CCE to store depletion (Petersen & Berridge, 1994; Parekh et al. 1997). Furthermore, changes in membrane potential and therefore the electrical driving force for Ca2+ entry could result in apparently graded Ca2+ influx (Parekh et al. 1997). It was suggested (Parekh et al. 1997; Huang & Putney, 1998) that multiple sets of intracellular Ca2+ stores with different sensitivities to Ca2+-depleting agents (IP3, thapsigargin, ionomycin) and different depletion kinetics could explain the complex relationship between store depletion and ICRAC activation. In these models a specialized subfraction of the endoplasmic reticulum would be responsible for the control of CCE or ICRAC activity.
In our study we took advantage of the fact that Mn2+ can substitute for Ca2+ ions as charge carrier through the CRAC channel (Merritt et al. 1989; Fasolato et al. 1993; Parekh & Penner, 1997). The degree of quenching of fura-2 fluorescence at a Ca2+-insensitive excitation wavelength provides an elegant method to measure changes of cytoplasmic [Mn2+] arising from unidirectional flux of Mn2+ through the CCE pathway in endothelial cells (Klishin et al. 1998). By exciting fura-2 fluorescence at a Ca2+-sensitive wavelength (380 nm) as well, the method is able to measure [Ca2+]i simultaneously with Mn2+ entry. Using this technique we have provided evidence that in pulmonary artery endothelial cells CCE is graded with the level of store filling over a wide range of depletion (Figs 1, 2, 3 and 6). Furthermore, a small degree of depletion was not sufficient to activate CCE (Figs 5 and 6). A possible interpretation of this observation is that activation of CCE might be a threshold-dependent phenomenon which requires a certain degree of Ca2+ depletion before it occurs. In RBL-1 cells for example, the observations of a threshold level of depletion required for activation of ICRAC are contradictory and inconclusive. Hofer et al. (1998a) observed store-operated Ca2+ influx following very small drops in store [Ca2+], suggesting the absence of a significant threshold level of depletion necessary for activation of ICRAC. Huang & Putney (1998) found a delay between store depletion by IP3 or thapsigargin and activation of ICRAC, but not after depletion with the Ca2+ ionophore ionomycin. The data of Parekh et al. (1997), also obtained in RBL-1 cells, suggest a threshold-dependent, all-or-none process, because no activation of ICRAC was observed at IP3 concentrations which typically led to a significant release of Ca2+ from intracellular stores. Unfortunately, in this study ICRAC activation was compared with data of IP3-dependent Ca2+ release obtained in a different laboratory (Meyer & Stryer, 1990) and therefore its conclusions may be somewhat limited.
Although the present and previous studies reveal substantial discrepancies regarding the quantitative relationship between store depletion and CCE activity as well as the requirement of a threshold level of depletion for activation of CCE, they largely agree on the fact that store depletion is not required to be maximal to fully activate Ca2+ entry. In our study maximal activation of CCE was seen under conditions where subsequent stimulation with agonist could enable further release of Ca2+ (Figs 1B, 5A and 6B), indicating that the stores were not completely emptied. This is consistent with the findings by Huang & Putney (1998) and Hofer et al. (1998a) that ICRAC could be fully activated in RBL-1 cells with only partially depleted Ca2+ stores. In mouse neuroblastoma (N1E 115) cells also, a store depletion-dependent Ca2+ current was observed to be maximally activated at times when the content of releasable Ca2+ pools was reduced to only 39 % (Mathes & Thompson, 1995). Full activation of CCE before complete depletion of intracellular Ca2+ stores can be interpreted as a safety mechanism that prevents potentially disastrous consequences for a multitude of Ca2+-dependent cellular functions that would arise from exhaustion of intracellular Ca2+ reserves. Replenishing of the Ca2+ reservoirs long before Ca2+ stores run empty prevents breakdown of such Ca2+-dependent processes.
