

Abstract
To address questions of whether long-term depression (LTD) in the visual cortex is expressed in pre- or postsynaptic sites, whether brain-derived neurotrophic factor (BDNF) exerts its LTD-blocking action without involvement of GABAergic inhibition, and whether the action of BDNF is pre- or postsynaptic, we observed excitatory postsynaptic currents (EPSCs) from solitary neurones cultured on glial microislands. In this preparation GABAergic inhibition is not involved and a group of synapses (autapses) which generate evoked EPSCs is thought to be the same as those generating spontaneous EPSCs.
A short depolarising voltage step to the soma generated Na+ spikes which were followed by autaptic EPSCs. When this somatic activation was paired with prolonged depolarisation for 100 ms to −30 mV and repeated at 1 Hz for 5 min, LTD was induced in all of the nine cells tested. Then, the frequency of spontaneous EPSCs decreased, but the amplitude did not change, suggesting that the site of LTD expression is presynaptic.
Application of BDNF at 50 ng ml−1 blocked the depression of evoked EPSCs and the decrease in the frequency of spontaneous EPSCs. An inhibitor for receptor tyrosine kinases, K252a, antagonised the action of BDNF, suggesting an involvement of BDNF receptors, TrkB.
These results suggest that BDNF prevents low-frequency inputs from inducing LTD of excitatory synaptic transmission through presynaptic mechanisms in the developing visual cortex.
Long-term depression (LTD) of synaptic transmission, a counterpart of long-term potentiation (LTP), can be induced in the developing visual cortex (Tsumoto & Suda, 1979; Artola & Singer, 1990; Kirkwood et al. 1993) and is supposed to provide a basis for experience-dependent modifiability of visual cortical neurones during the critical period of postnatal development (Tsumoto, 1992; Singer, 1995; Katz & Shatz, 1996). In contrast to LTP, however, mechanisms underlying LTD have not been fully explored in the developing visual cortex. For example, there has been no systematic approach to the classical, pre- versus postsynaptic issue, although there is a short report suggesting the presynaptic expression of LTD (Torii et al. 1997). To address such an issue, cortical slice preparations in which most studies have been carried out in the past two decades are not always appropriate, because activation of single input fibres, which is necessary for a precise analysis, is extremely difficult and the origins of spontaneous synaptic events which are to be analysed simultaneously with evoked synaptic activities are not necessarily the same as those of the latter. To overcome these difficulties, we used solitary neurones in culture obtained from visual cortex of neonatal rats (Furshpan et al. 1976; Bekkers & Stevens, 1991; Segal, 1991; Kimura et al. 1997). In this sort of preparation we could easily activate a single presynaptic cell which is also postsynaptic, and could record spontaneous and evoked synaptic (autaptic) activities which originate from the same group of synapses (autapses).
A class of neurotrophins, brain-derived neurotrophic factor (BDNF), has been reported to enhance excitatory synaptic transmission in various nervous systems and further to play a role in LTP in hippocampus and developing visual cortex (Lohof et al. 1993; Leßmann et al. 1994; Kang & Schuman, 1995; Korte et al. 1995; Levine et al. 1995; Figurov et al. 1996; Stoop & Poo, 1996; Patterson et al. 1996; Akaneya et al. 1997; Carmignoto et al. 1997; Scharfman, 1997; Leßmann & Heumann, 1998; Li et al. 1998; for reviews see Thoenen, 1995; Cellerino & Maffei, 1996; McAllister et al. 1999). In the visual cortex of young rats it has been reported that BDNF blocks the induction of LTD of excitatory synaptic transmission which otherwise is induced by low-frequency stimulation (LFS) of afferents at 1 Hz for 10–15 min (Akaneya et al. 1996; Huber et al. 1998; Kinoshita et al. 1999). There is a possibility, however, that such an LTD-blocking action of BDNF might be due not to direct action on excitatory transmission but to indirect action through a blockade of inhibition, because BDNF was reported to suppress GABAergic inhibitory transmission rather than to enhance excitatory transmission in hippocampus (Tanaka et al. 1997; Frerking et al. 1998) and inhibition is suggested to be involved in the induction of LTD (Artola & Singer, 1990; see Tsumoto, 1992).
To test this possibility and further to address the question of whether the site of the LTD expression and of the LTD-blocking action of BDNF is pre- or postsynaptic, we used preparations of excitatory solitary neurones in which GABAergic inhibition is not involved and spontaneous activities are generated at the same synapses (autapses) as those of evoked activities. We found that LTD of excitatory transmission is induced probably through presynaptic mechanisms, and that BDNF prevents LFS from inducing LTD by a direct action on excitatory synapses. Some of the results in this paper have been presented in abstract form (Kumura et al. 1999).
METHODS
Culture preparation
Sprague-Dawley rats (postnatal day 0–1) were anaesthetised with ketamine (> 50 mg kg−1, i.p.), and then killed by cervical dislocation. The experimental procedures met the regulations of the Animal Care Committee of Osaka University Medical School. Solitary neurones were cultured by using conventional microisland methods (Kimura et al. 1997). A piece of visual cortex was removed from the brain, enzymatically dissociated with papain (20 U ml−1), and triturated with a fire-polished glass pipette. Neurones were plated on previously prepared glial islands grown on collagen dots placed on an agarose sheet, and were grown in a solution based on Eagle's minimal essential medium supplemented with 5 % rat serum.
A whole-cell patch electrode (resistance, 5–10 MΩ) was used to record synaptic currents in a voltage-clamp mode (Fig. 1A). The neurones were clamped at −70 mV unless otherwise noted for recording excitatory postsynaptic currents (EPSCs) with a patch-clamp amplifier (Axopatch 200A, Axon Instruments). A short voltage step from the holding potential to 0 mV for 2 ms was applied to elicit a somatic Na+ spike. The perfusing solution, unless otherwise noted, contained the following (mM): NaCl, 150; KCl, 4; CaCl2, 2; MgSO4, 1; Hepes, 10; glucose, 10 (pH 7.4). Glycine (10 μM) was added to the medium. Osmolality was adjusted to 320–325 mosmol by adding sucrose when necessary. Neurotrophins and drugs such as kynurenic acid, a non-selective antagonist for ionotropic glutamate receptors, and 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX), a selective antagonist for glutamate receptors of the α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) type, were applied with a local perfusion system equipped with a Y-shaped tube. The electrode solutions mostly used were as follows (mM): caesium methane sulfonate, 130; Hepes, 10; CsCl, 10; EGTA, 0.5; MgATP, 5; and Na2GTP, 1.
