

Abstract
ClC-3 encodes a volume-regulated Cl− channel (ICl,vol) in heart. We studied the regulation of native and recombinant cardiac ICl,vol by intracellular cyclic AMP (cAMPi).
Symmetrical high Cl− concentrations were used to effectively separate outwardly rectifying ICl,vol from other non-rectifying Cl− currents, such as the cystic fibrosis transmembrane conductance regulator (CFTR) and Ca2+-activated Cl− currents (ICl,CFTR and ICl,Ca, respectively), which are concomitantly expressed in cardiac myocytes.
8-Bromo-cyclic AMP (8-Br-cAMP) significantly inhibited ICl,vol in most guinea-pig atrial myocytes. In ≈30 % of the atrial myocytes examined, 8-Br-cAMP increased macroscopic Cl− currents. However, the 8-Br-cAMP-stimulated difference currents exhibited a linear current-voltage (I–V) relation, consistent with activation of ICl,CFTR, not ICl,vol.
In canine atrial myocytes, isoprenaline (1 μM) consistently reduced ICl,vol in Ca2+-free hypotonic bath solutions with strong intracellular Ca2+ () buffering. In Ca2+-containing hypotonic bath solutions with weak buffering, however, isoprenaline increased net macroscopic Cl− currents. Isoprenaline-stimulated difference currents were not outwardly rectifying, consistent with activation of ICl,Ca, not ICl,vol.
In NIH/3T3 cells transfected with gpClC-3 (the gene encoding ICl,vol), 8-Br-cAMP consistently inhibited ICl,ClC-3. These effects were prevented by a protein kinase A (PKA) inhibitor, KT5720, or by mutation of a single consensus protein kinase C (PKC) phosphorylation site (S51A) on the N-terminus of ClC-3, which also mediates PKC inhibition of ICl,ClC-3.
We conclude that cAMPi causes inhibition of ICl,vol in mammalian heart due to cross-phosphorylation of the same PKC consensus site on ClC-3 by PKA. Our results suggest that contamination of macroscopic ICl,vol by ICl,CFTR and/or ICl,Ca may account for some of the inconsistent and controversial effects of cAMPi on ICl,vol previously reported in native cardiac myocytes.
A volume-regulated chloride current (ICl,vol) is widely expressed in cardiac and many other tissues of a variety of species (Sorota, 1992; Hagiwara et al. 1992; Tseng, 1992; Vandenberg et al. 1994; Oz & Sorota, 1995; Hall et al. 1995; Duan et al. 1995; Strange et al. 1996; Vandenberg et al. 1996; Okada, 1997; Duan et al. 1997a). Several unique properties of ICl,vol distinguish these channels from other types of Cl− channels: outward rectification with symmetrical high Cl− concentrations, time-dependent inactivation at positive potentials, higher permeability to I− than Cl−, and an intermediate single-channel conductance of 28–60 pS (Strange et al. 1996; Vandenberg et al. 1996; Okada, 1997). Recent evidence suggests that ClC-3, a member of the ClC Cl− channel family, may be responsible for native ICl,vol in many mammalian cells (Kawasaki et al. 1994; Duan et al. 1997b, 1999; Yamazaki et al. 1998; von Weikersthal et al. 1999). In heart, ICl,vol plays an important role in the regulation of cell volume homeostasis and electrical activity (Vandenberg et al. 1996; Hiraoka et al. 1998; Hume et al. 2000). ICl,vol function is known to be regulated by phosphorylation and dephosphorylation (Hall et al. 1995; Okada, 1997) and we recently described the role of protein kinase C (PKC) phosphorylation in the regulation of native ICl,vol in heart and expressed ClC-3 channels by changes in cell volume (Duan et al. 1999). The present study was designed to examine the regulation of native ICl,vol and expressed ClC-3 channels by intracellular cyclic AMP (cAMPi).
In many cardiac cells, elevation of cAMPi is known to activate a distinct class of Cl− channels through protein kinase A (PKA) phosphorylation (ICl,PKA) (Harvey & Hume, 1989; Bahinski et al. 1989; Gadsby et al. 1995; Hume et al. 2000). In contrast to ICl,vol, ICl,PKA exhibits a linear current-voltage relation in symmetrical high Cl−, and is generated by small (8-12 pS) unitary conductance channels encoded by the CFTR gene (Gadsby et al. 1995; Hart et al. 1996). Whether or not cardiac ICl,vol can also be regulated by cAMPi and cAMP-dependent PKA phosphorylation is controversial (Sorota, 1992; Hagiwara et al. 1992; Tseng, 1992; Oz & Sorota, 1995; Hall et al. 1995; Du & Sorota, 1997). In canine ventricular myocytes, ICl,vol has been reported to be insensitive to the PKA inhibitor H-89 (Tseng, 1992), and the stretch-induced Cl− current in rabbit heart has also been reported to be insensitive to a specific peptide inhibitor of PKA (Hagiwara et al. 1992). In contrast, in cultured chick cardiac myocytes, ICl,vol was inhibited by forskolin, and activation of ICl,vol during cell swelling was prevented when the pipette filling solution contained cAMP and isobutylmethylxanthine in the presence of okadaic acid (Hall et al. 1995). An increase in cAMPi levels by isoprenaline or forskolin was reported to stimulate ICl,vol in canine atrial myocytes (Sorota, 1992) and in human atrial and ventricular myocytes (Oz & Sorota, 1995), although these effects may be complex, with some cells responding with stimulation, inhibition, no response, or a biphasic stimulation and inhibition (Du & Sorota, 1997). Finally, in guinea-pig atrial and ventricular myocytes, no consistent effects attributable to cAMPi on ICl,vol were observed (Vandenberg et al. 1994).
At this time, no clear explanation is available to account for these reported disparate effects of cAMPi on ICl,vol. It is possible, however, that some of these inconsistencies might arise due to the combined complex effects of cAMPi and PKA on different types of Cl− channels (in addition to ICl,vol) which may be concomitantly expressed in the same myocyte. For example, many cardiac myocytes that express ICl,vol also express CFTR Cl− currents (ICl,CFTR) and/or Ca2+-activated Cl− currents (ICl,Ca) (Harvey, 1996; Hiraoka et al. 1998; Hume et al. 2000), making it difficult to distinguish and interpret the effects of cAMP stimulation on channel subtypes. This problem is accentuated when membrane currents are only studied using asymmetric Cl− gradients, since ICl,vol, ICl,CFTR and ICl,Ca all exhibit indistinguishable outwardly rectifying macroscopic current-voltage (I–V) relations under these conditions (Harvey, 1996; Hume et al. 2000). In the present study, we used symmetrical high Cl− concentrations to effectively separate the outwardly rectifying ICl,vol from other possible contaminating Cl− currents like ICl,CFTR and ICl,Ca (which have linear I–V relations in symmetrical high Cl−) and re-examined the cAMPi regulation of native ICl,vol in guinea-pig and canine cardiac myocytes. We also examined cAMPi regulation of the cloned ICl,vol in gpClC-3-transfected NIH/3T3 cells. Under these conditions, we find that elevation of cAMPi consistently causes inhibition, not stimulation, of outwardly rectifying ICl,vol exhibited by both native and recombinant channels. Finally, experiments employing site-directed mutagenesis of the ClC-3 gene product, identify one primary phosphorylation site involved in the inhibitory modulation of ICl,vol by both PKC and PKA phosphorylation.