Kinetics of CCE during [Ca2+]i oscillations
Several studies have shown clearly that agonist-induced [Ca2+]i oscillations not only depend on cyclical release and uptake of Ca2+ from and into intracellular stores, but also rely on Ca2+ influx (for references see e.g. Berridge, 1995; Parekh & Penner, 1997). Because the periodic release of Ca2+ during [Ca2+]i oscillations at least transiently reduces Ca2+ store content and regular [Ca2+]i oscillations require extracellular Ca2+, it seems conceivable that CCE is directly involved in [Ca2+]i oscillations. There is, however, also evidence that receptor-activated [Ca2+]i oscillations might be associated with enhanced Ca2+ entry trough a store-independent pathway (Shuttleworth & Thompson, 1998). If CCE participates in [Ca2+]i oscillations, it would raise the question of whether the rate of Ca2+ entry through the CCE pathway is oscillatory, either in or out of phase with oscillations of [Ca2+]i. In this regard available data are conflicting. In an earlier study on vascular endothelial cells (Jacob, 1990) no evidence was found for oscillatory Ca2+ entry. In contrast, oscillatory behaviour of Ca2+ entry was shown in pancreatic acinar cells (Loessberg et al. 1991), lymphocytes (Dolmetsch & Lewis, 1994) and kidney-derived MDCK-F cells (Wojnowski et al. 1994). In these studies [Ca2+]i oscillations required periodic activation of depletion-dependent Ca2+ entry as a result of periodic Ca2+ release and fluctuations of store content. In contrast, there is also evidence that capacitative Ca2+ entry itself can oscillate in cells with impaired Ca2+ stores, producing oscillatory changes of [Ca2+]i. This has been observed in lymphocytes (Lewis & Cahalan, 1989), parotid acinar cells (Foskett & Wong, 1994) and HeLa cells (Missiaen et al. 1994). A likely mediator for the periodic inhibition of CCE during [Ca2+]i oscillations could be Ca2+ itself, since it has been shown that ICRAC is inactivated by Ca2+ (Hoth & Penner, 1992). The feedback inhibition of CCE by intracellular Ca2+ has been suggested to underlie oscillations of Ca2+ entry in the absence of intracellular Ca2+ release (Hoth & Penner, 1992; Missiaen et al. 1994). Our results from vascular endothelial cells suggest the involvement of CCE in [Ca2+]i oscillations. Figure 3A shows that the rate of Mn2+ entry increased with each Ca2+ spike, until after a few spikes it became maximal. These results strongly suggest that the oscillations led to a progressive loss of store Ca2+ and the subsequent activation of CCE. Furthermore, it appears that in CPAE cells oscillations of Ca2+ entry observed during low frequency [Ca2+]i oscillations (Fig. 3B) are not the result of a negative feedback mechanism involving a Ca2+-dependent inhibition of CCE. The observation, that the rate of Mn2+ entry decreased at a point in time when [Ca2+]i had reached basal levels (Fig. 3B), suggests that refilling of the stores is the critical factor that leads to deactivation of CCE and therefore determines periodic changes in CCE activity during [Ca2+]i oscillations.
Deactivation of capacitative Ca2+ entry
The mechanism that switches off store depletion-dependent Ca2+ influx is largely unknown. In our experiments, following agonist-induced release of Ca2+ from the ER, CCE was activated rapidly and deactivated slowly (Fig. 4). In fact, at the temporal resolution of our measurements of Mn2+ entry, no significant delay between agonist-induced rise in [Ca2+]i and change in the rate of Mn2+ entry was observed (e.g. Fig. 1). This suggests, that following stimulation of Ca2+ release, a sufficient degree of store depletion occurred rapidly, leading to a rapid activation of CCE. In contrast, depending on the degree of store depletion, CCE remained activated for minutes after termination of release and even after cytosolic Ca2+ had declined to basal levels (Fig. 4). This slow turn off of CCE, compared to its activation, has also been observed in RBL-1 cells (Hofer et al. 1998a). In our study we conclude from the slow time course of CCE deactivation and the correlation between degree of depletion and the time required for CCE deactivation (Fig. 4), that refilling of the Ca2+ stores itself was the most time consuming process (an observation previously made in vascular smooth muscle cells; see Blatter, 1995) and the limiting step for the termination of CCE in our experiments. Under the premise that refilling of depleted Ca2+ stores constitutes the time limiting step for successive release of Ca2+ from stores, it is possible that CCE not only participates in [Ca2+]i oscillations by providing a source for [Ca2+]i elevations, but also determines their temporal pattern and frequency.