Figure 1. Schema showing preparations of a solitary neurone to which a whole-cell patch electrode was attached (A) and stimulus procedures (B).
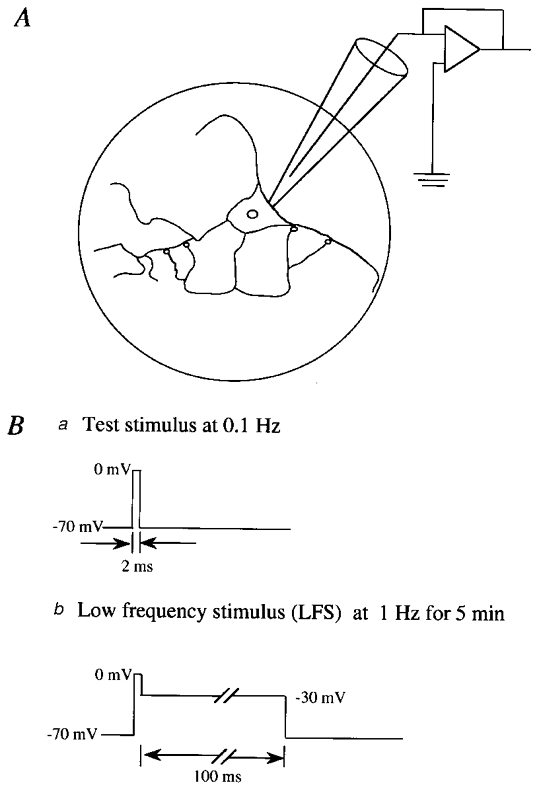
A, schematic depiction of glial microisland (circle) and a solitary neurone grown on the island. Autapses are schematically indicated by small circles. Ba, test stimuli were given to the soma every 10 s. Bb, conditioning stimuli were paired with the depolarisation to −30 mV for 100 ms and repeated at 1 Hz for 5 min.
The amplitude of stimulus-evoked EPSCs was evaluated by subtracting the mean value of the baseline for 5 ms just before the stimulus artifact from the peak value of the EPSCs. For spontaneous or miniature EPSCs (mEPSCs), data were searched with the use of an event detection program that is a built-in component of AxoGraph 3.5 (Axon Instruments). The detection of mEPSCs met the following criteria: (1) the amplitude of peaks is higher than 2 pA and a threshold of 4 times the standard deviation (s.d.) of the baseline noise level, and (2) peaks have a faster rise than decay phase. The difference between the baseline (2 ms immediately before the onset of each event) and the peak (average of 3 points) was then measured manually. Baseline noise was calculated as the difference between the mean of the baseline period (5 ms) and an average of three points 1 ms apart from the end of the baseline period.
Application of BDNF and drugs
In most of the experiments, recombinant human BDNF (provided by Sumitomo Pharmaceutical Co., Ltd, Japan), or K252a (provided by Kyowa Hakko Co., Ltd, Japan), was applied to neurones through the perfusion medium 20 min before the initiation of LFS. BDNF was prepared at a concentration of 500 μg ml−1 containing 0.1 % bovine serum albumin in undiluted Ca2+-, Mg2+-free phosphate-buffered saline solution (PBS). Then it was diluted to 50 ng ml−1 with the perfusion medium just prior to recordings. For application of K252a, it was prepared to a final concentration of 200 or 300 nM with dimethyl sulfoxide (DMSO) as vehicle of which the final concentration was 0.01 % (v/v). To prevent possible adhesion of the neurotrophin to the wall of the perfusion system, the inner walls of the reservoir and tubes connecting with the recording chamber were coated by silicon 2–3 days prior to recording.
RESULTS
Whole-cell patch-clamp recordings were carried out from solitary neurones in culture. As reported previously (Kimura et al. 1997), a short voltage step applied to the soma through a whole-cell pipette elicited a Na+ spike, which in turn produced synaptic currents in most of the neurones tested (see specimen records of Figs 2, 4, 5 and 6). In the present study we did not include neurones in which synaptic currents were not unambiguously judged as EPSCs on the following bases: they were blocked by kynurenic acid or CNQX and their reversal potentials were near 0 mV. To check possible changes in recording conditions and membrane properties of cells under observation, we continuously monitored series and input resistances. If these values changed by more than 20 % of the initial control, the recordings were stopped and the data were deleted from the analysis. Forty-one neurones which were recorded for longer than 35 min satisfied these criteria. In nine neurones to which BDNF or K252a was not applied, activation of the soma at 0.1 Hz elicited autaptic EPSCs with mean peak latency and amplitude of 6.6 ± 1.2 ms (s.d.) and 998 ± 514 pA, respectively, at a membrane potential of −70 mV.
Figure 2. Induction of LTD in solitary neurones.
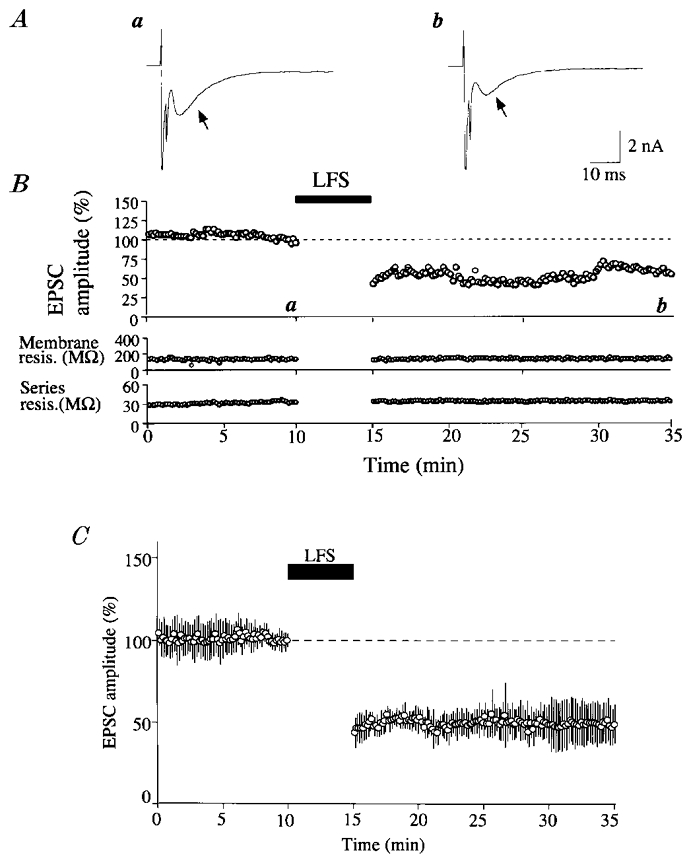
A, an example of autaptic EPSCs recorded from a solitary neurone at the time point indicated by corresponding letters in the upper graph in B. Arrows indicate autaptic EPSCs which follow stimulus artifacts and sharp Na+ spikes. B, peak amplitude of EPSCs plotted against time (top). The value is expressed as the percentage of the mean of 20 responses before LFS. The time during which LFS was applied to the cell is indicated by the horizontal bar. In the lower two graphs, the membrane resistance of the cell and the series resistance of the electrode are plotted against time. C, time course of the mean EPSC amplitude for 9 cells. For each cell, the amplitude of EPSCs was calculated as in B. Vertical bars indicate twice the s.d. In all the 9 cells the membrane potential was kept at −70 mV throughout recordings.