METHODS
Preparation of single cardiac myocytes
Single cardiac myocytes were obtained from guinea-pig and dog hearts using techniques as described previously (Collier et al. 1996; Yamazaki & Hume, 1997; Duan et al. 1997b, 1999). Briefly, guinea-pigs (350-450 g) were killed by a blow to the neck, and the hearts were quickly removed. The aorta was cannulated, and the heart was retrogradely perfused first with a modified Hepes-buffered Tyrode solution at 37°C, then with a nominally Ca2+-free Tyrode solution (until the heart ceased to beat), and finally with the same solution containing 0.1-0.3 mg ml−1 collagenase (Type II, Sigma) and 1.0 % bovine serum albumin for 10 min. The left atrium was removed and further dissected into small pieces, and cell dissociation was achieved by gentle mechanical agitation in the high-K+ cell storage solution.
Adult mongrel dogs of either sex were anaesthetized with pentobarbital sodium (45 mg kg−1i.v.) and their hearts were quickly removed and also perfused in the Langendorff mode. Atrial and ventricular myocytes were isolated in the same way as for guinea-pigs except for the enzyme concentration and duration of treatment. The heart was perfused with 0.5-1.5 mg ml−1 collagenase for 15–30 min. All cells studied were rod shaped, exhibited clear cross striations, and lacked any visible blebs under isotonic conditions. Data from blebbed cells after hypotonic cell swelling were not included in the final analysis.
Functional expression of guinea-pig cardiac ClC-3 in NIH/3T3 cells
Full-length gpClC-3 was ligated to the mammalian expression vector pZeoSV (Invitrogen) and transfected into NIH/3T3 cells by electroporation as previously described (Duan et al. 1997b, 1999). For transient transfections, dishes were transfected with an appropriate combination of CD8 (a lymphocyte cell surface antigen) in the πH3-CD8 plasmid construct as a marker for transfection (4 μg) and gpClC-3 in the pZeoSV vector (20 μg). Transfected cells were identified by their binding to CD8-coated beads (M-450 CD8; Dyna-beads, Dynal Inc., Oslo, Norway). Cells were subcultured on glass coverslips for electrophysiological recordings.
Site-directed mutagenesis
The serine at position 51 was altered by an S51 to alanine site-specific mutation introduced into gpClC-3 cDNA as previously described (Duan et al. 1999). The mutation was confirmed by nucleotide sequencing of both strands of the mutated cDNA.
Electrophysiological techniques
The whole-cell patch-clamp technique (Hamill et al. 1981) was used to record currents in isolated guinea-pig and canine cardiac myocytes and gpClC-3-transfected NIH/3T3 cells. Patch pipettes were made from borosilicate glass capillaries and had a tip resistance of 2–5 MΩ. Ag-AgCl wires were immersed in the bath and pipette solutions and connected to a patch-clamp amplifier (Axopatch-1D, Axon Instruments). A 3 M KCl-agar bridge between the bath and the Ag-AgCl reference electrode was used to minimize changes in liquid junctional potential during some experiments. To obtain whole-cell Cl−I–V relations, cells were held at the holding potential (0, -40 or -60 mV) and test potentials (400 or 150 ms) were applied from -100 to +120 mV in 20 mV increments at an interval of 5 s (voltage-clamp protocol is shown in each figure). In some cases, I–V relations were measured using depolarizing voltage ramps applied at a rate of 0.2 V s−1 following a voltage step from 0 mV to -100 mV. To examine time-dependent changes in current amplitude before and after different interventions, cells were clamped from a holding potential of 0 mV to hyperpolarizing potential of -100 mV for 100 ms, back to 0 mV for 10 ms, and then to a depolarizing potential of +100 mV for 100 ms. The same hyperpolarizing and depolarizing pulses were imposed every 30 s. Currents were filtered at a frequency of 1 kHz and digitized on-line at 5 kHz using an IBM PC/AT compatible computer and pCLAMP 6 software (Axon Instruments). Experiments were performed at room temperature (22-24°C).
Solutions and drugs
The modified Tyrode solution for cell isolation contained (mM): NaCl 126, KCl 5.4, CaCl2 2.0, MgCl2 1.0, NaH2PO4 0.33, glucose 10 and Hepes 10; pH was adjusted to 7.4 with NaOH. The high-K+ cell storage solution contained (mM): KCl 20, KH2PO4 10, glucose 10, L-glutamic acid 70, β-hydroxybutyric acid 10, taurine 10 and EGTA 10; along with 1 % alubumin, pH 7.4 (KOH). Osmolality was adjusted to 300 mosmol (kg H2O)−1 by adding mannitol.
Bath and pipette solutions were chosen to facilitate Cl− current recording. Hypotonic (220 mosmol (kg H2O)−1, measured by freezing point depression, Osmomette; Precision Systems Inc.) bath solution for recordings in cardiac myocytes contained (mM): NaCl 90, MgCl2 1.0, BaCl2 3.0, NaH2PO4 0.33, CsCl 10, Hepes 10 and glucose 5.5; pH = 7.4, [Cl−]o = 108 mM. The isotonic bath solutions were the same as the hypotonic bath solutions except that the osmolality was adjusted to 300 mosmol (kg H2O)−1 by adding mannitol. Pipette solution for recordings in cardiac myocytes contained (mM): NMDG-Cl 108, BAPTA 10, Mg-ATP 5 and Hepes 10; pH = 7.4, 300 mosmol (kg H2O)−1, [Cl−]i = 108 mM. In experiments using Ca2+-containing bath solution, 1 mM BaCl2 in the bath solutions was replaced by equimolar CaCl2, and BAPTA in pipette solutions was replaced by 2 mM EGTA.
Hypotonic (250 mosmol (kg H2O)−1) bath solution for whole-cell current recordings in NIH/3T3 cells contained (mM): NaCl 125, MgCl2 2.5, CaCl2 2.5 and Hepes 10; pH = 7.2, [Cl−]o = 135 mM. Osmolality was adjusted to 300 mosmol (kg H2O)−1 by adding mannitol for isotonic bath solution. Pipette solution for recordings in NIH/3T3 cells contained (mM): NMDG-Cl 135, EGTA 2, Mg-ATP 5 and Hepes 10; pH = 7.2, 300 mosmol (kg H2O)−1), [Cl−]i = 135 mM.
Isoprenaline and 8-Br-cAMP (both from Sigma, St Louis, MO, USA) were freshly dissolved in the bath solution. KT5720 (Calbiochem, San Diego, CA, USA) was prepared as a 1 mM stock solution in DMSO. The stock solution was diluted to the desired final concentration immediately before use. The final concentration of DMSO was < 0.1 %, which, by itself, did not affect Cl− currents. All other chemicals were purchased from Sigma.
Data analysis
All data are expressed as means ±s.e.m. Statistical analysis was performed using Student's t test; a two-tailed probability (P) of < 0.05 was taken to indicate statistical significance.
