

Abstract
Using single cell microfluorometry to monitor changes in bulk Ca2+ concentration ([Ca2+]bulk) and the whole-cell configuration of the patch clamp technique to measure K+ currents (voltage clamp) and membrane potential (current clamp), the mechanisms of histamine-induced Ca2+ oscillations in the umbilical vein endothelial cell-derived cell line EA.hy926 were studied.
In single cells, histamine (10 μm) evoked sinusoidal Ca2+ oscillations in low extracellular Ca2+ concentrations ([Ca2+]o= 10–30 μm). In contrast, histamine did not initiate Ca2+ oscillations either in the absence of extracellular Ca2+ (10 μm EGTA) or in the presence of 2.5 mM extracellular Ca2+.
Ca2+ oscillations were accompanied by rhythmic activation of Ca2+-activated K+ (KCa) channels and membrane hyperpolarization of 18.1 ± 3.9 mV. Hence, cell depolarization with 70 mM extracellular K+ or the inhibition of non-selective cation channels (NSCCs) and KCa channels by 10 μm Loe 908 and 10 mM tetrabutylammonium prevented histamine-evoked Ca2+ oscillations.
Preventing Na+-Ca2+ exchange (NCX) by 10 μm 2′,4′-dichlorobenzamil, or removal of extracellular Na+, abolished histamine-induced Ca2+ oscillations. Lowering the extracellular Na+ concentration and thus promoting the reversed mode of NCX (3Na+ out and 1Ca2+ in) increased the amplitude and frequency of histamine-induced Ca2+ oscillations by 25 and 13 %, respectively. Hence, in the absence of extracellular Ca2+, 10 μm histamine induced an elevation of intracellular Na+ concentration in certain subplasmalemmal domains.
The inhibitor of sarco/endoplasmic reticulum Ca2+-ATPase (SERCA) 2,5-di-tert-butyl-1,4-benzo-hydroquinone (15 μm) prevented histamine-induced Ca2+ oscillations. In addition, blockage of ryanodine-sensitive Ca2+ release (RsCR) by 25 μm ryanodine blunted Ca2+ oscillations.
In endothelial cells that were treated for 16 h with 10 μm nocodazole to collapse the superficial endoplasmic reticulum (sER), no histamine-induced Ca2+ oscillations were found.
We conclude that in low [Ca2+]o conditions histamine-induced Ca2+ oscillations depend on transmembrane Na+ loading through NSCCs that leads to Ca2+ entry via NCX. Cation influx is controlled by KCa channel activity that triggers membrane hyperpolarization and, thus, provides the driving force for cation influx. Hence, the Ca2+ entering needs to be sequestrated via SERCA into sER to become released by RsCR to evoke Ca2+ spiking. These data further support our previous work on localized Ca2+ signalling as a key phenomenon in endothelial Ca2+ homeostasis.
An elevation in intracellular free Ca2+ concentration plays a crucial role in the regulation of endothelial vascular function (Graier et al. 1994). Remarkably, in certain endothelial cells, such as those from the human umbilical vein (HUVEC), the Ca2+ response initiated by agonists that operate through the phosphoinositide pathway (Rink & Jacob, 1989; Jacob, 1991) and/or mechanical stress (Hoyer et al. 1998) can be of oscillatory patterns. The exact mechanisms contributing to these Ca2+ oscillations are unknown, but oscillatory changes in IP3-receptor sensitivity (Hajnoczky & Thomas, 1997), time-resolved Ca2+ release from different Ca2+ pools (Pozzan et al. 1988; Krause et al. 1989; Berridge, 1991), rhythmic entry of Ca2+ through non-selective cation channels (Laskey et al. 1992), changes in cellular morphology (Kraus et al. 1996) and the involvement of sarco/endoplasmic reticulum Ca2+-ATPases (SERCA) (Foskett & Wong 1991, 1992; Uneyama et al. 1998) have been discussed. Despite its clear contribution to endothelial Ca2+ signalling (Sage et al. 1991; Paltauf-Doburzynska et al. 1998, 1999; Dömötör et al. 1999) the involvement of Na+-Ca2+ exchange (NCX) in Ca2+ oscillation has not been demonstrated so far. Besides their regulatory role in cell function, the oscillatory nature of changes in the intracellular Ca2+ concentration has been correlated with regulation of gene expression (Dolmetsch & Lewis, 1994; Berridge, 1997; Meldolesi, 1998). Recently, we have described insulated Ca2+ elevation in the subplasmalemmal area of endothelial cells that is controlled by a functional coupling of certain Ca2+ shuttling proteins (subplasmalemmal Ca2+ control unit, SCCU; Graier et al. 1998; Paltauf-Doburzynska et al. 1998, 1999). Besides spatial Ca2+ elevation, the SCCU was described to control the activity of constitutive nitric oxide synthase (Paltauf-Doburzynska et al. 1998) and KCa channel activity (Frieden & Graier, 2000), thus indicating its contribution to the regulation of cell function. Based on this new principle of the SCCU in endothelial cells freshly isolated from the bovine left circumflex artery and the umbilical vein endothelial cell-derived cell line EA.hy926, this study is designed to reconsider the mechanisms of Ca2+ oscillations and to find out whether or not the SCCU contributes to agonist-evoked Ca2+ oscillations in endothelial cells.
METHODS
Materials
Cell culture chemicals were obtained from Life Technologies, Vienna, Austria. The acetoxymethyl ester form of fura-2 (fura-2 AM), ER-Traker Blue-White DPX, 2′,4′-dichlorobenzamil and SBFI AM were purchased from Molecular Probes, Eubio, Vienna, Austria, and fetal calf serum (FCS) was from PAA Laboratories, Linz, Austria. Ryanodine was purchased from Calbiochem or Alexis, Eubio, Vienna, Austria. Buffer salts were from Merck, Darmstadt and all other materials (e.g. nocodazole, histamine and tetrabutylammonium) were from Sigma, Vienna, Austria. The human umbilical vein endothelial cell line, EA.hy926 (Edgell et al. 1983), was a gift from Dr Cora-Jean S. Edgell, Pathology Department, University of North Carolina, Chapel Hill, NC, USA.
Cell culture and cell preparation
The endothelial cell line EA.hy926 was grown in Dulbecco's minimum essential medium (DMEM) containing 10 % FCS, 5 mM D-glucose and 1 % HAT (5 mM hypoxanthine, 20 μm aminopterine, 0.8 mM thymidine). To prepare the cells for experiments, cultured cells were washed twice with Ca2+-free DMEM, incubated with 0.05 % trypsin and 0.02 % EDTA for 1–2 min, centrifuged (5 min at 400 g) and resuspended in storage buffer (SB: DMEM containing 2 % horse serum and 0.1 % of a vitamin mixture and 0.2 % essential amino acids). All experiments were conducted at room temperature (approx. 22°C).
Intracellular Ca2+ measurement
For measuring the bulk Ca2+ concentration ([Ca2+]bulk), conventional fura-2 experiments were performed in single endothelial cells as described previously (Graier et al. 1995, 1998). Briefly, suspended cells in SB (see above) were loaded for 45 min at room temperature in the dark with 2 μm fura-2 AM, centrifuged, washed twice and resuspended in SB. After an equilibration period of 10 min, a drop of the cell suspension was transferred into a glass-bottomed experimental chamber and the cells were allowed to reattach for approximately 2 min before a constant superfusion of 2 ml min−1 was switched on. Cells were perfused for 2 min in Hepes buffer solution (HBS-Ca2+) containing (mM): 145 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2 and 10 Hepes-acid, pH adjusted to 7.4, followed by an incubation in Hepes buffer (HBS), with no added Ca2+, containing (mM): 145 NaCl, 5 KCl, 1 MgCl2 and 10 Hepes-acid, pH adjusted to 7.4 (contains approximately 10–30 μm free Ca2+), in nominal Ca2+-free Hepes buffer (HBS-EGTA) containing (mM): 145 NaCl, 5 KCl, 1 MgCl2, 10−5 M EGTA and 10 Hepes-acid, pH adjusted to 7.4 or in low Na+, no added Ca2+, Hepes buffer (19HBS) containing (mM): 19 NaCl, 5 KCl, 1 MgCl2, 126 choline chloride, 10 Hepes-acid, pH 7.4 as indicated. [Ca2+]bulk was monitored by sampling fluorescence intensity (F) at 510 nm emission alternately at 360 nm (i.e. Ca2+-insensitive wavelength) and 380 nm (i.e. Ca2+-sensitive wavelength) excitation. As previously described (Graier et al. 1995), fluorescence intensity for each pair of excitation-emission wavelengths was converted to analog by an optical processor and registered by a PC running AxoBASIC 1.0 (Axon Instruments, Foster City, CA, USA). In view of the reported errors of the [Ca2+]bulk calibration in our system and the general uncertainties of the calibration techniques (Graier et al. 1995, 1998), [Ca2+]bulk is expressed in ratio units (F360/F380).