Figure 4. No induction of LTD by LFS when BDNF was applied to neurones.

A, an example of autaptic EPSCs recorded at the time point indicated by corresponding letters in the upper graph in B. Other conventions are the same as in Fig. 2A. B, top: the peak amplitude of EPSCs plotted against time. The value is expressed as the percentage of the mean of 20 responses before LFS. The application of BDNF was started 20 min before the initiation of the recording. The time during which LFS was applied to the cell is indicated by the filled, horizontal bar. Other conventions are the same as in Fig. 2B.
Figure 5. Induction of LTD without changes in the amplitude of mEPSCs during the application of K252a.
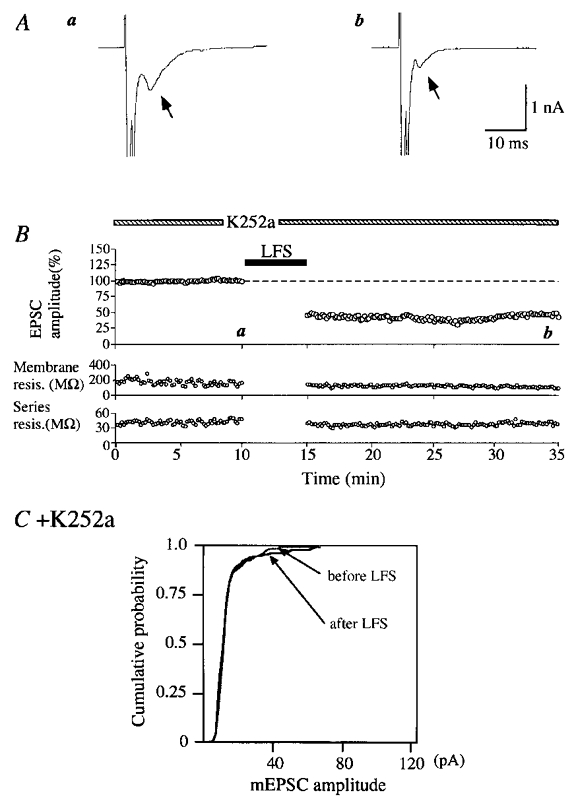
A, an example of autaptic EPSCs recorded at the time point indicated by corresponding letters in the upper graph in B. Other conventions are the same as in Fig. 2A. B, peak amplitudes of EPSCs plotted against time (top). The application of K252a at a concentration of 200 nM was started 20 min before the initiation of the recording. Other conventions are the same as in Fig. 2B. C, cumulative plots of the amplitude distribution of mEPSCs just before and 15–20 min after LFS. Other conventions are the same as in Fig. 3D.
Figure 6. Induction of LTD without change in the amplitude of mEPSCs during the co-application of BDNF and K252a.
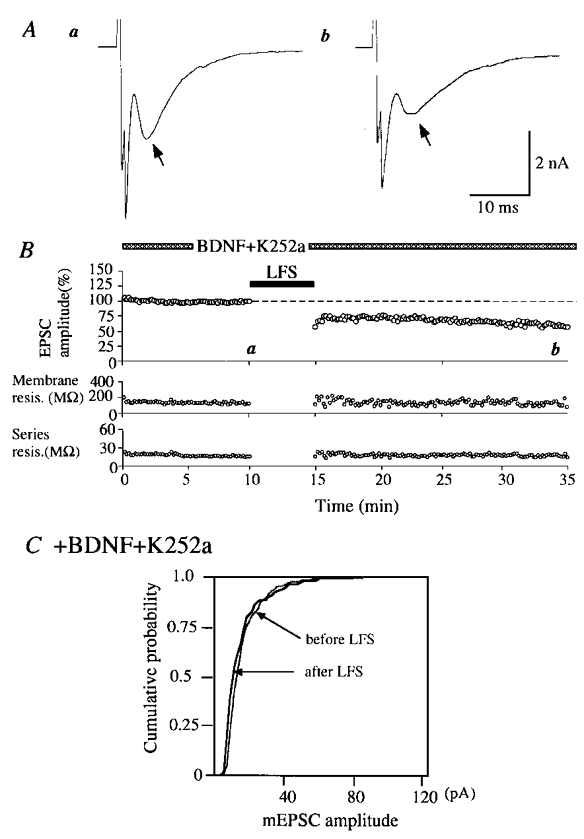
BDNF was applied to this cell at a concentration of 50 ng ml−1 together with K252a at 200 nM. A, an example of autaptic EPSCs recorded at the time point indicated by corresponding letters in the upper graph in B. Other conventions are the same as in Fig. 2A. B, peak amplitudes of EPSCs plotted against time (top). The co-application of BDNF (50 ng ml−1) and K252a (200 nM) was started 20 min before the initiation of the recording. Other conventions are the same as in Fig. 2B. C, cumulative plots of the amplitude distribution of mEPSCs just before and 15–20 min after LFS. Other conventions are the same as in Fig. 3D.
