

Abstract
Whilst protein tyrosine kinase (PTK) activity can modulate expressed GABAA receptors in cell culture, the physiological consequences on synaptic GABAA receptors are unknown. This was examined using whole-cell recording of bicuculline-sensitive mIPSCs in Purkinje cells (PCs) in cerebellar slices.
Postsynaptic application of a peptide activator of the non-receptor PTK Src (Src-peptide) enhanced mIPSC amplitudes by 39 % in the presence of brain-derived neurotrophic factor (BDNF) only; neurotrophin-3 (NT-3) was ineffective in this regard. Thus Src and TrkB (the receptor for BDNF) can physiologically interact to modulate synaptic GABAA receptors.
In the presence of BDNF, pharmacological activation of metabotrophic glutamate receptor subtype 1 (mGluR1) by (S)-3,5-dihydrophenylglycine (3,5-DHPG) also lead to a 32 % enhancement of mIPSCs. This enhancement was blocked by intracellular dialysis of PCs with PP1, a selective inhibitor of Src.
It is concluded that, whilst GABAA receptors are not constitutively regulated by endogenous PTK activity in PCs, co-activation of TrkB by BDNF and Src by mGluR1 is required to modulate GABAergic synapses in PCs.
Whilst GABAA receptor function is regulated, inter alia, by protein kinase-mediated phosphorylation (Moss & Smart, 1996), little is known regarding such regulation of synaptic GABAA receptors. As with serine/threonine protein kinases, PTK activity is necessary for maintaining both native and recombinant GABAA receptor function (Moss et al. 1995; Wan et al. 1997a; Jassar et al. 1997). Specifically, phosphorylation of the γ2L GABAA receptor subunit leads to a functional modification of the receptor (Moss et al. 1995). Until now, however, no evidence has been provided for regulation of synaptic GABAA receptor function by PTK activity.
Tyrosine kinases fall into two broad categories, the non-receptor PTKs and the receptor PTKs. The latter are membrane bound enzymes, activated by the binding of their cognate ligands, e.g. the neurotrophins (NTs). Receptor PTK activation has been implicated in neuronal growth, differentiation, and synaptic transmission in the hippocampus and cortex (Theonen, 1995; Lessmann, 1998). In the cerebellum, however, no data were available on the possible role of NTs in synaptic transmission. Cerebellar Purkinje cells (PCs), whilst lacking the NT receptor TrkA, express TrkB (Barbacid, 1994) and TrkC (Ernfors et al. 1992; Tessarollo et al. 1993). Moreover, PC survival and development requires TrkB (Schwartz et al. 1997) and TrkC (Mount et al. 1994) activation. Thus whilst NTs are known to regulate neuronal plasticity in PCs, a role for NTs in the regulation of synaptic transmission in PCs is unknown. The experiments reported herein thus describe the effects of NTs (BDNF and NT-3) on inhibitory, GABAergic synapses of cerebellar PCs. The data suggest that BDNF acts as a permissive factor in the regulation of synaptic GABAA receptors by mGluR1 activation of the non-receptor PTK Src.
METHODS
General methods to obtain cerebellar slices have been described previously (Llano et al. 1991). Briefly, thin (180–200 μm) sagittal slices were cut from cerebella of young rats (13–16 days old), which had been decapitated following cervical dislocation, in ice-cold, oxygenated (5 % O2-95 % CO2), bicarbonate-buffered (pH 7.3) external solution (BBS). BBS contained (mM): 125 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 1.25 NaH2PO4, 26 NaHCO3 and 10 glucose. Slices were superfused at a rate of 1–1.5 ml min−1, at room temperature, with BBS containing the AMPA receptor antagonist 2,3-dihydroxy-6-nitro-7-sulphamoyl-benzoquinozaline (NBQX, 10 μM; Tocris Cookson, Bristol, UK) and the Na+ channel blocker TTX (400 nM; Sigma, Missouri, USA). PCs were visually identified and tight-seal, whole cell recorded by the prescribed procedure for this neuronal type (capacitance cancelled and series resistance partially compensated (50–75 %); Llano et al. 1991). Patch pipettes of between 2.2 and 2.7 MΩ resistance with