Intracellular Na+ measurement
Intracellular free Na+ concentration was measured using SBFI as previously described (Graier et al. 1998). Briefly, suspended cells (see above) were loaded with 20 μm SBFI and 0.1 % Pluronic acid for 2 h at 37°C in the dark. After being washed twice, the cells were equilibrated for 15 min in SB. One drop of the cell suspension was transferred into a glass-bottomed experimental chamber and the cells were allowed to reattach for approximately 2 min before a constant superfusion of 2 ml min−1 was switched on. Experiments were performed using a deconvolution microscope (Graier et al. 1998; Paltauf-Doburzynksa et al. 1998, 1999). For measurements of the intracellular Na+ concentration ([Na+]i), cells were superfused with HBS-EGTA and spatial fluorescence of SBFI was monitored using a 340HT15 excitation filter and a 525DF20 emission filter. Images were collected every 10 s in the middle depth of the cells using a CFI Plan Fluor ×40 oil immersion objective (NA 1.3; 0.171 μm per pixel, Nikon, Vienna, Austria). Out-of-focus fluorescence was calculated with a computer algorithm using a point spread function that was obtained for the dye and cells on this equipment. Due to the considerable photobleaching of the SBFI in our system, data are expressed as the ratio of certain subplasmalemmal and bulk fluorescence intensities (Fsub/Fbulk). Unfortunately, no calibration of spatial Na+ concentration could be performed due to the dramatic effect of the ionophores necessary for calibration (gramicidin, ouabain and monensin).
Electrophysiological recordings
A drop of EA.hy926 cells in suspension (see above) was transferred into the glass-bottomed experimental chamber. After the cells were allowed to reattach for approximately 2 min the constant superfusion of 2 ml min−1 was switched on. Membrane potentials and membrane currents were recorded using the whole-cell configuration of the patch clamp technique (Hamill et al. 1981) as described (Frieden et al. 1999). Borosilicate glass pipettes were pulled with a Narishige puller (Narishige Co. Ltd, Tokyo, Japan), fire-polished and had a resistance of 3–6 MΩ. Patch clamp recordings were made using a PC-501A amplifier (Warner Instrument Corporation, Dixwell Avenue, Hamden, CT, USA); currents were filtered with a low-pass filter at 0.1 kHz, digitized by a PP-50LAB converter (Warner Instrument Corporation) and sampled on a PC running AxoBASIC 1.0 (Axon Instruments). Voltage ramps (1.8 s; -80 to +65 mV) were repeatedly applied in order to determine the current-voltage relationship. In current clamp experiments the membrane potential was set to a value between -20 and -30 mV. The pipette solution contained (mM): 130 KCl, 1 MgCl2, 5 MgATP, 0.2 Na3GTP, 10 Hepes (pH adjusted to 7.2 with NaOH). For the standard bath solution HBS-Ca2+ was used. For experiments HBS was used as indicated above.
Simultaneous ion current and [Ca2+]bulk measurements
Suspended EA.hy926 cells were loaded with fura-2 AM as described under ‘Intracellular Ca2+ measurement’. A drop of the cell suspension (see above) was transferred into the glass-bottomed experimental chamber. After the cells were allowed to reattach for approximately 2 min the constant superfusion of 2 ml min−1 was switched on. Simultaneous measurements of ion currents and [Ca2+]bulk were performed according to Sturek 1991et al.a, b. The instrumentation combines the microfluorometer described above and a patch clamp set-up, including a patch clamp amplifier with a 1 MΩ headstage (PC-501A, Warner Instrument Corp.), bessel filter (model 900-C9L8L, Frequency Devices, Haverhill, MA, USA), A/D converter (PP-50LAB, Warner Instrument Corp.) and a micromanipulator (WR-60, Narishige Co. Ltd). The digitalized signals for ion currents and fluorescence intensity at 510 nm emission at the corresponding excitation at 360 and 380 nm were registered by a PC running AxoBASIC 1.0 (Axon Instruments).
Statistics
Analysis of variance (ANOVA) was performed and statistical significance of differences was evaluated using Scheffé‘s F test. The level of significance was defined as P < 0.05.
RESULTS
Stimulation of single endothelial cells from the HUVEC-derived cell line EA.hy926 with 10 μm histamine in HBS (no added Ca2+, no EGTA) initiated an initial peak followed by sinusoidal Ca2+ oscillations in 57 out of 69 cells (Fig. 1A). The oscillations strictly correlated with the presence of the agonist and immediately faded after removal of the agonist (data not shown). In nominal Ca2+-free solution (EGTA-containing buffer; HBS-EGTA) the initial peak occurred while no rhythmic oscillations were found (Fig. 1B; n = 17). Hence, bulk Ca2+ concentration ([Ca2+]bulk) returned to baseline within 5.0 ± 0.2 min even in the presence of histamine. In contrast, in 2.5 mm Ca2+-containing buffer (HBS-Ca2+) 10 μm histamine yielded a sustained plateau phase following the initial peak (Fig. 1C; n = 13).
Figure 1. Histamine-induced changes in free bulk Ca2+ concentration in single cells from the human umbilical vein endothelial cell-derived cell line EA.hy926.

The cells were loaded for 45 min at room temperature in the dark with 2 μmfura-2 AM, centrifuged and resuspended in SB. After 10 min equilibration time, [Ca2+]bulk was monitored in a microfluorometer and expressed as a ratio of fluorescence intensities at 360 and 380 nm (F360/F380) as described in Methods. Representative tracings of the effect of 10 μm histamine in: A, HBS (i.e. no Ca2+ added, no EGTA, n = 57); B, HBS-EGTA (i.e. no Ca2+ added, 10 μm EGTA, n = 17); and C, HBS-Ca2+ (2.5 mM Ca2+ added, n = 13) are shown.
Involvement of cell membrane ion channels and transporters in Ca2+ oscillations
Our data that chelating extracellular Ca2+ by EGTA abolished histamine-evoked Ca2+ oscillations indicate the need for Ca2+ influx for Ca2+ oscillations. On the other hand, since cation (e.g. Ca2+, Na+) influx in endothelial cells critically depends on the driving force provided by membrane hyperpolarization due to the activation of KCa channels (Colden-Stanfield et al. 1987), the activity of these channels and changes in membrane potential during stimulation with histamine were monitored using the whole-cell configuration of the patch clamp technique.
Ca2+ permeable, non-selective cation channels
In the presence of 10 μm Loe 908, an inhibitor of non-selective cation channels, 10 μm histamine failed to initiate bulk Ca2+ oscillations (n = 16; data not shown), while in the absence of Loe 908 Ca2+ oscillations were observed in 7 out of 9 cells in these particular experiments. While Loe 908 did not affect basal [Ca2+]bulk (data not shown), addition of 10 μm Loe 908 to cells that were stimulated with 10 μm histamine in HBS, inhibited Ca2+ oscillations within 1 min (Fig. 2; n = 6).
Figure 2. Effect of Loe 908, an inhibitor of non-selective cation channels, on histamine-evoked Ca2+ oscillations in single EA.hy926 cells.

After loading the cells with fura-2 AM, cells were stimulated in HBS with 10 μm histamine. As indicated 10 μm Loe 908 was added to the superfusion (n = 6). The figure shows a representative tracing from 6 similar experiments. If the cells were stimulated in the presence of 10 μm Loe 908 no Ca2+ oscillations occurred (n = 16; data not shown). In contrast, in the absence of Loe 908, in 7 out of 9 experiments Ca2+ oscillations could be observed.