Induction of LTD of autaptic EPSCs in solitary neurones
Initially we attempted to induce LTD of autaptic EPSCs by a repetition of the somatic activation for 2 ms at 1 Hz for 5–10 min in six cells which otherwise were kept at −70 mV. However, this activation turned out to be ineffective in all of the six cells. Then, we found that LTD was induced reliably when the somatic activation was followed by depolarisation of the soma to −30 mV for 100 ms and this pairing was repeated at 1 Hz for 5 min (Fig. 1B). An example of LTD of EPSCs is shown in Fig. 2A and B. In this neurone the mean peak amplitude and latency of control EPSCs before LFS was 3083 pA and 6.4 ms, respectively. After test shocks at 0.1 Hz were confirmed to elicit EPSCs in the stable condition for 10 min, the somatic activation was paired with the prolonged depolarisation to −30 mV and repeated at 1 Hz for 5 min. Immediately after stopping this LFS the amplitude of EPSCs reduced to 43.3 % of the control value and seemed to recover slightly for a few minutes. Thereafter, however, it did not recover and remained depressed during the observation period of 20 min. This depression of EPSCs was not caused by a deterioration of the neuronal and recording conditions, because the membrane and series resistances were very stable (Fig. 2B, bottom two graphs). Mean EPSC amplitudes for the nine cells are plotted against time in Fig. 2C. It is obvious that LTD was induced by LFS. The peak amplitude of EPSCs 20 min after LFS was 49.6 ± 8.6 % of the control before LFS.
We then attempted to see whether the frequency, amplitude or both of mEPSCs was changed after the induction of LTD. As reported previously (Kimura et al. 1997), we could see spontaneous EPSCs which were generated without somatic activation. In the neurone shown in Fig. 3, the mean frequency of such EPSCs was 7.5 events s−1. These events could be identified as mEPSCs from the criteria reported previously (Kimura et al. 1997). In part of the present experiments this was confirmed by the observation that spontaneous EPSCs were not detectably changed by the application of tetrodotoxin (1 μM). As seen in the lower trace of Fig. 3A, the frequency of mEPSCs was reduced after LFS. This is obvious in Fig. 3B in which the frequency of mEPSCs is plotted against time. On the other hand, the peak amplitude of mEPSCs was not significantly changed after LFS. Figure 3C shows the amplitude distribution histograms just before and 18–20 min after LFS. No significant difference was seen in the amplitude distribution between the two histograms. This was confirmed by plotting the cumulative probability of amplitudes of mEPSCs before and after LFS (Fig. 3D). Statistical analysis with the Kolmogorov-Smirnov test indicated no significant difference between the two distributions (P > 0.05).
Figure 3. Decrease in the frequency of mEPSCs but not in their amplitude after LFS.
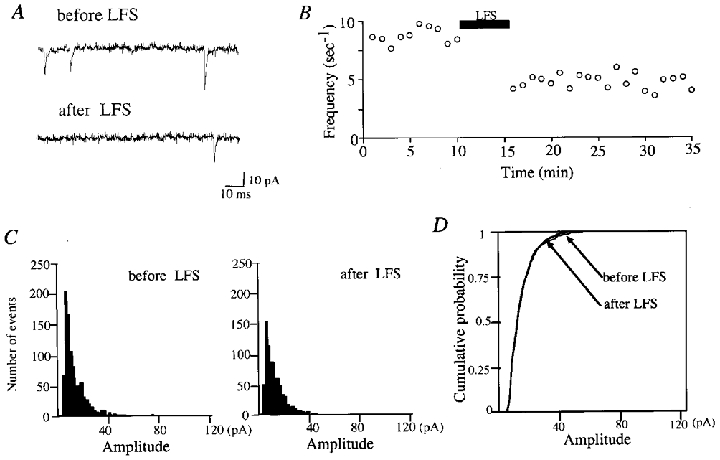
A, representative traces with spontaneous events before and after LFS. The lower trace was recorded 18 min after LFS. B, time course of frequency of mEPSCs. The frequency was measured every 60 s. LFS was applied during the period indicated by the horizontal bar. C, amplitude distribution of mEPSCs just before (left) and 18–20 min after (right) LFS. The total number of events in the left and right histograms is 950 and 720, respectively. D, cumulative plots of the amplitude distribution shown in C. Cumulative probability along the ordinate indicates the probability of observing mEPSCs with peak amplitudes smaller than or equal to a given value.
Prevention of LTD by BDNF
Next, we attempted to see whether BDNF could block the induction of LTD. For this test BDNF was applied to solitary neurones at a concentration of 50 ng ml−1. This concentration of BDNF did not significantly change the peak amplitude of evoked EPSCs: the mean amplitude of EPSCs of the eight cells to which BDNF was applied was 1042 ± 565 pA (s.d.), which was not significantly different (Student's unpaired t test, P = 0.17) from that of neurones without BDNF (998 ± 514 pA). Also, BDNF did not significantly change mEPSCs: the mean amplitude and frequency of mEPSCs of the former eight cells were 14.9 ± 5.3 pA and 9.3 ± 0.9 events s−1, respectively, which were not significantly different (unpaired t test, P = 0.58 and 0.10, respectively) from the respective values of the nine control neurones (13.6 ± 4.3 pA and 7.5 ± 2.7 events s−1). In the present study, we did not systematically increase the concentration of BDNF because we were not interested in the action of BDNF on basal synaptic transmission at 0.1 Hz, but rather in its action on LTD.
BDNF at a concentration of 50 ng ml−1 was found to block LTD which should have been induced by paired LFS. An example of this finding is shown in Fig. 4. Twenty minutes after the initiation of BDNF application, paired LFS was given to this neurone at 1 Hz for 5 min, but it turned out to be ineffective. After cessation of the LFS, the peak amplitude of EPSCs was not significantly reduced (Fig. 4A, record b). The time course of EPSC amplitude is shown together with those of membrane and series resistances in Fig. 4B. The amplitude of EPSCs seemed to be slightly reduced after the LFS, but there was no significant difference between the values before (2996 ± 160 pA) and 15–20 min after LFS (2852 ± 170 pA).
The amplitude of evoked EPSCs after LFS expressed as a percentage of that before LFS for the eight cells to which BDNF was applied is plotted in the second column of Fig. 7. LTD was not induced by LFS except in one cell in which the difference in amplitude from the control value before LFS was significant (paired t test, P < 0.05). The mean value of the EPSC amplitude for the eight cells was 86.7 ± 6.0 %. Without BDNF, LFS of the same parameters induced significant LTD in all of the nine cells (the leftmost column of Fig. 7). The mean amplitude for the nine cells was 49.6 ± 8.6 %, as mentioned above. The difference between these two values was statistically significant (analysis of variance followed by Scheffé's post hoc multiple comparison test, P < 0.001).