a Cs+-based internal solution were used. The internal solution consisted of (mM): 150 CsCl, 10 Hepes, 1 EGTA, 0.1 CaCl2, 4.6 MgCl2, 4.6 Na-ATP, 0.4 Na-GTP, pH set to 7.3 with KOH. PCs were monitored at a holding potential of -60 mV using an EPC-9 patch-clamp amplifier (HEKA Electronics, Germany) running Pulse control and acquisition software (HEKA Electronics). The sampling rate was 4 kHz, with a Bessel filter set at 0.8 kHz. Miniature IPSCs were acquired in 3 min recording blocks, ensuring that, throughout the course of an experiment, series resistance could be regularly monitored between acquisition protocols. Typical starting values for series resistance upon break-in ranged between 4 and 15 MΩ; during an experiment series resistance did not significantly change (data not shown). Lavendustin A (1 mM), 4-amino-5-(4-methylphenyl)-7-(t-butyl)pyrazole[3,4-d]pyrimidine (PP1, 10 μM) and Src-peptide (1 mM) were all dissolved into the internal solution from their stock solutions just prior to use (these three inhibitors were obtained from Calbiochem-Novabiochem, CA, USA). Following 9 min baseline recording (i.e. 3 acquisition protocols), BDNF and NT-3 (Sigma, MO, USA) were applied to the PC via a puffer pipette. Stock BDNF and NT-3 were freshly prepared every few days in phosphate-buffered saline (PBS), and diluted to the final concentration of 50 ng ml−1 in BBS. In order to maximize any PTK effects, vanadate (100 μM) was included in the internal solution in those experiments in which PTK (receptor or non-receptor) activators were employed. Note that vanadate was not included in the internal solution in experiments where (S)-3,5-dihydroxyphenylglycine (3,5-DHPG) was applied, since a possible mechanism of Src activation is through Ca2+-dependent protein tyrosine phosphatase (PTP) activity, which may otherwise have been compromised. Miniature IPSCs were detected and analysed off-line using custom-written Igor (Wavemetrics, USA)-based routines. Statistical deviations away from mean values are expressed ±s.e.m. (standard error of the mean), with n being the number of experiments. The statistical significance of the difference of the mean of two samples was calculated using the Mann-Whitney U test.
RESULTS
Properties of GABAA receptor-mediated mIPSCs
Whole cell, patch-clamp recording of bicuculline (30 μM)-sensitive (data not shown), GABAA receptor-mediated mIPSCs were monitored over a 25 min recording period from PCs. Whilst displaying a large degree of variability in amplitude (coefficient of variation, 0.87 ± 0.02), mIPSCs were stable over time, with no significant difference (P > 0.1, n = 8) in amplitude, frequency, rise time, or half-width, between control (3–5 min post break-in) and test (21–23 min) recording periods (Table 1). No significant correlation (P > 0.1) existed between rise times or half-widths and mIPSC amplitudes (Kendall's correlation coefficient (τ) < 0.6 in all cases), indicating that dendritic filtering did not shape mIPSC kinetics (Jonas et al. 1993). This mIPSC stationarity over time thus enabled the role of the PTK signalling system in the regulation of synaptic GABAA receptors to be examined.
Table 1. Mean mIPSC kinetic parameters.
| Amplitude (pA) | Frequency (HZ) | Rise time (ms) | Half-width (ms) | ||
|---|---|---|---|---|---|
| Control (8) | c | 155.79 ± 33.83 | 4.17 ± 0.81 | 3.71 ± 0.23 | 9.54 ± 0.77 |
| t | 135.70 ± 22.62 | 3.90 ± 0.64 | 3.91 ± 0.21 | 10.84 ± 0.84 | |
| Staurosporine (5) | c | 96.67 ± 9.74 | 6.55 ± 1.26 | 3.91 ± 0.80 | 12.98 ± 0.97 |
| t | 78.36 ± 6.66 | 6.04 ± 1.40 | 3.74 ± 0.62 | 15.03 ± 1.27 | |
| Lavendustin A (4) | c | 82.81 ± 12.87 | 5.94 ± 1.32 | 4.02 ± 1.18 | 10.33 ± 0.73 |
| t | 90.53 ± 7.42 | 4.99 ± 0.77 | 3.34 ± 0.61 | 9.98 ± 0.24 | |
| Vanadate (5) | c | 100.51 ± 8.65 | 8.07 ± 0.95 | 4.01 ± 0.58 | 10.8 ± 1.44 |
| t | 101.56 ± 10.04 | 6.76 ± 1.37 | 4.13 ± 0.56 | 11.88 ± 0.96 | |
| Src-peptide (7) | c | 108.35 ± 20.34 | 6.01 ± 1.19 | 3.55 ± 0.19 | 10.34 ± 0.75 |
| t | 103.35 ± 14.60 | 4.83 ± 0.92 | 3.23 ± 0.18 | 10.27 ± 0.46 | |
