

Abstract
A digital imaging microscope with fura-2 as the Ca2+ indicator was used to determine the sources for the rise in intracellular calcium concentration ([Ca2+]i) that occurs when the membrane in a cell-attached patch is stretched. Unitary ionic currents from stretch-activated channels and [Ca2+]i images were recorded simultaneously.
When suction was applied to the patch pipette to stretch a patch of membrane, Ca2+-permeable cation channels (stretch-activated channels) opened and a global increase in [Ca2+]i occurred, as well as a greater focal increase in the vicinity of the patch pipette. The global changes in [Ca2+]i occurred only when stretch-activated currents were sufficient to cause membrane depolarization, as indicated by the reduction in amplitude of the unitary currents.
When Ca2+ was present only in the pipette solution, just the focal change in [Ca2+]i was obtained. This focal change was not seen when the contribution from Ca2+ stores was eliminated using caffeine and ryanodine.
These results suggest that the opening of stretch-activated channels allows ions, including Ca2+, to enter the cell. The entry of positive charge triggers the influx of Ca2+ into the cell by causing membrane depolarization, which presumably activates voltage-gated Ca2+ channels. The entry of Ca2+ through stretch-activated channels is also amplified by Ca2+ release from internal stores. This amplification appears to be greater than that obtained by activation of whole-cell Ca2+ currents. These multiple pathways whereby membrane stretch causes a rise in [Ca2+]i may play a role in stretch-induced contraction, which is a characteristic of many smooth muscle tissues.
While stretching smooth muscle has been known for quite some time to induce contraction (Bülbring, 1955; Burnstock & Prosser, 1960; Fay, 1975; Himpens & Somlyo, 1988), the mechanism responsible for linking stretch to contraction is not known. There are a number of possibilities for the initial mechano-sensor involved in this process. For example, stretch-activated non-selective cation channels (Kirber et al. 1988; Davis et al. 1992; Wellner & Isenberg, 1993) and volume-regulated chloride channels (Nelson, 1998; Yamazaki et al. 1998) are two of many possible mechano-sensors. Here we consider the role of stretch-activated non-selective cation channels, which are evident from recordings of both whole-cell (Davis et al. 1992; Wellner & Isenberg, 1994, 1995; Setoguchi et al. 1997) and single channel currents (e.g. Kirber et al. 1988; Davis et al. 1992; Wellner & Isenberg, 1993), in various types of smooth muscle. This type of channel is also found in a wide variety of other cell types (see Morris, 1990; Sachs, 1992).
Opening of these stretch-activated non-selective cation channels in smooth muscle in response to membrane stretch presumably leads to contraction by elevating intracellular Ca2+ concentration ([Ca2+]i), but exactly how opening of these channels might cause [Ca2+]i to rise remains to be determined. For example, positive charge flowing into the cell resulting from opening of these channels might cause membrane depolarization and a consequent activation of voltage-gated Ca2+ channels. However, in addition, or perhaps alternatively, opening of these stretch-activated cation channels, which have been shown to be permeable to Ca2+ (Kirber et al. 1988), might allow enough Ca2+ to enter the cell to directly elevate [Ca2+]i sufficiently for contraction to occur and/or to trigger Ca2+ release from internal stores. Moreover, the local rise in [Ca2+]i might also spread from the region of the cell which is stretched to other regions by triggering additional release from these stores. As each of these mechanisms allows specific predictions to be made about the relationship between the area of the cell membrane subject to stretch and the time course and spatial extent of [Ca2+]i changes, we investigated the mechanisms linking membrane stretch to a rise in [Ca2+]i using a combination of [Ca2+]i imaging and patch-clamp methods.
We provide evidence consistent with two mechanisms whereby stretch-activated channels increase [Ca2+]i: by activation of voltage-gated Ca2+ channels via membrane depolarization and by significant amplification of stretch-activated channel Ca2+ influx by internal Ca2+ stores. Preliminary reports of some of our findings have been published in abstract form (Guerrero et al. 1993, 1994b; Kirber et al. 1994, 1995).
METHODS
Experimental preparation
Single smooth muscle cells were isolated from the stomach of the toad Bufo marinus by procedures described previously (Lassignal et al. 1986; Fay et al. 1992). Toads were killed by decapitation according to the guidelines of the Animal Care Committee of the University of Massachusetts Medical School and tissue was collected. When recording stretch-activated channels, cells were loaded with the acetoxymethylester of fura-2 (fura-2 AM) by incubating them for at least 1 h at room temperature in approximately 200 nM fura-2 AM in the final dissociation solution. A small aliquot of cells in this solution was then added to the bathing solutions indicated below. Agents such as caffeine or Gd3+ in the bathing solution were usually applied to the cell by pressure ejection from a micropipette (Lassignal et al. 1986). See the inset to Fig. 5B for the recording and application configuration.
Figure 5. When the cell is treated so as to deplete Ca2+ from internal stores, the focal changes in [Ca2+]i resulting from opening of stretch-activated channels are diminished.
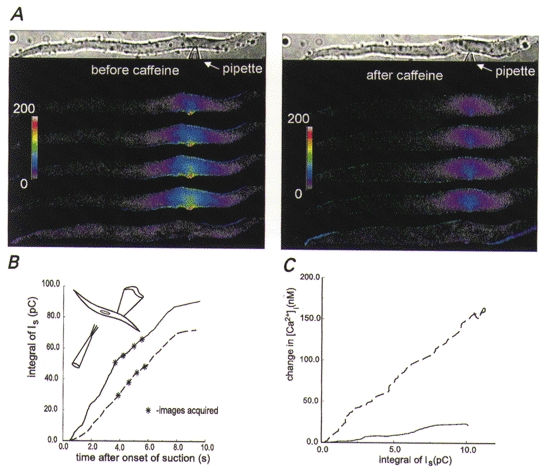
A, pseudocoloured images of changes in [Ca2+]i obtained by local stretching of the membrane of a smooth muscle cell before (left panel) and about 2 min after (right panel) caffeine (20 mM) was applied to the cell to induce Ca2+ release from stores. The cell was bathed in the usual solution except that it was devoid of Ca2+ (0 Ca2+, 0.2 mM BAPTA) but contained 100 nM thapsigargin to block reuptake of Ca2+ by the sarcoplasmic reticulum Ca2+ pumps. The pipette solution contained physiological levels of Ca2+. In order to facilitate comparisons of the responses to membrane stretch, the increment in [Ca2+]i above pre-stretch levels is displayed. As expected, this cell contracted transiently in response to the release of Ca2+ induced by caffeine. When the cell re-extended and the [Ca2+]i returned to resting levels, the patch of membrane was again stretched to produce the images on the right, now with the stores presumably depleted, showing a diminished focal change in [Ca2+]i. The optical configuration was as in Fig. 1 except for the addition of a liquid-filled light guide, and the experimental configuration is shown in the inset in B. The colour bar provides a linear scale for the change in [Ca2+]i from 0 to 200 nM. B, time course of the cumulative (integrated) stretch-activated currents for the cell shown in A. The dashed and continuous lines show the integrated stretch-activated channel currents (total charge influx) before and after caffeine application, respectively. Note that even though the integrated current induced by membrane stretch is slightly greater after caffeine application, the [Ca2+]i changes are smaller. C, prolonged exposure of a different smooth muscle cell to a Ca2+-free bathing solution decreases the change in [Ca2+]i due to stretch-activated current. The dashed and continuous curves show, respectively, the relationship between the cumulative stretch-activated current and changes in [Ca2+]i observed after 5 and 30 min of exposure to a Ca2+-free bathing solution containing 200 μM BAPTA. The vertical distance between the curves at any point along the x-axis represents the decrease in the change in [Ca2+]i due to a given total charge influx, presumably because of the gradual loss of store amplification. Ca2+ was present at the usual levels in the patch pipette solution. [Ca2+]i changes were recorded from a 30 μm long region of the cell centred around the patch pipette using the FDWM at a frequency of 150 Hz throughout the 3.2 s that the membrane was stretched.