The slow deactivation of CCE also represents an apparent contradiction to the observation that ICRAC is rapidly inactivated by Ca2+ itself (Hoth & Penner, 1992). In the present study, as well as in many other studies, it has been shown that store-operated Ca2+ entry can be maintained over prolonged periods of times despite substantial elevations of [Ca2+]i. It has been suggested (for review see Barritt, 1998) that the [Ca2+] in the restricted subplasmalemmal space, rather than bulk cytoplasmic [Ca2+], where peripheral ER elements and the plasma membrane are found in close vicinity, is crucial for the regulation of CCE. In the proposed model (Barritt, 1998) distinct structural arrangements of ER Ca2+ pumps and Ca2+ channels could maintain low levels of [Ca2+] in subplasmalemmal microdomains and therefore keep CCE activated. There is indeed increasing evidence that important Ca2+ regulatory mechanisms are organized in such microdomains. It has been shown for example that in endothelial cells vectorial Ca2+ release and Ca2+ entry into the subplasmalemmal space, as well as local cytoplasmic Ca2+ buffering capacities, can profoundly affect Ca2+ homeostasis and Ca2+ signalling (e.g. Cabello & Schilling, 1993; Graier et al. 1998). The importance of cytoplasmic Ca2+ buffering capacity for the refilling process has also been demonstrated in other non-excitable cells (Hofer et al. 1998b). Furthermore, we have demonstrated previously that CCE is controlled by a subcellularly restricted mechanism (Hüser et al. 1999). This observation further emphasizes the importance of subplasmalemmal microdomains or subplasmalemmal Ca2+ control units (Graier et al. 1998) for cellular [Ca2+]i regulation, including the control of CCE.
Acknowledgments
We thank Drs K. S. Ginsburg and S. L. Lipsius, as well as K. A. Sheehan, for critically reading the manuscript, and R. L. Gulling for expert technical help. This work was supported by the National Heart, Lung, and Blood Institute (HL-51941) and by grants from the American Heart Association National Center (L.A.B.) and Metropolitan Chicago (A.K.). L.A.B. is an Established Investigator of the American Heart Association.
References
- Adams DJ. Ionic channels in vascular endothelial cells. Trends Cardiovascular Medicine. 1994;4:18–26. doi: 10.1016/1050-1738(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Barritt GJ. Does a decrease in subplasmalemmal Ca2+ explain how store-operated Ca2+ channels are opened? Cell Calcium. 1998;23:65–75. doi: 10.1016/s0143-4160(98)90075-6. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–11. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Microdomains and elemental events in calcium signalling. Cell Calcium. 1996;20:95–96. doi: 10.1016/s0143-4160(96)90098-6. [DOI] [PubMed] [Google Scholar]
- Blatter LA. Depletion and filling of intracellular calcium stores in vascular smooth muscle cells. American Journal of Physiology. 1995;268:C503–512. doi: 10.1152/ajpcell.1995.268.2.C503. [DOI] [PubMed] [Google Scholar]
- Cabello OA, Schilling WP. Vectorial Ca2+ flux from the extracellular space to the endoplasmic reticulum via a restricted cytoplasmic compartment regulates inositol 1,4,5-trisphosphate-stimulated Ca2+ release from internal stores in vascular endothelial cells. Biochemical Journal. 1993;295:357–366. doi: 10.1042/bj2950357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Csutora P, Su Z, Kim HY, Bugrim A, Cunningham KW, Nuccitelli R, Keizer JE, Hanley MR, Blalock JE, Marchase RB. Calcium influx factor is synthesized by yeast and mammalian cells depleted of organellar calcium stores. Proceedings of the National Academy of Sciences of the USA. 1999;96:121–126. doi: 10.1073/pnas.96.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS. Signalling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. Journal of General Physiology. 1994;103:365–388. doi: 10.1085/jgp.103.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fasolato C, Hoth M, Penner R. Multiple mechanisms of manganese-induced quenching of fura-2 fluorescence in rat mast cells. Pflügers Archiv. 1993;423:225–231. doi: 10.1007/BF00374399. [DOI] [PubMed] [Google Scholar]