Figure 7. Peak amplitude of evoked EPSCs expressed as a percentage of that of control EPSCs before LFS.
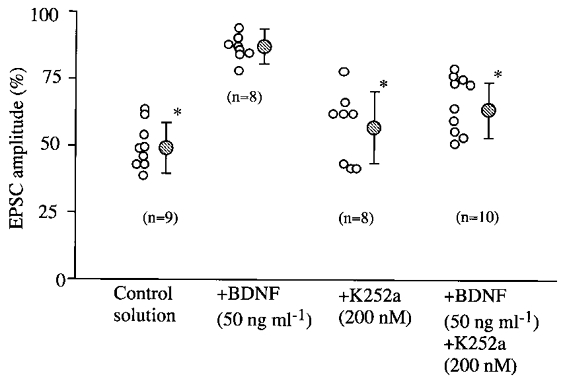
The ordinate represents the mean amplitude of EPSCs calculated from 30 consecutive responses for 15–20 min after LFS expressed as a percentage of that of another 20 EPSCs just before LFS. Shaded circles with vertical bars represent means and twice the s.d. for the same group of cells, respectively. The number of cells used for each test is shown in parentheses in each column. Asterisks indicate significant difference from the control value before LFS at P < 0.01 (paired t test).
Blockade of the BDNF action by an inhibitor for Trk receptor tyrosine kinases
In the next series of experiments, we tested whether K252a could antagonise the blocking action of BDNF on LTD. K252a is a membrane-permeant inhibitor for Trk receptor tyrosine kinases that is reported not to affect other tyrosine kinases at a concentration of 200 nM, and thus is an effective blocking agent of biological actions of BDNF and other neurotrophins (Knüssel & Hefti, 1992; Tapley et al. 1992). As reported previously in visual cortical slices (Akaneya et al. 1996, 1997), application of K252a at this concentration did not significantly change evoked and mEPSCs of solitary neurones. The mean amplitude of evoked EPSCs for the eight cells to which K252a was applied was 751 ± 412 pA, which was not significantly (unpaired t test, P = 0.29) different from that of the nine control cells.
An example of records during the application of K252a is shown in Fig. 5A. In the medium containing K252a at 200 nM a voltage step to 0 mV for 2 ms generated Na+ spikes which were followed by EPSCs with the peak amplitude of 968 ± 10 pA (Fig. 5Aa). After LFS the amplitude of EPSCs was reduced to 45.4 % of the control value and remained depressed for the whole observation period (Fig. 5B, upper graph). During such a strong depression of evoked EPSCs the frequency of mEPSCs was also reduced to 5.2 events s−1 from the pre-LFS value of 7.2 events s−1. On the other hand, the distribution of the peak amplitude of mEPSCs was not significantly changed after LFS (Fig. 5C; Kolmogorov-Smirnov test, P > 0.05). The magnitude of LTD of evoked EPSCs during the application of K252a for the eight cells was not significantly different from that in the control solution (third column of Fig. 7).
Then, we tested whether K252a could antagonise the action of BDNF. When K252a was co-applied with BDNF, LFS paired with the prolonged depolarisation to −30 mV induced LTD of EPSCs. An example of this finding is shown in Fig. 6. In this case, the peak amplitude of EPSCs 15–20 min after the paired LFS decreased to 69.7 ± 9.9 % of the control. The frequency of mEPSCs also decreased from 7.2 to 3.9 events s−1. These results suggest that the LTD-blocking action of BDNF is mediated through an activation of Trk receptor tyrosine kinases. Regarding the amplitude of mEPSCs, its distribution was not significantly (Kolmogorov-Smirnov test, P > 0.05) changed after the LFS (Fig. 6C).
A summary of the change in peak amplitude of evoked EPSCs after LFS in the four sets of tests is shown in Fig. 7. As mentioned above, LFS did not induce LTD in seven of the eight neurones to which BDNF was applied (second column of Fig. 7). It is also obvious that the mean values obtained during the application of K252a alone (57.0 ± 13.3 %) and the co-application of BDNF and K252a (66.1 ± 10.6 %) were significantly smaller than that with BDNF alone (analysis of variance followed by post hoc multiple comparison test, P < 0.001 and 0.005, respectively). The degree of LTD in the co-application test was also significantly different from that in the control (analysis of variance followed by post hoc multiple comparison test, P < 0.01). This suggests that the effect of BDNF might be mediated in part through a mechanism other than activation of Trk receptors. It is to be noted, however, that the mean value of EPSC amplitude in the co-application test was significantly smaller than that in the test with BDNF alone, as mentioned above.
Measurements of mEPSC amplitude and frequency are summarised in Fig. 8. The ratios of the mean amplitude of mEPSCs after LFS to that before LFS was similarly around 1.0 among the four different conditions (Fig. 8A), indicating that the amplitude of mEPSCs was not significantly changed in any condition. On the other hand, the mean frequency of mEPSCs was significantly (P < 0.01, paired t test) decreased after LFS except for the condition in which BDNF alone was applied to solitary neurones. These results suggest that LFS induces LTD through presynaptic mechanisms if there is not enough BDNF or if its action through Trk receptor tyrosine kinases is blocked.
Figure 8. Ratio of mean amplitude (A) and frequency (B) of mEPSCs after LFS to the respective control value before LFS.
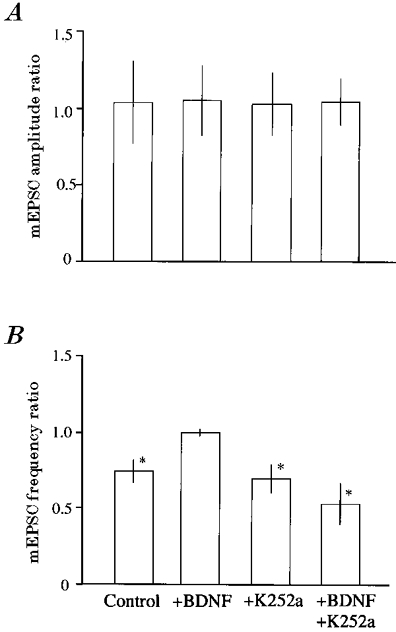
Vertical bars represent twice the s.d. of each mean. Data are the ratios of each value of mEPSCs for 18–20 min after LFS to that just before LFS. The numbers of cells used for each test are the same as in Fig. 7. Asterisks indicate significant difference from the control value before LFS at P < 0.01 (paired t test).