Calcium measurements: high-speed digital imaging
Pairs of images of the fura-2 fluorescence emission (500 nm long pass filter) excited at 351 and 380 nm were acquired with an argon ion laser (Coherent Inc., Santa Clara, CA, USA) illuminated microscope system described previously (Isenberg et al. 1996; Etter et al. 1996). Exposures at each wavelength were typically 15 ms, the time between the two images in each pair was about 4 ms, and the time between image pairs was 100–500 ms. All images were background and dark current subtracted. In order to compensate for minor inhomogeneities in illumination due to interference patterns generated by passing the coherent light through the optics of the microscope, [Ca2+]i was computed by measuring the fluorescence ratio and comparing it to the ratio at each point in the cell measured at rest. Assuming that [Ca2+]i at rest is uniformly distributed within the cell and using a value for the resting [Ca2+]i of 50 nM, the change in [Ca2+]i in response to stretch-activated channel activity was computed (Isenberg et al. 1996; Etter et al. 1996) from:
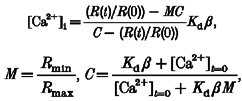 |
where R(t) is the ratio of the fluorescence excited at 351 nm to that excited at 380 nm. R(0) is the resting ratio (t = 0); Kd = 200 nM; β, Rmin and Rmax are constants according to Grynkiewicz et al. (1985). In addition, for some experiments a liquid-filled light guide was inserted in the illuminating pathway to provide more even illumination. The resting value of 50 nM for [Ca2+]i was based on values from fura-2 measurements obtained in the Ca2+-free bathing solution (see below) used in this study (46.8 ± 3.7 nM; n = 12) and obtained previously under voltage clamp in a 1.8 mM Ca2+-containing bathing solution (Guerrero et al. 1994a).
The increase in [Ca2+]i over the resting level beneath the patch pipette and at a longitudinal distance of 30 μm from the pipette was obtained by measuring (usually in the image with the largest increase in [Ca2+]i) the fluorescence ratio in a 3 μm × 3 μm area (comprising 100 pixels with a pixel size of 0.3 μm) of the cell at each location and subtracting the estimated resting [Ca2+]i of 50 nM. All measurements are expressed as the mean ± s.e.m., with n referring to the number of cells.
Fura-2 fluorescence images of cells where Ca2+ was present in the pipette and bathing solution were analysed for those cells where membrane stretch caused an elevation in the [Ca2+]i of at least 11 nM above the resting [Ca2+]i. This minimal required change in Ca2+ was chosen as it corresponds to 2.5 times the standard deviation of the mean change in [Ca2+]i measured in 12 cells 30 μm away from the pipette when there was Ca2+ present only in the pipette solution, a situation where no Ca2+ entry occurs other than through the membrane directly under the patch pipette. For some cells, stretching the membrane caused little or no obvious change in the [Ca2+]i. The absence of a response might reflect the health of the cell affecting the resting potential and/or the state of the internal stores, or perhaps the location of the membrane patch in the patch pipette.
Calcium measurements: photometric measurements of fluorescence ratios
Some experiments were carried out using a fast dual-wavelength microfluorimeter (FDWM) for recording the spatial average of the changes in [Ca2+]i with higher time resolution than could be obtained with digital imaging. Details of this technique have been described previously (Yagi et al. 1988; Guerrero et al. 1994a, c). Essentially, fura-2 fluorescence (centred at 510 nm) was collected from a masked area of the cell illuminated by a rotating filter wheel containing transmissive partitions passing either mainly 340 nm or mainly 380 nm light.
When we used this technique to determine the effect of internal stores on the increase in [Ca2+]i resulting from activation of voltage-gated Ca2+ channels, we plotted the time course of the actual change in [Ca2+]i measured with fura-2 against the integral of the inward current over time (proportional to the expected change in [Ca2+]i) obtained from the whole-cell current record. The slope (in nM pC−1) provided a measure of the relationship between Ca2+ influx and the rise in [Ca2+]i. Voltage-gated Ca2+ channels were activated by an 80 mV depolarizing voltage jump from a holding potential of −80 mV using conditions described previously (Guerrero et al. 1994c). The integral of the inward current was obtained using the steady-state current at the end of the depolarizing voltage jump as a baseline (K+ channels were blocked with internal Cs+ and external TEA+). For experiments where ryanodine was added to the pipette solution (100 μM) at least 5 min passed after breaking into the cell before data were collected. This was shown to be sufficient to remove the effect of caffeine on Ca2+ release from intracellular stores (Guerrero et al. 1994a, c; Xu et al. 1994).
A similar relationship was obtained between the measured changes in [Ca2+]i and the integral of the stretch-activated inward current. But, because the changes in [Ca2+]i occurred more slowly in this case, we corrected the increase in [Ca2+]i for Ca2+ removal (or extrusion) from the cytosol using a removal half-time (t1/2) of 1.7 s (Guerrero et al. 1994c). These experiments with stretch-activated channel currents (recorded from cell-attached patches) were carried out with Ca2+ in the patch pipette and either Ca2+ absent from the bath or with Gd3+ present.
When we used the FDWM to follow the continued rise in [Ca2+]i that sometimes occurred after stretch-activated channel activity ceased, the end of the rise was taken as the time where the rate of change of [Ca2+]i was statistically zero, based on it being less than 2.5 times the standard deviation of the noise measured in the absence of suction. These calculations were not corrected for Ca2+ removal, which would have caused the rise to be even longer. Thus, our calculations here provided an underestimate of the time course of the rise.
Recording of stretch-activated currents
Stretch-activated channel openings in cell-attached patches were obtained by applying negative pressure (suction) to the patch pipette (Kirber et al. 1988). Suction, which was monitored with a Sensym pressure transducer (Sunnyvale, CA, USA), was continually adjusted using visual feedback of stretch-activated channel activity in order to keep the activity relatively constant and at a low enough level to prevent the cell from contracting, which could also lead to excision of the patch. Thus, peak [Ca2+]i changes observed were near 300 nM, a level which usually prevented contraction. Patch pipette and bathing solutions usually contained (mM): 127 NaCl, 3 KCl, 1 MgCl2, 10 Hepes (pH 7.4) and either 1.8–1.9 CaCl2, or 0 CaCl2 and 0.2 BAPTA, designated, respectively, as Ca2+-containing (physiological concentrations) or 0 Ca2+ (Ca2+-free) solutions. Because we used cell-attached patches for these experiments, the actual patch membrane potential was not known, although it could be estimated from the amplitude of the unitary currents. Experiments were usually carried out at patch membrane potentials 100 mV more negative than the resting potential (unknown) of the cell to increase the driving force and therefore the magnitude of the unitary stretch-activated currents. While in most of these experiments we have added driving force to the inward currents in order to maximize the effects as well as provide clearer recordings of unitary currents, we have also checked the focal response (physiological concentrations of Ca2+ in the pipette only) with the pipette potential set at 0 mV (therefore, with the patch at the resting potential) and obtained qualitatively consistent but smaller changes in [Ca2+]i. To assure ourselves that excision of the patch did not occur, at times we either ruptured the patch to see if the cell contracted when there was Ca2+ in the pipette or checked for inward stretch-activated currents when the pipette potential was set to 0 mV.
These experiments were carried out with cell-attached patches under conditions where the membrane potential was not held constant. Currents passing through stretch-activated channels were often sufficient to cause a change in the whole-cell membrane potential with these high resistance (over a gigaohm) smooth muscle cells. Since the amplitude of the unitary current, i = γ(Vm – Vp – Vc) (where γ is the unitary conductance, Vm is the cell membrane potential, Vp is the pipette potential, Vc is the stretch-activated channel reversal or null potential which is near 0 mV, and (Vm – Vp) is the patch membrane potential), this change in cell membrane potential would be reflected in the recorded unitary current amplitude for the cell-attached patch recording configuration used here (see Fenwick et al. 1982; Barry & Lynch, 1991).