RESULTS
Effects of 8-Br-cAMP on ICl,vol in guinea-pig atrial cells
We first studied the effect of an increase in intracellular cAMP (cAMPi) on ICl,vol in guinea-pig atrial myocytes, since: (i) ICl,Ca is lacking in these myocytes (Sipido et al. 1995); (ii) the majority (90 %) of these myocytes express ICl,vol while only ∼10-21 % myocytes may express ICl,CFTR (Vandenberg et al. 1994; James et al. 1996); and (iii) the gene encoding ICl,vol, gpClC-3, was originally cloned from guinea-pig heart (Duan et al. 1997b). As shown in Fig. 1, hypotonic cell swelling activated an outwardly rectifying Cl− current in guinea-pig atrial myocytes under symmetrical high Cl− conditions (Fig. 1D and G). The properties of this swelling-induced current in guinea-pig myocytes, including its Cl− dependence and pharmacological properties, have been described in more detail in a previous study from this laboratory (Duan et al. 1999). In the continued presence of the hypotonic bath solution, subsequent application of the membrane-permeable cAMP analogue 8-Br-cAMP (500 μM) to the superfusate significantly decreased Cl− current amplitudes (Fig. 1C and F). This inhibition was voltage independent (37 ± 6 % inhibition at +80 mV, 44 ± 9 % inhibition at -80 mV, n = 6, P = not significant (n.s.)). The 8-Br-cAMP-sensitive current was indistinguishable from ICl,vol and had an outwardly rectifying I–V relation with symmetrical high Cl− concentrations.
Figure 1. Effect of 8-Br-cAMP on ICl,vol in guinea-pig atrial cells.

Representative current traces from the guinea-pig atrial cells under isotonic (A), hypotonic (B) and hypotonic + 8-Br-cAMP (500 μM) (C) conditions are shown. D, difference currents induced by cell swelling. E, 8-Br-cAMP-sensitive currents. Voltage-clamp protocol is shown in the inset. The cell was first equilibrated with the isotonic solution, then exposed to the hypotonic solution. Thereafter, 8-Br-cAMP was applied. Mean I–V relations (n = 6) of ICl,vol under isotonic (^), hypotonic (□) and hypotonic + 8-Br-cAMP (▵) conditions are shown in F. I-V relations of hypotonic-induced currents (•) and 8-Br-cAMP-sensitive currents (▪) are shown in G and H, respectively. The rectification ratios at ±80 mV of the hypotonic-sensitive and the 8-Br-cAMP-sensitive difference currents were 2.50 ± 0.47 and 2.22 ± 0.54, respectively (P = n.s.).
To confirm that 8-Br-cAMP-induced inhibition of ICl,vol in guinea-pig atrial myocytes is indeed mediated by an increase in cAMPi activity, we simultaneously monitored the well-known stimulatory effect of cAMPi on L-type Ca2+ channels (Bean et al. 1984) by examining the effects of 8-Br-cAMP on Ba2+ currents (IBa) in the same cell. As shown in Fig. 2, when the cell was held at more negative potentials (-60 mV) to allow the activation of Ba2+ currents, hypotonic cell swelling resulted in the activation of ICl,vol, which overlapped with IBa. Addition of 8-Br-cAMP markedly increased the transient inward IBa while it significantly inhibited the cell swelling-activated outwardly rectifying ICl,vol.
Figure 2. Effect of 8-Br-cAMP on Ba2+ currents (IBa) and macroscopic Cl− currents in guinea-pig atrial cells.

Representative current traces from guinea-pig atrial cells under isotonic (A), hypotonic (B) and hypotonic + 8-Br-cAMP (500 μM) (C) conditions are shown. The cell was held at -60 mV to record IBa through the voltage-dependent Ca2+ channel and ICl,vol simultaneously (voltage-clamp protocol is shown in the inset).
In the majority (7/10, 70 %) of guinea-pig atrial myocytes examined, 8-Br-cAMP consistently caused inhibition of ICl,vol. However, in a small percentage (3/10, 30 %) of atrial myocytes, 8-Br-cAMP elicited an apparent increase in total macroscopic Cl− current amplitude. In contrast to the well-known outwardly rectifying properties of ICl,vol in symmetrical high Cl− (see Fig. 3D and G), the 8-Br-cAMP-induced difference currents in these cells, although reversing near the estimated value of ECl (0 mV; the equilibrium potential for Cl−), exhibited a linear I–V relation (Fig. 3E and H). Since ICl,CFTR is known to exhibit a linear I–V relation in symmetrical high Cl− solutions (Harvey et al. 1990) and a small percentage of guinea-pig atrial myocytes express ICl,CFTR (Vandenberg et al. 1994; James et al. 1996), it seems likely that the 8-Br-cAMP-induced Cl− currents recorded in these cells may be contaminated by activation of ICl,CFTR.
Figure 3. Increase in macroscopic Cl− currents by 8-Br-cAMP in guinea-pig atrial cells.
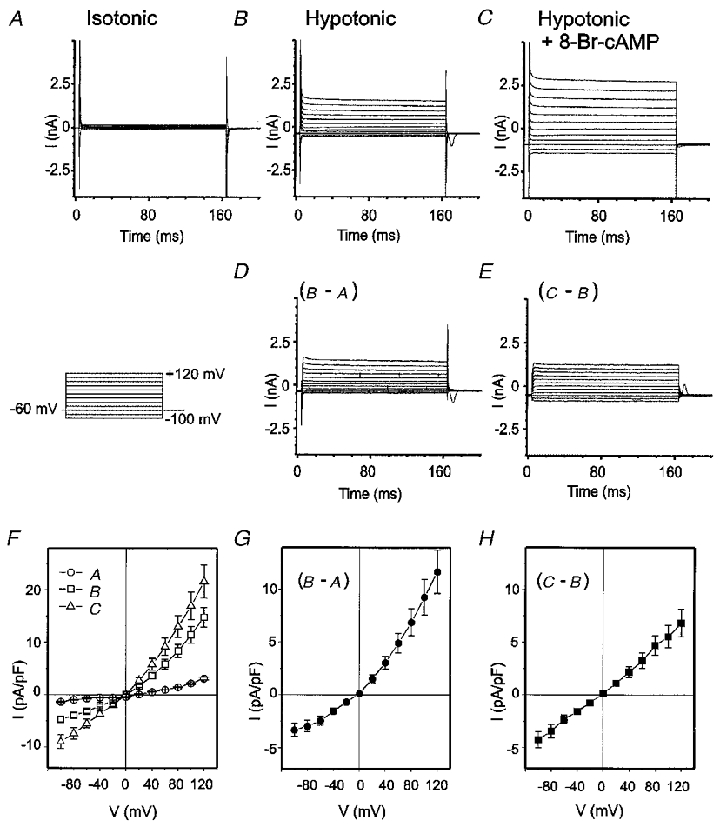
Representative current traces from the guinea-pig atrial cells under isotonic (A), hypotonic (B) and hypertonic + 8-Br-cAMP (500 μM) (C) conditions are shown. D, hypotonically induced currents. E, 8-Br-cAMP-induced currents. Mean I–V relations (n = 3) of macroscopic ICl under isotonic (^), hypotonic (□) and hypotonic + 8-Br-cAMP (▵) conditions are shown in F. I-V relations of hypotonically induced currents (•) and 8-Br-cAMP-induced currents (▪) are shown in G and H, respectively.