| BDNF (5) | c | 89.97 ± 21.90 | 6.11 ± 1.27 | 3.75 ± 0.08 | 10.63 ± 1.58 |
| t | 82.26 ± 20.51 | 4.56 ± 1.20 | 3.58 ± 0.12 | 11.78 ± 1.69 | |
| BDNF + Src-peptide (10) | c | 74.53 ± 14.64 | 5.57 ± 0.71 | 3.45 ± 0.22 | 11.88 ± 0.69 |
| t | 102.58 ± 20.75†† | 6.89 ± 1.61 | 3.23 ± 0.17 | 11.31 ± 1.04 | |
| NT-3 + Src-peptide (14) | c | 89.47 ± 7.25 | 5.81 ± 0.89 | 3.39 ± 0.06 | 10.28 ± 0.49 |
| t | 90.88 ± 7.55 | 5.2 ± 1.09 | 3.31 ± 0.07 | 10.13 ± 1.37 | |
| DHPG (8) | c | 86.90 ± 9.07 | 7.70 ± 1.49 | 3.58 ± 0.03 | 10.28 ± 0.49 |
| t | 79.98 ± 9.01 | 6.43 ± 1.13 | 3.57 ± 0.05 | 10.63 ± 1.50 | |
| DHPG + BDNF (9) | c | 81.47 ± 8.46 | 4.96 ± 0.57 | 3.53 ± 0.06 | 11.39 ± 1.28 |
| t | 106.56 ± 10.24† | 4.02 ± 0.67 | 3.47 ± 0.06 | 11.58 ± 1.75 | |
| DHPG + BDNF + PP1 (5) | c | 126.16 ± 10.08 | 8.72 ± 1.99 | 3.46 ± 0.14 | 10.30 ± 0.73 |
| t | 119.07 ± 10.25 | 7.65 ± 1.34 | 3.35 ± 0.08 | 11.85 ± 1.44 |
The data for each experimental manipulation are presented in 2 rows, the top of which represents the mean ±s.e.m. data obtained during control (c), the bottom of which similarly represents that obtained during test periods (t). Parameters emboldened are significantly different
(P < 0.005 or
P < 0.05).
Numbers in parentheses indicate the number of PCs within that experimental group (both control and test).
Constitutive PTK activity is not required for maintaining GABAA receptor-mediated mIPSC amplitudes
Bath application of staurosporine (1 μM), a PTK inhibitor at this concentration (Ruegg et al. 1989), had no significant affect on mIPSC parameters (P > 0.1, n = 5; Table 1). Intracellular dialysis with the selective PTK inhibitor lavendustin A (1 mM, n = 4; O'Dell et al. 1991) was also without effect (P > 0.1 for all mIPSC parameters; Table 1). Furthermore, intracellular dialysis with the PTP inhibitor sodium orthovanadate (100 μM, n = 5; Gordon, 1991) similarly failed to have any significant effects on mIPSC parameters (P > 0.1; Table 1). These data strongly suggest a lack of basal PTK-mediated regulation of synaptic GABAA receptors in PCs.
Modulation of GABAA receptor-mediated mIPSC amplitude requires a synergistic interaction between TrkB and Src
To test if endogenous Src activity could modulate synaptic GABAA receptors, Src-peptide, a selective phosphopeptide-activator of Src (Lancaster & Rogers, 1998) was intracellularly dialysed into PCs. No significant effect on mIPSCs (P > 0.1, for all mIPSC parameters, n = 7; Table 1) was observed.
NTs, by their selective binding to their receptors (nerve growth factor (NGF) to TrkA, BDNF and NT-4/5 to TrkB, and NT-3 to TrkC) activate the inherent tyrosine kinase capability of that receptor (Barbacid, 1994). To test if NTs could modulate synaptic GABAA receptor function, BDNF and NT-3 were exogenously applied by puffer pipette to the PCs under investigation. BDNF was applied at 50 ng ml−1, a concentration selective for TrkB activation (Rodriguez-Tébar et al. 1990). BDNF alone had no significant effect (P > 0.1, n = 5) on mIPSCs (Table 1) or upon PCs themselves (data not shown). When Src-peptide (1 mM) was included in the recording pipette, however, BDNF (50 ng ml−1) application quickly resulted (within 1–2 min of application) in a significant 39 ± 8 % increase (P < 0.001, n = 10) in mIPSC amplitude (Table 1 and Figs 1Aa-c and 1Ba), with no significant effect (P > 0.1) on either rise time or half-width (Table 1). Src-peptide/BDNF-enhanced mIPSCs were completely antagonized by bicuculline (30 μM; data not shown). Miniature IPSC frequency was unaffected by BDNF application (with or without postsynaptic Src-peptide dialysis; cf. Table 1 and Fig. 1Bb), indicating that any presynaptic effects of BDNF were negligible. Finally, NT-3 application (50 ng ml−1) had no significant effect (P > 0.1, n = 14) on mIPSCs with (Table 1 and Fig. 1C) or without (data not shown) Src-peptide included in the recording electrode.
Figure 1. An interaction between TrkB and Src enhances GABAA receptor-mediated mIPSC amplitudes.