DISCUSSION
LTD of autaptic EPSCs in solitary neurones
In the present study we observed evoked and spontaneous ‘autaptic’ currents of solitary neurones in culture. There are several lines of evidence indicating that autapses have the same properties as other synapses in hippocampus and visual cortex (Bekkers & Stevens, 1991; Kimura et al. 1997). Therefore, autaptic currents analysed in the present study correspond to evoked and miniature EPSCs observed in slice and in vivo preparations, as mentioned in Results. Thus, the present report is probably the first on neocortical LTD in solitary neurones in culture, although hippocampal LTD was reported to be induced in solitary neurones by 900 shocks at 5 Hz (Goda & Stevens, 1996). Also, the present results provide the straightforward evidence that LTD of excitatory synaptic transmission can be induced without any involvement of GABAergic inhibitory activity.
The next question addressed in the present study was the well known pre- versus postsynaptic issue. Traditionally this issue has been approached with the quantal analysis of evoked and miniature synaptic responses in slice preparations. There is a problem inherent in this approach, i.e. synapses which generate evoked and miniature responses are not necessarily identical. In solitary neurone preparations, on the other hand, synapses which generate mEPSCs are thought to be the same as those generating evoked EPSCs. Thus, the present results that the frequency of mEPSCs decreased after the induction of LTD whereas the distribution of peak amplitude did not change at all suggest that LTD of evoked EPSCs is accompanied by a decrease in release probability at presynaptic sites.
There is a possibility that the reduction of the frequency of mEPSCs might be due to a decrease in amplitude of mEPSCs and consequently small events which had been detected became lost in noise. This possibility seems unlikely, however, because the distribution pattern of mEPSC amplitude was almost the same, even after the frequency of mEPSCs was decreased markedly after LFS. Recently Lissin et al. (1999) reported that application of glutamate or an agonist for a subtype of glutamate receptors, AMPA, to cultured hippocampal neurones induced a redistribution of a subunit of AMPA receptors from synaptic to non-synaptic sites. They also observed a decrease in the frequency of mEPSCs without a significant decrease in the mean amplitude after the continuous application of 100 μM AMPA for 15 min. Thus, there is a possibility that the decrease in the frequency of mEPSCs without change in amplitude that we observed in solitary neurones is due to a redistribution of postsynaptic receptors. Although we cannot exclude this possibility in the present study, we would like to point out that their procedures to induce LTD, so called ‘chemically induced LTD’, were much less physiological than those used in the present study, and thus their results may not be directly applicable to the interpretation of the present ones. Also, our previous study using the technique of direct postsynaptic injection of a membrane-impermeant inhibitor of Trk receptors in visual cortical slices suggested that the LTD-blocking action of BDNF may take place at presynaptic sites (Kinoshita et al. 1999), as further discussed below.
The site at which BDNF exerts its action
Previous studies with immunohistochemistry reported that high-affinity receptors for BDNF, TrkB receptors, exist both at presynaptic axons and postsynaptic dendrites and soma in the cortex (Cabelli et al. 1996; Fryer et al. 1996; Yan et al. 1997). The present results that the decrease in frequency of mEPSCs after LFS was blocked by BDNF, although the amplitude was not changed, suggest that BDNF may exert its action on the expression mechanism of LTD in presynaptic sites, as mentioned above. This suggestion seems consistent with the previous study with whole-cell patch clamp recordings in slice preparations of visual cortex (Carmignoto et al. 1997; Kinoshita et al. 1999). Previous studies in other structures also suggest that BDNF acts on preynaptic sites so as to increase the release of transmitters (Lohof et al. 1993; Leßmann et al. 1994; Kang & Schuman, 1995; Stoop & Poo, 1996; Gottschalk et al. 1998; Leßmann & Heumann, 1998). Thus, it seems reasonable to state that BDNF prevents repetitive, low-frequency inputs from inducing a long-lasting decrease in transmitter release from presynaptic sites, as suggested previously (Kinoshita et al. 1999).
In slice preparations of hippocampus it was reported that BDNF suppressed GABAergic inhibition, but did not significantly change excitatory synaptic transmission (Tanaka et al. 1997; Frerking et al. 1998). This suggests that the potentiating or facilitatory effect of BDNF might be caused by the suppression of inhibition, although there are a few pieces of evidence against this possibility in cultured hippocampal neurones (Leßman & Heumann, 1998; Li et al. 1998). This raises the possibility that the LTD-blocking action of BDNF might be due to a modulation of GABAergic inhibition by BDNF, because inhibition is suggested to be involved in the induction of LTD (Artola & Singer, 1990; see Tsumoto, 1992). The present results obtained in excitatory solitary neurones provide direct evidence against this possibility, although we cannot rule out the modulation of inhibition by BDNF in slice preparations. In conclusion it is possible to state that BDNF exerts its LTD-blocking action without modification of GABAergic inhibition.
A physiological role of BDNF may be prevention of LTD rather than induction of LTP
In the present study we found that LFS paired with the somatic depolarisation became ineffective for inducing LTD during the application of BDNF at a concentration of 50 ng ml−1. This indicates that exogenously applied BDNF prevents LFS from inducing LTD of excitatory synaptic transmission. In visual cortical slices it was reported that BDNF at a concentration of 100 or 200 ng ml−1 potentiates excitatory synaptic transmission (Akaneya et al. 1997; Carmignoto et al. 1997). When its concentration was reduced to 20 ng ml−1, BDNF had no such potentiating action in sliced visual cortex (Akaneya et al. 1997). However, BDNF at this concentration was reported to have the blocking action on LTD of excitatory synaptic transmission (Akaneya et al. 1996; Kinoshita et al. 1999), as shown in the present study. Although the exact concentration of BDNF around synapses in the visual cortex in vivo is not known, the basal level of BDNF in cerebral cortical tissues of the rat was assessed as 7.5 ± 0.3 ng (g tissue)−1 at 2 months of age (Nawa et al. 1995) or 2.3 ± 0.2 ng (g tissue)−1 at 20 days of age (Katoh-Semba et al. 1997). Thus, the concentration of 20–50 ng ml−1 seems to be more physiological than that of 100–200 ng ml−1. Therefore, it seems possible to assume that the chance for BDNF to exert its LTD-blocking action may be higher than that of its potentiating action on excitatory synaptic transmission in the developing visual cortex.