Effects of isoprenaline on ICl,vol in canine atrial myocytes
Highly variable effects of cAMPi and PKA stimulation on ICl,vol have been reported in canine cardiac myocytes (Sorota, 1992; Tseng, 1992; Oz & Sorota, 1995; Du & Sorota, 1997). It is possible that the presence of mixed types of overlapping Cl− currents may be partially responsible for the reported variable effects of cAMPi and PKA stimulation on ICl,vol in canine atrial and/or ventricular myocytes. Contamination of macroscopic ICl,vol by ICl,CFTR as observed in some guinea-pig atrial myocytes is not expected, since most evidence to date has failed to show functional or molecular expression of CFTR Cl− channels in canine cardiac myocytes (Sorota et al. 1991; Horowitz et al. 1993; Harvey, 1996; Hume et al. 2000). However, these cells express ICl,Ca in significant abundance, which would become activated as cytoplasmic Ca2+ rises in response to elevation of cAMPi (Zygmunt, 1994; Collier & Hume, 1995; Yue et al. 1996). Therefore, it is possible that contamination from ICl,Ca could potentially confound any interpretation of the effects of cAMPi on ICl,vol. We, therefore, first performed experiments in canine atrial and ventricular myocytes under conditions where possible contamination of ICl,vol by ICl,Ca was minimized by strongly buffering [Ca2+]i and performing experiments in Ca2+-free bath (external) solutions.
Figure 4 shows the effects of isoprenaline on ICl,vol in a canine atrial myocyte under [Ca2+]o-free and high (10 mM) intracellular BAPTA conditions. While only small currents could be detected in isotonic solutions (Fig. 4A), subsequent exposure of the cell to hypotonic solutions activated substantial macroscopic Cl− currents (Fig. 4B). The hypotonically induced currents showed outward rectification and time-dependent inactivation at more positive potentials and had a reversal potential near the predicted value of ECl (0 mV) with symmetrical high Cl− concentrations (Fig. 4D and G). These properties are identical to those of cardiac ICl,vol previously described in canine atrial (Sorota, 1992), and ventricular myocytes (Tseng, 1992) and in the heart of guinea-pig and other species (Vandenberg et al. 1994; Shuba et al. 1996; Duan et al. 1999; Hume et al. 2000). Isoprenaline (1 μM), a β-adrenergic receptor agonist which elevates cAMPi, consistently decreased ICl,vol in the five different canine atrial myocytes examined (Fig. 4C and F). The inhibition by isoprenaline was voltage independent (48 ± 7 % inhibition at +80 mV vs. 39 ± 7 % inhibition at -80 mV, n = 5, P = n.s.). The isoprenaline-sensitive difference currents also showed time-dependent inactivation at positive potentials and had an outwardly rectifying I–V relation (Fig. 4E and H), confirming that the effects observed are due to inhibition of ICl,vol.
Figure 4. Effect of isoprenaline on ICl,vol in canine atrial cells in nominally Ca2+-free solutions.

Representative current traces from dog atrial cells under isotonic (A), hypotonic (B) and hypotonic + isoprenaline (1 μM) (C) conditions are shown. D, hypotonically induced currents. E, isoprenaline-sensitive currents. Mean I–V relations (n = 5) of ICl,vol under isotonic (^), hypotonic (□) and hypotonic + isoprenaline (▵) conditions are shown in F. I-V relations of hypotonically induced currents (•) and isoprenaline-sensitive currents (▪) are shown in G and H, respectively.
We also simultaneously verified the well-known stimulatory effect of isoprenaline on L-type Ca2+ channels in canine atrial cells using a ramp voltage-clamp protocol to simultaneously monitor the effects of isoprenaline on ICl,vol and IBa under [Ca2+]o-free and high (10 mM) intracellular BAPTA conditions. While isoprenaline (1 μM) inhibited the outwardly rectifying ICl,vol, it also caused a significant increase in the amplitude of IBa (data not shown).
We next tested the effects of isoprenaline on ICl in canine atrial myocytes using conditions (Ca2+ (1.0 mM)-containing bath solutions and low [Ca2+]i buffering) similar to those previously used in this preparation in which variable stimulatory and inhibitory effects of isoprenaline and forskolin on ICl,vol were reported (Sorota, 1992; Du & Sorota, 1997). However, we used symmetrical high Cl− concentrations in an attempt to distinguish effects of isoprenaline on ICl,vol from possible effects on other types of macroscopic Cl− currents, which may also be modulated by cAMPi. Figure 5 shows an example of the effect of isoprenaline on hypotonic cell swelling-activated Cl− currents in canine atrial myocytes recorded with a bath (external) solution containing 1.0 mM Ca2+ and the pipette (internal) solution containing 2 mM EGTA. Hypotonic cell swelling initially activated an outwardly rectifying Cl− current, but subsequent application of isoprenaline (1 μM) caused a larger increase in the amplitude of both inward and outward currents, and the increase was greater for inward currents compared with outward currents (Fig. 5C). Thus, the isoprenaline-induced difference currents were no longer outwardly rectifying, but actually exhibited some inward rectification, and failed to exhibit time-dependent inactivation at positive membrane potentials (Fig. 5E and H). Thus, these isoprenaline-induced difference currents cannot be attributed solely to an effect on ICl,vol, since in symmetrical high Cl− solutions, ICl,vol is outwardly rectifying and exhibits time-dependent inactivation at positive membrane potentials (cf. Figs 1 and 4). Similar effects of isoprenaline in canine atrial myocytes were also observed when the intracellular pipette solution contained 4 mM EGTA, suggesting that even this concentration of EGTA may not be enough to buffer [Ca2+]i when cells are stimulated with isoprenaline.
Figure 5. Effect of isoprenaline on macroscopic Cl− currents in canine atrial cells in Ca2+-containing solutions.

Representative current traces from dog atrial cells under isotonic (A), hypotonic (B) and hypotonic + isoprenaline (1 μM; C) conditions are shown. D, hypotonically induced currents. E, isoprenaline-induced currents. Mean I–V relations (n = 3) of macroscopic ICl under isotonic (^), hypotonic (□) and hypotonic + isoprenaline (▵) conditions are shown in F. I-V relations of hypotonically induced currents (•) and isoprenaline-induced currents (▪) are shown in G and H, respectively.
In summary, in canine atrial cells in Ca2+-free bath (external) solution and in the presence of strong intracellular Ca2+ buffering, isoprenaline consistently caused a monotonic decrease in ICl,vol in every cell examined, and the inhibited currents could be attributed to ICl,vol since the isoprenaline-induced difference currents were outwardly rectifying with symmetrical high Cl− concentrations. However, when 1.0 mM Ca2+ was included in the external bath solution and the intracellular Ca2+ buffer was changed from 10 mM BAPTA to 2–4 mM EGTA, we also observed a stimulatory effect of isoprenaline on ICl,vol, suggesting that the stimulatory effects caused by isoprenaline are Ca2+ dependent.
Effects of 8-Br-cAMP on recombinant ClC-3 channels expressed in NIH/3T3 cells
We next studied the regulation of ICl,vol by cAMP in a heterologous gene expression system in which the gene encoding ICl,vol cloned from guinea-pig heart, gpClC-3, was stably or transiently expressed in NIH/3T3 cells (Duan et al. 1997b, 1999). Quantitative RT-PCR has recently revealed significant expression of ClC-3 mRNA in both atrium and ventricles of canine heart suggesting that ClC-3 may be responsible for the native ICl,vol in this tissue as well (F. Britton, C. Rossow, D. Duan, J. R. Hume & B. Horowitz, unpublished observations).