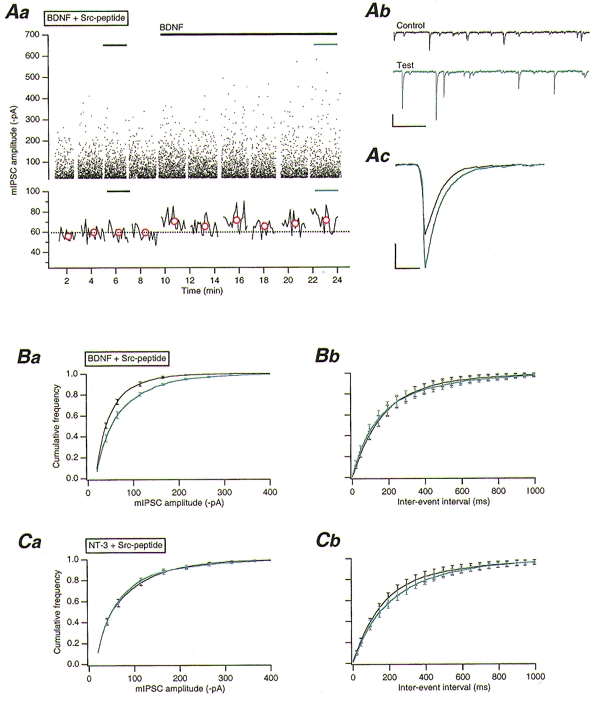
Aa, upper panel: plot of mIPSC amplitudes against time, with Src-peptide (1 mM) and orthovanadate (100 μM) included in the intracellular solution. BDNF (50 ng ml−1) application is indicated by the long black bar. The short black and blue bars in both the upper and lower panels indicate those data sets from which the mIPSCs illustrated in b and c were taken. Lower panel: running average of mIPSC amplitude against time (50 event bins). Red circles indicate the mean mIPSC amplitude of each corresponding data set in the upper panel. The dotted line indicates the overall mean control amplitude before BDNF application. Ab, example traces of mIPSC activity obtained from periods indicated in Aa. Scale bars, 100 pA and 500 ms. Ac, overlaid average mIPSCs obtained from all events in control (n = 843) and test (n = 813) periods illustrated in Aa. Scale bars, 20 pA and 20 ms. Ba, pooled (n = 10) cumulative frequency distributions for mIPSC amplitudes obtained before (black line) and during (blue line) BDNF application, as illustrated in Aa. Bb, pooled (n = 10) cumulative frequency distributions for mIPSC interevent intervals obtained before and during BDNF application. Ca, pooled (n = 14) cumulative frequency distributions for mIPSC amplitudes obtained before (black line) and during (blue line) NT-3 (50 ng ml−1) application. Cb, pooled (n = 14) cumulative frequency distributions for mIPSC inter-event intervals obtained before (black line) and during (blue line) NT-3 application.
mIPSC amplitudes are modulated through Src stimulation, via pharmacological activation of mGluR1, and TrkB
Pharmacological activation of mGluRs can activate neuronal PTKs (Siciliano et al. 1994). Long-term depression, a mGluR1-dependent form of synaptic plasticity at glutamatergic parallel fibre-PC synapses (Aiba et al. 1994), can be blocked by PTK inhibition (Boxall et al. 1996), suggesting that PTKs are important in PC synaptic physiology. Thus, whether mGluR1 activation could evoke a PTK-dependent modulation of synaptic GABAA receptors in PCs was tested by applying the selective mGluR1 agonist 3,5-DHPG (Schoepp et al. 1994).
Continuous bath application of 3,5-DHPG (10 μM) alone resulted in a slowly desensitizing (duration ∼260 s) inward current (mean peak amplitude 684.5 ± 100.1 pA, n = 8). 3,5-DHPG (10 μM) was itself without significant effect (P > 0.1, n = 8) on mIPSCs (Table 1), including mIPSC frequency. This observation is indicative of a lack of effect on presynaptic GABA release, contrary to previous findings employing the mGluR agonist trans-aminocyclopentane dicarboxylic acid (ACPD) (Llano & Marty, 1995). When BDNF (50 ng ml−1) was co-applied with 3,5-DHPG (BDNF application beginning at the peak of the agonist-induced inward current), a significant 32 ± 6 % increase (P < 0.001, n = 9) in mIPSC amplitude was observed (Fig. 2A and Ba); no significant effect (P > 0.1) on rise time, half-width, or frequency, was apparent (Table 1 and Fig. 2Bb). The 3,5-DHPG/BDNF-enhanced mIPSCs were completely antagonized by bicuculline (30 μM; data not shown).
Figure 2. Activation of Src via mGluR1, and TrkB stimulation by BDNF, enhances GABAA receptor-mediated mIPSCs.