- Fasolato C, Nilius B. Store depletion triggers the calcium release-activated calcium current (ICRAC) in macrovascular endothelial cells: a comparison with Jurkat and embryonic kidney cell lines. Pflügers Archiv. 1998;436:69–74. doi: 10.1007/s004240050605. [DOI] [PubMed] [Google Scholar]
- Foskett JK, Wong DC. [Ca2+]i inhibition of Ca2+ release-activated Ca2+ influx underlies agonist- and thapsigargin-induced [Ca2+]i oscillations in salivary acinar cells. Journal of Biological Chemistry. 1994;269:31525–31532. [PubMed] [Google Scholar]
- Graier WF, Paltauf-Doburzynska J, Hill BJF, Fleischhacker E, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. The Journal of Physiology. 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cell Biology. 1998a;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Landolfi B, Debellis L, Pozzan T, Curci S. Free [Ca2+] dynamics measured in agonist-sensitive stores of single living intact cells: a new look at the refilling process. EMBO Journal. 1998b;17:1986–1995. doi: 10.1093/emboj/17.7.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holda JR, Blatter LA. Capacitative calcium entry is inhibited in vascular endothelial cells by disruption of cytoskeletal microfilaments. FEBS Letters. 1997;403:191–196. doi: 10.1016/s0014-5793(97)00051-3. [DOI] [PubMed] [Google Scholar]
- Holda JR, Klishin A, Sedova M, Hüser J, Blatter LA. Capacitative calcium entry. News in Physiological Sciences. 1998;13:157–163. doi: 10.1152/physiologyonline.1998.13.4.157. [DOI] [PubMed] [Google Scholar]
- Hoth M, Penner R. Depletion of intracellular calcium stores activates a calcium current in mast cells. Nature. 1992;355:353–355. doi: 10.1038/355353a0. [DOI] [PubMed] [Google Scholar]
- Huang Y, Putney JW. Relationship between intracellular calcium store depletion and calcium release-activated calcium current in a mast cell line (RBL-1) Journal of Biological Chemistry. 1998;273:19554–19559. doi: 10.1074/jbc.273.31.19554. [DOI] [PubMed] [Google Scholar]
- Hüser J, Blatter LA. Elementary events of agonist-induced Ca2+ release in vascular endothelial cells. American Journal of Physiology. 1997;273:C1775–1782. doi: 10.1152/ajpcell.1997.273.5.C1775. [DOI] [PubMed] [Google Scholar]
- Hüser J, Holda JR, Kockskämper J, Blatter LA. Focal agonist stimulation results in spatially restricted Ca2+ release and capacitative Ca2+ entry in vascular endothelial cells. The Journal of Physiology. 1999;514:101–109. doi: 10.1111/j.1469-7793.1999.101af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob R. Agonist-stimulated divalent cation entry into single cultured human umbilical vein endothelial cells. The Journal of Physiology. 1990;412:55–77. doi: 10.1113/jphysiol.1990.sp017933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klishin A, Sedova M, Blatter LA. Time-dependent modulation of capacitative Ca2+ entry signals by plasma membrane Ca2+ pump in endothelium. American Journal of Physiology. 1998;274:C1117–1128. doi: 10.1152/ajpcell.1998.274.4.C1117. [DOI] [PubMed] [Google Scholar]
- Klishin A, Sedova M, Hüser J, Blatter LA. Capacitative Ca2+ entry is graded with depletion of intracellular Ca2+ stores in vascular endothelial cells. Biophysical Journal. 1999;76:A225. doi: 10.1111/j.1469-7793.2000.t01-3-00549.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis RS, Cahalan MD. Mitogen-induced oscillations of cytosolic Ca2+ and transmembrane Ca2+ current in human leukemic T cells. Cell Regulation. 1989;1:99–112. doi: 10.1091/mbc.1.1.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loessberg PA, Zhao H, Muallem S. Synchronized oscillation of Ca2+ entry and Ca2+ release in agonist-stimulated AR42J cells. Journal of Biological Chemistry. 1991;266:1363–1366. [PubMed] [Google Scholar]