KCa channels and membrane potential
In this study, KCa channel activity was used to monitor subplasmalemmal Ca2+ elevation. In HBS, histamine-induced elevation of [Ca2+]bulk was accompanied by an oscillatory activation of an outward current (Fig. 3A; n = 5 out of 5) that is due to stimulation of large-conductance Ca2+-activated K+ channels (BKCa; Frieden & Graier, 2000). In agreement with these findings, in current clamp experiments histamine was found to initiate oscillatory cell membrane hyperpolarization of 18.1 ± 3.9 mV in HBS (n = 5 out of 6; Fig. 3B). The mean resting membrane potential of the cells was -25.3 ± 5.1 mV (n = 9).
Figure 3. Histamine-induced activation of whole-cell currents (A) and membrane hyperpolarization (B) in EA.hy926 cells.

A, original recording of the oscillatory outward current (voltage clamp mode) stimulated by 10 μm histamine. The holding potential was -60 mV and repetitive voltage ramps (-80 to +65 mV) were applied throughout the whole recording. Histamine was applied in the bathing solution as indicated. Similar results were obtained in 5 out of 5 experiments. Detailed characterization of this current revealed that it is mainly due to KCa channel activity (Frieden & Graier, 2000). B, original tracing of histamine (10 μm)-induced change in the membrane potential recorded in the current clamp mode of the whole-cell method. Histamine was applied as indicated. Similar results were found in 5 out of 6 experiments.
To find out whether KCa channel activation-mediated membrane hyperpolarization contributes to Ca2+ oscillations in response to histamine in HBS, KCa channel activity was prevented by elevating extracellular K+ concentration ([K+]o) or by adding tetrabutylammonium (TBA). Under control conditions 10 μm histamine yielded sinusoidal Ca2+ oscillations in 7 out of 7 cells (data not shown). In contrast, in the presence of either 70 mM [K+]o (n = 12) or 10 mM TBA (n = 8) histamine failed to evoke Ca2+ oscillations (data not shown). Neither 70 mM [K+]o nor 10 mM TBA affected basal [Ca2+]bulk (data not shown).
Na+-Ca2+ exchange (NCX)
Inhibition of NCX activity by 10 μm 2′,4′-dichlorobenzamil prevented histamine-evoked Ca2+ oscillations (oscillations in 0 out of 13 cells; Fig. 4A), while in the accompanying experiments in the absence of 2′,4′-dichlorobenzamil Ca2+ oscillations were frequently found (7 out of 9; data not shown). In contrast to its effect on histamine-induced Ca2+ oscillations, 2′,4′-dichlorobenzamil did not affect basal [Ca2+]bulk (data not shown). In addition modulation of the equilibrium potential of the NCX (ENa-Ca) by decreasing the extracellular Na+ concentration ([Na+]o) to 19 mM (19HBS) increased the frequency and amplitude of histamine-induced Ca2+ oscillations compared with those found in HBS (n = 10; Fig. 4B) by 13 and 25 %, respectively (n = 16; P < 0.05; Fig. 4B). Hence in Na+-free solution (i.e. all extracellular Na+ has been substituted by choline) no histamine-induced Ca2+ oscillation could be found (n = 17; data not shown).
Figure 4. Histamine-evoked bulk Ca2+ signalling in single EA.hy926 cells in the presence of an inhibitor of Na+-Ca2+ exchange or low extracellular Na+ concentration.

A, representative tracing of the effect 10 μm histamine on fura-2-loaded endothelial cells in HBS in the presence of 10 μm 2′,4′-dichlorobenzamil (n = 13). In the absence of 2′,4′-dichlorobenzamil, histamine initiated, in 7 out of 9 experiments, Ca2+ oscillations in the accompanying control experiments (data not shown). B, single endothelial cells were stimulated with 10 μm histamine in HBS (continuous line; n = 10) or 19HBS (dashed line; n = 16).
To investigate the contribution of Na+ in more detail, the intracellular free sodium concentration ([Na+]i) was measured using SBFI in single endothelial cells using a high resolution deconvolution microscope (n = 11). In HBS no change in [Na+]i could be obtained (n = 12). However, when Na+ extrusion (NCX activity) was prevented by using a nominal Ca2+-free solution (i.e. HBS-EGTA), an increase in the SBFI fluorescence in certain subplasmalemmal domains was found in response to stimulation with 10 μm histamine (Fig. 5A). Thus, as shown in Fig. 5B, stimulation with 10 μm histamine in HBS-EGTA results in the slow Na+ loading of subplasmalemmal domains (n = 7). A similar effect on [Na+]i to that observed in HBS-EGTA was found in HBS in the presence of 10 μm 2′,4′-dichlorobenzamil (data not shown).
Figure 5. Histamine-evoked elevation in subplasmalemmal Na+ concentration in single EA.hy926 cells.

A, cultured endothelial cells were loaded with SBFI AM as described in Methods. Changes in intracellular Na+ concentration were monitored in the middle depth of the cells at 340 ± 15 nm excitation and 525 ± 20 nm emission in HBS-EGTA (nominal Ca2+-free solution to prevent any NCX activity) using a deconvolution microscope. Haze was removed after calculating the out-of-focus fluorescence with a computer algorithm using an experimental point spread function. Images represent the SBFI fluorescence in a representative cell at time point 1 min (image a) and after 5 min stimulation with 10 μm histamine (image b). To indicate changes in the spatial intracellular Na+ concentration the fluorescence intensity of image a was subtracted from image b as indicated on image b – a. Circles indicate the subplasmalemmal and perinuclear areas in which the ratio of fluorescence intensities was followed over time. B, statistical evaluation of the spatial changes in intracellular Na+ concentration in response to 10 μm histamine in EA.hy926 cells. To correct the photobleaching of SBFI, the ratio of the fluorescence intensities of given subplasmalemmal and perinuclear areas as indicated in A (Fsub/Fbulk) were calculated over time (n = 7).
Contribution of the filling state of intracellular Ca2+ stores for Ca2+ oscillations
To assess the importance of the filling state of intracellular Ca2+ stores for Ca2+ oscillations, the intracellular Ca2+ pools were depleted by repetitive histamine stimulations in HBS with or without a Ca2+ store refilling period in HBS-Ca2+. After the cell had been stimulated once with 10 μm histamine no further oscillations were found in response to a second stimulation with the agonist in HBS (Fig. 6A; n = 8). However, if the cells were exposed to 2.5 mM Ca2+-containing buffer (HBS-Ca2+) for 2 min after histamine, repetitive stimulation evoked identical Ca2+ oscillations to those obtained in response to the first addition of histamine (Fig. 6B; n = 6). The preserving effect of a short exposure to extracellular Ca2+ lasted for at least four repetitive stimulations within 30 min (data not shown).
Figure 6. The role of Ca2+ store refilling by Ca2+ entry for repetitive histamine stimulations and Ca2+ oscillations in single EA.hy926 cells.

A, in HBS, fura-2-loaded endothelial cells were exposed two times to 10 μm histamine as indicated. Within the histamine applications the cells remained in HBS (n = 8). B, as described for A, endothelial cells were repetitively stimulated with 10 μm histamine (H.) in HBS as indicated. Within two applications of histamine, the cells were superfused with HBS-Ca2+ (2.5 mM Ca2+-containing HBS) for 2 min. A and B, due to the long duration of the experimental protocols (35 min) every stimulation was recorded separately. Nevertheless, the tracings show representative recordings of one single cell (each) over the whole time indicated.
Role of endoplasmic reticulum Ca2+ shuttling proteins in Ca2+ oscillations
The data presented so far provide strong evidence for an involvement of membrane ion movements and intracellular Ca2+ stores for histamine-induced Ca2+ oscillations. To further elucidate the mechanisms of Ca2+ oscillations, the involvement of endoplasmic reticulum Ca2+ shuttling proteins (i.e. SERCA, ryanodine receptors) was investigated.