Stable or transient transfection of gpClC-3 into NIH/3T3 cells yields a basally active chloride conductance that is strongly modulated by cell volume (Fig. 6A and B). Properties of this expressed ICl,ClC-3 which resemble those reported for native ICl,vol in heart include: an outwardly rectifying unitary slope conductance of 40 pS, an anion selectivity of I− > Cl− > Asp−, inactivation at positive potentials, activation by extracellular hypotonicity, and inhibition by hypertonicity, by extracellular nucleotides, by phorbol esters, by stilbene derivatives, and by tamoxifen (Duan et al. 1997b). As shown in Fig. 6A, B and E, the expressed currents were outwardly rectifying under both isotonic and hypotonic conditions. 8-Br-cAMP (500 μM) consistently decreased the amplitude of ICl,ClC-3 in gpClC-3-transfected NIH/3T3 cells under hypotonic conditions (493 ± 20 vs. 334 ± 34 pA pF−1 at +80 mV (n = 7, P < 0.02) and -301 ± 15 vs. -195 ± 23 pA pF−1 at -80 mV (n = 7, P < 0.03) in the absence and presence of 8-Br-cAMP, respectively). Although the degree of rectification of ICl,ClC-3 in NIH/3T3 cells appeared to be somewhat less than that observed for ICl,vol in native cardiac myocytes, the difference in rectification ratios of ICl,vol in native guinea-pig atrial cells (2.50 ± 0.47, n = 6) and ICl,ClC-3 in NIH/3T3 cells (1.66 ± 0.21, n = 7) was not statistically significant (P = 0.113). As in native guinea-pig and canine myocytes (cf. Figs 1 and 4), the inhibition by 8-Br-cAMP was voltage independent (32 ± 7 % inhibition at +80 mV vs. 32 ± 7 % inhibition at -80 mV, n = 7, P = n.s.). The 8-Br-cAMP-sensitive currents (Fig. 6D and F) showed time-dependent inactivation at depolarizing potentials and had an outwardly rectifying I–V relation (rectification ratio = 1.62 ± 0.14, n = 7, P = n.s. vs. control), indicating that the cAMP-inhibited current can be attributed to ICl,ClC-3. No evidence for stimulation of ICl,ClC-3 was observed in any of the seven gpClC-3-transfected NIH/3T3 cells tested. To test whether these inhibitory effects of 8-Br-cAMP are due to activation of protein kinase A (PKA) in these experiments, we examined further the effects of a specific PKA inhibitor, KT5720 (Kase et al. 1987). As shown in Fig. 7A, pretreatment of 3T3 cells with KT5720 (200 nM) prevented the inhibition of ICl,ClC-3 by 500 μM 8-Br-cAMP (421 ± 58 vs. 452 ± 76 pA pF−1 at +80 mV (n = 4, P = n.s.) and -255 ± 45 vs. -256 ± 44 pA pF−1 at -80 mV (n = 4, P = n.s.) in the absence and presence of 8-Br-cAMP, respectively).
Figure 6. Effect of 8-Br-cAMP on recombinant ClC-3 channels expressed in NIH/3T3 cells.

Representative current traces from the ClC-3 expressed in NIH/3T3 cells under isotonic (A), hypotonic (B) and hypotonic + 8-Br-cAMP (500 μM) (C) are shown. D, 8-Br-cAMP-sensitive currents. Mean I–V relations (n = 7) of ICl,vol under hypotonic (^) and hypotonic + 8-Br-cAMP (□) conditions are shown in E. I-V relations of 8-Br-cAMP-sensitive currents (▵) are shown in F. The rectification ratios at ±80 mV of the hypotonic-sensitive and the 8-Br-cAMP-sensitive difference currents were 1.66 ± 0.21 and 1.62 ± 0.14, respectively (P = n.s.).
Figure 7. Both PKA inhibitor and site-directed mutation of the putative PKC phosphorylation site on the N-terminus of ClC-3 prevented the inhibitory effect of 8-Br-cAMP on ClC-3 channels.

A, whole-cell currents recorded from NIH/3T3 cells expressing wild-type ClC-3 under hypotonic conditions. Panels a and b show the representative currents in the presence of 200 nM KT5720 alone and KT5720 + 500 μM 8-Br-cAMP, respectively. Panel c shows the mean I–V curves from 4 different cells under conditions identical to those in panels a (^) and b (□). B, whole-cell currents recorded from NIH/3T3 cells expressing S51A ClC-3 mutant under hypotonic conditions. Panels a and b show the representative currents in the absence and presence of 500 μM 8-Br-cAMP, respectively. Panel c shows the mean I–V curves from 5 different cells under conditions identical to those in panels a (^) and b (□).
In a final series of experiments, we tested whether or not the inhibitory effects of 8-Br-cAMP on ICl,ClC-3 might be due to direct phosphorylation of the ClC-3 protein by PKA. One cytoplasmic consensus PKC phosphorylation site (serine 51 on the N-terminus) has recently been identified and functionally implicated in the regulation of ClC-3 channel activity by changes in cell volume (Duan et al. 1999). Since stimulation of PKA seems to mimic the inhibitory effects of PKC on ICl,ClC-3, and there is precedence showing that some putative phosphorylation sites often exhibit cross-recognition by different kinases (Kennelly & Krebs, 1991), we examined whether or not mutation of serine 51 to an alanine (S51A), which effectively eliminates PKC inhibition of ICl,ClC-3 (Duan et al. 1999), might also alter the ability of 8-Br-cAMP to inhibit ICl,ClC-3.
As previously reported, S51A ClC-3 channels expressed in NIH/3T3 cells were constitutively open under isotonic conditions and hypotonic cell swelling did not significantly change current amplitudes further (Duan et al. 1999). As shown in Fig. 7B, exposure of these S51A ClC-3-transfected NIH/3T3 cells to 8-Br-cAMP (500 μM) failed to produce any significant inhibition of ICl,ClC-3 in hypotonic solutions (530 ± 46 vs. 455 ± 43 pA pF−1 at +80 mV (n = 5, P = n.s.) and -373 ± 25 vs. -314 ± 37 pA pF−1 at -80 mV (n = 5, P = n.s.) in the absence and presence of 8-Br-cAMP, respectively). These results suggest that the observed weak inhibitory effects of 8-Br-cAMP on ICl,ClC-3 (Fig. 6) may be due to PKA phosphorylation of the same amino terminus serine residue (S51) that is predominantly phosphorylated by PKC, and also leads to inhibition of channel activity.
DISCUSSION
The regulation of cardiac ICl,vol by cAMPi has been a controversial issue (Sorota, 1992; Hagiwara et al. 1992; Tseng, 1992; Oz & Sorota, 1995; Hall et al. 1995; Du & Sorota, 1997). In the present study, we examined cAMPi regulation of native ICl,vol in atrial myocytes from guinea-pig and canine hearts and on recombinant ICl,vol encoded by gpClC-3, expressed in NIH/3T3 cells. In our experiments, we attempted to examine the effects of cAMPi on ICl,vol in isolation and avoid possible confounding influences due to contamination from multiple Cl− channel subtypes concomitantly expressed in the same cell. To this end, symmetrical high Cl− concentrations were used to separate outwardly rectifying ICl,vol from other possible contaminating Cl− currents (e.g. ICl,CFTR and ICl,Ca) which have linear I–V relations under these conditions. The results indicate that 8-Br-cAMP and agonists such as isoprenaline, which elevate cAMPi, consistently inhibit the outwardly rectifying ICl,vol in native cardiac myocytes from both guinea-pig and canine hearts. Furthermore, identical effects of cAMPi on recombinant ICl,vol were confirmed in gpClC-3-transfected NIH/3T3 cells, where the potential problem of multiple overlapping Cl− currents is mimimized.