RESULTS
Effect of membrane stretch on [Ca2+]i: general characteristics
When the membrane of a cell-attached patch is stretched, mechano-sensitive cation channels in the patch open (Fig. 1). With physiological levels of Ca2+ present in both the pipette and the bathing solutions, the opening of these stretch-activated channels causes [Ca2+]i to increase. Out of a sample of 16 cells, two cells exhibited only a global increase in [Ca2+]i. In the other 14 cells, however, a focal increase in [Ca2+]i occurred in the area near the tip of the patch pipette. In six of these 14 cells exhibiting a clear focal response, a global rise in [Ca2+]i also occurred, with the focal increase around the patch pipette being higher than that which occurred in the rest of the cell. The global changes in [Ca2+]i were observed only when depolarization of the plasma membrane occurred, as indicated by a reduction in the amplitude of the stretch-activated channel unitary currents (Fig. 1). These results may be explained as follows. The global [Ca2+]i changes were due to Ca2+ entry through voltage-gated Ca2+ channels outside of the region of the patch. Activation of these Ca2+ channels would be caused by depolarization of the cell membrane resulting from the influx of cations through the stretch-activated channels in the small patch of membrane which was being stretched. The focal changes in [Ca2+]i were due to the localized entry of Ca2+ through the stretch-activated channels which we have shown previously (Kirber et al. 1988) to be permeable to Ca2+. Several experiments were carried out to determine if this explanation could be correct.
Figure 1. Stretch-induced opening of stretch-activated channels in a localized region of the membrane causes both global and focal increases in [Ca2+]i.
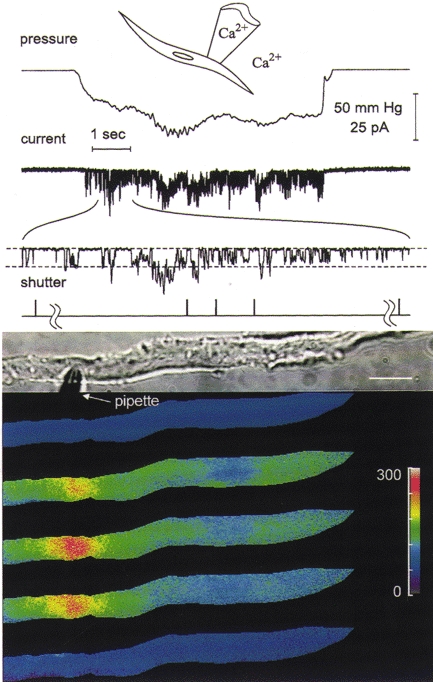
Images of [Ca2+]i from fura-2 AM-loaded cells were obtained using a UV laser-illuminated digital imaging microscope (Isenberg et al. 1996). The drawing at the top of this and the next three figures depicts the recording configuration and whether Ca2+ was present in the pipette or bathing solution. The traces shown above the images are from top to bottom: the output of a pressure transducer indicating the level of negative pressure or suction applied to the patch pipette, the stretch-activated channel currents obtained by application of suction to the patch pipette with an expanded ≈1 s section of the record below, and trigger pulses from the laser shutter(s) indicating when the images were obtained. To help follow the decline in unitary current amplitude with channel activity, dashed lines in the expanded section of the current trace are used to indicate the baseline and unitary current amplitude for the channels opening at the beginning of the expanded trace. The images of [Ca2+]i were obtained as previously described (Isenberg et al. 1996) and pseudocoloured, with the colour bar providing a linear scale for [Ca2+]i from 0 to 300 nM. A transmitted light image of the cell and pipette are shown above the pseudocoloured images (scale bar, ≈15 μm). A pair of dual wavelength images was acquired shortly before the onset of suction and the resting [Ca2+]i set to a value of 50 nM to give the top pseudocoloured image. Three sequential pairs of images were obtained during the application of suction, and a final pair of images was obtained approximately 30 s after cessation of suction and was used to generate the pseudocoloured image at the bottom. The hash marks in the shutter trace are used to set off the shutter pulses for the pair of images before the onset and after the cessation of suction. For this experiment the patch membrane potential was set to 100 mV more negative than the resting potential. The format used here is similar to the format used for the next four figures.
Mechanisms responsible for the changes in [Ca2+]i
When we removed Ca2+ from the patch pipette solution, stretching the patch still caused a rise in [Ca2+]i. However, in this case the increase in [Ca2+]i under the patch pipette was not significantly higher than that in other regions of the cell, as illustrated in Fig. 2. Similar results were obtained in four other cells. To obtain a measure of the extent of localization of the change in [Ca2+]i near the patch pipette, we determined the ratio of the change in [Ca2+]i over resting levels directly beneath the pipette to that 30 μm away along the longitudinal axis of the cell. With Ca2+ only in the bathing solution (for example, as in Fig. 2), we found the ratio to be 1.06 ± 0.04 (n = 5), consistent with the lack of localization seen in the image. By contrast, when Ca2+ was present in the pipette and the bathing solutions and when combined focal and global responses were obtained during membrane stretch (for example, as in Fig. 1), the average ratio of the change in [Ca2+]i at the location of the patch to that 30 μm away was 2.5 ± 0.5 (n = 6).
Figure 2. Opening of stretch-activated channels causes only a global increase in [Ca2+]i when the pipette solution at the extracellular surface of the channels is Ca2+ free (0 Ca2+, 0.2 mM BAPTA).
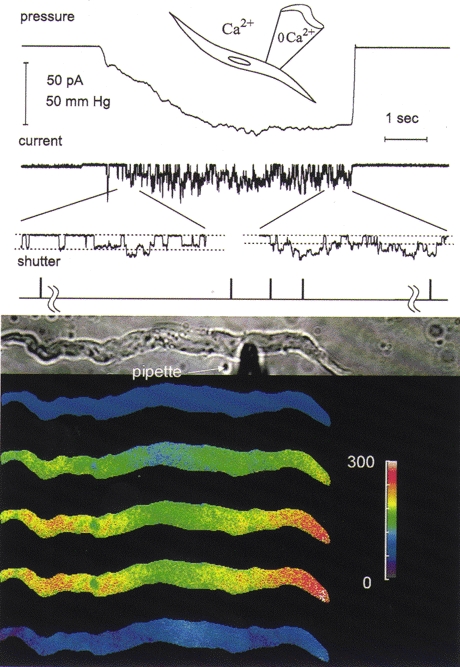
The results presented here are from the same type of experiment as in Fig. 1 except that Ca2+ was omitted from the pipette solution and 0.2 mM BAPTA was added to it. Note the reduction in the unitary current amplitude in the expanded current record (each ≈0.5 s in duration), indicative of membrane depolarization during continued membrane stretch. In the absence of Ca2+ in the pipette, stretch-activated unitary currents are larger (Kirber et al. 1988). The region near the centre of the cell where [Ca2+]i appears to rise less during the stretch and where after cessation of stretch [Ca2+]i remains slightly elevated is occupied by the nucleus. The colour bar provides a linear scale for [Ca2+]i from 0 to 300 nM.
For these toad stomach smooth muscle cells with a relatively high input resistance (over a gigaohm), the levels of current that could be obtained with the opening of stretch-activated channels in the cell-attached patch should cause sufficient whole-cell membrane depolarization so that the membrane potential would be in the range where measurable whole-cell Ca2+ currents are seen (Clapp et al. 1987). For example, for the cell shown in Fig. 2, we estimate from the channel conductance of 64 pS (Kirber et al. 1988) and the change in the amplitude of the unitary currents of 2 pA or more that membrane stretch produced at least a 30 mV depolarization. This depolarization from an estimated resting potential of −56 mV (Yamaguchi et al. 1988) would be sufficient to activate voltage-gated calcium channels in these cells (Clapp et al. 1987) which could then cause the observed global change in [Ca2+]i.