Endoplasmic reticulum Ca2+-ATPase (SERCA)
In addition to endoplasmic reticulum ryanodine receptors, the contribution of SERCA to SCCU activity was shown previously (Graier et al. 1998). Since we hypothesize that Ca2+ oscillations reflect SCCU activity, the role of SERCA in the occurrence of histamine-induced Ca2+ oscillations in HBS was studied. Thus, SERCA activity was inhibited by a preincubation for 3 min with 2,5-di-tert-butyl-1,4-benzohydroquinone (BHQ) before stimulation with histamine. BHQ (15 μm) transiently raised [Ca2+]bulk and prevented histamine-induced Ca2+ oscillations, while the initial peak in response to the addition of histamine was not affected (n = 12; Fig. 7A). In contrast, in control conditions histamine (10 μm) yielded Ca2+ oscillations in 8 out of 9 cells (data not shown).
Figure 7. Effect of inhibiting endoplasmic reticulum Ca2+-ATPase (SERCA; A) and ryanodine-sensitive Ca2+ release (RsCR; B) on histamine-induced Ca2+ oscillations of single EA.hy926 cells in HBS.

A, in the presence of the SERCA inhibitor 2,5-di-tert-butyl-1,4-benzohydroquinone (BHQ; 15 μm preincubated for 3 min) 10 μm histamine failed to initiate Ca2+ oscillations in all experiments performed (n = 12). The efficiency of BHQ to prevent SERCA is indicated by the transient elevation of [Ca2+]bulk during the preincubation period of 3 min. In the corresponding control experiments, 10 μm histamine induced, in 8 out of 9 cells, Ca2+ oscillations in HBS (data not shown). B, in single EA.hy926 cells changes in [Ca2+]bulk were monitored in the presence of 25 μm ryanodine, which prevented RsCR. Three minutes before starting the experiment cells were superfused with HBS with 25 μm ryanodine. At the time indicated, the cell was stimulated with 10 μm histamine in 19HBS. Under control conditions Ca2+ oscillations were observed in 11 out of 13 experiments (data not shown), while in the presence of ryanodine Ca2+ oscillations could only be obtained in 2 out of 12 experiments. The Ca2+ oscillations observed in the presence of ryanodine did not show sinusoidal kinetics and the amplitude was greatly reduced compared with the accompanying control experiments.
Ryanodine receptors
We have previously shown that ryanodine-sensitive Ca2+ release (RsCR) plays a major role in the elevation of subplasmalemmal Ca2+ concentration ([Ca2+]sub; Paltauf-Doburzynska et al. 1998). Therefore, the role of ryanodine-sensitive Ca2+ release (RsCR) in the occurrence of histamine-induced Ca2+ oscillations was studied. RsCR was inhibited by preincubation with 25 μm ryanodine. In control cells, histamine evoked Ca2+ oscillations in 11 out of 13 experiments in 19HBS (data not shown). In contrast, after inhibition of RsCR with 25 μm ryanodine, histamine-induced Ca2+ oscillations were only found in 2 out of 12 cells (Fig. 7B). Similar results were obtained in HBS (data not shown).
Involvement of superficial Ca2+ stores in endothelial Ca2+ oscillations
To investigate the importance of subplasmalemmal endoplasmic reticulum compartments (superficial endoplasmic reticulum, sER) the ER network was collapsed by a 16 h pretreatment with 10 μm nocodazole as described previously (Graier et al. 1998; Paltauf-Doburzynska et al. 1999). The collapse of ER domains by the nocodazole treatment was measured by changes in the distribution of the fluorescence of the highly selective ER marker, ER-Tracker Blue-White DPX as previously reported (Paltauf-Doburzynska et al. 1999). In agreement with our previous data on bovine left circumflex endothelial cells, the ratio of subplasmalemmal and perinuclear fluorescence of the ER marker decreased by treatment with 10 μm nocodazole for 16 h from 0.97 ± 0.12 (solvent; n = 12) to 0.42 ± 0.22 (nocodazole; n = 8, P < 0.05vs. solvent). In cells treated with the solvent alone, Ca2+ oscillations in response to 10 μm histamine in HBS were found in 11 out of 12 experiments (Fig. 8A). Pretreatment with nocodazole prevented histamine-evoked Ca2+ oscillations in all experiments (n = 12; Fig. 8B).
Figure 8. Effect of pretreatment with nocodazole on histamine-evoked Ca2+ oscillations.

EA.hy926 cells were pretreated for 16 h in DMEM with 0.2 % DMSO (solvent control; A) or 10 μm nocodazole (B). After fura-2 AM loading as described in Methods, endothelial cells were stimulated in HBS with 10 μm histamine in the absence of DMSO-nocodazole. In control cells, oscillations were measured in 11 out of 12 experiments. In nocodazole pretreated cells, no histamine-induced Ca2+ oscillations were found in 12 experiments.
Time correlation of oscillatory elevation of bulk Ca2+ with KCa channel activation
To verify the time correlation between activation of KCa channels and, thus, an increase in [Ca2+]sub with the elevation in [Ca2+]bulk, simultaneous recordings of ion currents and [Ca2+]bulk were performed. As shown in Fig. 9A, 10 μm histamine-evoked Ca2+ oscillations were accompanied by an oscillatory activation of KCa channels. Remarkably, changes in [Ca2+]bulk were sinusoidal, while KCa channel activity showed an instant maximal onset while it slowly decreased with time. A detailed analysis of the time needed to reach maximal current, and the elevation in [Ca2+]bulk, indicated that the elevation of [Ca2+]bulk peaked 2.52 ± 0.39 s (n = 7) after the current was maximal (Fig. 9B).
Figure 9. Time correlation of oscillatory elevation of [Ca2+]bulk with KCa channel activation in response to histamine in HBS.
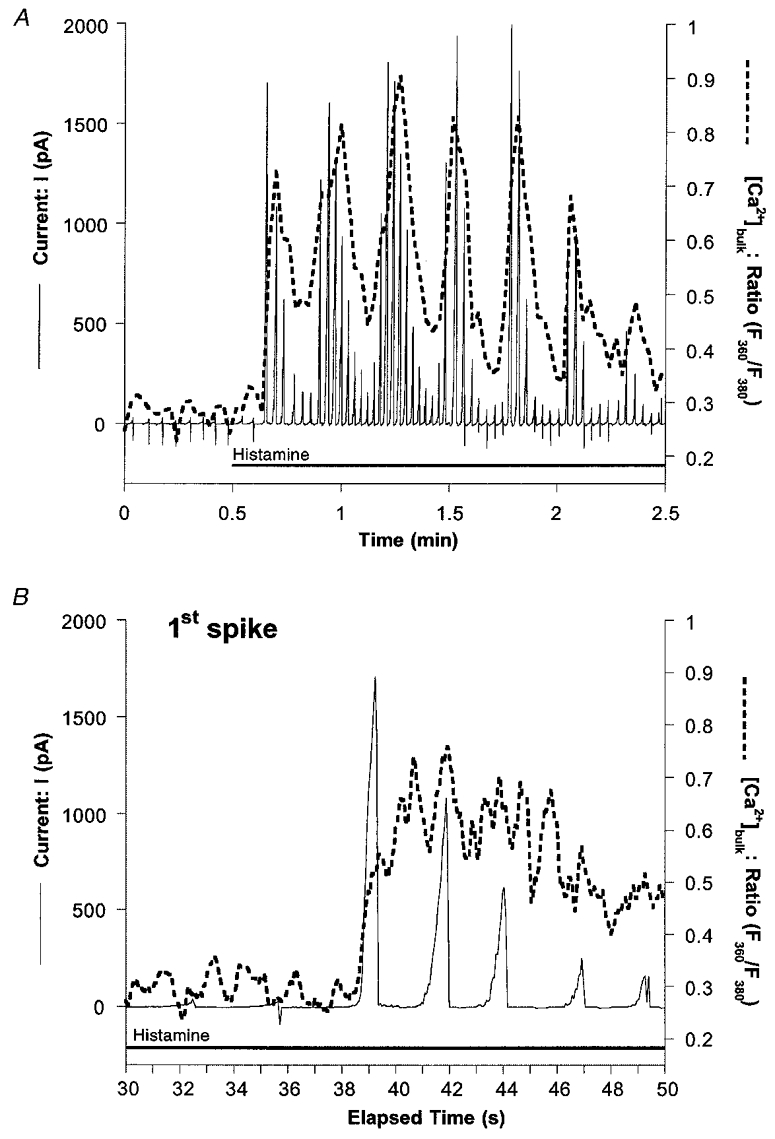
Changes in [Ca2+]bulk (dashed line) and membrane currents (continuous line) were simultaneously recorded in single EA.hy926 cells as described in Methods. A, original recording of the oscillatory outward current (voltage clamp mode) stimulated by 10 μm histamine in HBS as indicated. The holding potential was -30 mV and repetitive voltage ramps (-80 to +65 mV) were applied throughout the whole recording. Similar results were obtained in 3 out of 5 experiments. B, expanded time scale of the first spike shown in A to indicate the instant elevation of KCa channel activation compared with the sinusoidal kinetics of changes in [Ca2+]bulk.