Acknowledgments
This study is supported by a Grant-in-Aid for Scientific Research (07279102) from the Ministry of Education, Science, Sports and Culture of Japan and by a CREST program from Japan Science and Technology Corporation. We express many thanks to Sumitomo Pharmaceutical Co., Ltd for kind gifts of recombinant human BDNF.
References
- Akaneya Y, Tsumoto T, Hatanaka H. Long-term depression blocked by brain-derived neurotrophic factor in rat visual cortex. Journal of Neurophysiology. 1996;76:4198–4201. doi: 10.1152/jn.1996.76.6.4198. [DOI] [PubMed] [Google Scholar]
- Akaneya Y, Tsumoto T, Kinoshita S, Hatanaka H. Brain-derived neurotrophic factor enhances long-term potentiation in rat visual cortex. Journal of Neuroscience. 1997;17:6707–6716. doi: 10.1523/JNEUROSCI.17-17-06707.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artola A, Singer W. Different voltage-dependent thresholds for inducing long-term depression and long-term potentiation in slices of rat visual cortex. Nature. 1990;347:69–72. doi: 10.1038/347069a0. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Stevens CF. Excitatory and inhibitory autaptic currents in isolated hippocampal neurons maintained in cell culture. Proceedings of the National Academy of Sciences of the USA. 1991;88:7834–7838. doi: 10.1073/pnas.88.17.7834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabelli RJ, Allendoefer KL, Radeke MJ, Welcher AA, Feinstein SC, Shatz CJ. Changing patterns of expression and subcellular localization of TrkB in the developing visual system. Journal of Neuroscience. 1996;16:7965–7980. doi: 10.1523/JNEUROSCI.16-24-07965.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmignoto G, Pizzorusso T, Tia S, Vicini S. Brain-derived neurotrophic factor and nerve growth factor potentiate excitatory synaptic transmission in the rat visual cortex. The Journal of Physiology. 1997;498:153–164. doi: 10.1113/jphysiol.1997.sp021848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cellerino A, Maffei L. The action of neurotrophins in the development and plasticity of the visual cortex. Progress in Neurobiology. 1996;49:53–71. doi: 10.1016/0301-0082(96)00008-1. [DOI] [PubMed] [Google Scholar]
- Figurov A, Pozzo-Miller LD, Olafsson P, Wang T, Lu B. Regulation of synaptic responses to high-frequency stimulation and LTP by neurotrophins in the hippocampus. Nature. 1996;381:706–709. doi: 10.1038/381706a0. [DOI] [PubMed] [Google Scholar]
- Frerking M, Malenka RC, Nicoll RA. Brain-derived neurotrophic factor (BDNF) modulates inhibitory, but not excitatory, transmission in the CA1 region of the hippocampus. Journal of Neurophysiology. 1998;80:3383–3386. doi: 10.1152/jn.1998.80.6.3383. [DOI] [PubMed] [Google Scholar]
- Fryer RH, Kaplan DR, Feinstein SC, Radeke MJ, Grayson DR, Kromer LF. Developmental and mature expression of full-length and truncated TrkB receptors in the rat forebrain. Journal of Comparative Neurology. 1996;374:21–40. doi: 10.1002/(SICI)1096-9861(19961007)374:1<21::AID-CNE2>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Furshpan EJ, MacLeish PR, O'Lague PH, Potter DD. Chemical transmission between rat sympathetic neurons and cardiac myocytes developing in microcultures: Evidence for cholinergic, adrenergic, and dual-function neurons. Proceedings of the National Academy of Sciences of the USA. 1976;73:4225–4229. doi: 10.1073/pnas.73.11.4225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goda Y, Stevens CF. Long-term depression properties in a simple system. Neuron. 1996;16:103–111. doi: 10.1016/s0896-6273(00)80027-6. [DOI] [PubMed] [Google Scholar]
- Gottschalk W, Pozzo-Miller LD, Figurov A, Lu B. Presynaptic modulation of synaptic transmission and plasticity by brain-derived neutrotrophic factor in the developing hippocampus. Journal of Neuroscience. 1998;18:6830–6839. doi: 10.1523/JNEUROSCI.18-17-06830.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber KM, Sawtell NB, Bear MF. Brain-derived neurotrophic factor alters the synaptic modification threshold in visual cortex. Neuropharmacology. 1998;37:571–579. doi: 10.1016/s0028-3908(98)00050-1. [DOI] [PubMed] [Google Scholar]
- Kang H, Schuman EM. Long-lasting neurotrophin-induced enhancement of synaptic transmission in the adult hippocampus. Science. 1995;267:1658–1662. doi: 10.1126/science.7886457. [DOI] [PubMed] [Google Scholar]
- Katoh-Semba R, Takeuchi IK, Semba R, Kato K. Distribution of brain-derived neurotrophic factor in rats and its changes with development in the brain. Journal of Neurochemistry. 1997;69:34–42. doi: 10.1046/j.1471-4159.1997.69010034.x. [DOI] [PubMed] [Google Scholar]
- Katz LC, Shatz CJ. Synaptic activity and the construction of cortical circuits. Science. 1996;274:1133–1138. doi: 10.1126/science.274.5290.1133. [DOI] [PubMed] [Google Scholar]
- Kimura F, Otsu Y, Tsumoto T. Presynaptically silent synapses: Spontaneously active terminals without stimulus-evoked release demonstrated in cortical autapses. Journal of Neurophysiology. 1997;77:2805–2815. doi: 10.1152/jn.1997.77.5.2805. [DOI] [PubMed] [Google Scholar]
- Kinoshita S, Yasuda H, Taniguchi N, Katoh-Semba R, Hatanaka H, Tsumoto T. Brain-derived neurotrophic factor prevents low-frequency inputs from inducing long-term depression in the developing visual cortex. Journal of Neuroscience. 1999;19:2122–2130. doi: 10.1523/JNEUROSCI.19-06-02122.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkwood A, Dudek SM, Gold JT, Aizenman CD, Bear MF. Common forms of synaptic plasticity in the hippocampus and neocortex. Science. 1993;260:1518–1521. doi: 10.1126/science.8502997. [DOI] [PubMed] [Google Scholar]