When we removed Ca2+ from the bathing solution but kept it in the pipette solution (Fig. 3A and B), the global rise in [Ca2+]i seen following stretching of the cell membrane did not occur (even when there was evidence for depolarization), while the focal increase in the region of the pipette tip was still present. In this case the average increase in [Ca2+]i 30 μm away from the patch pipette (13.5 ± 1.3 nM; n = 12) was barely above fluctuations in the resting level for these cells (see the Methods section). When Ca2+ was included in the pipette and bathing solutions and evidence for depolarization was observed, as indicated by the decrease in unitary current, a combined focal and global response was characteristic (for example, see Fig. 1). In cells exhibiting this combined response, [Ca2+]i 30 μm away from the pipette rose significantly (94.1 ± 25 nM; n = 6; P < 0.001, Student's t test). With Ca2+ absent from the bath, the average ratio of the increase in [Ca2+]i near the pipette to that observed 30 μm away was 9.7 ± 1.5 (n = 12), considerably larger, as expected, than for those cells where global responses were obtained when Ca2+ was present everywhere.
Figure 3. Only a focal increase in [Ca2+]i occurs when stretch-activated channels open and Ca2+ is present in the pipette solution but the bathing solution is Ca2+ free (0 Ca2+, 0.2 mM BAPTA).
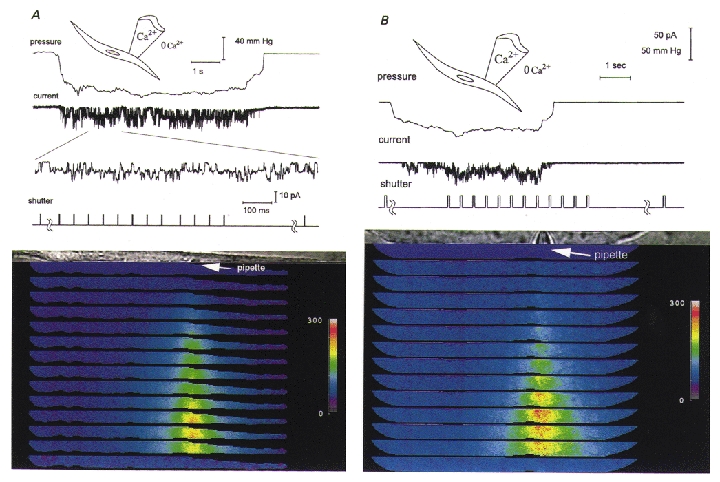
In order to follow [Ca2+]i changes with greater time resolution both during and after cessation of membrane stretch, the optics of the digital imaging microscope were reconfigured to allow the rapid acquisition of 12 rather than 3 image pairs by dividing the charge coupled device chip so that each of the images shown subtended an area of the field that is 150 mm × 6 μm rather than 150 mm × 22 μm as in Figs 1 and 2. In all other respects except for the absence of Ca2+ in the bathing solution, this experiment is similar to that depicted in Fig. 1, with the colour bars providing a linear scale for Ca2+ from 0 to 300 nM. Panels A and B are from two different cells. For panel A the set of images was taken from the time the channels opened, while for panel B the images were taken sometime after the channels opened but also during the period of time after channel activity ceased. Note in panel B the longer delay in the [Ca2+]i increase after the onset of channel activity and the increase in [Ca2+]i after membrane stretch and stretch-activated channel activity ceased (see text).
Similar results were obtained in another set of experiments using the FDWM, where a solution identical to the bathing solution but containing 100 μM Gd3+ was applied to the cell to block Ca2+ entry. This addition of Gd3+ to the Ca2+-containing bath solution (but not to the patch pipette solution) clearly decreased the rise in [Ca2+]i detected (n = 6; data not shown). This would be expected if Ca2+ influx across the membrane outside the patch pipette was contributing to the measured rise in [Ca2+]i. Gd3+ would block voltage-gated calcium channels in these cells (caption of Fig. 5 in Guerrero et al. 1994a), as well as stretch-activated non-selective cation channels (Yang & Sachs, 1989; Hisada et al. 1991; Wellner & Isenberg, 1993, 1994) in the cell membrane not isolated by the patch pipette.
When Ca2+ was present only in the patch pipette solution and we set the membrane potential of the cell-attached patch to positive potentials (120 mV more positive than the cell resting potential) to reverse the driving force for total cation influx and reduce that for Ca2+ influx, there was almost no change in the overall cell [Ca2+]i or in its spatial distribution compared to resting levels when suction was applied (Fig. 4). The change in [Ca2+]i in the region of the patch pipette was only 26.2 ± 5.8 nM (n = 6), which was significantly smaller (P < 0.01, t test) than the increase (126.6 ± 22.1 nM; n = 12) observed at a patch potential 100 mV more negative than the resting potential. Under this condition of reduced driving force for Ca2+ influx, the change in [Ca2+]i measured 30 μm away (8.7 ± 1.5 nM; n = 6) was almost the same as that measured at the patch pipette.
Figure 4. The focal increase in [Ca2+]i is nearly abolished when stretch-activated channels open with the inward driving force on Ca2+ greatly reduced.
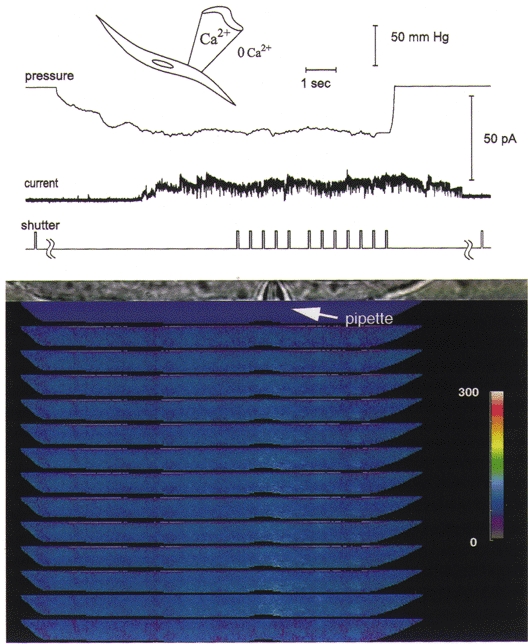
When the patch pipette potential is changed to −120 mV so that the patch membrane potential is 120 mV more positive than the resting potential, the stretch-activated channel currents are outward and the driving force for Ca2+ influx is greatly reduced. There is almost no change in [Ca2+]i under these conditions, indicating that an influx of Ca2+ is required for the observed focal increase at negative patch potentials. When the membrane potential was then returned to 100 mV more negative than the resting potential, the focal elevation in [Ca2+]i was again seen with the opening of stretch-activated channels, as shown in Fig. 3B. The outward currents would cause the whole-cell membrane potential to become more negative, decreasing the outward driving force for the unitary currents, so the unitary current amplitude declines with time. Because at positive patch potentials the stretch-activated channel openings are of longer duration (Kirber et al. 1988), this decline in amplitude is reflected in the unitary current while the channel is still open, giving rise to the decay in the current (see Barry & Lynch, 1991). In addition, there may be a contribution to the current record by stretch-activated K+ channels, which can continue to be active after membrane stretch has ceased (Ordway et al. 1995), as can, at times, the stretch-activated cation channels. The experimental conditions were the same as for Fig. 3. The colour bar provides a linear scale for [Ca2+]i from 0 to 300 nM.
It is unlikely that the stretch-activated channels outside the membrane contained within the patch pipette contribute to the global rise in [Ca2+]i. We base this conclusion on our findings from cell-attached patch experiments using the FDWM where calcium was present in both the pipette and bathing solutions. When the patch membrane potential was set at positive potentials (100 mV more positive than the resting potential), suction still caused stretch-activated channels to open (with outward currents), but there was essentially no change in [Ca2+]i. In the same cell, when the patch membrane potential was held at negative potentials a change in [Ca2+]i could be detected when the cell-attached patch was stretched (n = 5).