DISCUSSION
This study was designed to investigate the role of Ca2+ shuttling proteins and the superficial endoplasmic reticulum (sER) for histamine-evoked Ca2+ oscillations in the EA.hy926 cell line that is derived from umbilical vein endothelial cells (Edgell et al. 1983). In our previous work, we have extensively compared these cells with endothelial cells freshly isolated from the bovine left circumflex artery in terms of regulation and function of cytosolic and subplasmalemmal Ca2+ signalling and architectural organization of the ER network (Graier et al. 1998; Paltauf-Doburzynska et al. 1998, 1999). Here we obtained identical results in the EA.hy926 cell line to those in endothelial cells freshly isolated from the bovine left circumflex artery (Graier et al. 1998; Paltauf-Doburzynska et al. 1998, 1999). The primary finding of this work is that histamine-induced Ca2+ oscillations strictly depend on transmembrane ion flux through non-selective cation channels (NSCCs), the Na+-Ca2+ exchanger (NCX) and KCa channels, sequestration of Ca2+ into the sER via SERCA and intracellular Ca2+ release by IP3 receptors and ryanodine receptors (Fig. 10).
Figure 10. Schematic view of the proposed mechanism of histamine-induced Ca2+ oscillations in EA.hy926 cells.
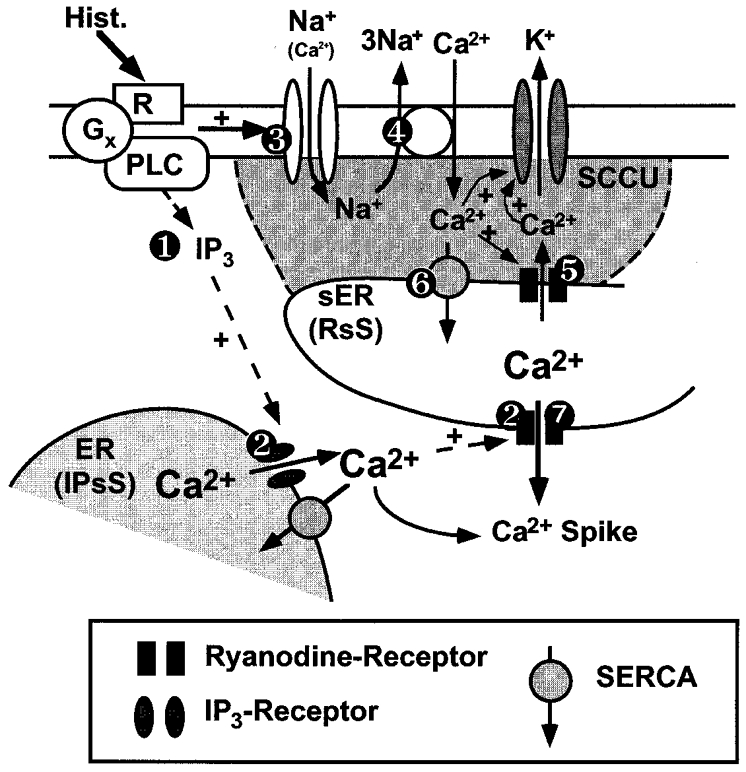
Histamine (Hist.) binds to its cell membrane receptor and activates via a G protein (Gx) phospholipase C (PLC), resulting in the generation of inositol 1,4,5-trisphosphate (IP3) that triggers depletion of IP3-sensitive Ca2+ stores (IPsS) ( ). This elevation of [Ca2+]bulk initiates Ca2+-induced Ca2+ release (CiCR) from the ryanodine-sensitive Ca2+ stores (RsS) (
). This elevation of [Ca2+]bulk initiates Ca2+-induced Ca2+ release (CiCR) from the ryanodine-sensitive Ca2+ stores (RsS) ( ). Hence, histamine, via yet still unknown mechanisms, stimulates non-selective cation channels (NSCCs) that favour the influx of Na+ under low [Ca2+]o conditions (i.e. HBS) (
). Hence, histamine, via yet still unknown mechanisms, stimulates non-selective cation channels (NSCCs) that favour the influx of Na+ under low [Ca2+]o conditions (i.e. HBS) ( ). As a result of such ‘Na+ loading’ in HBS via NSCCs Na+ is extruded via Na+-Ca2+ exchange (NCX) resulting in an influx of Ca2+ (
). As a result of such ‘Na+ loading’ in HBS via NSCCs Na+ is extruded via Na+-Ca2+ exchange (NCX) resulting in an influx of Ca2+ ( ). The Ca2+ entry via NCX initiates subplasmalemmal ryanodine-sensitive Ca2+ release (RsCR) and activates KCa channels (
). The Ca2+ entry via NCX initiates subplasmalemmal ryanodine-sensitive Ca2+ release (RsCR) and activates KCa channels ( ) that provide the driving force for ion influx through NSCCs. As a result of the increased [Ca2+]sub, sER Ca2+ pools (RsS) are refilled via SERCA (
) that provide the driving force for ion influx through NSCCs. As a result of the increased [Ca2+]sub, sER Ca2+ pools (RsS) are refilled via SERCA ( ) and become depleted via CiCR towards the perinuclear area by IP3-mediated Ca2+ release from IPsS (
) and become depleted via CiCR towards the perinuclear area by IP3-mediated Ca2+ release from IPsS ( ) that results in a Ca2+ spike.
) that results in a Ca2+ spike.
Histamine evoked sinusoidal oscillations in [Ca2+]bulk in HBS and 19HBS that contained approximately 10–30 μm free Ca2+, while in nominal Ca2+-free solution (HBS-EGTA) histamine did not produce any rhythmic oscillations in [Ca2+]bulk. On the other hand, in the presence of 2.5 mM extracellular Ca2+ (HBS-Ca2+) the elevation in [Ca2+]bulk in response to histamine was more sustained while no oscillatory characteristics were found. Interestingly, these differences occurred only in the second phase of the response to histamine, while the initial transient increase in [Ca2+]bulk was identical. These data indicate that a small influx of extracellular Ca2+ is essential for Ca2+ oscillations, while a large Ca2+ entry might overcome Ca2+ oscillations by a high, sustained elevation in [Ca2+]bulk. In agreement with these findings in EA.hy926 cells, bradykinin- and histamine-evoked Ca2+ oscillations have been shown to depend on the presence of extracellular Ca2+ in bovine aortic and arterial valve endothelial cells and HUVEC, respectively (Sage et al. 1989; Jacob, 1991; Laskey et al. 1992; Lynch et al. 1992). Hence, our data that Loe 908, a selective inhibitor of NSCCs (Encabo et al. 1996; Iwamuro et al. 1999), prevented Ca2+ oscillations in response to histamine are consistent with the findings in cultured bovine arterial valve endothelial cells that indicate that SKF 96365, another non-selective inhibitor of NSCCs, also abolished Ca2+ oscillations (Laskey et al. 1992). Thus these data indicate that histamine-evoked Ca2+ oscillations in EA.hy926 depend on ion influx through NSCCs.