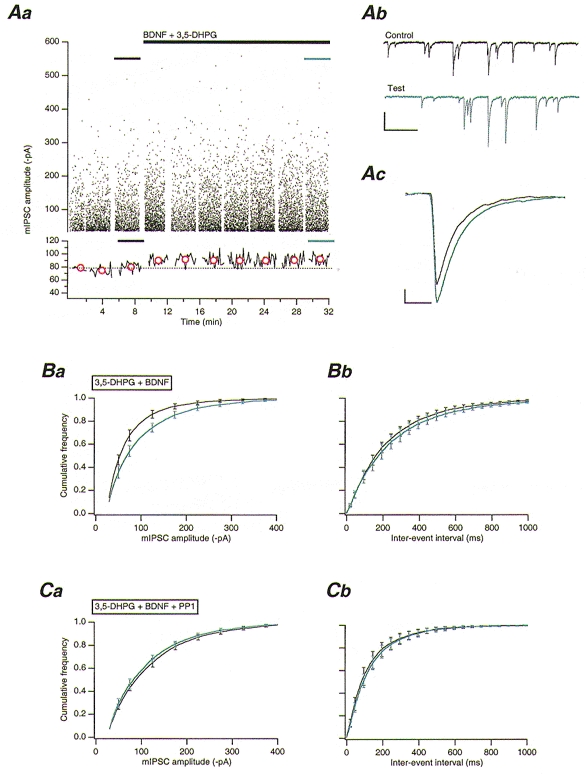
A, 3,5-DHPG application leads to a potentiation of mIPSCs in the presence of BDNF. Aa, upper panel: plot of mIPSC amplitudes against time. 3,5-DHPG (10 μM) and BDNF (50 ng ml−1) co-application is indicated by the long black bar. Lower panel: plot of running average of mIPSC amplitude against time (50 event bins). Red circles indicate the mean amplitude of all mIPSCs in each corresponding data set in the upper panel. The dotted line indicates the overall mean control amplitude before 3,5-DHPG/BDNF co-application. Ab, example traces of mIPSC activity obtained from periods indicated in Aa. Traces illustrate mIPSC activity before (black trace) and during (blue trace) 3,5-DHPG/BDNF application. Scale bars, 100 pA and 500 ms. Ac, overlaid average mIPSCs obtained from all events obtained before (n = 962), and during (n = 959) 3,5-DHPG/BDNF co-application. Scale bars, 20 pA and 20 ms. Ba, pooled (n = 9) cumulative frequency distributions for mIPSC amplitudes before (black line) and during (blue line) 3,5-DHPG/BDNF co-application. Bb, pooled (n = 9) cumulative frequency distributions for mIPSC inter-event intervals from PCs before (black line) and during (blue line) 3,5-DHPG/BDNF co-application. C, inhibition of Src by PP1 blocks the 3,5-DHPG/BDNF-mediated increase in mIPSC amplitude. Ca, pooled (n = 5) cumulative frequency distributions for mIPSC amplitudes before (black line) and during 3,5-DHPG/BDNF co-application (blue line). Note that PP1 (10 μM) was included in and orthovanadate excluded from the intracellular solution. Cb, pooled (n = 5) cumulative frequency distributions for mIPSC inter-event intervals before (black trace) and during 3,5-DHPG/BDNF co-application (blue trace).
Whether or not the 3,5-DHPG/BDNF-mediated enhancement in mIPSC amplitude was due to a mGluR1-dependent activation of Src was tested by intracellular dialysis with the selective Src inhibitor PP1 (Hanke et al. 1996). PP1 (10 μM) had no significant effect on either the 3,5-DHPG-mediated inward current (mean peak amplitude 440 ± 163 pA, n = 5;P > 0.1) or on mIPSCs themselves (data not shown). PP1 (10 μM) completely blocked the effects of 3,5-DHPG/BDNF co-application, however, with no significant changes in mIPSC amplitude, rise time, half-width, or frequency (P > 0.1, n = 5; Table 1 and Fig. 2Ca-b). Lavendustin A (1 mM) was similarly effective in inhibiting the 3,5-DHPG/ BDNF-mediated increase in mIPSC amplitude (n = 3; data not shown). These data strongly suggest that mGluR1 can activate Src in PCs.
DISCUSSION
In the presence of BDNF, intracellular dialysis of PCs with an activator of Src increased mIPSC amplitude, an observation suggestive of a postsynaptic interaction between the BDNF-activated NT receptor and Src. Given the lack of effects of various inhibitors of the PTK signalling pathway on mIPSCs, the data also suggest that no kinase-mediated constitutive regulation of synaptic GABAA receptors occurs in PCs. The data thus raise several questions regarding the nature of the NT receptor transducing the BDNF-mediated signal; the mechanism of interaction between this receptor and the non-receptor PTK; the nature of this non-receptor PTK; and the subsequent mechanism mediating the increase in mIPSC amplitude itself.