The results described above indicate that one mechanism whereby stretching the entire smooth muscle cell might trigger contraction is as follows: opening of stretch-activated non-selective cation channels, with the resulting influx of cations, causes sufficient depolarization to activate voltage-gated sarcolemmal Ca2+ channels, allowing extracellular Ca2+ to enter the cell (Kirber et al. 1988; Davis et al. 1992; Wellner & Isenberg, 1993, 1994).
Mechanism responsible for the larger focal change in [Ca2+]i – role for internal stores
While membrane depolarization can apparently account for the global increase in [Ca2+]i seen following localized application of stretch to a patch of smooth muscle cell membrane, it cannot explain why there is a larger focal increase directly beneath the pipette. Thus, a second mechanism, which depends on Ca2+ influx through the stretch-activated channels, must be involved. Might the local rise in [Ca2+]i simply reflect Ca2+ entry through the stretch-activated channel(s) or could this Ca2+ entry be amplified by additional Ca2+ released into the cytosol from internal stores, as suggested from indirect experiments in cardiac muscle (Sigurdson et al. 1992)?
The images in Fig. 3B provide an example of the possible existence of some amplification mechanism where [Ca2+]i was sometimes seen to rise after the cessation of suction when stretch-activated channels were closed (also seen in 4 other cells). Moreover, using the FDWM, we found that, in eight cells where we made the measurement and where Ca2+ was present in the pipette but not in the bath, [Ca2+]i continued to rise for an average of 450 ± 72 ms after closure of the last open channel before starting to return to its resting level.
In order to test more directly for the possible involvement of intracellular Ca2+ stores, cells were treated in a number of ways to deplete Ca2+ from internal stores (or block Ca2+ release), and [Ca2+]i changes were monitored following application of suction to a patch pipette with physiological levels of Ca2+ in the pipette solution. The bathing solution was devoid of Ca2+ to facilitate analysis of the mechanisms linking local Ca2+ entry to the focal rise in [Ca2+]i. After incubation of smooth muscle cells in 100 μM ryanodine and 0.5 mM caffeine for at least 30 min, to ensure that the contribution of internal stores was eliminated, stretching the membrane under the patch pipette failed to cause a rise in [Ca2+]i, although clear inward currents were elicited. In eight cells subject to treatment with ryanodine plus caffeine for 30–120 min, the average change in [Ca2+]i beneath the patch pipette was 4.9 ± 10 nM, whereas in control cells not treated with ryanodine and caffeine, [Ca2+]i increased significantly by 126.6 ± 22.1 nM (n = 12).
This absence of a detectable rise in [Ca2+]i following membrane stretch might be due to the inability of stores to amplify Ca2+ entry that occurs through stretch-activated channels or, alternatively, the stores, if empty, might take up Ca2+ that enters the cytosol through stretch-activated channels more rapidly. In order to test for this latter possibility, we carried out another series of experiments in the absence of external Ca2+, emptying the stores using a brief exposure to caffeine (20 mM) but now in the presence of (100 nM) thapsigargin, which we have shown quite rapidly inhibits the Ca2+ uptake by the sarcoplasmic reticulum in these cells (Steenbergen & Fay, 1996). The comparison of the response to stretch in a single smooth muscle cell before (Fig. 5A, left panel) and then after emptying of Ca2+ stores with a brief pulse of caffeine (Fig. 5A, right panel) indicates that, following emptying of internal stores, activation of inward currents by membrane stretch was markedly less effective in producing an increase in [Ca2+]i (Fig. 5B). A similar reduction in the change in [Ca2+]i induced by stretch-activated current was observed using the digital imaging microscope in another cell acutely treated with caffeine. When this experiment was carried out using the FDWM to obtain a more continuous and longer term record of [Ca2+]i changes, depletion of internal stores with caffeine in the presence of thapsigargin reduced the relationship between the change in [Ca2+]i and the integral of the stretch-activated current from 10.0 ± 0.16 to 3.3 ± 0.35 nM pC−1 (n = 3). This result suggests that, at least under our recording conditions with limited stretch-activated channel activity, viable internal Ca2+stores may be necessary for a significant stretch-activated channel-mediated increase in [Ca2+]i to occur when depolarization-induced Ca2+ influx is eliminated.
In order to obviate possible non-specific effects associated with agents acting on internal stores, we carried out a final series of experiments with the FDWM to determine the effect of store depletion on the stretch-induced rise in [Ca2+]i simply by determining the effect of membrane stretch shortly after the removal of Ca2+ from the bath (Ca2+ was present in the patch pipette solution) and then again after prolonged incubation and repeated stretch activation in Ca2+-free medium to deplete internal stores (Murray et al. 1975). The [Ca2+]i change caused by stretch-activated channel currents decreased with time when the cell was bathed in a Ca2+-free medium (Fig. 5C). That is, the efficacy of stretch-activated currents in increasing [Ca2+]i was reduced from 11.4 ± 0.7 to 3.0 ± 0.47 nM pC−1 (n = 3) when cells were placed in a bathing solution which was Ca2+-free for at least 30 min.
Stretch-activated Ca2+ entry and Ca2+ entry due to voltage-gated Ca2+ channels have quantitatively different effects on Ca2+ amplification
While Ca2+ entry through stretch-activated channels appears to be strongly amplified by Ca2+ release from internal stores, voltage-clamp experiments reveal that Ca2+ entry through voltage-gated Ca2+ channels seems to be only slightly amplified by this mechanism. Using the FDWM, with 100 μM ryanodine in the pipette, a condition that eliminates the effect of caffeine-sensitive internal stores completely (Guerrero et al. 1994a,c; Xu et al. 1994), the relationship between the [Ca2+]i increase and the integral of the whole-cell inward current (in nM pC−1) was not significantly affected during the initial 100 ms after a voltage jump from -80 to 0 mV (12.8 ± 1.5 nM pC−1 (n = 17) and 11.7 ± 1.0 nM pC−1 (n = 18) in the presence and absence, respectively, of ryanodine). This occurred even though the integral of the inward current in the presence of ryanodine was 22.7 % smaller than in the absence of ryanodine. Moreover, when 100 μM ryanodine was present in the pipette solution, the expected (calculated from the inward current) and measured (calculated from fura-2 fluorescence) changes in [Ca2+]i at 500 ms were virtually identical (−2 ± 2 %, n = 13) (an example is shown in the lower set of traces in Fig. 6B). However, in the absence of ryanodine, at 500 ms after the onset of the voltage jump when significant inactivation of the Ca2+ current had occurred (Fig. 6A), the measured change in [Ca2+]i was larger than that expected from the inward current by 20 ± 2 % (n = 15) (an example is shown in the upper set of traces in Fig. 6B).
Figure 6. The contribution of Ca2+ stores to the rise in [Ca2+]i due to whole-cell Ca2+ currents occurs with a delay and is smaller than for stretch-activated channel currents.