In endothelial cells, the cation influx through NSCCs depends on the activation of KCa channels that provides membrane hyperpolarization and, thus, the driving force for the ion influx through opened NSCCs (Busse et al. 1988; Lückhoff & Busse, 1990). In this study, the activity of KCa channels monitored by the whole-cell patch clamp technique (voltage clamp protocol; for detailed characterization see Frieden & Graier, 2000) and changes in membrane potential (current clamp protocol) showed oscillatory characteristics in response to histamine in HBS. In agreement with these findings on the EA.hy926 cell line, Laskey and co-workers (1992) reported rhythmic changes in membrane potential monitored with a potential-sensitive bisoxonol dye in oscillating bovine arterial valve endothelial cells. Moreover, a correlation between Ca2+ oscillations and rhythmic changes in membrane potential has been demonstrated in native endothelial cells in an excised rat aorta (Usachev et al. 1995). The importance of this rhythmic KCa channel activity/membrane hyperpolarization for histamine-evoked Ca2+ oscillations was further confirmed by our findings that increasing the extracellular K+ concentration to 70 mM to prevent K+ current abolished Ca2+ oscillations. While these data are in agreement with those of other groups who showed that membrane depolarization prevents Ca2+ oscillations in endothelial cells (Jacob, 1990; Carter & Bjaaland, 1991), they are not in agreement with those reported in HUVEC where 140 mM extracellular K+ failed to affect histamine-induced Ca2+ oscillations, although Ca2+ entry was required (Jacob et al. 1988). Nevertheless, our data indicate that histamine-induced Ca2+ oscillations in low Ca2+-containing solution depend on (oscillatory?) ion influx through store-operated channels (Groschner et al. 1994) and/or NSCCs (Nilius et al. 1993, 1997) that are triggered by and/or associated with a rhythmic stimulation of KCa channels evoking oscillating hyperpolarization.
Recently, a model for the regulation of spatial subplasmalemmal Ca2+ elevation by a so-called subplasmalemmal Ca2+ control unit was proposed (SCCU; Graier et al. 1998; Paltauf-Doburzynska et al. 1998). So far evidence has been provided that the SCCU controls the activation of constitutive nitric oxide synthase (Paltauf-Doburzynska et al. 1998), (re)filling and depletion of ryanodine-sensitive Ca2+ stores (Paltauf-Doburzynska et al. 1999), Ca2+ entry (Graier et al. 1998) and KCa channel activity (Frieden & Graier, 2000). In these studies the NCX activity plays a crucial role in the regulation of subplasmalemmal Ca2+ signalling in endothelial cells. Hence, our data that inhibition of NCX with 2′,4′-dichlorobenzamil or removal of extracellular Na+ prevented Ca2+ oscillations further suggest that the NCX also plays a crucial role in agonist-evoked Ca2+ oscillations. To evaluate the role of NCX in Ca2+ oscillations in low [Ca2+]o the presence of agonist-sensitive NSCCs in the endothelial cells has to be considered (Nilius et al. 1997). It is notable that, in the HUVEC-derived cell line used in this study, non-selective cation currents were described through the human TRP3 channels (Kamouchi et al. 1999a). Considering the rather low Ca2+ permeability of these channels compared with monovalent cations (PCa/PNa∼1.6, Kamouchi et al. 1999b) and the finding that removal of extracellular Ca2+ increased the current through this type of channel (Kamouchi et al. 1999b), one might expect Na+ loading under low [Ca2+]o conditions (Graier et al. 1995) that facilitates the switch of the NCX activity towards the reversed mode (i.e. 3Na+ out/1Ca2+ in). This is further supported by our findings that in the EA.hy926 cells Na+ loading in response to histamine could be observed at least in certain subplasmalemmal domains. Unfortunately, due to the technical limitations these experiments needed to be performed in conditions where the reversed mode of the NCX (i.e. 1Ca2+ in and 3Na+ out) was prevented by removal of extracellular Ca2+. However, similar data were obtained by direct inhibition of NCX with 2′,4′-dichlorobenzamil and the amplitude of histamine-evoked Ca2+ oscillations was greatly increased in low [Na+]o solution (19HBS), conditions in which the reversed mode of the NCX is pronounced. These data are in line with our assumption that under low [Ca2+]o conditions NCX might work in the reversed mode and might be responsible for Ca2+ influx and Na+ efflux.
In endothelial cells, agonist-induced Ca2+ oscillations have been often reported to depend on intracellular Ca2+ release by IP3 (Jacob, 1991). In agreement with these reports, the IP3-generating agonist histamine initiated Ca2+ oscillations in EA.hy926 cells. Interestingly, a repetitive stimulation failed to initiate Ca2+ oscillations in low Ca2+ solution (HBS), while a short exposure to HBS-Ca2+ restored the ability to observe Ca2+ oscillations in response to a repetitive histamine stimulation. These data further support our findings on the dependency of Ca2+ oscillations on extracellular Ca2+. Hence, the data suggest that to initiate Ca2+ spiking in these cells the IP3-sensitive Ca2+ pools need to be refilled by a large Ca2+ influx (in HBS-Ca2+: [Ca2+]o, 2.5 mM) to ensure a large Ca2+ release in response to the addition of the agonist, while only a small Ca2+ entry (in HBS: [Ca2+]o, 10–30 μm) is necessary to preserve oscillations once they have been initiated.
Nevertheless, to maintain Ca2+ oscillations refilling of Ca2+ pools seems to constitute a crucial event in Ca2+ oscillations, indicated by the inhibitory effect of the SERCA inhibitor BHQ on Ca2+ oscillations. In agreement with these findings SERCA has been shown to contribute to histamine-induced Ca2+ oscillation in human umbilical vein endothelial cells (Morgan & Jacob, 1998), the cell type from which the EA.hy926 are derived (Edgell et al. 1983). In contrast, bradykinin-induced Ca2+ oscillations in bovine arterial valve endothelial cells do not depend on the activity of SERCA or the NCX (Laskey et al. 1992), further supporting the hypothesis that the mechanisms for agonist-evoked Ca2+ oscillations vary between two given endothelial cells (Rink & Jacob, 1989). Alternatively, the distinct patterns/mechanisms of Ca2+ spiking in both reports might be due to the different agonists used (i.e. bradykinin and histamine) as has been recently shown for substance P and bradykinin on Ca2+-mediated KCa channel activation in porcine coronary endothelial cells (Frieden et al. 1999).
Besides the pivotal role of SERCA-mediated refilling of intracellular Ca2+ pools, our findings that an inhibition of ryanodine receptors prevented Ca2+ oscillations indicate that RsCR is involved in the rhythmic changes in [Ca2+]bulk after stimulation. This is in agreement with previous reports in rat chromaffin cells (Malgaroli et al. 1990) and mouse pancreatic acinar cells (Wakui et al. 1990).
Thus, histamine-induced Ca2+ oscillations in EA.hy926 cells represent a complex phenomenon that may include two different Ca2+ pools as suggested in pancreatic acinar cells (Wakui et al. 1990) and several others (Pozzan et al. 1988; Krause et al. 1989; Berridge, 1991). Convincingly, we have described evidence that in EA.hy926 cells the subplasmalemmal Ca2+ stores are sensitive to either IP3 (IP3-sensitive Ca2+ store, IPsS) or ryanodine (ryanodine-sensitive Ca2+ store, RsS; Paltauf-Doburzynska et al. 1999).
As summarized in Fig. 10, our data suggest that stimulation with histamine results in the generation of IP3 () that triggers the depletion of intracellular Ca2+ pools via IP3 and Ca2+-induced Ca2+ release (CiCR;). Hence, histamine, possibly via increasing [Ca2+]sub activates NSCCs (Baron et al. 1996) that, under low [Ca2+]o conditions favour the influx of Na+ (). As a result of such ‘Na+ loading’ in certain subplasmalemmal domains in low [Ca2+]o solution via NSCCs (TRP3?) Na+ is extruded via NCX resulting in an influx of Ca2+ (). The Ca2+ entry via NCX increases [Ca2+]sub, initiates subplasmalemmal RsCR (Paltauf-Doburzynska et al. 1998, 1999) and activates KCa channels (Frieden & Graier, 2000; ). Activation of KCa channels hyperpolarizes the cells and provides the driving force for cation influx via NSCCs. As a result of this increased [Ca2+]sub, sER Ca2+ pools (RsS) are refilled via SERCA () and, again, become depleted via CiCR, triggered by IP3-mediated Ca2+ release from IPsS resulting in an elevation in [Ca2+]bulk (). In agreement with this hypothesis, pulsatile intracellular Ca2+ release has been shown to be independent of fluctuations in IP3 concentration (Wakui et al. 1989).