Role of Src and TrkB in regulating synaptic GABAA receptor-mediated mIPSC amplitudes
Firstly, as only application of BDNF, and not NT-3, was effective in increasing mIPSC amplitude, this indicates that the relevant NT receptor activated by BDNF was TrkB and not TrkC (Barbacid, 1994). Indeed, at the concentration used in these experiments (50 ng ml−1), BDNF is selective for TrkB (Rodriguez-Tébar et al. 1990).
Secondly, the likely target for the peptide PTK activator was Src itself as, apart from the selectivity of the peptide for Src, PCs are enriched in the neuronal isoform of Src (Sugrue et al. 1990) and PP1 (a selective Src inhibitor) blocked the observed effects of the peptide. As Src was incapable of affecting mIPSCs unless TrkB was first activated by BDNF, some form of interaction between these two PTKs would be necessary for GABAA receptor modulation to occur. Indeed, a direct molecular interaction between a Src-family kinase (Fyn) and TrkB has recently been reported to directly modulate AMPA receptor expression (Narisawa-Saito et al. 1999).
Thirdly, since a Src/TrkB-dependent increase in mIPSC amplitude was postsynaptic in origin, possible explanations are constrained by considerations of synaptic GABAA receptor saturation following quantal transmitter release, a notion as yet unaddressed in PCs. Assuming either postsynaptic receptor saturation or non-saturation, an increase in mIPSC amplitude upon PTK activation would require a functional change in the receptors themselves, and/or an increase in the number of expressed postsynaptic receptors, and/or an increase in postsynaptic receptor clustering to be apparent. Evidence exists for possible PTK involvement in such mechanisms. Thus, Src-mediated phosphorylation of the γ2L subunit of GABAA receptors increases mean open time and open probability of the channels (Moss et al. 1995), which could result in an increase in mIPSC amplitude. The incorporation of functional GABAA receptors into hippocampal neuronal membranes following insulin-receptor PTK activation has been described also (Wan et al. 1997b). Moreover, this incorporation was dependent on the β2 subunit of the GABAA receptor (Wan et al. 1997b), a subunit expressed in PCs (Laurie et al. 1992) and which can be phosphorylated by Src (Wan et al. 1997a). Whilst the data presented herein provide no direct evidence for any one mechanism, the fast time course of the effect of BDNF on mIPSCs (within 60 s of application; cf. Fig. 2A) argues against a change in the number of expressed GABAA receptors.
Pharmacological activation of mGluR1 stimulates Src
3,5-DHPG, in the presence of BDNF, increased mIPSC amplitude, indicating that mGluR1-TrkB co-activation activates a signalling cascade similar to that of Src-TrkB. Metabotropic GluR1 receptors are coupled, via G-proteins, to phospholipase C and, thence, to the generation of inositol trisphosphate (IP3) and diacylglycerol (DAG). Src could be activated via a number of possible processes downstream of mGluR1 activation following an elevation in [Ca2+]i from IP3-sensitive stores (Boxall & Lancaster, 1998).
Implications for synaptic transmission at inhibitory synapses in PCs
Activation of parallel fibre-PC synapses leads to a mGluR1-dependent depression of parallel fibre-PC synaptic transmission that can be blocked by PTK inhibitors (Boxall et al. 1996). It is thus likely that Src could be stimulated by synaptic mGluR activation, a frequency-dependent phenomenon (Batchelor & Garthwaite, 1997). This would imply that GABAA receptors would be modulated only at high frequency glutamatergic, excitatory parallel fibre input. This would enable PCs to dynamically match their inhibitory inputs directly to the degree of parallel fibre-mediated excitation being received.
BDNF expression is developmentally regulated in the CNS, with maturing granule cells being the only cell type to express BDNF in the cerebellar cortex (Maisonpierre et al. 1990). As NT release can occur in an activity-dependent manner (Thoenen, 1995), hypothetically, BDNF could be released from parallel fibres following high frequency stimulation. Such a mechanism would allow PCs to match their inhibitory inputs to those of their excitatory ones upon synaptic activation of mGluRs at parallel fibre-PC synapses.
In conclusion, the data suggest that postsynaptic TrkB activation by BDNF is required for Src-mediated modulation of synaptic GABAA receptors in cerebellar PCs in vitro. TrkC activation by NT-3 is ineffective in this regard. The data also provide strong evidence for the activation of Src by mGluR1 stimulation, thereby leading to the modulation of synaptic GABAA receptors upon TrkB activation. The co-activation of TrkB and mGluR1 is thus critical for the Src-mediated modulation of synaptic GABAA receptors in PCs. Whether or not such a modulation occurs through a Src-mediated phosphorylation of the γ2L GABAA receptor subunit is, as yet, unknown.