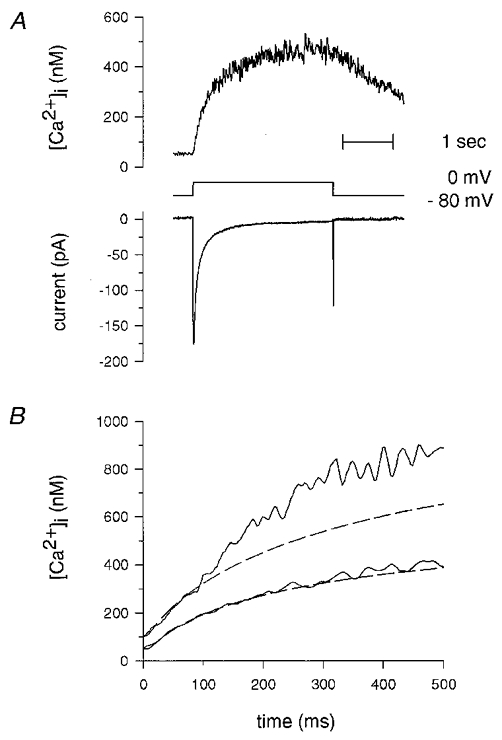
A, typical [Ca2+]i changes (upper trace) due to membrane depolarization to 0 mV from a holding potential of −80 mV (middle trace). The net inward current (lower trace) was obtained by subtracting the current obtained in the presence of 100 μM CdCl2 using the same voltage-clamp protocol. The experiment was carried out using the FDWM and simultaneous standard whole-cell current recording with CsCl in the pipette solution (see Guerrero et al. 1994c). A similar set of traces, but with 100 μM ryanodine in the pipette can be found in Fig. 1 of Guerrero et al. (1994c). B, assuming all of the inward current was carried by Ca2+, the ratio of the increase in [Ca2+]i to the integral of the Ca2+ current (slope, in nm pC−1) was obtained as described in the Methods section during the first 100 ms after the onset of depolarization from −80 mV to 0 mV. The integral of the Ca2+ current as a function of time was multiplied by its corresponding ratio (and added to the resting [Ca2+]i) to predict the resulting [Ca2+]i due to Ca2+ influx through voltage-gated Ca2+ channels for the time between 100 and 500 ms, and is plotted as dashed lines. The continuous lines show the actual [Ca2+]i measured with fura-2 using the FDWM. When ryanodine was present in the pipette solution, the inward current (and the slope, in nm pC−1, obtained during the first 100 ms) could predict the change in [Ca2+]i (lower set of traces). However, in the absence of ryanodine, the change in [Ca2+]i is higher than that predicted by the inward current (upper set of traces), in this case by 26 %. Representative traces are shown from two different cells with very similar slopes for the initial 100 ms after depolarization: 12.2 nm pC−1 in the presence (lower set of traces) and 13.1 nm pC−1 in the absence (upper set of traces) of ryanodine. The contribution of stores to the rise in [Ca2+]i due to Ca2+ currents appears to be proportionately less than that obtained with stretch-activated channel currents (compare with Fig. 5C).
On the other hand, a more significant effect of stores is obtained for the stretch-induced changes in [Ca2+]i. Using the FDWM in the absence of ryanodine in the bathing solution, the relationship between the [Ca2+]i increase and the integral of the stretch-activated current was 11.97 ± 1.4 nM pC−1 (n = 10), but it was reduced to 2.8 ± 0.3 nM pC−1 (n = 10) in the presence of ryanodine (100 μM). This suggests an amplification of as much as 325 % for the change in [Ca2+]i induced by stretch-activated currents.
DISCUSSION
Our results suggest that stretching of the toad stomach smooth muscle cell membrane increases [Ca2+]i by two major mechanisms. First, inward currents through stretch-activated channels can lead to depolarization of the whole-cell membrane, opening voltage-gated Ca2+ channels which in turn will cause a global change in [Ca2+]i. Second, Ca2+ influx through the stretch-activated channels themselves is amplified by triggering Ca2+ release from internal stores which can cause an elevation of [Ca2+]i in the region of the channels. The magnitude of the focal [Ca2+]i changes we observe will be a function of the total amount of Ca2+ entering the cell through stretch-activated channels, the rate of Ca2+ entry, and the state of the internal Ca2+ stores. The last will be affected by such factors as the time the cell was in a Ca2+-free bathing solution.
While the driving force for entry of Ca2+ through the stretch-activated channels in these experiments was considerably greater than expected in vivo, this mechanism for linking membrane stretch to a rise in [Ca2+]i nonetheless could be important physiologically. In the intact tissue, unlike the present experiments where only a patch of membrane is stretched, stretch is applied to the entire cell surface, which would be expected to result in significant whole-cell inward current through stretch-activated channels. (For examples of recordings of whole-cell current in smooth muscle cells see Wellner & Isenberg (1994, 1995) and Davis et al. (1992), where currents were obtained by stretching the cell membrane, and Setoguchi et al. (1997) who obtained whole-cell currents from inflation of the cell.) With these high-membrane-resistance smooth muscle cells, small steady net inward currents through stretch-activated channels could drive the membrane potential from the resting level into the appropriate range for steady voltage-gated Ca2+ channel window currents, thereby maintaining contraction. Moreover, Ca2+ stores, at least initially, could contribute to the elevation in [Ca2+]i. Our experiments with 0 Ca2+ or Gd3+ present in the bathing solutions and the observed requirement for membrane depolarization suggest that the global influx of Ca2+ most probably occurs through voltage-gated Ca2+ channels.
Role for internal Ca2+ stores in the elevation of [Ca2+]i from opening of stretch-activated channels
Our results clearly indicate that ryanodine-sensitive internal Ca2+ stores are involved in locally amplifying Ca2+ entry into the cytosol when localized stretches of cell membrane evoke low levels of stretch-activated channel activity in toad stomach smooth muscle cells. What is the linkage between membrane stretch and the release of Ca2+ from internal stores? Stretch of the membrane under conditions where little or no Ca2+ moves through stretch-activated channels is not sufficient to trigger a measurable local rise in [Ca2+]i, even when that stretch is accompanied by a significant inward current. Hence, membrane stretch does not appear to be linked to release of Ca2+ from internal stores by local changes in membrane potential, possible stretching of the sarcoplasmic reticulum membrane directly under the patch pipette, or the local generation of a second messenger acting on those stores independently of [Ca2+]i changes. Instead, it appears that stretching the plasma membrane triggers Ca2+ release from ryanodine-sensitive internal stores by a mechanism that at least in part requires a local rise in [Ca2+]i from Ca2+ passing through the stretch-activated channels.
Comparison of amplification of Ca2+ entry due to voltage-gated Ca2+ currents with that due to stretch-activated channel currents
Since Ca2+ entry through stretch-activated channels appears to be strongly amplified by ryanodine-sensitive Ca2+ release, it is unclear why the whole-cell current resulting from activation of voltage-gated Ca2+ channels caused a much smaller and consistently delayed amplification, on average about 20 % compared to between ∼200 % and ∼325 % for stretch-activated currents observed in the experiments using the FDWM (for an example of the latter see Fig. 5C).
One possibility for the difference in the degree of amplification is that the comparison being made is between the amplification that occurs with voltage-gated Ca2+ currents obtained from the whole-cell current recording configuration (most likely from Ca2+-induced Ca2+ release) and that due to unitary stretch-activated currents from a patch of membrane. The recording conditions could influence the results. First, unlike the recording of stretch-activated channel currents, for whole-cell recording of Ca2+ currents, Cs+ was present in the intracellular (pipette) solution to eliminate K+ currents and thereby allow isolation of the Ca2+ current. In guinea-pig coronary artery it has been suggested that Cs+ may block Ca2+-induced Ca2+ release from internal stores (Ganitkevich & Isenberg, 1995), and in rat cerebral artery only 30–50 % of the depolarization-induced Ca2+ transient could be attributed to Ca2+-induced Ca2+ release in the presence of Cs+ (Kamishima & McCarron, 1997), a percentage which, although higher, is in the range of the amplification we obtained for toad stomach. Therefore, the presence of Cs+ may have suppressed amplification in toad stomach smooth muscle cells. Second, more negative membrane potentials are associated with a higher gain of voltage-gated Ca2+ current-initiated Ca2+-induced Ca2+ release in rat heart and cerebral artery cells (Wier et al. 1994; Kamishima & McCarron, 1997), and for toad stomach cells the assessment of the degree of amplification was carried out at a more negative potential for stretch-activated channels and at 0 mV for the Ca2+ currents. Third, even when using the same whole-cell recording technique there is quite a bit of variability in the reported voltage-gated Ca2+ current-initiated Ca2+-induced Ca2+ release. There appears to be little or no Ca2+-induced Ca2+ release in equine airway smooth muscle (Fleischmann et al. 1996) or in rat portal vein (Kamishima & McCarron, 1996), but it appears to occur in rat cerebral arteries (Kamishima & McCarron, 1997), as discussed above, and in guinea-pig urinary bladder (Ganitkevich & Isenberg, 1992), vas deferens (Imaizumi et al. 1998) and mesenteric artery (Bolton & Gordienko, 1998). Therefore our observations in toad stomach cells may be in line with the variable amount of Ca2+-induced Ca2+ release found in different smooth muscle tissues.