Our hypothesis on the crucial role of subplasmalemmal Ca2+ stores is further supported by our findings that dislocation of the sER with nocodazole (Graier et al. 1998; Paltauf-Doburzynska et al. 1999) prevented Ca2+ oscillations. These findings are in agreement with the stochastic computer model of Kraus et al. (1996) that indicates the importance of the cell morphology for Ca2+ oscillations. Since collapsing the sER by nocodazole disturbs SCCU function (Graier et al. 1998; Paltauf-Doburzynska et al. 1998; Frieden & Graier, 2000), these data point to an involvement of SCCU in Ca2+ oscillations in EA.hy926 cells and suggest that the refilling of sER Ca2+ stores via SERCA depends on the morphological vicinity of the sER with certain cell membrane domains. This is in line with the report by Hofer et al. (1998) who showed that the refilling of Ca2+ stores does not require an elevation in [Ca2+]bulk in BHK-21 fibroblasts. Consistent with our suggestion, that SERCA-mediated Ca2+ store refilling depends on an insulated elevation of [Ca2+]sub, activation of KCa channels always precedes the elevation in [Ca2+]bulk. Moreover, the instant activation of the KCa channels may reflect the sudden RsCR (Fig. 10,). However, the rhythmic elevation of [Ca2+]bulk constitutes a ‘buffered’ phenomenon depending on the refilling of RsS (Fig. 10,).
Certainly, there are several points that need to be clarified to further understand agonist-induced Ca2+ oscillations. Based on our comparative studies of the EA.hy926 cells with native cells from the human umbilical vein and bovine coronary artery, we believe that this model has physiological significance to provide a better understanding of localized Ca2+ signalling in oscillating endothelial cells. Unfortunately, the spatial resolution of confocal/deconvolution microscopes does not allow Ca2+/Na+ measurements within the SCCU area and we need to target, for example, Ca2+-sensing proteins to the cell membrane to monitor highly localized Ca2+ signalling in this small area. Nevertheless, our data presented here provide evidence for an involvement of subplasmalemmal Ca2+ signalling in Ca2+ oscillations in endothelial cells. Our previous work (Graier et al. 1998; Paltauf-Doburzynska et al. 1998, 1999), the accompanying paper (Frieden & Graier, 2000) and the recent work of other groups (Hüser et al. 1999) strengthen the idea of localized Ca2+ signalling as a key phenomenon in endothelial Ca2+ homeostasis, the physiological consequences of which need further investigation.
Acknowledgments
We thank Mrs Beatrix Petschar for her excellent technical assistance. This work was supported by the Austrian Science Funds (P-12341-Med and SFB 714), the Austrian Nationalbank (P7542 and P7902), the Kamillo-Eisner Stiftung (Hergiswil, Switzerland), the Franz Lanyar Foundation and the Swiss National Funds (M.F.).
References
- Baron A, Frieden M, Chabaud F, Bény J-L. Ca2+-dependent non-selective cation and potassium channels activated by bradykinin in pig coronary artery endothelial cells. The Journal of Physiology. 1996;493:691–706. doi: 10.1113/jphysiol.1996.sp021415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Cytoplasmic calcium oscillations: a two pool model. Cell Calcium. 1991;12:63–72. doi: 10.1016/0143-4160(91)90009-4. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. The AM and FM of calcium signaling. Nature. 1997;386:759–760. doi: 10.1038/386759a0. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichter H, Lückhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. American Journal of Physiology. 1988;255:H965–969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- Carter TD, Bogle RG, Bjaaland T. Spiking of intracellular calcium ion concentration in single cultured pig aortic endothelial cells stimulated with ATP or bradykinin. Biochemical Journal. 1991;278:697–704. doi: 10.1042/bj2780697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL. Bradykinin-induced increases in cytosolic free calcium and ionic currents in bovine aortic endothelial cells. Circulation Research. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- Dolmetsch RE, Lewis RS. Signaling between intracellular Ca2+ stores and depletion-activated Ca2+ channels generates [Ca2+]i oscillations in T lymphocytes. Journal of General Physiology. 1994;103:365–388. doi: 10.1085/jgp.103.3.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dömötör E, Abbott NJ, Adam-Vizi V. Na+-Ca2+ exchange and its implications for calcium homeostasis in primary cultured rat brain microvascular endothelial cells. The Journal of Physiology. 1999;515:147–155. doi: 10.1111/j.1469-7793.1999.147ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgell CJS, McDonald CC, Graham JB. Permanent cell line expressing factor VIII related antigen established by hybridization. Proceedings of the National Academy of Sciences of the USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Encabo A, Romanin C, Birke FW, Kukovetz WR, Groschner K. Inhibition of a store-operated Ca2+ entry pathway in human endothelial cells by the isoquinoline derivative LOE 908. British Journal of Pharmacology. 1996;119:702–706. doi: 10.1111/j.1476-5381.1996.tb15729.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foskett JK, Wong D. Free cytosolic Ca2+ concentration oscillation in thapsigargin-treated parotid acinar cells are caffeine- and ryanodine-sensitive. Journal of Biological Chemistry. 1991;266:14535–14538. [PubMed] [Google Scholar]
- Foskett JK, Wong D. Calcium oscillation in parotid acinar cells induced by microsomal Ca2+-ATPase inhibition. American Journal of Physiology. 1992;262:C656–663. doi: 10.1152/ajpcell.1992.262.3.C656. [DOI] [PubMed] [Google Scholar]
- Frieden M, Graier WF. Subplasmalemmal ryanodine-sensitive Ca2+ release contributes to Ca2+-dependent K+ channel activation in a human umbilical vein endothelial cell line. The Journal of Physiology. 2000;524:715–724. doi: 10.1111/j.1469-7793.2000.00715.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieden M, Sollini M, Bény J-L. Substance P and bradykinin activate different types of KCa currents to hyperpolarize cultured porcine coronary artery endothelial cells. The Journal of Physiology. 1999;519:361–371. doi: 10.1111/j.1469-7793.1999.0361m.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Paltauf-Doburzynska J, Hill BJF, Fleischhacker E, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca2+ elevation beneath the cell membrane. The Journal of Physiology. 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. The Journal of Physiology. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Sturek M, Kukovetz WR. Ca2+ regulation and endothelial vascular function. Endothelium. 1994;1:223–236. [Google Scholar]
- Groschner K, Graier WF, Kukovetz WR. Histamine induces K+, Ca2+ and Cl− currents in human vascular endothelial cells – Role of ionic currents in stimulation of nitric oxide biosynthase. Circulation Research. 1994;75:304–314. doi: 10.1161/01.res.75.2.304. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Thomas AP. Minimal requirements for calcium oscillations driven by the IP3 receptor. EMBO Journal. 1997;16:3533–3543. doi: 10.1093/emboj/16.12.3533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Landolfi B, Debellis L, Pozzan T, Curci S. Free [Ca2+] dynamics measured in agonist-sensitive stores of single living intact cells: a new look at the refilling process. EMBO Journal. 1998;17:1986–1995. doi: 10.1093/emboj/17.7.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoyer J, Kohler R, Distler A. Mechanosensitive Ca2+ oscillations and STOC activation in endothelial cells. FASEB Journal. 1998;12:359–366. doi: 10.1096/fasebj.12.3.359. [DOI] [PubMed] [Google Scholar]