Acknowledgments
I would like to express my gratitude to Drs O. Caillard, M. Diana, L. Forti, I. Llano, A. Marty, C. Pouzat, D. Protti and C. Rosenmund for fruitful discussions. I am also greatly indebted to C. Pouzat for allowing me to use his Igor routines, and to O. Caillard for his help in preparing the figures. A.R.B. was an Alexander von Humboldt Fellow and a Marie Curie (TMR) Fellow of the EC.
References
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA, Tonegawa S. Deficient cerebellar long-term depression and impaired motor learning in mGluR1 mutant mice. Cell. 1994;79:377–388. [PubMed] [Google Scholar]
- Barbacid M. The Trk family of neurotrophin receptors. Journal of Neurobiology. 1994;25:1386–1403. doi: 10.1002/neu.480251107. [DOI] [PubMed] [Google Scholar]
- Batchelor AM, Garthwaite J. Frequency detection and temporally dispersed synaptic signal association through a metabotropic glutamate receptor pathway. Nature. 1997;385:74–77. doi: 10.1038/385074a0. [DOI] [PubMed] [Google Scholar]
- Boxall AR, Lancaster B. Tyrosine kinases and synaptic transmission. European Journal of Neuroscience. 1998;10:2–7. doi: 10.1046/j.1460-9568.1998.00009.x. [DOI] [PubMed] [Google Scholar]
- Boxall AR, Lancaster B, Garthwaite J. Tyrosine kinase is required for long-term depression in the cerebellum. Neuron. 1996;16:805–813. doi: 10.1016/s0896-6273(00)80100-2. [DOI] [PubMed] [Google Scholar]
- Ernfors P, Merlio J-P, Persson H. Cells expressing mRNA for neurotrophins and their receptors during embryonic rat development. European Journal of Neuroscience. 1992;4:1140–1158. doi: 10.1111/j.1460-9568.1992.tb00141.x. [DOI] [PubMed] [Google Scholar]
- Gordon JA. Use of vanadate as protein-phosphotyrosine phosphatase inhibitor. Methods in Enzymology. 1991;201:477–482. doi: 10.1016/0076-6879(91)01043-2. [DOI] [PubMed] [Google Scholar]
- Hanke JH, Gardner JP, Dow RL, Changelian PS, Brissette WH, Weringer EJ, Pollok BA, Connelly PA. Discovery of a novel, potent, and Src-family-selective tyrosine kinase inhibitor. Journal of Biological Chemistry. 1996;271:695–701. doi: 10.1074/jbc.271.2.695. [DOI] [PubMed] [Google Scholar]
- Jassar BS, Ostashewski PM, Jhamandas JH. GABAA receptor modulation by protein tyrosine kinase in the rat diagonal band of Broca. Brain Research. 1997;775:127–133. doi: 10.1016/s0006-8993(97)00892-5. [DOI] [PubMed] [Google Scholar]
- Jonas P, Major G, Sakmann B. Quantal components of unitary EPSCs at the mossy fiber synapse on CA3 pyramidal cells of rat hippocampus. The Journal of Physiology. 1993;472:615–663. doi: 10.1113/jphysiol.1993.sp019965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lancaster B, Rogers MV. A peptide activator of endogenous tyrosine kinase enhances synaptic currents mediated by NMDA receptors. European Journal of Neuroscience. 1998;10:2302–2308. doi: 10.1046/j.1460-9568.1998.00241.x. [DOI] [PubMed] [Google Scholar]
- Laurie DJ, Seeburg PH, Wisden W. The distribution of 13 GABAA receptor subunit mRNAs in the rat brain. II. Olfactory bulb and cerebellum. Journal of Neuroscience. 1992;12:1063–1076. doi: 10.1523/JNEUROSCI.12-03-01063.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessmann V. Neurotrophin-dependent modulation of glutamatergic synaptic transmission in the mammalian CNS. General Pharmacology. 1998;31:667–674. doi: 10.1016/s0306-3623(98)00190-6. [DOI] [PubMed] [Google Scholar]
- Llano I, Marty A. Presynaptic metabotropic glutamatergic regulation of inhibitory synapses in rat cerebellar slices. The Journal of Physiology. 1995;486:163–176. doi: 10.1113/jphysiol.1995.sp020800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llano I, Marty A, Armstrong CM, Konnerth A. Synaptic- and agonist-induced excitatory currents of Purkinje cells in rat cerebellar slices. The Journal of Physiology. 1991;434:183–213. doi: 10.1113/jphysiol.1991.sp018465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maisonpierre PC, Belluscio L, Friedman B, Alderson RF, Wiegand SJ, Furth ME, Lindsay RM, Yancopoulos GD. NT-3, BDNF, and NGF in the developing rat nervous system: parallel as well as reciprocal patterns of expression. Neuron. 1990;5:501–509. doi: 10.1016/0896-6273(90)90089-x. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Gorrie GH, Amato A, Smart TG. Modulation of GABAA receptors by tyrosine phosphorylation. Nature. 1995;377:344–348. doi: 10.1038/377344a0. [DOI] [PubMed] [Google Scholar]