Our results are most consistent with those of Hall et al. (1995) in chick cardiac myocytes, where increases in cAMPi caused inhibition and even prevented the activation of ICl,vol. Similar to our observations in the ClC-3-transfected NIH/3T3 cells (Fig. 7A), they also found that the inhibitory effect of cAMPi on ICl,vol in chick cardiac myocytes is due to a cAMP-PKA-dependent phosphorylation process. They proposed that the activation of ICl,vol in chick heart cells requires dephosphorylation of a cAMP-dependent phosphorylation site. We now provide molecular evidence that mutation of a single serine residue S51 at the N-terminus of ClC-3, which may be a phosphorylation consensus site for both PKC and PKA, abolished the inhibitory effect of 8-Br-cAMP (Fig. 7B).
Our results are in marked contrast to those reported by Sorota and colleagues (Sorota, 1992; Oz & Sorota, 1995; Du & Sorota, 1997) in canine and human atrial myocytes. They originally found that elevation of cAMPi stimulated ICl,vol (Sorota, 1992; Oz & Sorota, 1995) but later suggested that the effects of cAMPi could actually be more complicated with both inhibitory and stimulatory effects seen in different cells or even in the same cell (Du & Sorota, 1997). They further suggested that the inhibitory effects were due to PKA phosphorylation, but the stimulatory effects were mediated by a PKA-independent pathway. These experiments relied solely upon the use of ramp voltage-clamp protocols to record whole-cell currents under asymmetric Cl− gradients (low [Cl−]i). Unfortunately, ramp voltage-clamp protocols do not provide kinetic information to distinguish ICl,vol from other types of Cl− currents expressed in heart (Shuba et al. 1996) and with asymmetrical Cl− conditions, most cardiac Cl− currents exhibit nearly identical outwardly rectifying I–V relations (Harvey et al. 1990; Hume et al. 2000). Under these experimental conditions, therefore, contamination from multiple overlapping Cl− current subtypes concomitantly expressed in the same cell is possible. With symmetrical high Cl− concentrations, only ICl,vol remains outwardly rectifying, which effectively separates it from other possible contaminating Cl− currents (Strange et al. 1996; Shuba et al. 1996; Okada, 1997; Hume et al. 2000). Thus, in the present study, we used voltage step protocols to investigate the effects of isoprenaline on ICl,vol under symmetrical high Cl− conditions. We found that cell swelling-induced ICl,vol and the currents inhibited by elevation of cAMPi in both guinea-pig and canine atrial myocytes and in the gpClC-3-transfected NIH/3T3 cells were all outwardly rectifying. These currents also showed time-dependent inactivation at positive potentials. Since the cAMPi-inhibited currents have properties identical to ICl,vol and ICl,ClC-3, it is clear that they can be attributed to inhibition of ICl,vol. It is possible that an earlier report (Vandenberg et al. 1994) which failed to see inhibition of ICl,vol by isoprenaline might also be complicated by the presence of contaminating overlapping Cl− channel subtypes.
In a small percentage of guinea-pig cardiac myocytes, we observed some stimulation of whole-cell Cl− currents by 8-Br-cAMP. However, these currents showed no time-dependent inactivation and had linear I–V relations with symmetrical high Cl− concentrations. It is well known that cAMP activates ICl,CFTR in guinea-pig atrial and ventricular myocytes (Bahinski et al. 1989; Harvey et al. 1990; Vandenberg et al. 1994; Shuba et al. 1996). Thus, the currents induced by 8-Br-cAMP in a small percentage of guinea-pig atrial myocytes in the present study have properties most consistent with activation of ICl,CFTR. However, the effects actually observed in these cells are complicated and probably reflect the combined inhibitory effects of cAMPi on ICl,vol as well as the stimulatory effects of cAMPi on ICl,CFTR.
In canine atrial myocytes, stimulatory effects of isoprenaline on whole-cell Cl− currents were only observed in Ca2+-containing bath solutions and with weakly buffered. This is consistent with the results reported previously by Sorota and colleagues (Sorota, 1992; Du & Sorota, 1997) with a similar (4 mM) concentration of EGTA in the intracellular pipette solution but a higher [Ca2+]o (1.8 vs. 1.0 mM) in the bath solutions. In contrast, with Ca2+-free bath solutions and with strongly buffered, we found that isoprenaline consistently caused inhibition of the hypotonic cell swelling-activated Cl− currents. It is well known that isoprenaline causes a marked elevation of intracellular Ca2+ due to stimulation of L-type Ca2+ channels (Bean et al. 1984) and ICl,Ca can be activated as [Ca2+]i rises in response to an elevation of cAMPi (Zygmunt, 1994; Kawano et al. 1995; Zygmunt et al. 1998). It is also possible that exposure to hypotonic solutions alone, which has been reported to cause inhibition of Na+-Ca2+ exchange (Wright et al. 1995), could also contribute to a rise in [Ca2+]i in isolated cardiac myocytes. Although ICl,Ca was initially described as a transient outward current (Ito2) with a bell-shaped I–V relation (Zygmunt & Gibbons, 1992), later studies in canine ventricular myocytes found that ICl,Ca showed little or no voltage- or Ca2+-induced inactivation and is essentially time and voltage independent when intracellular Ca2+ is pseudo-clamped to constant levels (Zygmunt, 1994; Yue et al. 1996). In addition, ICl,Ca exhibits a linear I–V relation with symmetrical high Cl− (Kawano et al. 1995; Yamazaki et al. 1999). Thus, the isoprenaline-activated Cl− currents observed in canine atrial myocytes have properties most consistent with activation of ICl,Ca, although the current changes measured under these conditions probably reflect the combined inhibitory effects of cAMPi on ICl,vol as well as the stimulatory effects of cAMPi on ICl,Ca.
It is possible that the cAMPi-stimulated linear current observed in both guinea-pig and dog atrial myocytes might be due to modulation of ICl,vol if isoprenaline alters the voltage dependence of ICl,vol and linearizes its I–V relation. However, this possibility seems highly unlikely since such an effect was not observed in canine atrial myocytes in Ca2+-free bath (external) solutions with strongly buffered (Fig. 4), or in gpClC-3-transfected NIH/3T3 cells, a system with minimal contamination from ICl,CFTR and ICl,Ca. In these cells, 8-Br-cAMP caused only consistent inhibition and no alteration in the rectification and voltage dependence of ICl,ClC-3 (Fig. 6).
In addition, we demonstrated that the inhibitory effects of 8-Br-cAMP on ICl,ClC-3 are due to phosphorylation by PKA, since these effects were prevented by the specific PKA inhibitor KT5720. We also showed that this inhibition probably involves direct PKA phosphorylation of the ClC-3 protein itself as the S51A mutation abolished the inhibition by 8-Br-cAMP. While the serine at position 51 in gpClC-3 is within a perfect consensus PKC phosphorylation sequence (RINSKK), this sequence also represents a less well-utilised PKA phosphorylation sequence (R-X2-S, where X represents any amino acid) (Kennelly & Krebs, 1991). Thus, phosphorylation of S51 by either PKC or PKA appears to inhibit IClC-3, although the former effect appears to be more potent. The transcriptional expression of ClC-3 in both canine atrial and ventricular myocytes has recently been demonstrated (F. Britton, C. Rossow, D. Duan, J. R. Hume & B. Horowitz, unpublished observations), but further studies are required to determine if S51 plays a similar role to that in gpClC-3.