- Hüser J, Holda JR, Kockskämper J, Blatter LA. Focal agonist stimulation results in spatially restricted Ca2+ release and capacitative Ca2+ entry in bovine vascular endothelial cells. The Journal of Physiology. 1999;514:101–109. doi: 10.1111/j.1469-7793.1999.101af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamuro YMS, Zhang XF, Minowa T, Enoki T, Okamoto Y, Hasegawa H, Furutani H, Okazawa M, Ishikawa M, Hashimoto NMT. Activation of three types of voltage-independent Ca2+ channel in A7r5 cells by endothelin-1 as revealed by a novel Ca2+ channel blocker LOE 908. British Journal of Pharmacology. 1999;126:1107–1114. doi: 10.1038/sj.bjp.0702416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob R. Calcium oscillations in electrically non-excitable cells. Biochimica et Biophysica Acta. 1990;1052:427–438. doi: 10.1016/0167-4889(90)90152-4. [DOI] [PubMed] [Google Scholar]
- Jacob R. Calcium oscillations in endothelial cells. Cell Calcium. 1991;12:127–134. doi: 10.1016/0143-4160(91)90014-6. [DOI] [PubMed] [Google Scholar]
- Jacob R, Merrit JE, Hallam TJ, Rink TJ. Repetitive spikes in cytoplasmic calcium evoked by histamine in human endothelial cells. Nature. 1988;335:40–45. doi: 10.1038/335040a0. [DOI] [PubMed] [Google Scholar]
- Kamouchi M, Mamin A, Droogmans G, Nilius B. Nonselective cation channels in endothelial cells derived from human umbilical vein. Journal of Membrane Biology. 1999a;169:29–38. doi: 10.1007/pl00005898. [DOI] [PubMed] [Google Scholar]
- Kamouchi M, Philipp S, Flockerzi V, Wissenbach U, Mamin A, Raeymaekers L, Eggermont J, Droogmans G, Nilius B. Properties of heterologously expressed hTRP3 channels in bovine pulmonary artery endothelial cells. The Journal of Physiology. 1999b;518:345–358. doi: 10.1111/j.1469-7793.1999.0345p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krause KH, Pittet D, Volpe P, Pozzan T, Meldolesi J, Lew DP. Calciosome, a sarcoplasmic reticulum-like organelle involved in intracellular Ca2+-handling by non-muscle cells: studies in human neutrophils and HL-60 cells. Cell Calcium. 1989;10:351–361. doi: 10.1016/0143-4160(89)90061-4. [DOI] [PubMed] [Google Scholar]
- Kraus M, Wolf B, Wolf B. Crosstalk between cellular morphology and calcium oscillation patterns. Insights from a stochastic computer model. Cell Calcium. 1996;19:461–472. doi: 10.1016/s0143-4160(96)90055-x. [DOI] [PubMed] [Google Scholar]
- Laskey RE, Adams DJ, Cannell M, van Breemen C. Calcium entry-dependent oscillation of cytosolic calcium concentration in cultured endothelial cell monolayer. Proceedings of the National Academy of Sciences of the USA. 1992;89:1690–1694. doi: 10.1073/pnas.89.5.1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lückhoff A, Busse R. Calcium influx into endothelial cells and formation of endothelium-derived relaxing factor is controlled by the membrane potential. Pflügers Archiv. 1990;416:305–311. doi: 10.1007/BF00392067. [DOI] [PubMed] [Google Scholar]
- Lynch M, Gillespie J, Greenwell J, Johnson C. Intracellular calcium ‘signatures’ evoked by different agonists in isolated bovine aortic endothelial cells. Cell Calcium. 1992;13:227–233. doi: 10.1016/0143-4160(92)90011-g. [DOI] [PubMed] [Google Scholar]
- Malgaroli A, Fesce R, Meldolesi J. Spontaneous [Ca2+]i fluctuations in rat chromaffin cells do not require inositol 1,4,5-trisphosphate elevations but are generated by a caffeine- and ryanodine-sensitive intracellular Ca2+ store. Journal of Biological Chemistry. 1990;265:3005–3008. [PubMed] [Google Scholar]
- Meldolesi J. Oscillation, activation, expression. Nature. 1998;392:863–866. doi: 10.1038/31804. [DOI] [PubMed] [Google Scholar]
- Morgan AJ, Jacob R. Differential modulation of the phases of a Ca2+ spike by the store Ca2+-ATPase in human umbilical vein endothelial cells. The Journal of Physiology. 1998;513:83–101. doi: 10.1111/j.1469-7793.1998.083by.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilius B, Schwartz G, Oike M, Droogmans G. Histamine-activated, non-selective cation currents and Ca2+ transients in endothelial cells from human umbilical vein. Pflügers Archiv. 1993;424:285–293. doi: 10.1007/BF00384354. [DOI] [PubMed] [Google Scholar]
- Nilius B, Viana F, Droogmans G. Ion channels in vascular endothelium. Annual Review of Physiology. 1997;59:145–170. doi: 10.1146/annurev.physiol.59.1.145. [DOI] [PubMed] [Google Scholar]
- Paltauf-Doburzynska J, Frieden M, Graier WF. Mechanisms of Ca2+ store depletion in single endothelial cells in a Ca2+-free environment. Cell Calcium. 1999;25:345–353. doi: 10.1054/ceca.1999.0038. [DOI] [PubMed] [Google Scholar]
- Paltauf-Doburzynska J, Posch K, Paltauf G, Graier WF. Stealth ryanodine-sensitive Ca2+ release contributes to activity of capacitative Ca2+ entry and nitric oxide synthase in bovine endothelial cells. The Journal of Physiology. 1998;513:369–379. doi: 10.1111/j.1469-7793.1998.369bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T, Volpe P, Zorzato F, Bravin M, Krause KH, Lew DP, Hashimoto S, Bruno B, Meldolesi J. The Ins(1,4,5)P3-sensitive Ca2+ store of non-muscle cells: endoplasmic reticulum or calciosomes? Journal of Experimental Biology. 1988;139:181–193. doi: 10.1242/jeb.139.1.181. [DOI] [PubMed] [Google Scholar]
- Rink TJ, Jacob R. Calcium oscillations in non-excitable cells. Trends in Neurosciences. 1989;12:43–46. doi: 10.1016/0166-2236(89)90133-1. [DOI] [PubMed] [Google Scholar]
- Sage SO, Adams DJ, van Breemen C. Synchronized oscillations in cytoplasmic free calcium concentration in confluent bradykinin-stimulated bovine pulmonary artery endothelial cell monolayers. Journal of Biological Chemistry. 1989;264:6–9. [PubMed] [Google Scholar]
- Sage SO, van Breemen C, Cannell MB. Sodium-calcium exchange in cultured bovine pulmonary artery endothelial cells. The Journal of Physiology. 1991;440:569–580. doi: 10.1113/jphysiol.1991.sp018725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M, Caldwell WM, Humphrey DA, Wagner-Mann C. Methods for simultaneous voltage-clamp, microfluorimetry, and video of cells. I. Electronic and optical instrumentation. In: Sperelakis N, Kuriyama H, editors. Ion Channels of Vascular Smooth Muscle Cells and Endothelial Cells. New York: Elsevier; 1991a. pp. 239–267. [Google Scholar]
- Sturek M, Stehno-Bittle L, Obye PK. Methods for simultaneous voltage-clamp, microfluorimetry, and video of cells. II. Physiology. In: Sperelakis N, Kuriyama H, editors. Ion Channels of Vascular Smooth Muscle Cells and Endothelial Cells. New York: Elsevier; 1991b. pp. 269–294. [Google Scholar]
- Uneyama C, Uneyama H, Akaike N, Takahashi M. Cyclic GMP inhibits cytoplasmic Ca2+ oscillation by increasing Ca2+-ATPase activity in rat megakaryocytes. European Journal of Pharmacology. 1998;347:355–361. doi: 10.1016/s0014-2999(98)00123-x. [DOI] [PubMed] [Google Scholar]
- Usachev YM, Marchenko SM, Sage SO. Cytosolic calcium concentration in resting and stimulated endothelium of excised intact rat aorta. The Journal of Physiology. 1995;489:309–317. doi: 10.1113/jphysiol.1995.sp021052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakui M, Osipchuk YV, Petersen OH. Receptor-activated cytoplasmic Ca2+ spiking mediated by inositol trisphosphate is due to Ca2+-induced Ca2+ release. Cell. 1990;63:1025–1032. doi: 10.1016/0092-8674(90)90505-9. [DOI] [PubMed] [Google Scholar]
- Wakui M, Potter BVL, Petersen OH. Pulsatile intracellular calcium release does not depend on fluctuations in inositol trisphosphate concentration. Nature. 1989;339:317–320. doi: 10.1038/339317a0. [DOI] [PubMed] [Google Scholar]