- Moss SJ, Smart TG. Modulation of amino acid-gated ion channels by protein phosphorylation. International Review of Neurobiology. 1996;39:1–52. doi: 10.1016/s0074-7742(08)60662-5. [DOI] [PubMed] [Google Scholar]
- Mount HTJ, Dreyfus CF, Black IB. Neurotrophin-3 selectively increases cultured Purkinje cell survival. NeuroReport. 1994;5:2497–2500. doi: 10.1097/00001756-199412000-00023. [DOI] [PubMed] [Google Scholar]
- Narisawa-Saito M, Silva AJ, Yamaguchi T, Hayashi T, Yamamoto T, Nawa H. Growth factor-mediated Fyn signaling regulates α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptor expression in rodent neocortical neurons. Proceedings of the National Academy of Sciences of the USA. 1999;96:2461–2466. doi: 10.1073/pnas.96.5.2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Dell TJ, Kandell ER, Grant SN. Long-term potentiation in the hippocampus is blocked by tyrosine kinase inhibitors. Nature. 1991;353:558–560. doi: 10.1038/353558a0. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Tébar A, Dechant G, Barde Y-A. Binding of brain-derived neurotrophic factor to the nerve growth factor receptor. Neuron. 1990;4:487–492. doi: 10.1016/0896-6273(90)90107-q. [DOI] [PubMed] [Google Scholar]
- Ruegg UT, Burgess GM. Staurosporine, K-252 and UCN-01: potent but nonspecific inhibitors of protein kinases. Trends in Pharmacological Science. 1989;101:218–220. doi: 10.1016/0165-6147(89)90263-0. [DOI] [PubMed] [Google Scholar]
- Schoepp DD, Goldsworthy J, Johnson BG, Salhoff CR, Baker SR. 3,5-Dihydroxyphenylglycine is a highly selective agonist for phosphoinositide-linked metabotropic glutamate receptors in the rat hippocampus. Journal of Neurochemistry. 1994;63:769–772. doi: 10.1046/j.1471-4159.1994.63020769.x. [DOI] [PubMed] [Google Scholar]
- Schwartz PM, Borghesani PR, Levy RL, Pomeroy SL, Segal R. Abnormal cerebellar development and foliation in BDNF−/− mice reveals a role for neurotrophins in CNS patterning. Neuron. 1997;19:269–281. doi: 10.1016/s0896-6273(00)80938-1. [DOI] [PubMed] [Google Scholar]
- Siciliano JC, Gelman M, Girault J-A. Depolarization and neurotransmitters increase neuronal protein tyrosine phosphorylation. Journal of Neurochemistry. 1994;62:950–959. doi: 10.1046/j.1471-4159.1994.62030950.x. [DOI] [PubMed] [Google Scholar]
- Sugrue MM, Brugge JS, Marshak DR, Greengard P, Gustafson EL. Immunocytochemical localization of the neuronal isoform of the c-src gene product, pp60c-src(+), in rat brain. Journal of Neuroscience. 1990;10:2513–2527. doi: 10.1523/JNEUROSCI.10-08-02513.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tessarollo L, Tsoulfas P, Martin-Zanca D, Gilbert DJ, Jenkins NA, Copeland NG, Parada LF. trkC, a receptor for neurotrophin-3, is widely expressed in the developing nervous system and in non-neuronal tissues. Development. 1993;118:463–475. doi: 10.1242/dev.118.2.463. [DOI] [PubMed] [Google Scholar]
- Thoenen H. Neurotrophins and neuronal plasticity. Science. 1995;270:593–598. doi: 10.1126/science.270.5236.593. [DOI] [PubMed] [Google Scholar]
- Wan Q, Man HY, Braunton J, Wang W, Salter MW, Becker L, Wang YT. Modulation of GABAA receptor function by tyrosine phosphorylation of β subunits. Journal of Neuroscience. 1997a;17:5062–5069. doi: 10.1523/JNEUROSCI.17-13-05062.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan Q, Xiong ZG, Man HY, Ackerley CA, Braunton J, Lu WY, Becker LE, MacDonald JF, Wang YT. Recruitment of functional GABAA receptors to postsynaptic domains by insulin. Nature. 1997b;388:686–690. doi: 10.1038/41792. [DOI] [PubMed] [Google Scholar]