In summary, our results suggest that contamination of macroscopic ICl,vol by ICl,CFTR and/or ICl,Ca may account for some of the inconsistent and controversial effects of cAMPi on ICl,vol reported previously in native cardiac myocytes. When contributions from contaminating macroscopic Cl− currents are prevented, elevation of cAMPi consistently inhibits ICl,vol in both guinea-pig and canine native cardiac myocytes and inhibits recombinant ClC-3 channels expressed in NIH/3T3 cells. These experiments emphasize the potential pitfalls of assessing ICl,vol regulation using experimental conditions (asymmetric Cl− gradients with low [Cl−]i and voltage ramps) which do not allow reliable separation of overlapping macroscopic Cl− currents. In the case of ICl,vol, such a separation can easily be achieved using symmetrical high Cl− concentrations, since these channels are unique in retaining their outwardly rectifying properties under these conditions. It is tempting to speculate that similar complications due to the presence of multiple overlapping macroscopic Cl− currents, as illustrated in the present study, may also be at least partially responsible for recent controversies regarding PKC modulation of ICl,vol in heart (Duan et al. 1995, 1997b, 1999; Clemo & Baumgarten, 1998; Clemo et al. 1999; Du & Sorota, 1999) and other mammalian tissues (Okada, 1997), or even the reported sensitivity of ICl,vol to niflumic acid and other putative Cl− channel antagonists (Sorota, 1994) as well.
Acknowledgments
This study was supported by NIH grant HL52803. D.D. was supported by MRC Canada and then a Grant-in-Aid from the American Heart Association.
References
- Bahinski A, Nairn AC, Greengard P, Gadsby DC. Chloride conductance regulated by cyclic AMP-dependent protein kinase in cardiac myocytes. Nature. 1989;340:718–721. doi: 10.1038/340718a0. [DOI] [PubMed] [Google Scholar]
- Bean BP, Nowycky MC, Tsien RW. Beta-adrenergic modulation of calcium channels in frog ventricular heart cells. Nature. 1984;307:371–375. doi: 10.1038/307371a0. [DOI] [PubMed] [Google Scholar]
- Clemo HF, Baumgarten CM. Protein kinase C activation blocks ICl(swell) and causes myocyte swelling in a rabbit congestive heart failure model. Circulation. 1998;98:I-695. [Google Scholar]
- Clemo HF, Stambler BS, Baumgarten CM. Swelling-activated chloride current is persistently activated in ventricular myocytes from dogs with tachycardia-induced congestive heart failure. Circulation Research. 1999;84:157–165. doi: 10.1161/01.res.84.2.157. [DOI] [PubMed] [Google Scholar]
- Collier ML, Hume JR. Unitary chloride channels activated by protein kinase C in guinea pig ventricular myocytes. Circulation Research. 1995;76:317–324. doi: 10.1161/01.res.76.2.317. [DOI] [PubMed] [Google Scholar]
- Collier ML, Levesque PC, Kenyon JL, Hume JR. Unitary Cl− channels activated by cytoplasmic Ca2+ in canine ventricular myocytes. Circulation Research. 1996;78:936–944. doi: 10.1161/01.res.78.5.936. [DOI] [PubMed] [Google Scholar]
- Du XY, Sorota S. Modulation of dog atrial swelling-induced chloride current by cAMP: protein kinase A-dependent and -independent pathways. The Journal of Physiology. 1997;500:111–122. doi: 10.1113/jphysiol.1997.sp022003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du XY, Sorota S. Protein kinase C stimulates swelling-induced chloride current in canine atrial cells. Pflügers Archiv. 1999;437:227–234. doi: 10.1007/s004240050773. [DOI] [PubMed] [Google Scholar]
- Duan D, Cowley S, Horowitz B, Hume JR. A serine residue in ClC-3 links phosphorylation-dephosphorylation to chloride channel regulation by cell volume. Journal of General Physiology. 1999;113:57–70. doi: 10.1085/jgp.113.1.57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan D, Fermini B, Nattel S. Alpha-adrenergic control of volume-regulated Cl− currents in rabbit atrial myocytes. Characterization of a novel ionic regulatory mechanism. Circulation Research. 1995;77:379–393. doi: 10.1161/01.res.77.2.379. [DOI] [PubMed] [Google Scholar]
- Duan D, Hume JR, Nattel S. Evidence that outwardly rectifying Cl− channels underlie volume-regulated Cl− currents in heart. Circulation Research. 1997a;80:103–113. doi: 10.1161/01.res.80.1.103. [DOI] [PubMed] [Google Scholar]
- Duan D, Winter C, Cowley S, Hume JR, Horowitz B. Molecular identification of a volume-regulated chloride channel. Nature. 1997b;390:417–421. doi: 10.1038/37151. [DOI] [PubMed] [Google Scholar]
- Gadsby DC, Nagel G, Hwang TC. The CFTR chloride channel of mammalian heart. Annual Review of Physiology. 1995;57:387–416. doi: 10.1146/annurev.ph.57.030195.002131. [DOI] [PubMed] [Google Scholar]
- Hagiwara N, Masuda H, Shoda M, Irisawa H. Stretch-activated anion currents of rabbit cardiac myocytes. The Journal of Physiology. 1992;456:285–302. doi: 10.1113/jphysiol.1992.sp019337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall SK, Zhang J, Lieberman M. Cyclic AMP prevents activation of a swelling-induced chloride-sensitive conductance in chick heart cells. The Journal of Physiology. 1995;488:359–369. doi: 10.1113/jphysiol.1995.sp020972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hart P, Warth JD, Levesque PC, Collier ML, Geary Y, Horowitz B, Hume JR. Cystic fibrosis gene encodes a cAMP-dependent chloride channel in heart. Proceedings of the National Academy of Sciences of the USA. 1996;93:6343–6348. doi: 10.1073/pnas.93.13.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RD. Cardiac chloride currents. News in Physiological Sciences. 1996;11:175–181. [Google Scholar]
- Harvey RD, Clark CD, Hume JR. Chloride current in mammalian cardiac myocytes. Novel mechanism for autonomic regulation of action potential duration and resting membrane potential. Journal of General Physiology. 1990;95:1077–1102. doi: 10.1085/jgp.95.6.1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harvey RD, Hume JR. Autonomic regulation of a chloride current in heart. Science. 1989;244:983–985. doi: 10.1126/science.2543073. [DOI] [PubMed] [Google Scholar]
- Hiraoka M, Kawano S, Hirano Y, Furukawa T. Role of cardiac chloride currents in changes in action potential characteristics and arrhythmias. Cardiovascular Research. 1998;40:23–33. doi: 10.1016/s0008-6363(98)00173-4. [DOI] [PubMed] [Google Scholar]
- Horowitz B, Tsung SS, Hart P, Levesque PC, Hume JR. Alternative splicing of CFTR Cl− channels in heart. American Journal of Physiology. 1993;264:H2214–2220. doi: 10.1152/ajpheart.1993.264.6.H2214. [DOI] [PubMed] [Google Scholar]