- Knüssel B, Hefti F. K-252 compounds: modulator of neurotrophin signal transduction. Journal of Neurochemistry. 1992;59:1987–1996. doi: 10.1111/j.1471-4159.1992.tb10085.x. [DOI] [PubMed] [Google Scholar]
- Korte M, Carroll P, Wolf E, Brem G, Thoenen H, Bonhoeffer T. Hippocampal long-term potentiation is impaired in mice lacking brain-derived neurotrophic factor. Proceedings of the National Academy of Sciences of the USA. 1995;92:8856–8860. doi: 10.1073/pnas.92.19.8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumura E, Kimura F, Taniguchi N, Tsumoto T. Brain-derived neurotrophic factor prevents induction of long-term depression in cultured solitary neurons. Japanese The Journal of Physiology. 1999. supplement (in the Press) [DOI] [PMC free article] [PubMed]
- Leßmann V, Heumann R. Modulation of unitary glutamatergic synapses by neurotrophin-4/5 or brain-derived neurotrophic factor in hippocampal microcultures: presynaptic enhancement depends on pre-established paired-pulse facilitation. Neuroscience. 1998;86:399–413. doi: 10.1016/s0306-4522(98)00035-9. [DOI] [PubMed] [Google Scholar]
- Leßmann V, Kottmann K, Heumann R. BDNF and NT-4/5 enhance glutamatergic synaptic transmission in cultured hippocampal neurones. NeuroReport. 1994;6:21–25. doi: 10.1097/00001756-199412300-00007. [DOI] [PubMed] [Google Scholar]
- Levine ES, Dreyfus CF, Black IB, Plummer MR. Brain-derived neurotrophic factor rapidly enhances synaptic transmission in hippocampal neurons via postsynaptic tyrosine kinase receptor. Proceedings of the National Academy of Sciences of the USA. 1995;92:8074–8077. doi: 10.1073/pnas.92.17.8074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y-X, Zhang Y, Lester HA, Schuman EM, Davidson N. Enhancement of neurotransmitter release induced by brain-derived neurotrophic factor in cultured hippocampal neurons. Journal of Neuroscience. 1998;15:10231–10240. doi: 10.1523/JNEUROSCI.18-24-10231.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissin DV, Carroll RC, Nicoll RA, Malenka RC, Zastrow M. Rapid, activation-induced redistribution of ionotropic glutamate receptors in cultured hippocampal neurons. Journal of Neuroscience. 1999;19:1263–1272. doi: 10.1523/JNEUROSCI.19-04-01263.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lohof AM, Ip NY, Poo M-M. Potentiation of developing neuromuscular synapses by the neurotrophins NT-3 and BDNF. Nature. 1993;363:350–353. doi: 10.1038/363350a0. [DOI] [PubMed] [Google Scholar]
- McAllister AK, Katz LC, Lo DC. Neurotrophins and synaptic plasticity. Annual Review of Neuroscience. 1999;22:295–318. doi: 10.1146/annurev.neuro.22.1.295. [DOI] [PubMed] [Google Scholar]
- Nawa H, Carnahan J, Gall C. BDNF protein measured by a novel enzyme immunoassay in normal brain and after seizure: partial disagreement with mRNA levels. European Journal of Neuroscience. 1995;7:1527–1535. doi: 10.1111/j.1460-9568.1995.tb01148.x. [DOI] [PubMed] [Google Scholar]
- Patterson SL, Abel T, Deuel TAS, Martin KC, Rose JC, Kandel ER. Recombinant BDNF rescues deficits in basal synaptic transmission and hippocampal LTP in BDNF knockout mice. Neuron. 1996;16:1137–1145. doi: 10.1016/s0896-6273(00)80140-3. [DOI] [PubMed] [Google Scholar]
- Scharfman HE. Hyperexcitability in combined entorhinal/hippocampal slices of adult rat after exposure to brain-derived neurotrophic factor. Journal of Neurophysiology. 1997;78:1082–1095. doi: 10.1152/jn.1997.78.2.1082. [DOI] [PubMed] [Google Scholar]
- Segal M. Epileptiform activity in microcultures containing one excitatory hippocampal neuron. Journal of Neurophysiology. 1991;65:761–770. doi: 10.1152/jn.1991.65.4.761. [DOI] [PubMed] [Google Scholar]
- Singer W. Development and plasticity of cortical processing architectures. Science. 1995;270:758–764. doi: 10.1126/science.270.5237.758. [DOI] [PubMed] [Google Scholar]
- Stoop R, Poo M-M. Synaptic modulation by neurotrophic factors: differential and synergistic effects of brain-derived neurotrophic factor and ciliary neurotrophic factor. Journal of Neuroscience. 1996;16:3256–3264. doi: 10.1523/JNEUROSCI.16-10-03256.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T, Saito H, Matsuki N. Inhibition of GABAA synaptic responses by brain-derived neurotrophic factor (BDNF) in rat hippocampus. Journal of Neuroscience. 1997;17:2959–2966. doi: 10.1523/JNEUROSCI.17-09-02959.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapley P, Lamballe F, Barbacid M. K252a is a selective inhibitor of the tyrosine protein kinase activity of the trk family of oncogenes and neurotrophin receptors. Oncogene. 1992;7:371–381. [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Torii N, Tsumoto T, Uno L, Astrelin AV, Voronin LL. Quantal analysis suggests presynaptic involvement in expression of neocortical short- and long-term depression. Neuroscience. 1997;79:317–321. doi: 10.1016/s0306-4522(97)00129-2. [DOI] [PubMed] [Google Scholar]
- Tsumoto T. Long-term potentiation and long-term depression in the neocortex. Progress in Neurobiology. 1992;39:209–228. doi: 10.1016/0301-0082(92)90011-3. [DOI] [PubMed] [Google Scholar]
- Tsumoto T, Suda K. Cross-depression: an electrophysiological manifestation of binocular competition in the developing visual cortex. Brain Research. 1979;168:190–194. doi: 10.1016/0006-8993(79)90138-0. [DOI] [PubMed] [Google Scholar]
- Yan Q, Radeke MJ, Matheson CR, Talvenheimo J, Welcher AA, Feinstein SC. Immunocytochemical localization of TrkB in the central nervous system of the adult rat. Journal of Comparative Neurology. 1997;378:135–157. [PubMed] [Google Scholar]