- Madge L, Marshall ICB, Taylor CW. Delayed autoregulation of the Ca2+ signals resulting from capacitative Ca2+ entry in bovine pulmonary artery endothelial cells. The Journal of Physiology. 1997;498:351–369. doi: 10.1113/jphysiol.1997.sp021863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathes C, Thompson SH. The relationship between depletion of intracellular Ca2+ stores and activation of Ca2+ current by muscarinic receptors in neuroblastoma cells. Journal of General Physiology. 1995;106:975–993. doi: 10.1085/jgp.106.5.975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merritt JE, Jacob R, Hallam TJ. Use of manganese to discriminate between calcium influx and mobilization from internal stores in stimulated human neutrophils. Journal of Biological Chemistry. 1989;264:1522–1527. [PubMed] [Google Scholar]
- Meyer T, Stryer L. Transient calcium release by successive increments of inositol 1,4,5,-triphosphate. Proceedings of the National Academy of Sciences of the USA. 1990;87:3841–3845. doi: 10.1073/pnas.87.10.3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missiaen L, van Smedt H, Pary JB, Oike M, Casteels R. Kinetics of empty store-activated Ca2+ influx in HeLa cells. Journal of Biological Chemistry. 1994;269:5817–5823. [PubMed] [Google Scholar]
- Parekh AB, Fleig A, Penner R. The store-operated calcium current ICRAC: nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–930. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Patterson RL, van Rossum DB, Gill DL. Store-operated Ca2+ entry: evidence for a secretion-like coupling model. Cell. 1999;98:487–499. doi: 10.1016/s0092-8674(00)81977-7. [DOI] [PubMed] [Google Scholar]
- Petersen CCH, Berridge MJ. The regulation of capacitative calcium entry by calcium and protein kinase C in Xenopus oocytes. Journal of Biological Chemistry. 1994;269:32246–32253. [PubMed] [Google Scholar]
- Putney JW. Capacitative Calcium Entry. Austin, TX, USA: R. G. Landes Company; 1997. [Google Scholar]
- Shuttleworth TJ, Thompson JL. Muscarinic receptor activation of arachidonate-mediated Ca2+ entry in HEK293 cells is independent of phospholipase C. Journal of Biological Chemistry. 1998;273:32636–32643. doi: 10.1074/jbc.273.49.32636. [DOI] [PubMed] [Google Scholar]
- Wang Y, Shin WS, Kawaguchi H, Inukai M, Kato M, Sakamoto A, Uehara Y, Miyamoto M, Shimamoto N, Korenaga R, Ando J, Toyo-oka T. Contribution of sustained Ca2+ elevation for nitric oxide production in endothelial cells and subsequent modulation of Ca2+ transient in vascular smooth muscle cells in coculture. Journal of Biological Chemistry. 1996;271:5647–5655. doi: 10.1074/jbc.271.10.5647. [DOI] [PubMed] [Google Scholar]
- Wojnowski L, Schwab A, Hoyland J, Mason WT, Silbernagl S, Oberleitner H. Cytoplasmic Ca2+ determines the rate of Ca2+ entry into Mardin-Darby canine kidney-focus (MDCK-F) cells. Pflügers Archiv. 1994;426:95–100. doi: 10.1007/BF00374676. [DOI] [PubMed] [Google Scholar]
- Xu X, Star RA, Tortorici G, Muallem S. Depletion of intracellular Ca2+ stores activates nitric-oxide synthase to generate cGMP and regulate Ca2+ influx. Journal of Biological Chemistry. 1994;269:12645–12653. [PubMed] [Google Scholar]
- Yao Y, Ferrer-Montiel AV, Montal M, Tsien RY. Activation of store-operated Ca2+ current in Xenopus oocytes requires SNAP-25 but not a diffusible messenger. Cell. 1999;98:475–485. doi: 10.1016/s0092-8674(00)81976-5. [DOI] [PubMed] [Google Scholar]
- Zweifach A, Lewis RS. Calcium-dependent potentiation of store-operated calcium channels in T lymphocytes. Journal of General Physiology. 1996;107:597–610. doi: 10.1085/jgp.107.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]