- Hume JR, Duan D, Collier ML, Yamazaki J, Horowitz B. Anion transport in heart. Physiological Reviews. 2000;80:31–81. doi: 10.1152/physrev.2000.80.1.31. [DOI] [PubMed] [Google Scholar]
- James AF, Tominaga T, Okada Y, Tominaga M. Distribution of cAMP-activated chloride current and CFTR mRNA in the guinea pig heart. Circulation Research. 1996;79:201–207. doi: 10.1161/01.res.79.2.201. [DOI] [PubMed] [Google Scholar]
- Kase H, Iwahashi K, Nakanishi S, Matsuda Y, Yamada K, Takahashi M, Murakata C, Sato A, Kaneko M. K-252 compounds, novel and potent inhibitors of protein kinase C and cyclic nucleotide-dependent protein kinases. Biochemical and Biophysical Research Communications. 1987;142:436–440. doi: 10.1016/0006-291x(87)90293-2. [DOI] [PubMed] [Google Scholar]
- Kawano S, Hirayama Y, Hiraoka M. Activation mechanism of Ca2+-sensitive transient outward current in rabbit ventricular myocytes. The Journal of Physiology. 1995;486:593–604. doi: 10.1113/jphysiol.1995.sp020837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawasaki M, Uchida S, Monkawa T, Miyawaki A, Mikoshiba K, Marumo F, Sasaki S. Cloning and expression of a protein kinase C-regulated chloride channel abundantly expressed in rat brain neuronal cells. Neuron. 1994;12:597–604. doi: 10.1016/0896-6273(94)90215-1. [DOI] [PubMed] [Google Scholar]
- Kennelly PJ, Krebs EG. Consensus sequences as substrate specificity determinants for protein kinases and protein phosphatases. Journal of Biological Chemistry. 1991;266:15555–15558. [PubMed] [Google Scholar]
- Okada Y. Volume expansion-sensing outward-rectifier Cl− channel: fresh start to the molecular identity and volume sensor. American Journal of Physiology. 1997;273:C755–789. doi: 10.1152/ajpcell.1997.273.3.C755. [DOI] [PubMed] [Google Scholar]
- Oz MC, Sorota S. Forskolin stimulates swelling-induced chloride current, not cardiac cystic fibrosis transmembrane-conductance regulator current, in human cardiac myocytes. Circulation Research. 1995;76:1063–1070. doi: 10.1161/01.res.76.6.1063. [DOI] [PubMed] [Google Scholar]
- Shuba LM, Ogura T, McDonald TF. Kinetic evidence distinguishing volume-sensitive chloride current from other types in guinea-pig ventricular myocytes. The Journal of Physiology. 1996;491:69–80. doi: 10.1113/jphysiol.1996.sp021197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Callewaert G, Porciatti F, Vereecke J, Carmeliet E. [Ca2+]i-dependent membrane currents in guinea-pig ventricular cells in the absence of Na/Ca exchange. Pflügers Archiv. 1995;430:871–878. doi: 10.1007/BF00386189. [DOI] [PubMed] [Google Scholar]
- Sorota S. Swelling-induced chloride-sensitive current in canine atrial cells revealed by whole-cell patch-clamp method. Circulation Research. 1992;70:679–687. doi: 10.1161/01.res.70.4.679. [DOI] [PubMed] [Google Scholar]
- Sorota S. Pharmacologic properties of the swelling-induced chloride current of dog atrial myocytes. Journal of Cardiovascular Electrophysiology. 1994;5:1006–1016. doi: 10.1111/j.1540-8167.1994.tb01143.x. [DOI] [PubMed] [Google Scholar]
- Sorota S, Siegal MS, Hoffman BF. The isoproterenol-induced chloride current and cardiac resting potential. Journal of Molecular and Cellular Cardiology. 1991;23:1191–1198. doi: 10.1016/0022-2828(91)90207-3. [DOI] [PubMed] [Google Scholar]
- Strange K, Emma F, Jackson PS. Cellular and molecular physiology of volume-sensitive anion channels. American Journal of Physiology. 1996;270:C711–730. doi: 10.1152/ajpcell.1996.270.3.C711. [DOI] [PubMed] [Google Scholar]
- Tseng GN. Cell swelling increases membrane conductance of canine cardiac cells: evidence for a volume-sensitive Cl channel. American Journal of Physiology. 1992;262:C1056–1068. doi: 10.1152/ajpcell.1992.262.4.C1056. [DOI] [PubMed] [Google Scholar]
- Vandenberg JI, Rees SA, Wright AR, Powell T. Cell swelling and ion transport pathways in cardiac myocytes. Cardiovascular Research. 1996;32:85–97. [PubMed] [Google Scholar]
- Vandenberg JI, Yoshida A, Kirk K, Powell T. Swelling-activated and isoprenaline-activated chloride currents in guinea pig cardiac myocytes have distinct electrophysiology and pharmacology. Journal of General Physiology. 1994;104:997–1017. doi: 10.1085/jgp.104.6.997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Weikersthal SF, Barrand MA, Hladky SB. Functional and molecular characterization of a volume-sensitive chloride current in rat brain endothelial cells. The Journal of Physiology. 1999;516:75–84. doi: 10.1111/j.1469-7793.1999.075aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright AR, Rees SA, Vandenberg JI, Twist VW, Powell T. Extracellular osmotic pressure modulates sodium-calcium exchange in isolated guinea-pig ventricular myocytes. The Journal of Physiology. 1995;488:293–301. doi: 10.1113/jphysiol.1995.sp020967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Britton F, Collier ML, Horowitz B, Hume JR. Regulation of recombinant cardiac cystic fibrosis transmembrane conductance regulator chloride channels by protein kinase C. Biophysical Journal. 1999;76:1972–1987. doi: 10.1016/S0006-3495(99)77356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. The Journal of Physiology. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki J, Hume JR. Inhibitory effects of glibenclamide on cystic fibrosis transmembrane regulator, swelling-activated, and Ca(2+)-activated Cl− channels in mammalian cardiac myocytes. Circulation Research. 1997;81:101–109. doi: 10.1161/01.res.81.1.101. [DOI] [PubMed] [Google Scholar]
- Yue L, Feng J, Li GR, Nattel S. Transient outward and delayed rectifier currents in canine atrium: properties and role of isolation methods. American Journal of Physiology. 1996;270:H2157–2168. doi: 10.1152/ajpheart.1996.270.6.H2157. [DOI] [PubMed] [Google Scholar]
- Zygmunt AC. Intracellular calcium activates a chloride current in canine ventricular myocytes. American Journal of Physiology. 1994;267:H1984–1995. doi: 10.1152/ajpheart.1994.267.5.H1984. [DOI] [PubMed] [Google Scholar]
- Zygmunt AC, Gibbons WR. Properties of the calcium-activated chloride current in heart. Journal of General Physiology. 1992;99:391–414. doi: 10.1085/jgp.99.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zygmunt AC, Goodrow RJ, Weigel CM. INaCa and ICl(Ca) contribute to isoproterenol-induced delayed afterdepolarizations in midmyocardial cells. American Journal of Physiology. 1998;275:H1979–1992. doi: 10.1152/ajpheart.1998.275.6.H1979. [DOI] [PubMed] [Google Scholar]