Another possibility is that the sensitivity of the sarcoplasmic reticulum Ca2+ release process following membrane stretch may be enhanced by the generation of a diffusible substance (messenger molecule) which acts as a coagonist with Ca2+ in activating the release channels on the sarcoplasmic reticulum. There is increasing evidence that when cells are stretched, various second messengers (IP3, diacylglycerol, single chain lipids, etc.) can be formed and protein kinase C can be activated in a matter of seconds (Nakayama & Tanaka, 1993; Tanaka et al. 1994; Somlyo & Somlyo, 1994; Matsumoto et al. 1995; Persson et al. 1995; Ordway et al. 1995). Several of these factors acting on either ryanodine or IP3 receptors might be expected to enhance Ca2+ release from the sarcoplasmic reticulum. Production of such a compound(s) in response to stretch may help explain why, after membrane stretch (and stretch-activated channel activity) ceased, [Ca2+]i sometimes continued to rise for a period of time after closure of the last open channel (see Fig. 3B for an example).
The delay in the involvement of intracellular stores in amplifying Ca2+ entry through voltage-gated Ca2+ channels which is consistently seen in toad stomach smooth muscle cells is also seen (and is of about the same magnitude, 80 ms) in guinea-pig mesenteric artery (Bolton & Gordienko, 1998), but appears to be lacking in rat cerebral artery (Kamishima & McCarron, 1997) and is less than 20 ms in guinea-pig vas deferens (Imaizumi et al. 1998). Why this delay occurs, depending on the tissue type, is unclear.
For stretch-activated channels there is not a consistent delay in the amplification of Ca2+ entry; instead, it is much more variable and sometimes quite long (compare Fig. 3A with Fig. 3B). Some of this delay may be due to the required accumulation of a possible messenger molecule and/or [Ca2+]i. The state of the stores may also be affecting the delay, because when the focal Ca2+ changes were imaged, the cells were bathed in a Ca2+-free solution. Therefore, the stores might have been partially empty so that the initial influx of Ca2+ entering the cell through stretch-activated channels was diverted to fill the stores before release occurred. Another possibility is that the position of the cell-attached patch in the pipette might provide a variable distance for diffusion to the sarcoplasmic reticulum.
While additional experiments will have to be performed to determine the mechanism behind amplification for stretch-activated Ca2+ influx, the present experiments clearly demonstrate that the response of smooth muscle to membrane stretch involves entry of Ca2+ into the cytoplasm by two routes, apparently involving two different transduction pathways. The existence of these two mechanisms for Ca2+ entry, as well as the augmentation of this influx by release from intracellular stores, may have physiological significance by ensuring the robustness of the stretch-induced response in the face of neurogenic or other relaxing factors.
Acknowledgments
This work was supported by grant HL-14523 from NIH to F. S. Fay and J. J. Singer. We thank Kristine Perry, Jeff Carmichael, Rebecca McKinney and Brian Packard for their excellent technical assistance.
Fredric S. Fay died March 18, 1997.
A.G. and M.T.K. contributed equally to this work.
References
- Barry PH, Lynch JW. Liquid junction potentials and small cell effects in patch-clamp analysis. Journal of Membrane Biology. 1991;121:101–117. doi: 10.1007/BF01870526. [DOI] [PubMed] [Google Scholar]
- Bolton TB, Gordienko DV. Confocal imaging of calcium release events in single smooth muscle cells. Acta Physiologica Scandinavica. 1998;164:567–575. doi: 10.1046/j.1365-201X.1998.00464.x. [DOI] [PubMed] [Google Scholar]
- Bülbring E. Correlation between membrane potential, spike discharge and tension in smooth muscle. The Journal of Physiology. 1955;128:200–221. doi: 10.1113/jphysiol.1955.sp005299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burnstock G, Prosser CL. Responses of smooth muscles to quick stretch; relation of stretch to conduction. American Journal of Physiology. 1960;198:921–925. doi: 10.1152/ajplegacy.1960.198.5.921. [DOI] [PubMed] [Google Scholar]
- Clapp LH, Vivaudou MB, Walsh JV, Jr, Singer JJ. Acetylcholine increases voltage-activated Ca2+ current in freshly dissociated smooth muscle cells. Proceedings of the National Academy of Sciences of the USA. 1987;84:2092–2096. doi: 10.1073/pnas.84.7.2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis MJ, Donovitz JA, Hood JD. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. American Journal of Physiology. 1992;262:C1083–1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- Etter EF, Minta A, Poenie M, Fay FS. Near-membrane [Ca2+] transients resolved using the Ca2+ indicator FFP18. Proceedings of the National Academy of Sciences of the USA. 1996;93:5368–5373. doi: 10.1073/pnas.93.11.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay FS. Mechanical properties of single isolated smooth muscle cells. In: Worcel M, Vassort G, editors. Les Colloques de INSERM: Smooth Muscle Pharmacology and Physiology. Vol. 50. Paris: INSERM; 1975. pp. 327–342. [Google Scholar]
- Fay FS, Hoffman R, LeClair S, Merriam P. Preparation of individual smooth muscle cells from the stomach of Bufo marinus. Methods in Enzymology. 1992;85:284–292. doi: 10.1016/0076-6879(82)85027-1. [DOI] [PubMed] [Google Scholar]
- Fenwick EM, Marty A, Neher E. A patch-clamp study of bovine chromaffin cells and of their sensitivity to acetylcholine. The Journal of Physiology. 1982;331:577–597. doi: 10.1113/jphysiol.1982.sp014393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleischmann BK, Wang Y-X, Pring M, Kotlikoff MI. Voltage-dependent calcium currents and cytosolic calcium in equine airway myocytes. The Journal of Physiology. 1996;492:347–358. doi: 10.1113/jphysiol.1996.sp021313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganitkevich VYa, Isenberg G. Contribution of Ca2+-induced Ca2+ release to the [Ca2+]i transients in myocytes from guinea-pig urinary bladder. The Journal of Physiology. 1992;458:119–137. doi: 10.1113/jphysiol.1992.sp019409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganitkevich VYa, Isenberg G. Efficacy of peak Ca2+ currents (ICa) as trigger of sarcoplasmic reticulum Ca2+ release in myocytes from the guinea-pig coronary artery. The Journal of Physiology. 1995;484:287–306. doi: 10.1113/jphysiol.1995.sp020665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Guerrero A, Fay FS, Singer JJ. Caffeine activates a Ca2+-permeable, non-selective cation channel in smooth muscle cells. Journal of General Physiology. 1994a;104:375–394. doi: 10.1085/jgp.104.2.375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerrero A, Kirber MT, Singer JJ, Fay FS. Ryanodine sensitive amplification of Ca2+ signals in smooth muscle cells. Biophysical Journal. 1993;64:A153. [Google Scholar]
- Guerrero A, Kirber MT, Tuft RA, Fay FS, Singer JJ. Amplification of Ca2+ signals during stretch-activated channel activity in membrane patches in single smooth muscle cells. Biophysical Journal. 1994b;66:A167. [Google Scholar]
- Guerrero A, Singer JJ, Fay FS. Simultaneous measurement of Ca2+ release and influx into smooth muscle cells in response to caffeine. A novel approach for calculating the fraction of current carried by calcium. Journal of General Physiology. 1994c;104:395–422. doi: 10.1085/jgp.104.2.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Himpens B, Somlyo AP. Free-calcium and force transients during depolarization and pharmacomechanical coupling in guinea-pig smooth muscle. The Journal of Physiology. 1988;395:507–530. doi: 10.1113/jphysiol.1988.sp016932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hisada T, Ordway RW, Kirber MT, Singer JJ, Walsh JV., Jr Hyperpolarization-activated cationic channels in smooth muscle cells are stretch sensitive. Pflügers Archiv. 1991;417:493–499. doi: 10.1007/BF00370945. [DOI] [PubMed] [Google Scholar]
- Imaizumi Y, Torii Y, Ohi Y, Nagano N, Atsuki K, Yamamura H, Muraki K, Watanabe M, Bolton TB. Ca2+ images and K+ current during depolarization in smooth muscle cells of the guinea-pig vas deferens and urinary bladder. The Journal of Physiology. 1998;510:705–719. doi: 10.1111/j.1469-7793.1998.705bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isenberg G, Etter EF, Wendt-Gallitelli M-F, Schiefer A, Carrington WA, Tuft RA, Fay FS. Intrasarcomere [Ca2+] gradients in ventricular myocytes revealed by high speed digital imaging microscopy. Proceedings of the National Academy of Sciences of the USA. 1996;93:5413–5418. doi: 10.1073/pnas.93.11.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamishima T, McCarron JG. Depolarization-evoked increases in cytosolic calcium concentration in isolated smooth muscle cells of rat portal vein. The Journal of Physiology. 1996;492:61–74. doi: 10.1113/jphysiol.1996.sp021289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamishima T, McCarron JG. Regulation of the cytosolic Ca2+ concentration by Ca2+ stores in single smooth muscle cells from rat cerebral arteries. The Journal of Physiology. 1997;501:497–508. doi: 10.1111/j.1469-7793.1997.497bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirber MT, Guerrero A, Tuft RA, Bowman DS, Fay FS, Singer JJ. Ca2+ entry through stretch-activated channels is amplified by Ca2+ release from intracellular stores. Biophysical Journal. 1995;68:A393. [Google Scholar]
- Kirber MT, Guerrero A, Tuft RA, Fay FS, Singer JJ. Stretch-activated channels cause elevation of cytosolic Ca2+ by multiple pathways. Gastroenterology. 1994;107:1225. [Google Scholar]
- Kirber MT, Walsh JV, Jr, Singer JJ. Stretch-activated ion channels in smooth muscle: a mechanism for the initiation of stretch-induced contraction. Pflügers Archiv. 1988;412:339–345. doi: 10.1007/BF01907549. [DOI] [PubMed] [Google Scholar]
- Lassignal NL, Singer JJ, Walsh JV., Jr Multiple neuropeptides exert a direct effect on the same isolated single smooth muscle cell. American Journal of Physiology. 1986;250:C792–798. doi: 10.1152/ajpcell.1986.250.5.C792. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Baron CB, Coburn RF. Smooth muscle stretch-activated phospholipase C activity. American Journal of Physiology. 1995;268:C458–465. doi: 10.1152/ajpcell.1995.268.2.C458. [DOI] [PubMed] [Google Scholar]
- Morris CE. Mechanosensitive ion channels. Journal of Membrane Biology. 1990;113:93–107. doi: 10.1007/BF01872883. [DOI] [PubMed] [Google Scholar]
- Murray JJ, Reed PW, Fay FS. Contraction of isolated smooth muscle cells by ionophore A23187. Proceedings of the National Academy of Sciences of the USA. 1975;72:4459–4463. doi: 10.1073/pnas.72.11.4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Tanaka Y. Stretch-induced contraction and Ca2+ mobilization in vascular smooth muscle. Biological Signals. 1993;2:241–252. doi: 10.1159/000109505. [DOI] [PubMed] [Google Scholar]
- Nelson MT. Perspective: Bayliss, myogenic tone and volume-regulated chloride channels in arterial smooth muscle. The Journal of Physiology. 1998;507:629. doi: 10.1111/j.1469-7793.1998.629bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway RW, Petrou S, Kirber MT, Walsh JV, Jr, Singer JJ. Stretch activation of a toad stomach smooth muscle K+ channel may be mediated by fatty acids. The Journal of Physiology. 1995;484:331–337. doi: 10.1113/jphysiol.1995.sp020668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persson K, Sando JJ, Tuttle JB, Steers WD. Protein kinase C in cyclic stretch-induced nerve growth factor production by urinary tract smooth muscle cells. American Journal of Physiology. 1995;269:C1018–1024. doi: 10.1152/ajpcell.1995.269.4.C1018. [DOI] [PubMed] [Google Scholar]
- Sachs F. Stretch-sensitive ion channels: an update. In: Corey DP, Roper SD, editors. Sensory Transduction, Society of General Physiologists Series. Vol. 47. New York: Rockefeller University Press; 1992. pp. 241–260. [PubMed] [Google Scholar]
- Setoguchi M, Ohya Y, Abe I, Fujishima M. Stretch-activated whole-cell currents in smooth muscle cells from mesenteric resistance artery of guinea-pig. The Journal of Physiology. 1997;501:343–353. doi: 10.1111/j.1469-7793.1997.343bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdson W, Ruknudin A, Sachs F. Calcium imaging of mechanically induced fluxes in tissue-cultured chick heart: role of stretch-activated ion channels. American Journal of Physiology. 1992;262:H1110–1115. doi: 10.1152/ajpheart.1992.262.4.H1110. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Steenbergen JM, Fay FS. The quantal nature of calcium release to caffeine in single smooth muscle cells results from activation of the sarcoplasmic reticulum Ca2+-ATPase. Journal of Biological Chemistry. 1996;271:1821–1824. doi: 10.1074/jbc.271.4.1821. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Hata S, Ishiro H, Ishii K, Nakayama K. Quick stretch increases the production of inositol 1,4,5-trisphosphate (IP3) in porcine coronary artery. Life Sciences. 1994;55:227–235. doi: 10.1016/0024-3205(94)00884-1. [DOI] [PubMed] [Google Scholar]
- Wellner M-C, Isenberg G. Properties of stretch-activated channels in myocytes from the guinea-pig urinary bladder. The Journal of Physiology. 1993;466:213–227. [PMC free article] [PubMed] [Google Scholar]
- Wellner M-C, Isenberg G. Stretch effects on whole-cell currents of guinea-pig urinary bladder myocytes. The Journal of Physiology. 1994;480:439–448. doi: 10.1113/jphysiol.1994.sp020373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellner M-C, Isenberg G. cAMP accelerates the decay of stretch-activated inward currents in guinea-pig urinary bladder myocytes. The Journal of Physiology. 1995;482:141–156. doi: 10.1113/jphysiol.1995.sp020505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wier WG, Egan TM, López-López JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. The Journal of Physiology. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Lai FA, Cohn A, Etter E, Guerrero A, Fay FS, Meissner G. Evidence for a Ca2+-gated ryanodine-sensitive Ca2+ release channel in visceral smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1994;91:3294–3298. doi: 10.1073/pnas.91.8.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagi S, Becker PL, Fay FS. Relationship between force and Ca2+ concentration in smooth muscle as revealed by measurements on single cells. Proceedings of the National Academy of Sciences of the USA. 1988;85:4109–4113. doi: 10.1073/pnas.85.11.4109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamaguchi H, Honeyman TW, Fay FS. β-Adrenergic actions on membrane electrical properties of dissociated smooth muscle cells. American Journal of Physiology. 1988;254:C423–431. doi: 10.1152/ajpcell.1988.254.3.C423. [DOI] [PubMed] [Google Scholar]
- Yamazaki J, Duan D, Janiak R, Kuenzli K, Horowitz B, Hume JR. Functional and molecular expression of volume-regulated chloride channels in canine vascular smooth muscle cells. The Journal of Physiology. 1998;507:729–736. doi: 10.1111/j.1469-7793.1998.729bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X-C, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium ions. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]